

ABSTRACT
Xenophagy, also known as antibacterial autophagy, functions as a crucial defense system that can utilize intracellular pattern recognition sensors, such as NLRP4, to recognize and selectively eliminate bacterial pathogens. However, little is known about how NLRP4 regulates xenophagy. Here, we report that NLRP4 binds ARHGDIA (Rho GDP dissociation inhibitor α) to regulate Rho GTPase signaling and facilitate actin-mediated xenophagy. Specifically, NLRP4 is recruited to Group A Streptococcus (GAS) and colocalizes with GAS-containing autophagosome-like vacuoles (GcAVs), where it regulates ARHGDIA-Rho GTPase recruitment to promote autophagosome formation. The interaction between NLRP4, ARHGDIA, and Rho GTPases is regulated by ARHGDIA Tyr156 phosphorylation, which acts as a gate to induce Rho-mediated xenophagy. Moreover, ARHGDIA and Rho GTPase are involved in actin-mediated ATG9A recruitment to phagophores, facilitating elongation to form autophagosomes. Collectively, these findings demonstrate that NLRP4 functions as a Rho receptor complex to direct actin dynamics regulating xenophagy.
KEYWORDS: ARHGDIA, autophagy, Group A Streptococcus, NLRP4, xenophagy
Introduction
Macroautophagy/autophagy is a regulated process of self-degradation in which double-membrane phagophores sequester portions of the cytoplasm or other cellular components, mature into autophagosomes, and deliver the contents to the lysosome for degradation. Although autophagy is primarily a nonselective bulk degradation system, it has been shown to specifically target damaged organelles, protein aggregates, mitochondria, and invading bacteria.1,2 This selectivity for bacterial components plays an important role for both phagocytic and nonphagocytic cells of the innate immune system, and utilizes various strategies to recognize and target pathogens.3,4 For example, multiple autophagy cargo receptors—such as SQSTM1/p62, CALCOCO2, OPTN (optineurin), and diacylglycerol—promote the autophagic targeting of Salmonella Typhimurium,5,6 whereas Group A Streptococcus (GAS) is specifically recognized by CALCOCO2.7,8 Alternatively, Shigella flexneri and Listeria monocytogenes, which create phagocytic membrane remnants, are recognized by the ubiquitin-SQSTM1 pathway,9,10 while the ATG5 binding protein TECPR1 is involved in the recognition of S. flexneri, S. Typhimurium, and GAS.11,12
In addition to autophagy cargo receptors, NLRs and NOD-like receptors also facilitate bacterial pathogen recognition and the induction of autophagy.13-16 NLRs are a family of intracellular pattern recognition sensors known to regulate several host responses, including inflammation, apoptosis, and autophagy.3,17 NOD1 and NOD2 recognition receptors interact with the autophagy factor ATG16L1 and recruit it to the plasma membrane, resulting in the enhanced incorporation of S. flexneri into phagophores.13 Similarly, Jounai et al. showed that NLRP4 (NLR family pyrin domain containing 4) promotes autophagy in response to GAS infection15 through its interaction with BECN1—an essential factor for the initiation of canonical autophagy. Therefore, NLRs also play crucial roles in the induction of autophagy against bacterial infection via canonical autophagy-related (ATG) proteins.
Autophagosomes are formed by dynamic rearrangements of cellular membrane structures induced by the formation of a phagophore, and are subsequently trafficked to the lysosome for degradation.18,19 In mammalian cells, the ATG1/ULK complex is responsible for the initiation of autophagy,20 whereas class III phosphatidylinositol 3-kinase (PtdIns3K) complexes containing BECN1, ATG14, and PIK3C3/VPS34 localize to the phagophore and facilitate further maturation.21 In addition to these ATG proteins, several regulators of actin dynamics and membrane trafficking, such as RAB and ARF, are reported to be involved in the formation of the phagophore or autophagosome;22-27 and delivery and fusion of cargo-containing autophagosomes to the lysosome involves the microtubule motor dynein, TOM1, MYO6, RAB7, and a SNARE complex containing STX17.28-31 However, in case of bacterial infection, various pathogens have evolved mechanisms to block membrane trafficking and cytoskeleton dynamics in host cells to potentiate their intracellular survival. While the host cytoskeleton plays a crucial and multifaceted role in bacteria-induced autophagy,32 the means by which the host cell mediates the cytoskeletal changes necessary for antibacterial autophagy remains unclear. Furthermore, several small phagophores must fuse to form the large vacuoles sufficient to engulf large bacteria, and this mechanism is thought to be regulated by an uncharacterized process distinct from that of canonical autophagy.33,34
In this study, we examined the roles of NLRP4 in autophagosomal regulation during GAS infection. Notably, NLRP4 localized to GAS-containing autophagosome-like vacuoles (GcAVs), where it interacted with ARHGDIA and subsequently recruited RHOA and CDC42 to the GcAVs to promote GcAV formation. Moreover, the NLRP4-ARHGDIA-Rho cascade regulates ATG9A trafficking via actin to facilitate phagophore elongation. These findings illustrate a novel pathway in which intracellular sensors control membrane dynamics via the regulation of cytoskeletal effectors in response to bacterial infection.
Results
NLRP4 complex is recruited to invading GAS and localizes to autophagosomes
To examine NLRP4 involvement in the regulation of GcAV membranes, we transiently expressed GFP-NLRP4 and mCherry-LC3 (an autophagic membrane marker) in HeLa cells and observed changes in their localization upon GAS infection, and found that NLRP4 clearly colocalized with LC3 surrounding the invading GAS (Fig. 1A). To confirm this finding, we investigated the localization of GFP-NLRP4 with endogenous LC3 and again observed NLRP4 at GAS-capturing LC3-positive structures (Fig. S1A). We also examined NLRP10 localization during this process because NLRP4 binds NLRP10 to function as a receptor complex.15 As expected, GFP-NLRP10 also colocalized with these LC3-positive structures (Fig. S1B). GAS secrete streptolysin O (SLO) to disrupt the endosomal membrane and become a target of autophagy.35 To examine the possibility that the NLRP4 complex is recruited to intact GAS-containing endosomes, we infected HeLa cells with isogenic SLO-negative mutant GAS and examined NLRP4 and GAS localization. As shown in Fig. 1B, GFP-NLRP4 exhibited transient colocalization at 2–3 h postinfection, and we rarely observed GFP-NLRP4 signals surrounding SLO-mutants, suggesting that NLRP4 was recruited to GAS in response to SLO-mediated endosomal membrane damage and escape into the cytosol.
Figure 1.
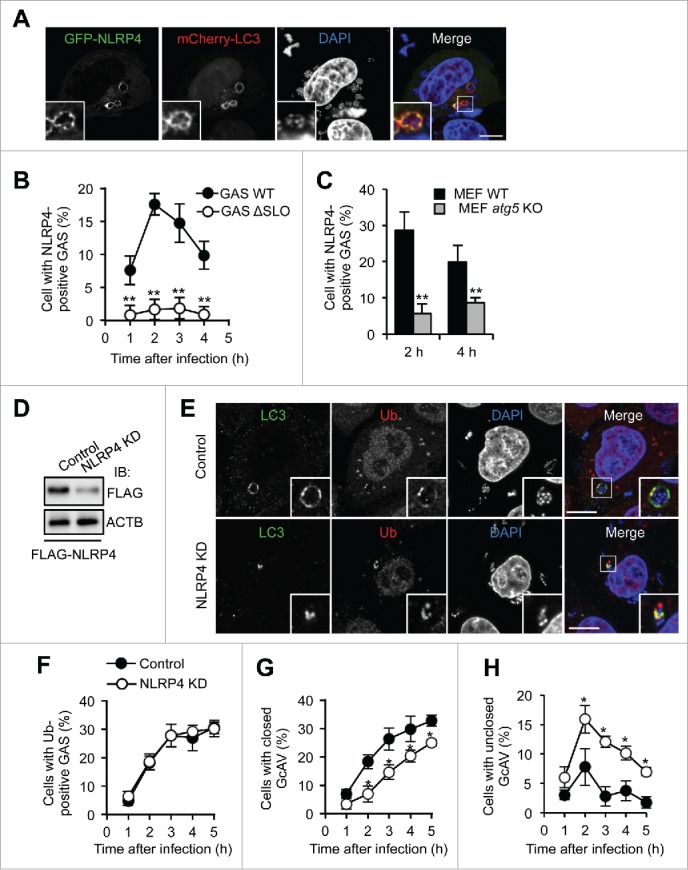
NLRP4 is recruited to GAS and facilitates GcAV formation. (A) HeLa cells expressing GFP-NLRP4 and mCherry-LC3 fusion proteins were infected with Group A Streptococcus (GAS) for 2 h. Cellular and bacterial DNA was stained with DAPI. (B) Percentage cells with NLRP4-positive GAS. HeLa cells expressing GFP-NLRP4 were infected with GAS wild-type (WT) or isogenic SLO-mutant (ΔSLO) for the indicated times. (C) Percentage cells with NLRP4-positive GAS. MEF cells expressing GFP-NLRP4 were infected with GAS for 2 or 4 h. (D) Immunoblot analysis of NLRP4 knockdown in HeLa cells. (E) HeLa cells transfected with either miR-Control (Control) or miR-NLRP4 (NLRP4 KD) were infected with GAS for 2 h. LC3 and ubiquitin were immunostained. Scale bars: 10 μm. (F-H) Quantification of cells with ubiquitin-positive GAS (F), with closed GcAV (G), and with unclosed GcAV (H). HeLa cells expressing mCherry-LC3 were infected with GAS. Data in (B, C, F-H) represent the mean ± SD (n > 100 cells, *P < 0.05, **P < 0.01) from 3 independent experiments.
To determine whether NLRP4 was localized to GAS or autophagosomes, we used atg5-knockout mouse embryonic fibroblasts (MEFs). ATG5 is critical for the LC3 conjugation system and GcAV formation.35,36 NLRP4 could be found around bacteria, even in atg5-knockout cells (Fig. S1C); however, the numbers of cells showing NLRP4-positive bacteria were considerably decreased in atg5-knockout cells compared with that in wild-type cells (Fig. 1C). Therefore, NLRP4 was recruited to both bacteria (endosomal membrane) and phagophores, and the phagophore membrane may act as a scaffold to stably tether NLRP4. Collectively, these data demonstrated that the NLRP4 complex was recruited to invading GAS and colocalized with the LC3-positive membrane.
To test whether the NLRP4 complex targeted damaged endosomal membranes or bacteria, we observed NLRP4 when cells were exposed to hypertonic conditions, which triggered sterile endosomal membrane damage, or after treatment with the lysosomotropic compound l-leucyl-l-leucine methyl ester (LLOMe) to disrupt the lysosomal membrane. GFP-NLRP4 was not colocalized with mCherry-LGALS3 (a damaged membrane marker; Fig. S2A), suggesting that NLRP4 was not recruited only to damaged endosomal membranes. In addition, GFP-NLRP4 was partially associated with Staphylococcus aureus, but not with S. Typhimurium (Fig. S2B and S2C), implying that NLRP4 sensed bacterial components rich in Gram-positive bacteria.
NLRP4 is required for phagophore elongation to fully envelop GAS
Next, we tested whether NLRP4 was involved in autophagosome formation against GAS using immunofluorescence microscopy. Although LC3-positive vacuoles clearly surrounded ubiquitin-positive GAS in control cells, LC3 signals were limited in a portion of ubiquitin-coated bacteria in NLRP4-knockdown cells (Fig. 1D and E). To further confirm these results, we examined the mCherry-LC3 signals during GAS infection. Representative microscopy images (Fig. S3) showed that most LC3-positive membranes targeting GAS did not fully envelop bacteria in NLRP4-knockdown cells. We next quantified the percentages of cells that harbored ubiquitin-positive GAS and closed or unclosed GcAVs. NLRP4 knockdown did not affect the recruitment of ubiquitin (Fig. 1F). Consistent with the microscopy images, closed GcAVs decreased, whereas unclosed GcAVs increased (Fig. 1G and H). Therefore, these findings suggested that NLRP4 was involved in the elongation or closing of phagophores targeting GAS.
ARHGDIA interacts with the NLRP4 complex and is recruited to GAS
To identify NLRP4-interacting proteins involved in GcAV formation, we performed screening using a yeast 2-hybrid system with a human cDNA library. The NLRP4 NACHT domain was used as bait since it is reported to be essential for GAS autophagy.15 As a result of the screening, 109 colonies were grown on selective medium, and 43 colonies produced positive reactions in the β-galactosidase assay. DNA sequencing of the 43 positive colonies resulted in 8 displaying an identical cDNA sequence (Table S1). To screen for NLRP4-interacting proteins involved in GcAV regulation, we expressed the 8 candidate proteins in GAS-infected HeLa cells and assessed their colocalization with NLRP4 and LC3. Of the 8 candidate proteins, ARHGDIA (Rho GDP dissociation inhibitor α) showed a strong colocalization with both NLRP4 and LC3 (Fig. 2A and B). Specifically, ARHGDIA was distributed in the cytosol of non-infected cells and recruited to intracellular GAS along with NLRP4 upon infection (Fig. 2A). This finding was also confirmed by ARHGDIA-specific antibody staining (Fig. S4A). We also found that although nearly 25% of wild-type MEFs showed ARHGDIA-positive GAS, only about 5% of atg5-knockout MEFs harbored ARHGDIA-positive GAS at 2 h (Fig. 2C), suggesting that ARHGDIA was recruited to GAS and associated with LC3-positive membranes.
Figure 2.
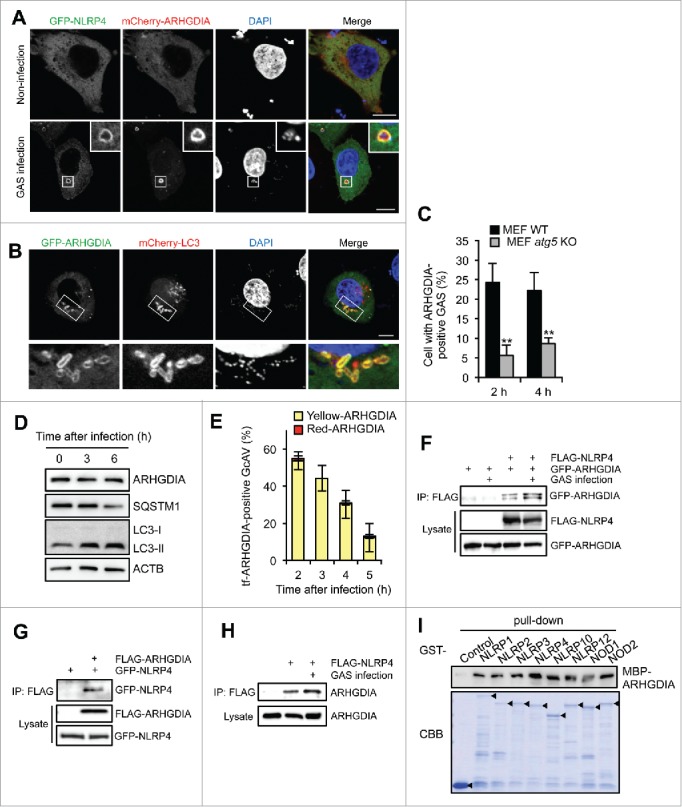
ARHGDIA interacts with NLRP4 and colocalizes with GcAV. (A) HeLa cells expressing GFP-NLRP4 and mCherry-ARHGDIA were either non-infected or infected with GAS for 2 h. (B) HeLa cells expressing GFP-ARHGDIA and mCherry-LC3 were infected with GAS for 2 h. Scale bars: 10 μm. (C) Percentage cells with ARHGDIA-positive GAS. MEF cells expressing GFP-ARHGDIA were infected GAS for 2 or 4 h. Colocalization frequencies of GAS with ARHGDI signals were manually determined. Data represent the mean ± SD (n > 100 cells, **P < 0.01) from 3 independent experiments. (D) HeLa cells were infected with GAS for the indicated times and subjected to immunoblotting with the indicated antibodies. (E) Quantification of GFP-mCherry-ARHGDIA (tf-ARHGDIA)-positive GcAVs at the indicated time point after infection. Data represent the mean ± SD (n > 50 GcAVs in each experiment) from 3 independent experiments. (F) HeLa cells transfected with FLAG-NLRP4, GFP-ARHGDIA, or empty vector controls were infected with GAS. Cell lysates were immunoprecipitated (IP) with anti-FLAG antibody and immunoblotted with anti-GFP antibody. Total lysates served as input controls. (G) FLAG IPs in HeLa cells transfected with FLAG-ARHGDIA, GFP-NLRP4, or empty vector controls infected with GAS. (H) Lysates from HeLa cells transfected with FLAG-NLRP4 or empty vector and infected with GAS were immunoprecipitated with anti-FLAG antibody and immunoblotted with anti-ARHGDIA antibody. (I) In vitro affinity-isolation assay between MBP-ARHGDIA and GST-NLRPs. Beads carrying the indicated purified GST fusion protein were incubated with lysates from E. coli expressing MBP-ARHGDIA. CBB, Coomassie Briliant Blue.
LC3 can be localized to open phagophores, closed double-membrane autophagosomes, and lysosome-fused autolysosomes; thus, GcAVs may also be classified into these 3 autophagic structures.25 To identify the structures in which ARHGDIA is present, we examined the localization of ARHGDIA with ATG5 (a phagophore marker) or LAMP1 (a lysosomal marker). GFP-ARHGDIA colocalized with ATG5 but not LAMP1 in GAS-infected HeLa cells (Fig. S4B and S4C), suggesting that ARHGDIA localized to early autophagosomal structures (phagophores and autophagosomes), but not matured autolysosomes.
Since ARHGDIA was observed in early LC3-positive structures and subsequently lost in autolysosomes, there were 2 possibilities: i) ARHGDI was degraded via lysosomal fusion and/or ii) ARHGDI dissociated from the membrane. Thus, we first examined the amount of endogenous ARHGDIA but it was not clearly changed, even after bacterial challenge (LC3-II increased with time after infection), whereas the SQSTM1 receptor protein decreased (Fig. 2D). In addition, we used GFP-mCherry-ARHGDIA (tf-ARHGDIA) to examine whether ARHGDIA was delivered to the lysosomal low pH environment. As shown in Fig. 2E, tf-ARHGDIA on GcAVs was not changed from yellow to red but disappeared at a later time after infection. Thus, although we could not exclude the possibility that a portion of ARHGDIA was degraded inside autophagosomes, it was suggested that ARHGDIA dissociates from phagophores and autophagosomes, rather than being degraded following lysosomal fusion.
We also performed co-immunoprecipitation analyses to confirm the ARHGDIA and NLRP4 interaction (Fig. 2F and G). As expected, endogenous ARHGDIA precipitated with FLAG-NLRP4 (Fig. 2H), and this interaction was enhanced in GAS-infected cells (Fig. 2F and H). Since the NACHT domain is relatively conserved among NLR family proteins, we investigated whether ARHGDIA specifically bound to NLRP4 by in vitro affinity isolation assays. As shown in Fig. 2I, MBP-ARHGDIA directly interacted with all GST-NLRPs examined. We then observed the localization of these NLRPs during GAS infection. In addition to NLRP4 and NLRP10, NOD2 was also rarely accumulated around GAS in an SLO-dependent manner (Fig. S5). GFP-NOD1 and GFP-NLRP3 surrounded SLO mutant GAS, indicating that these NLRPs could localize to GAS-containing endosomes. Others were not observed around GAS (Fig. S5). Therefore, these data suggested that NLRP4 and NLRP10 specifically functioned with ARHGDIA against invading GAS.
To examine whether ARHGDIA also associates with starvation-induced autophagy, we observed the ARHGDIA localization during starvation. ARHGDIA did not colocalize with LC3-puncta induced by starvation (Fig. S6), suggesting that ARHGDI is not involved in canonical autophagy.
NLRP4 is required for ARHGDIA recruitment to GAS and GcAV
ARHGDIA regulates the Rho family GTPases by extracting family members from membranes and solubilizing them in the cytosol, and are suggested to be involved in Rho GTPase localization.37,38 Knockdown of NLRP4 significantly decreased the number of ARHGDIA-positive GAS (Fig. 3A and B), suggesting that NLRP4 was required for ARHGDIA recruitment to the region around GAS. If NLRP4 regulates ARHGDIA localization, deletion of the NLRP4 interaction domain may also prevent the ARHGDIA recruitment. To examine this hypothesis, we constructed a series of GST-tagged ARHGDIA domain deletion mutants (Fig. 3C). ARHGDIA consists of 2 domains: a flexible, ∼70-amino acid N-terminal domain and a folded, 134-residue immunoglobulin-like C-terminal domain. GST immunoprecipitation showed that the C-terminal domain of ARHGDIA interacted with NLRP4 (Fig. 3D). We then examined the GcAV localization of ARHGDIA N- and C-terminal deletion mutants (ARHGDIA-ΔN and ARHGDIA-ΔC, respectively) in GAS-infected HeLa cells. We found that the ARHGDIA-ΔN mutant displayed normal GcAV localization; however, this was abolished with the NLRP4 binding domain-deleted ARHGDIA-ΔC mutant (Fig. 3E and F). Thus, these results demonstrated that ARHGDIA was recruited to GAS and localized with GcAV via its interaction with NLRP4.
Figure 3.
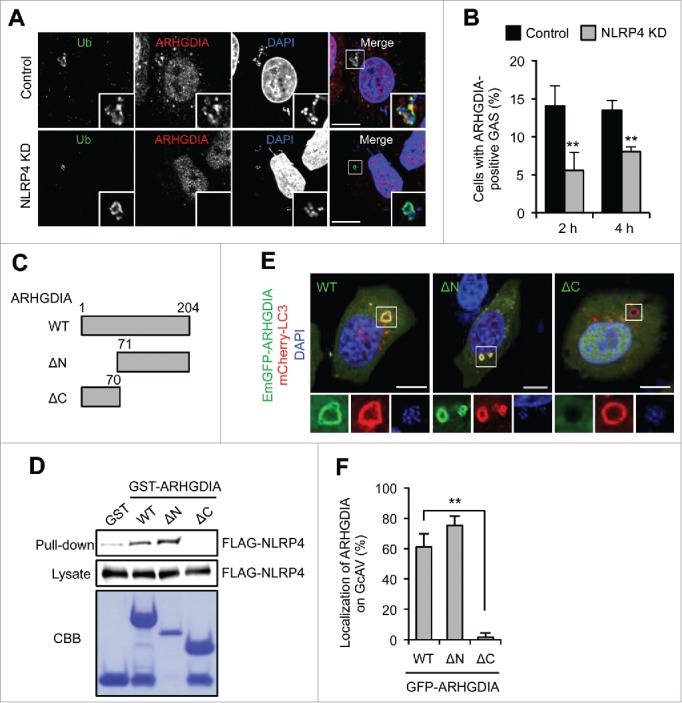
NLRP4 is required for the recruitment of ARHGDIA to GcAV. (A) HeLa cells expressing either miR-Control (Control) or miR-NLRP4 (NLRP4 KD) were infected with GAS for 2 h. ARHGDIA and ubiquitin were immunostained. Cellular and bacterial DNA were stained with DAPI. Scale bars: 10 μm. (B) Percentages of cells with ARHGDIA-positive GAS were quantified at 2 and 4 h after infection. Data represent the mean ± SD (n > 100 cells, **P < 0.01) from 3 independent experiments. (C) Schematic representation of the ARHGDIA domain structure. (D) FLAG-NLRP4 binding to each GST-ARHGDIA-domain deletion mutant. Beads carrying the indicated purified GST fusion protein were incubated with lysates from FLAG-NLRP4-expressing HeLa cells infected with GAS. (E) HeLa cells expressing mCherry-LC3 and either GFP-ARHGDIA (WT), GFP-ARHGDIA-ΔN or GFP- ARHGDIA-ΔC were infected with GAS for 2 h. Scale bars: 10 μm. (F) ARHGDIA-positive GcAV quantification. Data represent the mean ± SD (n > 50 GcAVs, **P < 0.01) from 3 independent experiments.
ARHGDIA is involved in GcAV formation
Cytosolic GAS are coated with ubiquitin, recognized by SQSTM1, and then selectively targeted by phagophores.39 To clarify the role of ARHGDIA in GAS infection-induced autophagy, we examined the recruitment of autophagic recognition components to GAS in ARHGDIA-knockdown or control HeLa cells (Fig. 4A). Analysis of GAS invasion efficiency in ARHGDIA-knockdown cells revealed no significant differences in entry rates between knockdown and control cells (6.8 ± 0.6% and 7.2 ± 0.8%, respectively; P = 0.49). Next, to determine whether ARHGDIA-knockdown affected the recruitment of well-known recognition factors in xenophagy, we observed the localizations of ubiquitin and SQSTM1 in GAS-infected cells. The percentages of cells with ubiquitin- or SQSTM1-positive GAS were not changed by ARHGDIA-knockdown at all time points examined (Fig. 4B and C), suggesting that ARHGDIA was not involved in recognition though ubiquitin and the receptor protein SQSTM1. However, similar to the results in NLRP4-knockdown cells, LC3 signals did not clearly surround GAS, and GcAV-positive cells decreased in ARHGDIA-knockdown cells (Fig. 4C). These results suggest that ARHGDIA knockdown inhibited GcAV formation.
Figure 4.
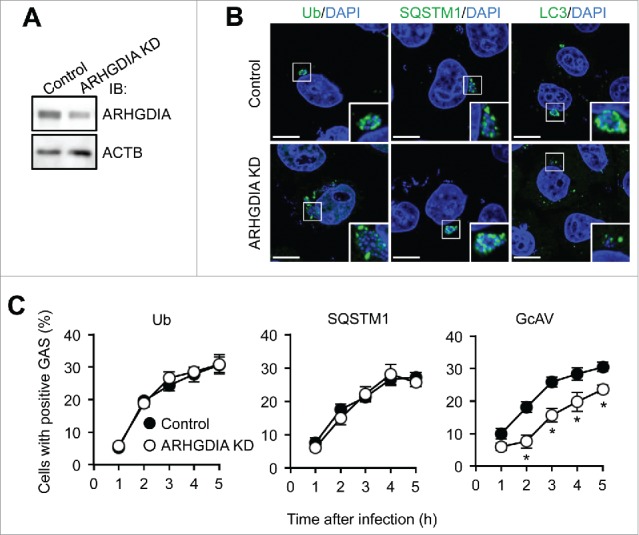
ARHGDIA regulates GcAV formation. (A) Immunoblot analysis of ARHGDIA-knockdown in HeLa cells. (B) HeLa cells expressing either miR-Control (Control) or miR-ARHGDIA (ARHGDIA KD) were infected with GAS for 2 h. Ubiquitin, SQSTM1, and LC3 were immunostained. DNA was stained with DAPI. Scale bars: 10 μm. (C) Quantification of cells with Ub-, SQSTM1-, or GcAV-positive GAS. Data represent the mean ± SD (n > 100 cells, *P < 0.05) from 3 independent experiments.
We also investigated the effects of ARHGDIA depletion on starvation-induced autophagosome formation. The numbers of autophagosomes were not changed by knockdown of ARHGDIA in both regular and starvation media (Fig. S7A and S7B). Consistent with the above results, ARHGDI is likely specific for the GcAV formation process.
RHOA and CDC42 regulate GcAV formation
We next examined whether Rho GTPases were involved in GcAV formation via their interaction with ARHGDIA. Especially, we found that the Rho GTPases RHOA and CDC42, but not RAC1, clearly colocalized with GcAVs based on confocal microscopy (Fig. 5A). To address whether the recruitment of Rho GTPases to GcAVs is dependent on ARHGDIA, we compared the GcAV localization rates of RHOA and CDC42 in ARHGDIA-knockdown cells to control counterparts. This analysis revealed that ARHGDIA knockdown decreased RHOA and CDC42 colocalization in GcAVs (Fig. 5B). These results suggest that ARHGDIA is involved in the recruitment of RHOA and CDC42 to GcAVs during infection.
Figure 5.
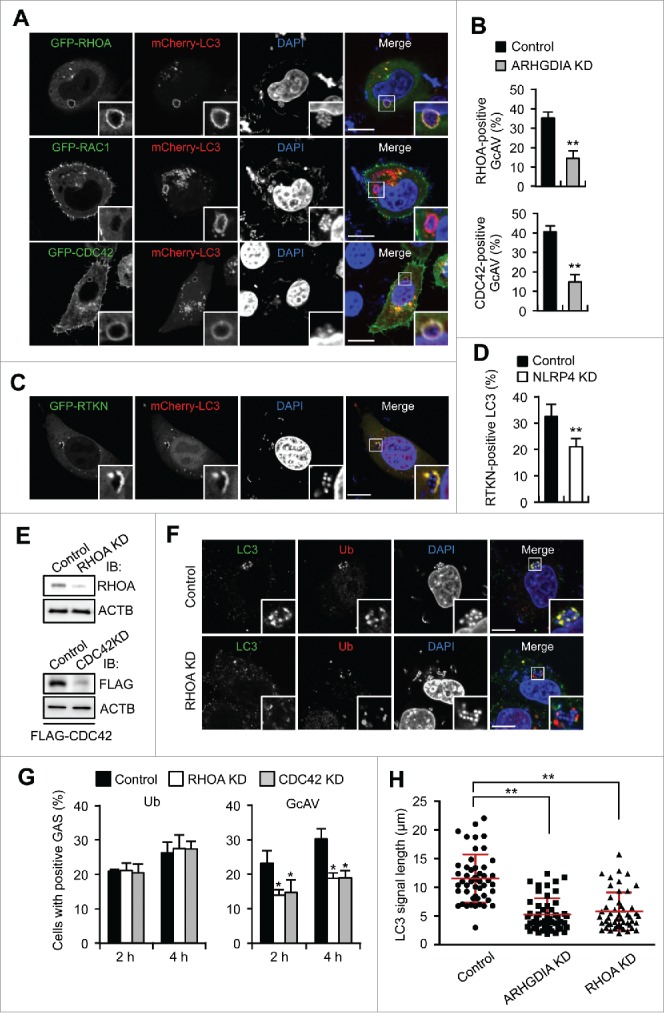
RHOA and CDC42 regulate GcAV formation. (A) HeLa cells expressing mCherry-LC3 and either GFP-RHOA, GFP-RAC1, or GFP-CDC42 were infected with GAS for 2 h. Cellular and bacterial DNA are stained with DAPI. Scale bars: 10 μm. (B) Quantification of RHOA- or CDC42-positive GcAVs in control or ARHGDIA knockdown cells in HeLa cells transfected with mCherry-LC3 and GFP-RHOA or GFP- CDC42, and miR-Control or miR-ARHGDIA infected with GAS for 2 h. (C) HeLa cells expressing GFP-RTKN and mCherry- LC3 were infected with GAS for 2 h. (D) Quantification of RTKN-positive GcAVs in NLRP4 knockdown or control HeLa cells transfected with mCherry-LC3 following GAS infection for 2 h. (E) Immunoblot analysis of RHOA or CDC42-knockdown HeLa cells. (F) HeLa cells transfected with miR-Control or miR-RHOA were infected with GAS for 2 h, fixed and immunostained with anti-LC3 and anti-ubiquitin antibodies. (G) Quantifications of ubiquitin- or GcAV positive cells in RHOA or CDC42 knockdown cells. (H) HeLa cells expressing GFP-LC3 and either miR-Control, miR-ARHGDIA, or miR-RHOA were infected with GAS for 4 h. Quantification of the length of LC3-positive structures surrounding GAS (n > 60 LC3 signals in each condition). Red bars represent the mean ± SD.
Rho GTPases are recruited to target membrane compartment and subsequently dissociate from ARHGDIA to acquire an activated GTP-bound form capable of regulating intracellular signaling pathways. To examine the activation status of the Rho proteins on GcAV, we expressed GFP-RTKN—an effector that specifically binds activated RHOA—and observed its localization in GAS-infected cells. GFP-RTKN clearly colocalized with GcAVs (Fig. 5C). Consistent with the effects of ARHGDIA-knockdown on Rho-positive GcAVs, NLRP4 depletion significantly decreased the number of RTKN-positive GcAVs (Fig. 5D). Therefore, these data suggest that RHOA is activated on GcAVs in an NLRP4-dependent manner. RHOA knockdown decreased LC3 signals around ubiquitin-positive bacteria without affecting ubiquitin recruitment (Fig. 5E–G), and the number of cells showing GcAVs decreased following RHOA or CDC42 knockdown (Fig. 5G). Moreover, to confirm that Rho protein activation is required for GcAV formation, we overexpressed the GDP-bound form of RHOA (RHOAT19N) or CDC42 (CDC42T17N) and measured the GcAV formation efficiency. Expression of wild-type (WT) RHOA or CDC42 showed no apparent effects on GcAV formation efficiencies; however, this was significantly decreased with RHOAT19N or CDC42T17N overexpression (Fig. S8A and S8B). Taken together, our results demonstrate that RHOA and CDC42 are recruited to GcAVs via the NLRP4-ARHGDIA complex, activated on the membrane, and facilitate GcAV formation.
Inhibition of the NLRP4-ARHGDIA-Rho cascades accumulated unclosed, incomplete LC3-positive membranes (Fig. 1E, S3, 4B, and 5F). We then measured the lengths of LC3 signals surrounding bacteria and found that knockdown of ARHGDIA or RHOA shortened the LC3 signal length, suggesting that the NLRP4-ARHGDIA-Rho cascade facilitated phagophore elongation to envelop large bacterial cells and to form complete GcAVs (Fig. 5H).
ARHGDIA Tyr156 phosphorylation is important for its incorporation in the GcAV-NLRP4 complex
The interaction of Rho GTPases with ARHGDIA is precisely regulated by various mechanisms.38,40 For example, the concurrent phosphorylation of ARHGDIA on 2 sites (Ser101 and Ser174) by PAK1 (p21 [RAC1] activated kinase 1) leads to a selective release of RAC1 from ARHGDIA complexes, whereas ARHGDIA Tyr156 phosphorylation by SRC promotes the dissociation of RHOA, RAC1, and CDC42.41,42 Thus, we mutated Tyr27, Ser34, Ser101, Tyr133, and Tyr156 in ARHGDIA to Glu or Ala to mimic the phosphorylated or unphosphorylated amino acids, respectively, and observed their respective effects on ARHGDIA localization in response to GAS infection. Interestingly, WT and most GFP-ARHGDIA mutants dissipated from GcAV approximately 5 h post-infection (Fig. S9), whereas ARHGDIAY156A continued to localize even at 5 h (Fig. 6A and B). We also found that the ARHGDIAY156E phosphomimetic substitution impaired its localization to GcAV at 2 h (Fig. 6B), suggesting that ARHGDIA Tyr156 phosphorylation regulates GcAV-cytosol cycling.
Figure 6.
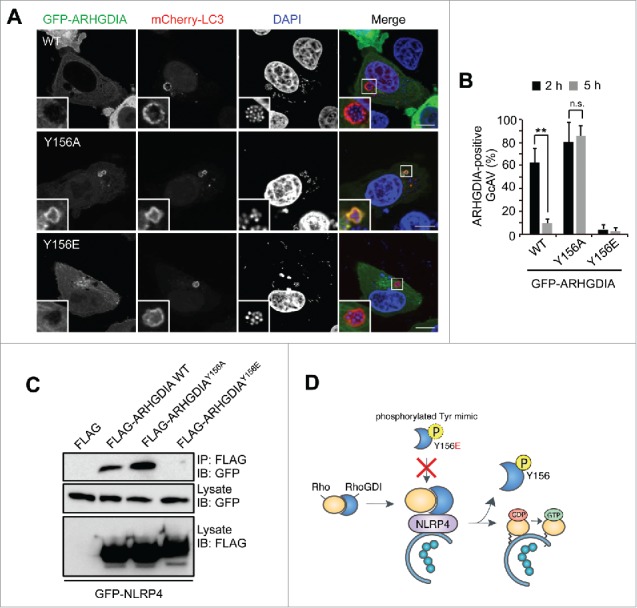
ARHGDIA Tyr156 phosphorylation status is important for its association with NLRP4 and GcAVs. (A) Representative images of ARHGDIA mutant recruitment to GcAV. HeLa cells expressing mCherry-LC3 and either GFP-ARHGDIA, GFP-ARHGDIAY156A, or GFP-ARHGDIAY156E were infected with GAS for 5 h. Scale bars: 10 μm. (B) Quantification of GFP-ARHGDIAs-positive GcAVs at the indicated time point after infection. Data represent the mean ± SD (n > 50 GcAVs, ** P < 0.01) of 3 independent experiments. (C) Immunoprecipitation from HeLa cells transfected with EmGFP-NLRP4 and either empty vector, FLAG-ARHGDIA, FLAG-ARHGDIAY156A, or FLAG-ARHGDIAY156E. (D) A model for the association of ARHGDIA with NLRP4 on GcAVs.
We then examined the effect of ARHGDIA Tyr156 phosphorylation on its interaction with NLRP4. Particularly, NLRP4 precipitated with FLAG-ARHGDIA WT and FLAG-ARHGDIAY156A, but not FLAG-ARHGDIAY156E (Fig. 6C), indicating that NLRP4 binds ARHGDIA in the absence of Tyr156 phosphorylation. Collectively, these data suggest that the ARHGDIA complex is recruited to GcAV via NLRP4 and subsequently dissociates upon phosphorylation at Tyr156 to release activated Rho proteins (Fig. 6D).
ARHGDIA and Rho regulated actin-mediated ATG9A trafficking
Rho family proteins are key regulators of the actin cytoskeleton. The actin cytoskeleton is thought to be required for proper ATG9A trafficking to phagophores.43 We then observed ATG9A localization during GAS infection. GFP-ATG9A clearly surrounded GAS and colocalized with LC3 (Fig. 7A). To examine the role of actin in ATG9A recruitment, we used drugs (latrunculin B, CK-666, and jasplakinolide) that affect actin polymerization. Latrunculin B and CK-666 (an ARP2/3 inhibitor) depolymerize the actin cytoskeleton, whereas jasplakinolide immobilizes the actin cytoskeleton. Although we examined the effects of these drugs on cell health, these drugs showed negligible effects on cell viability during GAS infection (Fig. S10). In cells treated with latrunculin B, CK-666, and jasplakinolide, ATG9A-positive GAS decreased (Fig. 7A and B), and unclosed autophagosomes (i.e., phagophores) appeared. These data suggested that ATG9A was recruited to phagophores targeted against GAS via an actin-dependent mechanism. We next tested whether ARHGDIA and Rho proteins regulated ATG9A trafficking and found that recruitment of ATG9A to GAS was suppressed by depletion of ARHGDIA, RHOA, or CDC42 (Fig. 7C and D). Finally, we examined the involvement of ATG9A in GcAV formation. As expected, ATG9A knockdown inhibited the formation of complete GcAVs and decreased the length of LC3-positive signals associated with intracellular GAS (Fig. 7E–G). Taken together, these findings demonstrated that Rho-mediated actin dynamics regulated ATG9A trafficking to promote phagophore elongation.
Figure 7.
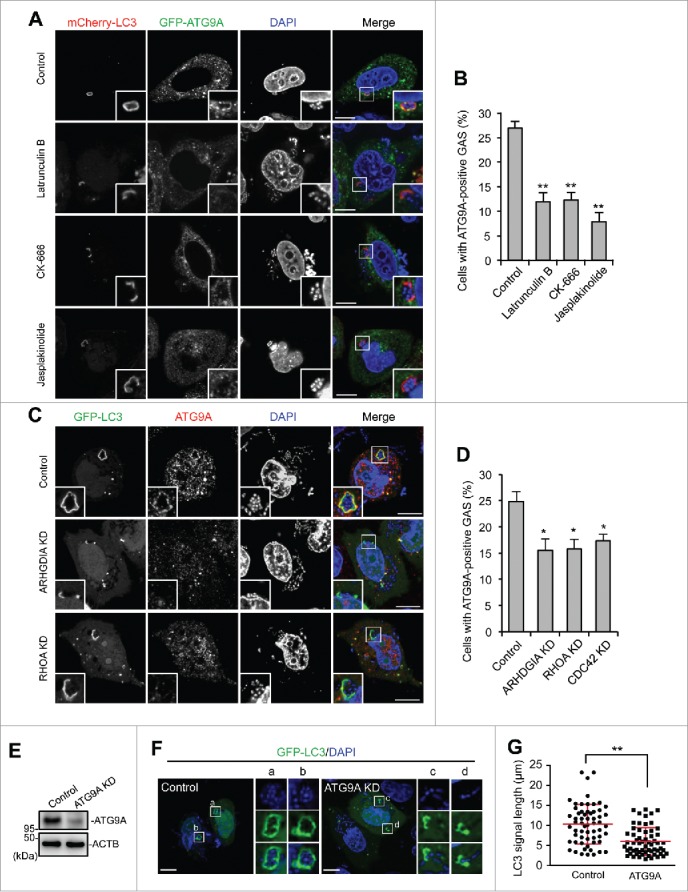
ARHGDIA and RHO proteins are involved in actin-mediated ATG9A recruitment to GcAVs. ((A)and B) HeLa cells expressing mCherry-LC3 and GFP-ATG9A were infected with GAS and added with drugs affecting actin polymerization at 1 h after infection; latrunculin B (500 nM, CK-666 (50 mM), and jasplakinolide (200 nM). At 4 h after infection, cells were fixed and the percentages of cells with ATG9A-positive GAS were quantified (B). Data represent the mean ± SD (n > 100 cells, ** P < 0.01) of 3 independent experiments. (C) HeLa cells expressing GFP-LC3 and either miR-Control, miR-RHOA, or miR-CDC42 were infected with GAS. ATG9A was immunostained. (D) The percentages of cells with ATG9A-positive GAS were quantified. Data represent the mean ± SD (n > 100 cells, * P < 0.05) of 3 independent experiments. (E) Immunoblot analysis of ATG9A-knockdown HeLa cells. ((F)and G) HeLa cells expressing GFP-LC3 and either miR-Control or miR-ATG9A were infected with GAS for 4 h. Representative images of LC3-positive GAS (F) and quantification of the length of LC3-signals (G). Individual LC3-signals were measured using ImageJ software (n > 60 LC3-signals in each condition). Red bars represent the mean ± SD.
Discussion
NLRs act as a sensor for invading bacteria and activate autophagy signaling via an interaction with ATG proteins.44 Recent studies have established that the host cytoskeleton plays a pivotal role in antibacterial autophagy;27 however, little is known about the relationship between NLRs and cytoskeletal regulators during this process. In this study, we found that NLRP4 directly associates with Rho cytoskeletal regulators to regulate autophagosome formation through actin-mediated ATG9A trafficking (Fig. 8).
Figure 8.
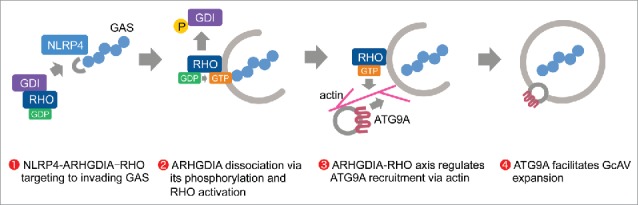
Model of NLRP4-ARHGDIA-Rho signaling regulating autophagosome formation during GAS infection.
First, we identified ARHGDIA as an NLRP4 interactor. ARHGDIA and NLRP localized to intracellular GAS and LC3. Knockdown analyses showed that ARHGDIA was involved with Rho family member localization and GcAV formation, particularly the elongation process of phagophore membranes to fully envelop bacteria. Therefore, our results link NLR-mediated bacterial sensing to autophagosome formation via Rho cytoskeletal regulators. Whereas the ubiquitin-mediated recognition system is well known to connect invading pathogens with autophagic vacuole structure, further analysis of NLR-Rho complex function in autophagy would shed light on the specific mechanisms of selective autophagy. Interestingly, ARHGDIA directly binds to various NLRPs. Since only the NLRP4-NLRP10 complex is frequently recruited to GAS exposed to cytoplasm, this complex would mainly function during GAS infection. Given that endosomes are platforms for NOD1 and NOD2 signaling in dendritic cells and that Rho and actin polymerization are thought to be involved in NLRP-mediated inflammasome assembly, pyroptosis, and IL1B/IL-1β production,45,46 NLRP-ARHGDIA-Rho may be a common connection point in NLRP signaling.
Since ARHGDIA localization to intracellular GAS was reduced by NLRP4 knockdown or ATG5 knockout and dependent on its C-terminal interaction with NLRP4, we proposed a model wherein NLRP4 recruits ARHGDIA to GAS, and the complex then associates with the autophagic membrane. However, the means by which NLRP4 identifies bacterial components and localizes to the autophagic membrane during GAS infection remains unclear. NLRs recognize cytosolic bacterial components as a first step in the innate immune response. Specifically, NOD1 detects bacteria by interacting with peptidoglycans from endosomal Gram-negative bacteria to induce autophagy and inflammatory responses in epithelial cells,16 whereas NOD2 plays the same function in innate immune cells,47 illustrating that endosomes are a platform for bacterial recognition and immune response.48 In this study, we showed that the NLRP4 complex was recruited to GAS exposed to cytosol and S. aureus, but not to sterile damaged endosomal membranes or Gram-negative S. Typhimurium. Therefore, NLRP4 may also sense bacterial components that are rich in gram-positive bacteria. Although we focused on NLRP4 in this study, NLRP10 is also recruited to invading GAS and forms a complex with NLRP4. NLRP10 lacks the putative ligand-binding leucine-rich-repeat domain, and thus has been postulated to act as an adaptor or regulator rather than a sensor.49 Further analysis of NLRP10-interacting factors could explain how the NLRP4-NLRP10 complex associates with autophagic membranes.
ARHGDIA functions physiologically in the cytosol where it forms soluble complexes via a bipartite interaction with Rho GTPases. Considering the above function of ARHGDIA, it is likely that ARHGDIA functions to shuttle Rho GTPases between different membranes. However, the mechanism by which RHOA-RHGDIA complexes are delivered to appropriate target membrane compartments and their subsequent dissociation remain unclear.37,40 Nevertheless, several models of Rho protein membrane delivery by ARHGDIA have been proposed, including: i) electrostatic attraction between ARHGDIA and the target membrane, ii) the restricted recruitment of the RHOA-ARHGDIA complex via membrane-bound receptors, and iii) complex destabilization by displacement factors, such as NGFR (nerve growth factor receptor),50 or ARHGDIA phosphorylation. In this study, we found that ARHGDIA localized to GAS-capturing autophagic membranes through its interaction with NLRP4, and dissociated from the GcAV before lysosomal fusion. Notably, phospho-null mutant ARHGDIAY156A failed to dissociate from GcAV, whereas the ARHGDIAY156E phospho-mimic impaired GcAV localization and NLRP4 interaction. Moreover, depletion of NLRP4 or ARHGDIA suppressed Rho GTPase recruitment to GcAV. Collectively, these data suggest that ARHGDIA is recruited to GcAV via the NLRP4 complex, and the membrane-cytosol cycle and Rho GTPase activation on GcAVs is regulated by ARHGDIA phosphorylation at Tyr156. Although our data demonstrated Rho's interaction with the NLRP4 complex, other reports have also identified associations between various NLRs and Rho proteins;51,52 thus, NLRs seem to be closely connected with the Rho GTPase signaling pathways.
Moreover, our data revealed that Rho GTPases is involved in actin-mediated ATG9A trafficking to phagophores directed against bacteria. The importance of the actin cytoskeleton by various autophagic stimuli has already reported by several studies.23 For instance, a functional actin cytoskeleton and the Arp2/3 (actin-related protein 2/3) complex are involved in Atg9 anterograde transport to the phagophore assembly site and selective autophagy in yeast.32,53,54 Mammalian cells also require actin to regulate ATG9A trafficking.43 Actin filaments colocalize with omegasomes (another type of autophagosome precursor) and are involved in the initial membrane remodeling necessary for autophagosome formation upon starvation-induced autophagy.55 Lee et al.56 also reported that the actin cytoskeleton is required for efficient autophagosome-lysosome fusion in the selective autophagy of protein aggregates and damaged mitochondria. Furthermore, Tumbarello et al.29 described that autophagosome maturation and lysosome fusion requires the docking of MYO6 (myosin VI), an actin-based motor protein, to autophagy receptors including CALCOCO2 and OPTN. Taken together, these studies suggest that the actin cytoskeleton plays a crucial role throughout autophagy.
In summary, there is evidence that NLRs contribute to the induction of autophagy in response to bacterial invasion. Our data demonstrate that the NLRP4 complex interacts with cytoskeletal effectors to regulate the autophagic process, and thus give insight into the mechanism by which selective autophagy efficiently defends cells against invading bacteria.
Materials and methods
Bacterial strain
The Group A Streptococcus strain JRS4 (M6+ F1+), Staphylococcus aureus strain COL, and Salmonella enterica Typhimurium strain LT2 were grown in Todd-Hewitt broth (BD Diagnostic Systems, 249240) supplemented with 0.2% yeast extract (Nacalai Tesque, 15838–45) as described previously.35
Cell culture and transfection
The HeLa cell line was maintained in Dulbecco's modified Eagle's medium (Nacalai Tesque, 18459–64) supplemented with 10% fetal bovine serum (Gibco, 26140079) and 50 g/mL gentamicin (Nacalai Tesque, 11980–14) in a 5% CO2 incubator at 37°C. Transfections were performed with polyethylenimine (Polyscience, 23966–2) or Lipofectamine 3000 (Invitrogen Corporation, L3000001).
Antibodies and other reagents
The following primary antibodies were used: rabbit anti-ARHGDIA (Cell Signaling Technology, 2564S), mouse anti-GAL4 (RK5C1; Santa Cruz Biotechnology, sc-510), mouse anti-LAMP1 (H4A3; Santa Cruz Biotechnology, sc-20011), mouse anti-FLAG M2 (Sigma-Aldrich, A2220), mouse anti-GFP (GF200; Nacalai Tesque, 04363–24), rabbit anti-ACTB (13E5; Cell Signaling Technology, 4970), mouse anti-polyubiquitin (FK2; Nippon Bio-Test Laboratories, 0918–2), mouse anti-SQSTM1 (D-3; Santa Cruz Biotechnology, sc-28359), rabbit anti-SQSTM1 (H-290; Santa Cruz Biotechnology, sc-25575), rabbit anti-ubiquitin (Dako, Z 0458), mouse anti-LC3 (LC3–1703; Cosmo Bio, CAC-CTB-LC3–2-IC), and rabbit anti-LC3B (D11; Cell Signaling Technology, 3868), rabbit anti-RHOA (67B9; Cell Signaling Technology, 2117), mouse anti-ATG9A (abcam, ab108338). Secondary antibodies anti-mouse or anti-rabbit IgG conjugated with Alexa Fluor 488, 594, and 647 were purchased from Invitrogen Corporation (A11001, A11008, A11005, A11012, A21235, and A21244). Latrunculin B (BML-T110–0001; used at 500 nM) and CK-666 (ALX-270–506-M002; used at 50 μM) were from Enzo Life Sciences. Jasplakinolide (sc-202191; used at 200 nM) was from Santa Cruz Biotechnology. A lactate dehydrogenase (LDH) cytotoxicity assay kit (Takara Bio Inc., MK401) was used to assess the cytotoxicity.
Plasmids
Human NLRP4, NLRP10, ARHGDIA, RHOA, RAC1, CDC42, RTKN, LGALS3, and ATG9A cDNAs were amplified by PCR from KYSE or HeLa total mRNA and cloned into pcDNA-6.2/C-EmGFP-DEST (C-terminal tagged, Invitrogen Corporation, V35520), pcDNA-6.2/N-EmGFP-DEST (N-terminal tagged, Invitrogen Corporation, V35620), using Gateway cloning technology as described previously.25 pcDNA-6.2/C-3xFLAG-DEST (C-terminal tagged), pcDNA-6.2/N-3xFLAG-DEST (N-terminal tagged), pcDNA-6.2/N-mCherry-DEST (N-terminal tagged) vectors were made by replacement from pcDNA-6.2-N-EmGFP to the 3xFLAG corresponding oligonucleotide or mCherry gene fragment (kindly provided from Dr. Tsien) for compatible with the Gateway system, respectively. Plasmids encoding human NLRP1, NLRR2, NLRP3, and NLRP12 were purchased from InvivoGen (puno1-hnalp1a, puno1-hnalp2, puno1-hnalp3, puno1-hnalp10, and puno1-hnalp12). Double-stranded miRNAs were ligated into pcDNA-6.2-GW/miR according to the manufacturer's instructions (Invitrogen Corporation, K493500). pcDNA-6.2-GW/miR-Neg was used as an miRNA-Control (Invitrogen Corporation, K493500). Plasmids were transfected into HeLa cells as described above. Knockdowns were confirmed by western blot analysis. Site-directed mutagenesis of ARHGDIA, RHOA, and CDC42 was performed using a PrimeSTAR Mutagenesis Basal kit (Takara Bio Inc., R046A).
Yeast 2-hybrid assay
The bait plasmid pGBKT7-NLRP4-NACHT domain was transformed into HF7c yeast cells (Clontech, 630489) with lithium acetate. Cells were grown in synthetic defined medium lacking tryptophan. Cells were then transformed with a human MATCHMAKER cDNA library (Takara Bio Inc., 638821) constructed in a pACT2 (GAL4 activation domain) vector (Clontech, 638822). The yeast colonies were selected on medium lacking tryptophan, leucine, and histidine over a period of 5 d. Candidate colonies were picked, expanded, and further examined by a β-galactosidase assay.
Fluorescence microscopy
Harvested cells were washed with phosphate-buffered saline (PBS; 137 mM NaCl, 2.7 mM KCl, 8.1 mM Na2HPO4, 1.47 mM KH2PO4, pH 7.4), fixed with 4% paraformaldehyde in PBS for 20 min, and then permeabilized with 0.1% Triton (Nacalai Tesque, 35501–15) in PBS for 10 min. After washing with PBS, the cells were incubated in skim milk blocking buffer (5% skim milk, 2.5% goat serum [Sigma-Aldrich, G9023], 2.5% donkey serum [Millipore, S30], and 0.1% gelatin [BD Diagnostic Systems, 214340] in PBS) or bovine serum albumin (BSA) blocking solution (2% BSA [Sigma-Aldrich, A4503] and 0.02% sodium azide in PBS) for 1 h. For immunostaining, cells were incubated with primary antibodies diluted 1:100 in blocking solution at room temperature for 1 h, washed with PBS, and then probed with secondary antibody. DAPI (Nacalai Tesque, 11034–56) was diluted in blocking buffer and used to stain bacterial and cellular DNA. Fluorescence images were acquired with a confocal FV1000 laser-scanning microscope (Olympus).
Immunoprecipitation
Cells were harvested, washed with PBS, and lysed for 30 min on ice in lysis buffer containing 10 mM Tris-HCl, pH 7.4, 150 mM NaCl, 10 mM MgCl2, 1 mM EDTA, 1 % Triton X-100 (Nacalai Tesque, 35501–15), and proteinase inhibitor cocktail (Nacalai Tesque, 25955–11). Lysates were then centrifuged, and supernatants were pre-cleared for 30 min at 4°C with protein A Sepharose 4B (GE Healthcare Life Sciences, 17–1279–03). After a brief centrifugation (600 x g, 1 min), supernatants were incubated and with anti-FLAG antibody for 2 h at 4°C. Protein A Sepharose beads were then added and mixed for another 1 h. Immunoprecipitates were collected by brief centrifugation, washed 5 times with lysis buffer, and analyzed by immunoblotting as described previously.35
GST affinity isolation
GST fusion proteins constructed in pGEX-6P-1 (GE Healthcare Life Sciences, 28954648) were transformed into Escherichia coli BL21 (DE3) cells, which were then cultured at 37°C in LB medium supplemented with 100 μg/mL ampicillin, and induced for 3 h at 37°C with 0.5 mM isopropyl β-d-thiogalactopyranoside (Nacalai Tesque, 07496–91). Cells were harvested by centrifugation, washed with PBS, lysed in lysis buffer containing 40 mM Tris-HCl, pH 7.5, 5 mM EDTA, 0.5 % Triton X-100, sonicated, and cleared by centrifugation. The resulting supernatant was incubated with Glutathione Sepharose 4 Fast Flow (GE Healthcare Life Sciences, 17513201) for 2 h at 4°C. Beads were then washed 3 times with lysis buffer, and incubated with lysates from HeLa cells expressing the appropriate protein or from E. coli expressing MBP-tagged protein at 4°C for 2 h. HeLa cells were lysed for 30 min on ice in 500 µL buffer containing 50 mM HEPES, pH 7.4, 250 mM NaCl, 10 mM MgCl2, 1 % Triton X-100, and proteinase inhibitor cocktail for use in this assay. Protein-bound beads were washed 5 times with HEPES buffer, and analyzed by immunoblotting.
Bacterial infection
Bacterial infection was performed as described previously.35 Briefly, bacteria were added to cell cultures at a multiplicity of infection of 100 without antibiotics for 1 h. Infected cells were washed with PBS, and antibiotic (100 g/mL gentamicin) was added to the medium to kill extracellular bacteria.
Statistical analysis
Colocalization and GcAV formation were manually quantified by direct visualization via confocal microscopy. Unless indicated otherwise, at least 50 GcAVs or 100 GAS-infected cells were counted per condition for each experiment. All values represent the mean ± SD. Data were analyzed using 2-tailed Student t test or ANOVA. P < 0.05 was considered to be statistically significant, and are marked *P < 0.05, **P < 0.01, and ***P < 0.001. ns, not significant.
Supplementary Material
Abbreviations
- BSA
bovine serum albumin
- CBB
Coomassie briliant blue, GAS, Group A Streptococcus
- DAPI
4′,6-diamidino-2-phenylindole
- GcAV
GAS-containing autophagosome-like vacuole
- LLOMe
lysosomotropic compound L-leucyl-L-leucine methyl ester
- MAP1LC3B/LC3B
microtubule-associated protein 1 light chain 3 β (a mammalian ortholog of yeast Atg8)
- MEF
mouse embryonic fibroblast
- NLR
nucleotide-binding domain leucine-rich repeat containing receptor
- NLRP4
NLR family pyrin domain containing 4
- SLO
streptolysin O.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- [1].Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069-75. doi: 10.1038/nature06639. PMID:18305538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy. 2011;7:279-96. doi: 10.4161/auto.7.3.14487. PMID:21189453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469:323-35. doi: 10.1038/nature09782. PMID:21248839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Deretic V. Autophagy in infection. Curr Opin Cell Biol. 2010;22:252-62. doi: 10.1016/j.ceb.2009.12.009. PMID:20116986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Boyle KB, Randow F. The role of 'eat-me' signals and autophagy cargo receptors in innate immunity. Curr Opin Microbiol. 2013;16:339-48. doi: 10.1016/j.mib.2013.03.010. PMID:23623150 [DOI] [PubMed] [Google Scholar]
- [6].Randow F, Munz C. Autophagy in the regulation of pathogen replication and adaptive immunity. Trends Immunol. 2012;33:475-87. doi: 10.1016/j.it.2012.06.003. PMID:22796170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Thurston TL, Ryzhakov G, Bloor S, von Muhlinen N, Randow F. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat Immunol. 2009;10:1215-21. doi: 10.1038/ni.1800. PMID:19820708 [DOI] [PubMed] [Google Scholar]
- [8].von Muhlinen N, Thurston T, Ryzhakov G, Bloor S, Randow F. NDP52, a novel autophagy receptor for ubiquitin-decorated cytosolic bacteria. Autophagy. 2010;6:288-9. doi: 10.4161/auto.6.2.11118. PMID:20104023 [DOI] [PubMed] [Google Scholar]
- [9].Yoshikawa Y, Ogawa M, Hain T, Yoshida M, Fukumatsu M, Kim M, Mimuro H, Nakagawa I, Yanagawa T, Ishii T, et al.. Listeria monocytogenes ActA-mediated escape from autophagic recognition. Nat Cell Biol. 2009;11:1233-40. doi: 10.1038/ncb1967. PMID:19749745 [DOI] [PubMed] [Google Scholar]
- [10].Mostowy S, Sancho-Shimizu V, Hamon MA, Simeone R, Brosch R, Johansen T, Cossart P. p62 and NDP52 proteins target intracytosolic Shigella and Listeria to different autophagy pathways. J Biol Chem. 2011;286:26987-95. doi: 10.1074/jbc.M111.223610. PMID:21646350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ogawa M, Yoshikawa Y, Kobayashi T, Mimuro H, Fukumatsu M, Kiga K, Piao Z, Ashida H, Yoshida M, Kakuta S, et al.. A Tecpr1-dependent selective autophagy pathway targets bacterial pathogens. Cell Host Microbe. 2011;9:376-89. doi: 10.1016/j.chom.2011.04.010. PMID:21575909 [DOI] [PubMed] [Google Scholar]
- [12].Ogawa M, Yoshikawa Y, Mimuro H, Hain T, Chakraborty T, Sasakawa C. Autophagy targeting of Listeria monocytogenes and the bacterial countermeasure. Autophagy. 2011;7:310-4. doi: 10.4161/auto.7.3.14581. PMID:21193840 [DOI] [PubMed] [Google Scholar]
- [13].Travassos LH, Carneiro LA, Ramjeet M, Hussey S, Kim YG, Magalhaes JG, Yuan L, Soares F, Chea E, Le Bourhis L, et al.. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol. 2010;11:55-62. doi: 10.1038/ni.1823. PMID:19898471 [DOI] [PubMed] [Google Scholar]
- [14].Homer CR, Richmond AL, Rebert NA, Achkar JP, McDonald C. ATG16L1 and NOD2 interact in an autophagy-dependent antibacterial pathway implicated in Crohn's disease pathogenesis. Gastroenterology. 2010;139:1630-41, 41.e1-2. doi: 10.1053/j.gastro.2010.07.006. PMID:20637199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Jounai N, Kobiyama K, Shiina M, Ogata K, Ishii KJ, Takeshita F. NLRP4 negatively regulates autophagic processes through an association with beclin1. J Immunol. 2011;186:1646-55. doi: 10.4049/jimmunol.1001654. PMID:21209283 [DOI] [PubMed] [Google Scholar]
- [16].Irving AT, Mimuro H, Kufer TA, Lo C, Wheeler R, Turner LJ, Thomas BJ, Malosse C, Gantier MP, Casillas LN, et al.. The immune receptor NOD1 and Kinase RIP2 interact with bacterial peptidoglycan on early endosomes to promote autophagy and inflammatory signaling. Cell Host Microbe. 2014;15:623-35. doi: 10.1016/j.chom.2014.04.001. PMID:24746552 [DOI] [PubMed] [Google Scholar]
- [17].Franchi L, Park JH, Shaw MH, Marina-Garcia N, Chen G, Kim YG, Núñez G. Intracellular NOD-like receptors in innate immunity, infection and disease. Cell Microbiol. 2008;10:1-8. PMID:17944960 [DOI] [PubMed] [Google Scholar]
- [18].Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol. 2007;9:1102-9. doi: 10.1038/ncb1007-1102. PMID:17909521 [DOI] [PubMed] [Google Scholar]
- [19].Noda T, Yoshimori T. Molecular basis of canonical and bactericidal autophagy. Int Immunol. 2009;21:1199-204. doi: 10.1093/intimm/dxp088. PMID:19737785 [DOI] [PubMed] [Google Scholar]
- [20].Mizushima N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr Opin Cell Biol. 2010;22:132-9. doi: 10.1016/j.ceb.2009.12.004. PMID:20056399 [DOI] [PubMed] [Google Scholar]
- [21].Simonsen A, Tooze SA. Coordination of membrane events during autophagy by multiple class III PI3-kinase complexes. J Cell Biol. 2009;186:773-82. doi: 10.1083/jcb.200907014. PMID:19797076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Chua CE, Gan BQ, Tang BL. Involvement of members of the Rab family and related small GTPases in autophagosome formation and maturation. Cell Mol Life Sci. 2011;68:3349-58. doi: 10.1007/s00018-011-0748-9. PMID:21687989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Bento CF, Puri C, Moreau K, Rubinsztein DC. The role of membrane-trafficking small GTPases in the regulation of autophagy. J Cell Sci. 2013;126:1059-69. doi: 10.1242/jcs.123075. PMID:23620509 [DOI] [PubMed] [Google Scholar]
- [24].Ao X, Zou L, Wu Y. Regulation of autophagy by the Rab GTPase network. Cell Death Differ. 2014;21(3):348-58. doi: 10.1038/cdd.2013.187. PMID:24440914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Nozawa T, Aikawa C, Goda A, Maruyama F, Hamada S, Nakagawa I. The small GTPases Rab9A and Rab23 function at distinct steps in autophagy during Group A Streptococcus infection. Cell Microbiol. 2012;14:1149-65. doi: 10.1111/j.1462-5822.2012.01792.x. PMID:22452336 [DOI] [PubMed] [Google Scholar]
- [26].Haobam B, Nozawa T, Minowa-Nozawa A, Tanaka M, Oda S, Watanabe T, Aikawa C, Maruyama F, Nakagawa I. Rab17-mediated recycling endosomes contribute to autophagosome formation in response to Group A Streptococcus invasion. Cell Microbiol. 2014;16:1806-21. doi: 10.1111/cmi.12329. PMID:25052408 [DOI] [PubMed] [Google Scholar]
- [27].Mostowy S. Multiple roles of the cytoskeleton in bacterial autophagy. PLoS Pathog. 2014;10:e1004409. doi: 10.1371/journal.ppat.1004409. PMID:25412422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kimura S, Noda T, Yoshimori T. Dynein-dependent movement of autophagosomes mediates efficient encounters with lysosomes. Cell Struct Funct. 2008;33:109-22. doi: 10.1247/csf.08005. PMID:18388399 [DOI] [PubMed] [Google Scholar]
- [29].Tumbarello DA, Waxse BJ, Arden SD, Bright NA, Kendrick-Jones J, Buss F. Autophagy receptors link myosin VI to autophagosomes to mediate Tom1-dependent autophagosome maturation and fusion with the lysosome. Nat Cell Biol. 2012;14:1024-35. doi: 10.1038/ncb2589. PMID:23023224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Pankiv S, Alemu EA, Brech A, Bruun JA, Lamark T, Overvatn A, Bjørkøy G, Johansen T. FYCO1 is a Rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end-directed vesicle transport. J Cell Biol. 2010;188:253-69. doi: 10.1083/jcb.200907015. PMID:20100911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Itakura E, Kishi-Itakura C, Mizushima N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell. 2012;151:1256-69. doi: 10.1016/j.cell.2012.11.001. PMID:23217709 [DOI] [PubMed] [Google Scholar]
- [32].Monastyrska I, Rieter E, Klionsky DJ, Reggiori F. Multiple roles of the cytoskeleton in autophagy. Biol Rev Camb Philos Soc. 2009;84:431-48. doi: 10.1111/j.1469-185X.2009.00082.x. PMID:19659885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Yamaguchi H, Nakagawa I, Yamamoto A, Amano A, Noda T, Yoshimori T. An initial step of GAS-containing autophagosome-like vacuoles formation requires Rab7. PLoS Pathog. 2009;5:e1000670. doi: 10.1371/journal.ppat.1000670. PMID:19956673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Huett A, Ng A, Cao Z, Kuballa P, Komatsu M, Daly MJ, Podolsky DK, Xavier RJ. A novel hybrid yeast-human network analysis reveals an essential role for FNBP1L in antibacterial autophagy. J Immunol. 2009;182:4917-30. doi: 10.4049/jimmunol.0803050. PMID:19342671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Nakagawa I, Amano A, Mizushima N, Yamamoto A, Yamaguchi H, Kamimoto T, Nara A, Funao J, Nakata M, Tsuda K, et al.. Autophagy defends cells against invading group A Streptococcus. Science. 2004;306:1037-40. doi: 10.1126/science.1103966. PMID:15528445 [DOI] [PubMed] [Google Scholar]
- [36].Oda S, Nozawa T, Nozawa-Minowa A, Tanaka M, Aikawa C, Harada H, Nakagawa I. Golgi-Resident GTPase Rab30 Promotes the Biogenesis of Pathogen-Containing Autophagosomes. PLoS One. 2016;11:e0147061. doi: 10.1371/journal.pone.0147061. PMID:26771875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Dovas A, Couchman JR. RhoGDI: multiple functions in the regulation of Rho family GTPase activities. Biochem J. 2005;390:1-9. doi: 10.1042/BJ20050104. PMID:16083425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Garcia-Mata R, Boulter E, Burridge K. The 'invisible hand': regulation of RHO GTPases by RHOGDIs. Nat Rev Mol Cell Biol. 2011;12:493-504. doi: 10.1038/nrm3153. PMID:21779026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].O'Seaghdha M, Wessels MR. Streptolysin O and its co-toxin NAD-glycohydrolase protect group A Streptococcus from Xenophagic killing. PLoS Pathog. 2013;9:e1003394. doi: 10.1371/journal.ppat.1003394. PMID:23762025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Dransart E, Olofsson B, Cherfils J. RhoGDIs revisited: novel roles in Rho regulation. Traffic (Copenhagen, Denmark). 2005;6:957-66. doi: 10.1111/j.1600-0854.2005.00335.x. PMID:16190977 [DOI] [PubMed] [Google Scholar]
- [41].DerMardirossian C, Schnelzer A, Bokoch GM. Phosphorylation of RhoGDI by Pak1 mediates dissociation of Rac GTPase. Mol Cell. 2004;15:117-27. doi: 10.1016/j.molcel.2004.05.019. PMID:15225553 [DOI] [PubMed] [Google Scholar]
- [42].DerMardirossian C, Rocklin G, Seo JY, Bokoch GM. Phosphorylation of RhoGDI by Src regulates Rho GTPase binding and cytosol-membrane cycling. Mol Biol Cell. 2006;17:4760-8. doi: 10.1091/mbc.E06-06-0533. PMID:16943322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Moreau K, Ghislat G, Hochfeld W, Renna M, Zavodszky E, Runwal G, Puri C, Lee S, Siddiqi F, Menzies FM, et al.. Transcriptional regulation of Annexin A2 promotes starvation-induced autophagy. Nat Commun. 2015;6:8045. doi: 10.1038/ncomms9045. PMID:26289944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol. 2013;13:722-37. doi: 10.1038/nri3532. PMID:24064518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Man SM, Ekpenyong A, Tourlomousis P, Achouri S, Cammarota E, Hughes K, Rizzo A, Ng G, Wright JA, Cicuta P, et al.. Actin polymerization as a key innate immune effector mechanism to control Salmonella infection. Proc Natl Acad Sci U S A. 2014;111:17588-93. doi: 10.1073/pnas.1419925111. PMID:25422455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Eitel J, Meixenberger K, van Laak C, Orlovski C, Hocke A, Schmeck B, Hippenstiel S, N'Guessan PD, Suttorp N, Opitz B. Rac1 regulates the NLRP3 inflammasome which mediates IL-1beta production in Chlamydophila pneumoniae infected human mononuclear cells. PLoS One. 2012;7:e30379. doi: 10.1371/journal.pone.0030379. PMID:22276187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Nakamura N, Lill JR, Phung Q, Jiang Z, Bakalarski C, de Maziere A, Klumperman J, Schlatter M, Delamarre L, Mellman I. Endosomes are specialized platforms for bacterial sensing and NOD2 signalling. Nature. 2014;509:240-4. doi: 10.1038/nature13133. PMID:24695226 [DOI] [PubMed] [Google Scholar]
- [48].Bonham KS, Kagan JC. Endosomes as platforms for NOD-like receptor signaling. Cell Host Microbe. 2014;15:523-5. doi: 10.1016/j.chom.2014.05.001. PMID:24832447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Damm A, Lautz K, Kufer TA. Roles of NLRP10 in innate and adaptive immunity. Microbes Infect Institut Pasteur. 2013;15:516-23. doi: 10.1016/j.micinf.2013.03.008 [DOI] [PubMed] [Google Scholar]
- [50].Yamashita T, Tohyama M. The p75 receptor acts as a displacement factor that releases Rho from Rho-GDI. Nat Neurosci. 2003;6:461-7. PMID:12692556 [DOI] [PubMed] [Google Scholar]
- [51].Eitel J, Krull M, Hocke AC, N'Guessan PD, Zahlten J, Schmeck B, Slevogt H, Hippenstiel S, Suttorp N, Opitz B. Beta-PIX and Rac1 GTPase mediate trafficking and negative regulation of NOD2. J Immunol. 2008;181:2664-71. doi: 10.4049/jimmunol.181.4.2664. PMID:18684957 [DOI] [PubMed] [Google Scholar]
- [52].Keestra AM, Winter MG, Auburger JJ, Frassle SP, Xavier MN, Winter SE, Kim A, Poon V, Ravesloot MM, Waldenmaier JF, et al.. Manipulation of small Rho GTPases is a pathogen-induced process detected by NOD1. Nature. 2013;496:233-7. doi: 10.1038/nature12025. PMID:23542589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Monastyrska I, He C, Geng J, Hoppe AD, Li Z, Klionsky DJ. Arp2 links autophagic machinery with the actin cytoskeleton. Mol Biol Cell. 2008;19:1962-75. doi: 10.1091/mbc.E07-09-0892. PMID:18287533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Reggiori F, Monastyrska I, Shintani T, Klionsky DJ. The actin cytoskeleton is required for selective types of autophagy, but not nonspecific autophagy, in the yeast Saccharomyces cerevisiae. Mol Biol Cell. 2005;16:5843-56. doi: 10.1091/mbc.E05-07-0629. PMID:16221887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Aguilera MO, Beron W, Colombo MI. The actin cytoskeleton participates in the early events of autophagosome formation upon starvation induced autophagy. Autophagy. 2012;8:1590-603. doi: 10.4161/auto.21459. PMID:22863730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Lee JY, Koga H, Kawaguchi Y, Tang W, Wong E, Gao YS, Pandey UB, Kaushik S, Tresse E, Lu J, et al.. HDAC6 controls autophagosome `maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. 2010;29:969-80. doi: 10.1038/emboj.2009.405. PMID:20075865 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.