

ABSTRACT
Macroautophagy/autophagy is increasingly recognized as an important regulator of myocardial ischemia-reperfusion (MI-R) injury. However, whether and how diabetes may alter autophagy in response to MI-R remains unknown. Deficiency of ADIPOQ, a cardioprotective molecule, markedly increases MI-R injury. However, the role of diabetic hypoadiponectinemia in cardiac autophagy alteration after MI-R is unclear. Utilizing normal control (NC), high-fat-diet-induced diabetes, and Adipoq knockout (adipoq−/−) mice, we demonstrated that autophagosome formation was modestly inhibited and autophagosome clearance was markedly impaired in the diabetic heart subjected to MI-R. adipoq−/− largely reproduced the phenotypic alterations observed in the ischemic-reperfused diabetic heart. Treatment of diabetic and adipoq−/− mice with AdipoRon, a novel ADIPOR (adiponectin receptor) agonist, stimulated autophagosome formation, markedly increased autophagosome clearance, reduced infarct size, and improved cardiac function (P < 0.01 vs vehicle). Mechanistically, AdipoRon caused significant phosphorylation of AMPK-BECN1 (Ser93/Thr119)-class III PtdIns3K (Ser164) and enhanced lysosome protein LAMP2 expression both in vivo and in isolated adult cardiomyocytes. Pharmacological AMPK inhibition or genetic Prkaa2 mutation abolished AdipoRon-induced BECN1 (Ser93/Thr119)-PtdIns3K (Ser164) phosphorylation and AdipoRon-stimulated autophagosome formation. However, AdipoRon-induced LAMP2 expression, AdipoRon-stimulated autophagosome clearance, and AdipoRon-suppressed superoxide generation were not affected by AMPK inhibition. Treatment with MnTMPyP (a superoxide scavenger) increased LAMP2 expression and stimulated autophagosome clearance in simulated ischemic-reperfused cardiomyocytes. However, no additive effect between AdipoRon and MnTMPyP was observed. Collectively, these results demonstrate that hypoadiponectinemia impairs autophagic flux, contributing to enhanced MI-R injury in the diabetic state. ADIPOR activation restores AMPK-mediated autophagosome formation and antioxidant-mediated autophagosome clearance, representing a novel intervention effective against MI-R injury in diabetic conditions.
KEYWORDS: adipokines, adiponectin receptor, autophagy, diabetes, myocardial ischemia-reperfusion injury
Although improved reperfusion strategies have decreased mortality in nondiabetic patients after myocardial infarction (MI), both the prevalence and severity of post-MI heart failure continually escalates in type 2 diabetics. Mechanisms leading to exacerbated post-MI remodeling and poor outcome in diabetic patients are incompletely understood, and effective therapeutic interventions are limited.
Autophagy is an intracellular process mediating protein degradation, organelle turnover, and cytoplasmic component recycling during nutrient starvation-driven cellular stress.1 Abundant evidence indicates that autophagy is essential for normal maintenance, repair, and adaptation of the heart over a lifetime.2,3 The impact of diabetes, a disease affecting > 20 million people in the U.S., upon cardiac autophagy has been a target of extensive research in recent years.4 However, the role of autophagy in diabetic cardiovascular complications remains undefined as either excess or inhibited autophagic activity in diabetic cardiomyopathy has been reported.4-6 Mechanisms underlying the diverse role of autophagy in diabetic cardiovascular injury remain unclear.
The role of autophagy in MI-R injury and ischemic heart failure has been extensively investigated in recent years. Consistent evidence demonstrates that autophagy is protective during ischemic cardiac injury.7-9 However, the role of autophagy in myocardial ischemia-reperfusion (MI-R) injury is controversial, and both adaptive and detrimental effects have been reported.1,8,9 Recent studies suggest that impaired autophagic flux (not merely autophagosome formation) is critical in reperfusion injury.8,10 It is well recognized that diabetic patients endure greater ischemic heart disease-associated morbidity compared with nondiabetics, with poorer outcome even after successful reperfusion.11 However, whether and how type 2 diabetes may alter cardiac autophagic flux in response to MI-R, thus contributing to accelerated cardiomyocyte injury, has surprisingly not been deeply investigated.
ADIPOQ (adiponectin, C1Q and collagen domain containing) is a novel adipokine that maintains insulin responsiveness, increases glucose and free fatty acid utilization, stimulates mitochondrial biogenesis, and inhibits the inflammatory response.12,13 Hypoadiponectinemia during diabetes is correlated with increased risk of acute MI, as well as worse cardiac functional recovery after MI-R.14,15 However, whether and how hypoadiponectinemia may contribute to diabetic cardiomyocyte autophagic flux alteration in response to MI-R has not been previously investigated.
Utilizing normal control (NC), high-fat-diet induced diabetes (DB), and Adipoq knockout (adipoq−/−) mice, the current study addressed 4 critical questions. First, whether diabetes may alter the formation and/or clearance of autophagosomes following MI-R was determined. Second, whether hypoadiponectinemia may contribute to impaired autophagic flux in the diabetic heart was clarified. Third, whether activation of ADIPOR1 (adiponectin receptor 1) and ADIPOR2 with a small molecule ADIPOR1/ADIPOR2 agonist (AdipoRon) may reverse diabetes-induced pathological alteration of autophagic flux was determined.16 Finally, the molecular mechanisms responsible for ADIPOR activation-mediated autophagic flux were investigated.
Results
Modestly decreased autophagosome formation and severely inhibited autophagosome clearance in diabetic mice heart subjected to MI-R
Ten wk of a high-fat diet caused significant weight gain and increased fasting serum glucose and insulin levels, characteristics of type 2 diabetes (Fig. S1A, Fig. S1B and Fig, S1C). To understand the effect of diabetes upon MI-R-induced autophagic flux, 2 different methods were used. First, the MAP1LC3B:ACTC1 (microtubule-associated protein 1 light chain 3 β:actin, α, cardiac muscle 1) ratio was determined with chloroquine (+CQ) or without (-CQ) pretreatment. As summarized in Fig. 1A (upper panel), the MAP1LC3B:ACTC1 ratio was modestly increased in the diabetic heart (sham MI-R animals) without CQ treatment. CQ pretreatment significantly (NC, 1.73-fold, P < 0.01) and modestly (DB, 1.29-fold, P < 0.05) increased the MAP1LC3B:ACTC1 ratio in the non-ischemic heart (Fig. 1A, upper panel). No significant difference in the MAP1LC3B:ACTC1 ratio was observed between NC and DB heart after MI-R without CQ pretreatment (Fig. 1A, lower panel, P > 0.5). In the NC heart subjected to MI-R, CQ pretreatment modestly (1.23-fold, P < 0.05) increased the MAP1LC3B:ACTC1 ratio. However, CQ pretreatment failed (1.05-fold, P > 0.05) to increase the MAP1LC3B:ACTC1 ratio in the DB heart after MI-R (Fig. 1A, lower panel), indicating severely impaired autophagic flux in these animals.
Figure 1.
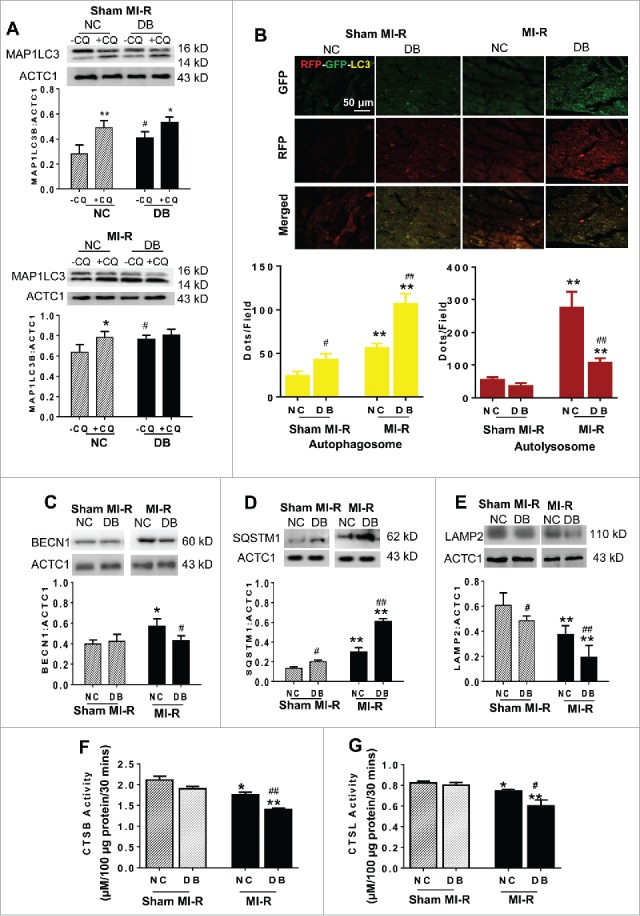
Determination of cardiomyocyte autophagy and lysosome function in high-fat-induced diabetic (DB) and age-matched normal control (NC) mice by MAP1LC3B:ACTC1 ratio (A), RFP-GFP-Map1lc3 transgenic mice (B), BECN1 expression (C), SQSTM1 expression (D), LAMP2 expression (E), CTSB activity (F), and CTSL activity (G). Yellow puncta in B represent autophagosomes, red-only puncta represent autolysosomes. NC or DB mice were subjected to sham MI-R (coronary artery not occluded) or 30 min coronary occlusion followed by 3 h of reperfusion (MI-R). N = 6–8 mice/group. Data were analyzed by 2-way ANOVA followed by the Tukey post hoc test for pairwise comparisons. *P < 0.05, **P < 0.01 between -CQ and +CQ (A) or between sham MI-R and MI-R (B-G) in the same animal group (NC or DB); #P < 0.05, ##P < 0.01 between NC and DB animals.
To more quantitatively assess autophagic flux, cardiomyocyte-specific RFP-GFP-Map1lc3b mice were used and autophagosome (yellow puncta) and autolysosome (red-only puncta) were determined. The total number of yellow puncta was 1.9-fold greater in the diabetic heart compared with control heart after MI-R (Fig. 1B). However, MI-R-induced autophagosome increase was comparable in the NC (2.33-fold) and DB (2.47-fold) groups as autophagosome formation was significantly increased in the diabetic heart without MI-R (sham MI-R, Fig. 1B). In the control group, the number of red-only puncta was significantly increased after MI-R compared with sham MI-R (5.09-fold, P < 0.01). However, the number of red puncta in the diabetic heart only modestly increased compared with sham MI-R. A markedly significant difference in autolysosome formation in response to MI-R was observed between the DB and NC groups (Fig. 1B).
The above results demonstrate that diabetes caused a 1.73-fold reduction in MI-R-induced autophagosome clearance (red-only puncta) compared with control. However, no significance difference was observed in the MAP1LC3B:ACTC1 ratio between NC and DB after MI-R (Fig. 1A, lower panel). Moreover, autophagosome (yellow puncta) increase in response to MI-R was similar between these 2 groups (fold change over respective sham MI-R). These results suggest that diabetes not only markedly blocked cardiac autophagosome clearance after MI-R, but may also inhibited autophagosome formation. To obtain direct evidence supporting this notion, additional experiments were performed. Similar to a recent report,10 we demonstrated the expression level of BECN1 (Beclin 1, autophagy related), a critical molecule in autophagosome formation, was significantly increased in the control group after MI-R. However, no significant change was observed in BECN1 expression in the diabetic heart after MI-R. A significant difference in BECN1 expression in response to MI-R was observed between the control and diabetic group (Fig. 1C). Moreover, MI-R-induced accumulation of SQSTM1 (sequestosome 1; a protein sequestered within autophagosomes for lysosomal degradation) was further augmented in the DB (3.1-fold) compared with NC (2.2-fold) (Fig. 1D). Additionally, MI-R-induced reduction in expression levels of LAMP2 (lysosomal-associated membrane protein 2, a critical determinant of autophagosome-lysosome fusion) was significantly exaggerated in the DB (66%) compared with NC (42%) groups (Fig. 1E, P < 0.01). Finally, although no significant difference in CTSB (cathepsin B) and CTSL (cathepsin L) activity was observed between the NC and DB hearts in the sham MI-R condition, inhibition of CTSB and CTSL by MI-R was significantly exaggerated in the DB heart (Fig. 1F, Fig. 1G, P < 0.05). Taken together, these results demonstrate that type 2 diabetes attenuated autophagosome formation and markedly blocked autophagosome clearance after MI-R.
ADIPOQ/adiponectin deficiency largely reproduced the autophagic phenotype as observed in diabetic animals in response to MI-R
Consistent with reports from others and our own laboratory, plasma ADIPOQ levels were significantly reduced 10 wk after a high-fat-diet (Fig. S1D). To determine whether decreased ADIPOQ contributes to the altered autophagic flux observed in diabetic animals, the MAP1LC3B:ACTC1 ratio (+/−CQ), expression levels of BECN1, SQSTM1 and LAMP2, and CTSB and CTSL activity were determined in WT and adipoq−/− hearts. In sham MI-R heart, CQ-pretreatment significantly (WT: 1.92-fold, P < 0.01) and modestly (adipoq−/−: 1.35-fold, P < 0.05) increased the MAP1LC3B:ACTC1 ratio (Fig. 1A, left panel). In MI-R heart, CQ-pretreatment modestly increased the MAP1LC3B:ACTC1 ratio in WT heart (1.28-fold, P < 0.05). However, CQ-pretreatment failed to increase the MAP1LC3B:ACTC1 ratio in adipoq−/− heart (Fig. 2A, right panel). No significant difference in BECN1 (Fig. 2B) and LAMP2 (Fig. 2C last panel) expression levels was observed between WT and adipoq−/− without MI-R (sham MI-R). However, SQSTM1 expression (Fig. 2C, middle panel) was significantly increased and both CTSB and CTSL activity was significantly reduced in the adipoq−/− heart, even without MI-R (Fig. 2D). In adipoq−/− mice subjected to MI-R, BECN1 expression was significantly reduced (P < 0.01, Fig. 2B), SQSTM1 expression was further increased (P < 0.01, Fig. 2C), LAMP2 expression was further reduced (P < 0.01, Fig. 2C), and CTSB and CTSL activity was further decreased (P < 0.01, Fig. 2D) compared with WT subjected to MI-R.
Figure 2.
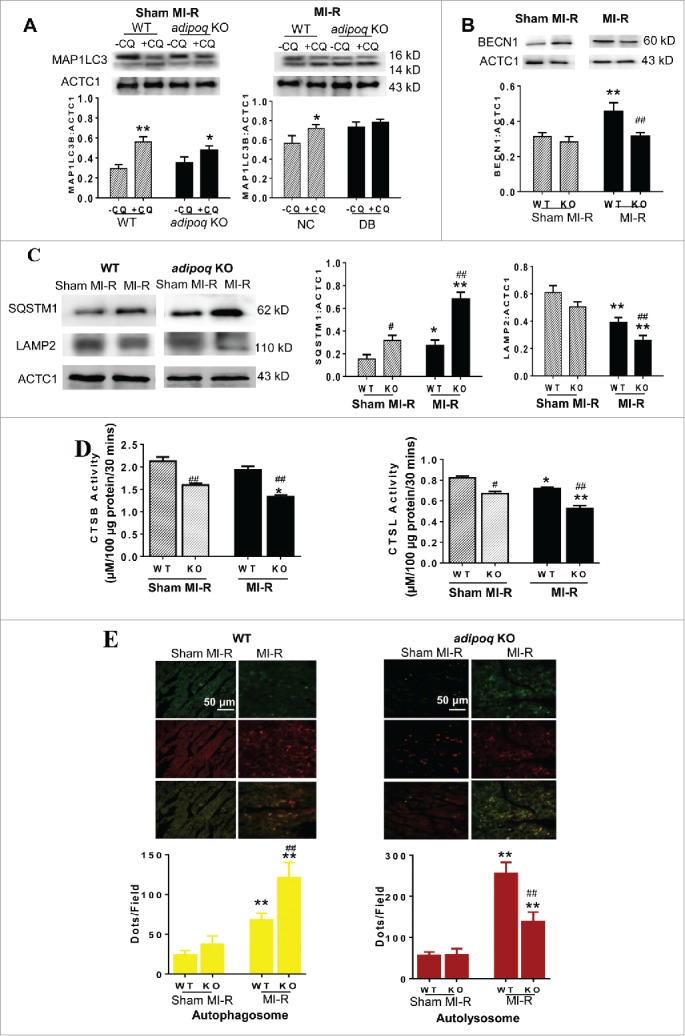
Assessment of cardiomyocyte autophagy and lysosome function in WT littermate (WT) or adipoq/adiponectin knockout (adipoq−/−) by MAP1LC3B:ACTC1 ratio (A), BECN1 expression (B), SQSTM1 and LAMP2 expression (C), CTSB and CTSL activity (D), and RFP-GFP-Map1lc3b transgenic mice (E). Yellow puncta in (E) represent autophagosomes, red-only puncta represent autolysosomes. WT or adipoq−/− mice were subjected to sham MI-R or MI-R (30 min/3 h). N = 6–8 mice/group. Data were analyzed by 2-way ANOVA followed by the Tukey post hoc test for pairwise comparisons. *P < 0.05, **P < 0.01 between -CQ and +CQ (A) or between sham MI-R and MI-R (B-E) in the same animal group (WT or adipoq−/−); #P < 0.05, ##P < 0.01 between WT and adipoq−/− animals.
To more quantitatively assess autophagic flux, adipoq−/− RFP-GFP-Map1lc3b transgenic mice were produced and autophagosome and autolysosome numbers were determined. No significant difference in autophagosome formation (Fig. 2E, yellow puncta and yellow bars) and autolysosome formation (Fig. 2E, red-only puncta and red bars) was observed between WT and adipoq−/− without MI-R (sham MI-R). These results slightly differ from those presented in Fig. 1B, wherein autophagic flux was modestly impaired in diabetic mice even before MI-R. However, the autophagic flux alterations observed in the diabetic heart after MI-R were largely reproduced in the adipoq−/− heart. Compared to WT, autophagosome formation was significantly increased (Fig. 2E, yellow) and autolysosome formation was markedly inhibited (Fig. 2E, red) in adipoq−/− mice. These results suggest hypoadiponectinemia in diabetic mice may contribute to impaired autophagic flux following MI-R.
ADIPOR activation improved autophagic flux in the diabetic heart after MI-R
To obtain more evidence supporting the idea that the reduced ADIPOQ level in diabetes contributes to impaired autophagic flux after MI-R, diabetic animals were treated with AdipoRon, a novel small ADIPOR agonist with demonstrated metabolic benefit in diabetic animals.16 As summarized in Fig. 3A (left panel), treatment of NC mice with AdipoRon significantly increased the MAP1LC3B:ACTC1 ratio in both -CQ and +CQ groups. Compared to vehicle-treated animals, treatment of DB mice with AdipoRon modestly increased the MAP1LC3B:ACTC1 ratio without CQ pretreatment (P < 0.05). More importantly, AdipoRon treatment restored autophagic flux in diabetic mice subjected to MI-R, as evidenced by significantly increased MAP1LC3B:ACTC1 ratio in +CQ animals (Fig. 3A, right panel). Moreover, administration of AdipoRon shortly before reperfusion increased BECN1 expression and reduced the SQSTM1 level to the same levels in both control and diabetic groups (Fig. 3B). As the expression level of BECN1 was decreased and SQSTM1 levels was increased in the vehicle-treated diabetic group compared with the vehicle-treated control group, a preferential protective effect of AdipoRon in the diabetic group was observed (P < 0.05 in NC group, P < 0.01 in DB group). Finally, treatment of NC and DB mice with AdipoRon significantly restored CTSB and CTSL activity after MI-R (Fig. 3C).
Figure 3.
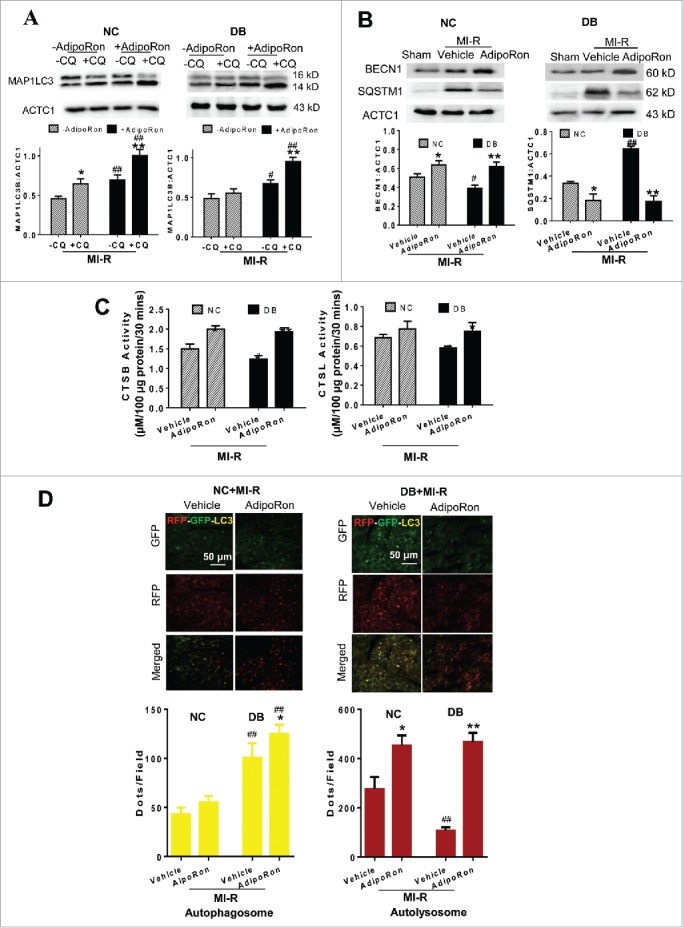
Effect of AdipoRon, a small molecule ADIPOR agonist, upon cardiomyocyte autophagy and lysosome function in age-matched normal control (NC) or high-fat-induced diabetic (DB) mice by BECN1 and SQSTM1 expression (A,B), CTSB and CTSL activity (C), and RFP-GFP-Map1lc3b transgenic mice (D). Yellow puncta in (D) represent autophagosomes, red-only puncta represent autolysosomes. NC or DB mice were subjected to sham MI-R, MI-R. Ten min before reperfusion, animals were randomized to receive either vehicle or AdipoRon (intravenous bolus injection). N = 8–10 mice/group. Data were analyzed by 2-way ANOVA followed by the Tukey post hoc test for pairwise comparisons. *P < 0.05, **P < 0.01 between -CQ and +CQ (A) or between vehicle and AdipoRon treated MI-R animals (B-D) in the same animal group (NC or DB); #P < 0.05, ##P < 0.01 between NC and DB animal with the same treatment (vehicle or AdipoRon).
To obtain more evidence supporting a conclusion that AdipoRon restores autophagic flux in diabetic animals, the effect of AdipoRon upon autophagosome and autolysosome formation was determined in RFP-GFP-Map1lc3b animals. As summarized in Fig. 3D, treatment with AdipoRon slightly increased autophagosome formation (yellow puncta) and markedly increased autolysosome formation (red-only puncta) in diabetic MI-R hearts. Taken together, these results demonstrate that activation of ADIPOR increased expression levels of MAP1LC3 and LAMP2, stimulated autophagosome formation, and promoted autophagosome clearance. AdipoRon may thus facilitate both autophagosome formation and autophagosome clearance.
AMP-activated protein kinase (AMPK) is essential for AdipoRon-stimulated autophagosome formation but not requisite for AdipoRon-stimulated autophagosome clearance
Having demonstrated that ADIPOR activation significantly increased autophagosome formation and augmented autophagosome clearance, we next determined the molecular mechanisms mediating this protective effect in diabetic mice. A proven downstream factor mediating the metabolic actions of ADIPOQ, AMPK is a recognized upstream multisubunit protein activating autophagic signaling.17 We therefore first determined whether AMPK may play a significant role in ADIPOR activation-induced autophagic flux in diabetic mice. As summarized in Fig. 4A, treatment with AdipoRon shortly before reperfusion resulted in significant phosphorylation of AMPK, and increased its activity as assessed by ACACA (acetyl-coenzyme A carboxylase α) phosphorylation (Fig. 4B). To determine whether AdipoRon-induced AMPK phosphorylation may activate autophagy signaling, phosphorylation levels of multiple autophagy regulating molecules were screened. Treatment with AdipoRon had no significant effect upon the phosphorylation levels of ULK1 (unc-51 like kinase 1; Ser317/Ser77), BECN1 (Ser14), ULK1 (Ser757) and BECN1 (S295) (data not shown). However, phosphorylation levels of the class III phosphatidylinositol 3-kinase (PtdIns3K) catalytic subunit PIK3C3/VPS34 (Ser164), BECN1 (Ser93),and BECN1 (Thr119) were significantly increased in AdipoRon-treated animals (Fig. 4B, Fig. 4C), and PtdIns3K activity was significantly increased (Fig. 4D).
Figure 4.
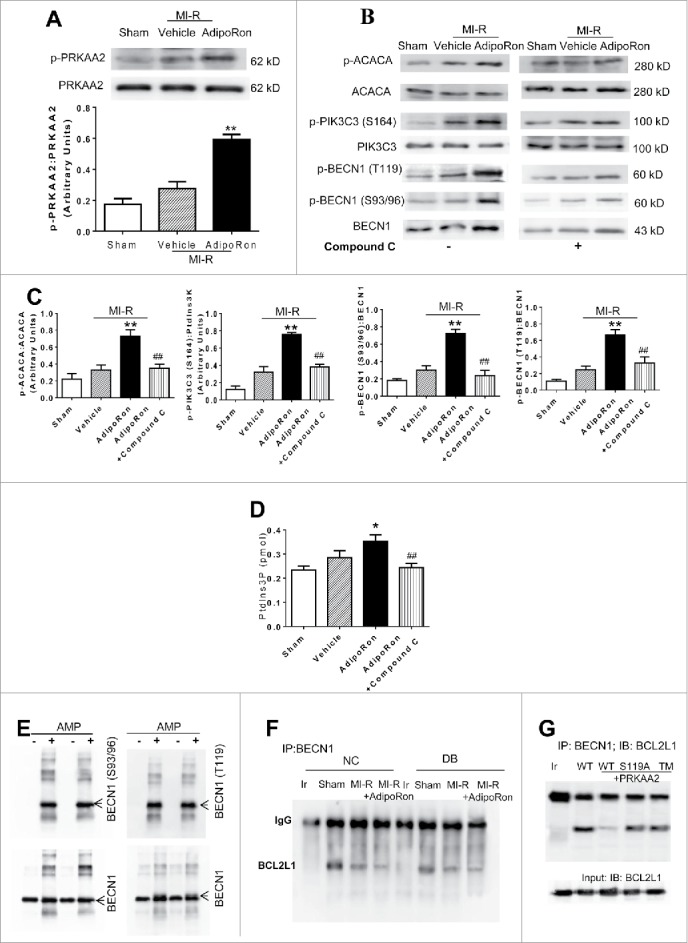
Determination of the signaling mechanisms involved in AdipoRon-mediated regulation of cardiomyocyte autophagosome formation in high-fat-induced diabetic mice. AdipoRon caused significant PRKAA2 phosphorylation (A). Representative western blots (B) and densitometry analysis (C) showing AdipoRon caused significant phosphorylation of ACACA, PIK3C3 (Ser164), BECN1 (Ser93) and BECN1 (Thr119), an effect blocked by compound C (intravenously administered 10 min before AdipoRon), an PRKAA inhibitor. AdipoRon caused significant activation of PtdIns3K, an effect blocked by compound C (D). An in vitro kinase assay demonstrated that activation of PRKAA2 by AMP caused significant BECN1 phosphorylation at Ser93 as well as Thr119 (E: Western blot). AdipoRon promoted BECN1-BCL2L1 dissociation in NC and DB mice (F). DB mice were subjected to sham MI-R or MI-R. Ten min before reperfusion, animals were randomized to receive either vehicle or AdipoRon (intravenous bolus injection). (G) Determination of PRKAA2-induced BECN1 phosphorylation at different sites upon BECN1-BCL2L1 interaction. (A-D) N = 7–8 mice/group; (E-G) representative results from 3–4 repeated experiments. Data were analyzed by one-way ANOVA followed by the Tukey post hoc test for pairwise comparisons. *P < 0.05, **P < 0.01 between vehicle and AdipoRon-treated MI-R animals; ##P < 0.01 between AdipoRon-treated animals with or without compound C pretreatment (C,D).
To determine a cause and effect relationship between AMPK activation and PIK3C3 (Ser164)-BECN1 (Ser93/Thr119) phosphorylation, animals were treated with compound C (an AMPK inhibitor) 10 min before AdipoRon administration. As illustrated in Fig. 4B and summarized in Fig. 4C, pretreatment with compound C abolished AdipoRon-induced PIK3C3 and BECN1 phosphorylation, and blocked PtdIns3K activation (Fig. 4D). To obtain more evidence that AMPK phosphorylated BECN1 at S93 as well as T119, an in vitro kinase reaction followed by mass spectrophotometry (MS) and western blot analysis were performed (using BECN1 as substrate and AMP as an AMPK activator). As illustrated in Fig. S2-S3, MS analysis revealed that AMPK indeed phosphorylates BECN1 at S93 as well as T119. This observation was confirmed by the western blot analysis presented in Fig. 4E.
To further validate our finding that AdipoRon promoted phosphorylation of BECN1 at Thr119, the effect of AdipoRon upon BECN1-BCL2L1 (BCL2-like 1) interaction was evaluated. Our experimental results demonstrated that AdipoRon indeed promoted the dissociation of BECN1 from BCL2L1 (Fig. 4F). Finally, to support the notion that AMPK-induced BECN1 phosphorylation at Thr119 was responsible for BECN1-BCL2L1 dissociation, WT or mutated BECN1 were phosphorylated by AMPK and BECN1-BCL2L1 interaction was determined. As illustrated in Fig. 4G, addition of activated AMPK significantly reduced BECN1-BCL2L1 interaction, an effect blocked by either T119A or S93,S96,T119A (triple mutation, TM) mutation.
To obtain direct evidence that AdipoRon stimulates autophagy via AMPK activation, the effect of compound C upon autophagic flux was determined by using cardiomyocyte-specific RFP-GFP-Map1lc3b mice. As illustrated in Fig. 5A, AdipoRon-stimulated autophagosome formation (yellow puncta) was abolished by compound C pre-treatment, demonstrating the critical role of AMPK in AdipoRon-induced autophagosome formation. Somewhat to our surprise, AdipoRon-stimulated autolysosome abundance (red-only puncta) was unaffected by compound C, suggesting AdipoRon promoted autophagosome clearance in an AMPK-independent manner. To obtain more evidence supporting this notion, the effect of compound C upon AdipoRon-mediated regulation of SQSTM1-LAMP2 expression was determined. Neither AdipoRon's inhibitory effect upon SQSTM1 accumulation (Fig. 5B), nor AdipoRon's promotive effect upon LAMP2 expression (Fig. 5C), were affected by pre-treatment with compound C.
Figure 5.
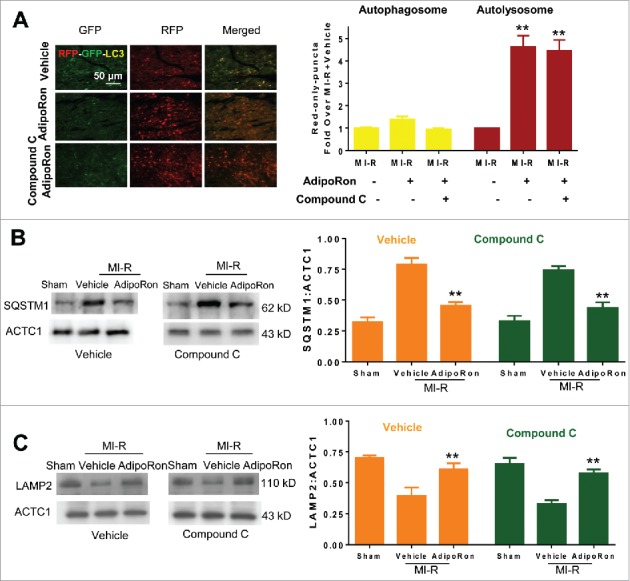
Determination of the signaling mechanisms involved in AdipoRon-mediated regulation of cardiomyocyte autolysosome formation in high-fat-induced diabetic mice. (A) Compound C pre-treatment abolished the effect of AdipoRon upon autophagosome formation (yellow puncta) but had no effect upon AdipoRon-induced autolysosome formation (red-only puncta). (B,C) Compound C pre-treatment had no effect upon SQSTM1 and LAMP2 expression in response to AdipoRon treatment. DB mice were subjected to sham MI-R. Ten min before reperfusion, animals were randomized to receive either vehicle or AdipoRon (intravenous bolus injection). Compound C was intravenously administered 10 min before AdipoRon. N = 7–8 mice/group. Data were analyzed by one-way ANOVA followed by the Tukey post hoc test for pairwise comparisons. **P < 0.01 between AdipoRon-treated animals with or without compound C pretreatment.
To obtain more conclusive results supporting the differential role of AMPK in AdipoRon regulation of autophagosome formation and autophagosome clearance, adult cardiomyocytes were isolated from WT or cardiomyocyte-specific Prkaa2 dominant negative (DN) overexpression mice and subjected to simulated MI-R. Similar to our in vivo findings, AdipoRon treatment resulted in significant phosphorylation of PIK3C3 (Ser164)-BECN1 (Thr119) in WT cardiomyocytes (Fig. 6A), but not in Prkaa2 DN cardiomyocytes (Fig. 6B). However, AdipoRon's promotive effect upon LAMP2 expression was unchanged in Prkaa2 DN cardiomyocytes (Fig. 6A, Fig. 6B).
Figure 6.
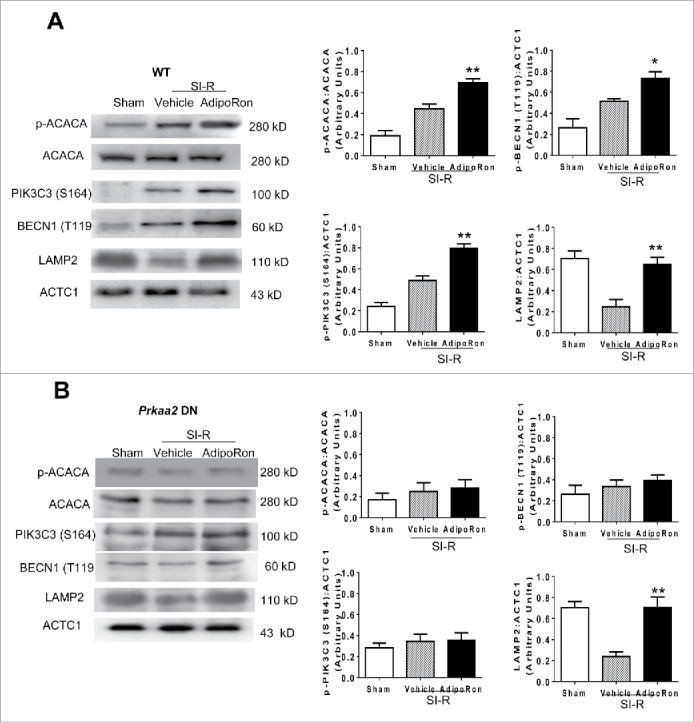
In vitro assessment of PRKAA2 involvement in AdipoRon-stimulated autophagic flux. Adult cardiomyocytes isolated from WT or Prkaa2 DN mice were subjected to 3 h of simulated ischemia and 6 h of simulated reperfusion. At the time of reperfusion, cells were treated with vehicle or AdipoRon. (A) AdipoRon caused significant phosphorylation of ACACA, PIK3C3 (Ser164) and BECN1 (T119), and increased LAMP2 expression in cardiomyocytes from WT mice. (B) AdipoRon-induced phosphorylation of ACACA, PIK3C3 (Ser164), and BECN1 (Thr119) was abolished in cardiomyocytes from Prkaa2 DN mice; however, ADIPOR-induced LAMP2 expression was unaffected. N = 14–16 dishes from at least 5 mice/group. Data were analyzed by one-way ANOVA followed by the Tukey post hoc test for pairwise comparisons. *P < 0.05, **P < 0.01 between vehicle and AdipoRon-treated cells.
AdipoRon facilitates autophagosome clearance through an AMPK-independent antioxidant effect
We previously reported that ADIPOQ provides cardioprotection partially via AMPK-independent antioxidant action.18 Moreover, a recent study demonstrated that reperfusion blocks autophagosome clearance largely due to reactive oxygen species (ROS) overproduction.10 We thus reasoned that AdipoRon may promote autophagosome clearance by reducing ROS production. Three lines of evidence support our hypothesis. First, in vivo experimental results demonstrated that AdipoRon significantly reduced superoxide concentration in the ischemic-reperfused diabetic heart. Pre-treatment with compound C did not affect AdipoRon's anti-oxidant action (Fig. 7A). Second, adult cardiomyocytes were isolated from WT or adipor1−/− adipor2+/− mice, and subjected to simulated ischemia-reperfusion (SI-R). At the time of reperfusion, cells were treated with MnTMPyP (a cell permeable superoxide scavenger, 50 μM),10 AdipoRon, or MnTMPyP+AdipoRon. Treatment with either MnTMPyP or AdipoRon in WT cardiomyocytes significantly increased LAMP2 and reduced SQSTM1 expression (Fig. 7B). However, the combination treatment of AdipoRon and MnTMPyP yielded no additive effect. Moreover, Adipor knockout abolished the effect of AdipoRon but not MnTMPyP, indicating AdipoRon achieved its autophagosome clearance effect by ADIPOR activation. Third, treatment with either AdipoRon or MnTMPyP alone significantly increased red-only puncta in neonatal rat cardiomyocytes subjected to SI-R. However, treatment with both AdipoRon and MnTMPyP yielded no additive effect (Fig. 7C).
Figure 7.
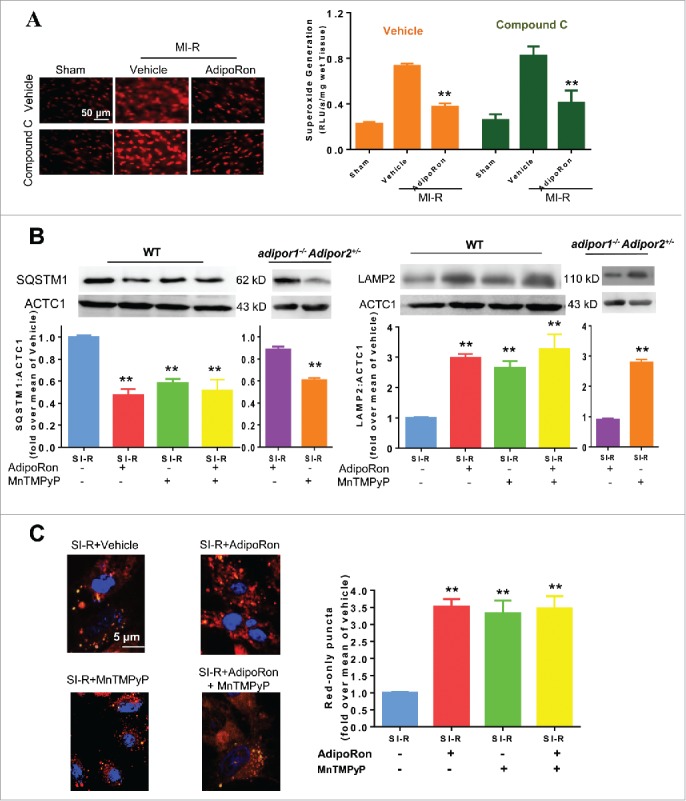
AdipoRon facilitates autophagosome clearance through an AMPK-independent antioxidant effect. (A) AdipoRon significantly reduced superoxide concentration in the ischemic-reperfused diabetic heart, an effect unaffected by compound C pretreatment. (B) Adult cardiomyocytes were isolated from WT or adipor1−/− Adipor2+/− mice, and subjected to SI-R. At the time of reperfusion, cells were treated with MnTMPyP, AdipoRon, or MnTMPyP+AdipoRon. Treatment with either MnTMPyP or AdipoRon in WT cardiomyocytes significantly increased LAMP2 and reduced SQSTM1 expression. However, the combination treatment of AdipoRon and MnTMPyP yielded no additive effect. Adiporknockout abolished the effect of AdipoRon but not MnTMPyP. (C) treatment with either AdipoRon or MnTMPyP alone significantly increased red-only puncta in neonatal rat cardiomyocytes subjected to SI-R. However, treatment with both AdipoRon and MnTMPyP yielded no additive effect. N = 6 for the in vivo experiment presented in panel (A); N = 14–16 dishes from at least 5 mice/group for in vitro experiments presented in panels ((B)and C). Data were analyzed by one-way ANOVA followed by the Tukey post hoc test for pairwise comparisons. *P < 0.05, **P < 0.01 between vehicle and AdipoRon treated animals (A) and cells (B, C).
AdipoRon significantly reduced infarct size and improved cardiac function in diabetic mice
In a final step to demonstrate that the autophagic-promoting effect of AdipoRon can translate to sustained cardioprotection, cardiac function and infarct size were determined 24 h after reperfusion. As illustrated in Fig. 8A and summarized in Fig. 8B, MI-R significantly reduced LVEF in control, diabetic, and adipoq−/− mice (with the most severe cardiac dysfunction observed in the diabetic group). Treatment with AdipoRon restored LVEF to a similar level in all 3 groups. Moreover, AdipoRon treatment significantly reduced myocardial infarct size in control, diabetic, and adipoq−/− mice. Although the infarct size in diabetic and adipoq−/− mice was larger in comparison to control, no significant difference in infarct size was observed in AdipoRon-treated groups (Fig. 8C).
Figure 8.
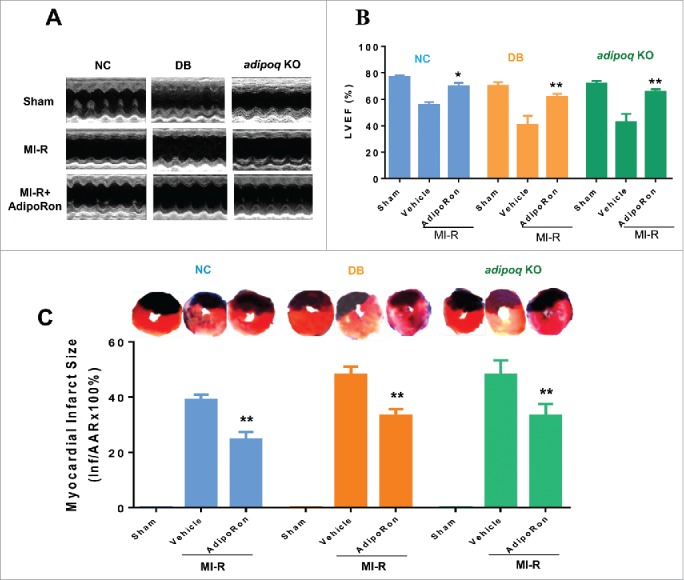
Effect of AdipoRon upon cardiac function and infarct size after MI-R. NC, DB, and adipoq−/− mice were subjected to 30 min of MI followed by 24 h of reperfusion. Ten min before reperfusion, mice were randomized to receive vehicle or AdipoRon intravenous injection. (A) Representative records of echocardiography. (B) Left ventricular ejection fraction (LVEF) in NC, DB and adipoq−/− mice subjected to sham MI-R, MI-R and MI-R treated with vehicle or AdipoRon. (C) Myocardial infarct size determined by Evans blue-TTC double-staining protocol demonstrating that AdipoRon significantly reduced infarct size in NC, DB, and adipoq−/− mice. N = 12–15 mice/group. Data were analyzed by one-way ANOVA followed by the Tukey post hoc test for pairwise comparisons. *P < 0.05, **P < 0.01 between vehicle and AdipoRon-treated animals. Inf/AARx100%: Infarct size/Area-At-Risk x 100%.
Discussion
Although cardiomyocyte autophagic alterations in diabetic cardiomyopathy and cardiac ischemia-reperfusion injury in nondiabetic animals have been a hot topic of investigation in recent years,4,19 the definitive role of autophagy in cardiac injury associated with these pathological conditions remains controversial.1 This is partly due to the inappropriate autophagic evaluation methods used in early studies, resulting in diverse interpretation.8 Moreover, although it is well recognized that ischemic heart disease is the primary cause of mortality in type 2 diabetic patients, the specific impact of diabetes upon cardiomyocyte autophagic flux after MI-R has not been determined. We have made several important observations in this study. First, we have provided clear evidence that type 2 diabetes severely impaired autophagic flux in the ischemic-reperfused heart via mechanisms involving both autophagosome formation and autophagosome clearance. Specifically, compared with the nondiabetic heart, autophagosome clearance was further inhibited in the diabetic heart subjected to MI-R, as evidenced by decreased LAMP2 expression, increased SQSTM1 accumulation, and decreased autolysosome abundance. Moreover, contrary to nondiabetic hearts manifesting increased autophagosome formation following MI-R, the autophagosome formation process was impaired in the diabetic heart following MI-R. As such, ischemia followed by reperfusion in the nondiabetic heart impaired autophagosome flux primarily by inhibiting autophagosome clearance, whereas ischemia followed by reperfusion in the diabetic heart impaired autophagic flux by exacerbating impaired autophagosome clearance atop reduced autophagosome formation.
These results are important for the following 2 reasons: First, to the best of our knowledge, this is the first study demonstrating that type 2 diabetes places the heart in “double-trouble” concerning autophagic flux, suggesting that promoting both autophagosome clearance and formation are necessary to achieve the optimal protective effect in diabetic patients having MI-R. Second, these results identified a likely explanation for previously reported controversial results regarding the impact of diabetes upon autophagy during the development of diabetic cardiomyopathy.6,20,21 If autophagy is assessed by MAP1LC3B:ACTC1 ratio (Fig. 1A), one would conclude that diabetes has no significant effect upon autophagy in the ischemic-reperfused heart. However, if autophagy is assessed by MAP1LC3 immunofluorescent puncta (Fig. 1B), one would conclude that diabetes markedly increases autophagy in MI-R heart. In contrast, when autophagy is assessed by combining MAP1LC3B:ACTC1 ratio with the abundance of autophagosomes-autolysosomes, one may finally conclude that type 2 diabetes impairs both autophagosome clearance and autophagosome formation.
Second, we have demonstrated that ADIPOQ is an important cardiomyocyte autophagy regulator and that hypoadiponectinemia in diabetes contributes to defective cardiomyocyte autophagy, particularly after ischemia-reperfusion. This conclusion is supported by 2 lines of evidence. First, the autophagic flux alterations observed in the diabetic heart after MI-R were largely reproduced in the adipoq−/− heart. Second, reactivation of ADIPOR with a small molecular ADIPOR agonist preferentially restored autophagic flux in the ischemic-reperfused diabetic heart. Although the cardiovascular protective effects of ADIPOQ are well accepted, and the contribution of hypoadiponectinemia to diabetic cardiovascular injury is well demonstrated in experimental and clinical investigations, the role of ADIPOQ in cardiomyocyte autophagy regulation has only been recognized in recent years. Although currently available evidence consistently supports the cardioprotective role of ADIPOQ-regulated autophagy flux,22-25 the specific regulatory role of ADIPOQ in autophagic flux remains unclear as both inhibitory and promotive effects have been reported. Specifically, transgenic overexpression of ADIPOR1 protects against high-fat-diet induced cardiomyopathy via cardiomyocyte autophagy inhibition,22 and ADIPOQ administration reduces excessive ROS-induced cardiomyocyte injury by inhibiting ROS-induced cardiomyocyte autophagy.23 In contrast, pressure overload-induced cardiac autophagy is significantly inhibited in adipoq−/−, and administration of ADIPOQ in vivo or in vitro restores autophagy flux and attenuates cardiomyocyte injury.24 Moreover, Adipoq deficiency has been reported to aggravate high-fat-diet induced cardiac hypertrophy and contractile dysfunction via myocardial autophagy inhibition.25 These results indicate that the specific regulatory role of ADIPOQ in cardiomyocyte autophagy likely depends upon ambient pathological conditions. Given that ischemic heart disease is the most significant cause of death in diabetic patients, clarification of the specific role of ADIPOQ in the ischemic-reperfused diabetic heart in our current study is not only scientifically significant, but also clinically important. It should be noted that autophagic flux is significantly inhibited in the diabetic heart even before MI-R, wherein no significant changes were observed in adipoq−/− mice during basal conditions. These results are consistent with previous studies demonstrating that significant phenotypic alterations develop in adipoq−/− mice only after pathological stress.26,27
Third, we have provided the first evidence that ADIPOR activation with an orally active ADIPOR1/ADIPOR2 agonist restores cardiomyocyte autophagic flux by activating AMPK-mediated autophagosome formation and promoting AMPK-independent anti-oxidative mediated autophagosome clearance. Abnormal MTORC1 (mechanistic target of rapamycin [serine/threonine kinase] complex 1) activation and excessive ROS production, the 2 most important pathological mediators inhibiting autophagosome formation and clearance, co-exists in the diabetic heart.4,5,19,28 Recent studies demonstrated that AMPK, an enzyme critical in cellular metabolism, activates autophagy.17 AMPK inhibits the MTORC1 complex by phosphorylation of TSC2 and direct phosphorylation of RPTOR (regulatory-associated protein of MTOR, complex 1), resulting in activation of the TSC1/2 (tuberous sclerosis 1/2) complex and inactivation of MTORC1.29 Additionally, AMPK phosphorylates several other signaling proteins critical for autophagosome formation, including ULK1, BECN1, and PIK3C3.4,30,31
Considerable evidence exists that defective AMPK signaling is causatively related to impaired autophagy in organs adversely affected by diabetes, including the heart.4,25,31 Our current study demonstrated that AdipoRon significant activated AMPK, and increased phosphorylation levels of PIK3C3 (Ser164) and BECN1 (Ser93/Thr119), 2 phosphorylative modifications known to increase BECN1-PIK3C3-ATG14 interaction and autophagosome formation.32,33 Genetic inhibition of Adipor1 and Adipor2 (siRNA), pharmacological inhibition of AMPK or genetic inhibition of Prkaa2 abolished AdipoRon-induced PIK3C3 (Ser164) and BECN1 (Ser93/Thr119) phosphorylation, and inhibited the promotive effect of AdipoRon upon autophagosome formation. These results demonstrated that AdipoRon restores autophagosome formation in ischemic-reperfused diabetic heart via an ADIPOR1/ADIPOR2-AMPK signaling mechanism. Concomitantly, our study demonstrated that AMPK inhibition neither blocked the anti-oxidant nor promotive effect of AdipoRon upon autophagosome clearance. Moreover, our in vitro experiments performed in Prkaa2 dominant negative cardiomyocytes demonstrated that treatment with AdipoRon or MnTMPyP restored autophagosome clearance in SI-R cardiomyocytes. However, no additive effect was observed when cells were treated with both AdipoRon and MnTMPyP. These results indicate that activation of adiponectin receptors promoted cardiomyocyte autophagosome clearance in an AMPK-independent, anti-oxidative stress-mediated mechanism. This conclusion is supported by a recent study demonstrating that reperfusion impairs myocardial autophagosome clearance due to increased ROS production and subsequent inhibition of LAMP2 expression.10 Moreover, our results demonstrating AdipoRon stimulates autophagosome formation through AMPK-dependent signaling, and restores autophagosome clearance through AMPK-independent mechanisms, is also consistent with a previous study demonstrating cardiomyocyte autophagy is regulated by AMPK-dependent signaling during ischemia, whereas impaired autophagic flux during reperfusion (likely due to ROS production) is AMPK independent.9
As recent work consistently demonstrates that cardiac autophagy is inhibited in obesity and diabetes, considerable effort has been made to determine whether reactivation of autophagy may prevent or reduce the cardiovascular abnormalities in the diabetic heart.4 Restoration of autophagic flux by Akt2 (thymoma viral proto-oncogene 2) deletion or reactivation of autophagosome formation by Mif knockout improve cardiac function in high-fat-diet-induced cardiomyopathy.34,35 However, genetic manipulation has limited clinical application. Rapamycin is the most studied MTOR inhibitor; considerable literature supports its beneficial effect against defective autophagy. However, rapamycin may not be an ideal chronic treatment, particularly in diabetic patients, because it can disrupt MTORC2 (which promotes cardiomyocyte survival) and aggravate the insulin resistance status.36 Finally, metformin activates AMPK and promotes autophagosome formation. However, promoting autophagosome formation alone without concomitant restoration of autophagosome clearance may aggravate reperfusion injury by increasing cardiomyocyte apoptosis.10 Ideally, molecules promoting autophagy flux by interfering with multiple aspects of the autophagic machinery would be the most effective agents reactivating diabetic heart autophagic flux after MI-R. Although ADIPOQ supplementation may restore autophagic flux in the diabetic heart, the clinical application of recombinant proteins is limited due to high cost and undesirable administration routes. Our current study, demonstrating AdipoRon administration shortly before reperfusion reactivates diabetes-induced defective autophagic flux in the ischemic-reperfused heart by promoting autophagosome clearance as well as their formation, is therefore important from a translational point of view.37 These results suggest that development of novel, more selective and less toxic small molecule ADIPOR agonists may hold promise of an effective therapy against diabetes-induced cardiac injury.
Materials and methods
Animals and reagents
Adult male wild-type C57BL/6 mice (WT), adipoq−/− (8195, Jackson Laboratory, Bar Harbor, ME), cardiomyocyte-specific Prkaa2/α2 (protein kinase, AMP-activated, α 2 catalytic subunit) subunit mutant transgenic mice (Prkaa2 DN, Dr. R Tian, University of Washington),38 and cardiomyocyte-specific red fluorescent protein (RFP)-green fluorescent protein (GFP)-Map1lc3b transgenic mice (RFP-GFP-Map1lc3b, Drs. Joseph Hill and Zhao Wang, University of Texas Southwestern Medical Center39) were fed either a normal or high-fat (D12492, Research Diets, Inc.) diet for 10 wk. All animal studies were approved by the Institutional Animal Care and Use Committee at Thomas Jefferson University.
AdipoRon (509104) was purchased from Calbiochem. Antibodies against p-ULK (S317) (12753), PIK3C3 (4263), MAP1LC3A/B (12741), GAPDH (2118), BECN1 (3495), SQSTM1 (5114), BCL2L1 (2764), p-BECN1 (Ser93/96; 12476), AMPK (2603), p-AMPK (2531) and IgG horseradish peroxidase (HRP)-conjugated secondary antibodies (7074; 7076) were purchased from Cell Signaling Technology. Antibodies against p-PIK3C3 (Ser164; bs5581R) were obtained from Bioss Inc., and p-BECN1 (Thr119; ABC118) were purchased from EMD Millipore. Antibodies against LAMP1 (OAAB13754) and LAMP2 (OAAB06712) were purchased from Aviva Systems Biology. ACTC1 (A7811) and compound C (171260) was purchased from Sigma-Aldrich. Collagenase type B/D (11088807001; 11088858001) was purchased from R&D system, Inc. The PtdIns3K kinase activity assay kit was purchased from Echelon (PtdIns3K activity assay kit, K-3000). The CTSB activity assay kit (CBA001) was purchased from Millipore and CTSL activity assay kit (ab65306) was purchased from Abcam.
Myocardial ischemia-reperfusion
Mice were anesthetized with 2% isoflurane. MI-R was induced by temporarily exteriorizing the heart via a left thoracic incision. A 6–0 silk suture slipknot around the left anterior descending coronary artery was tied. Twenty min after MI, animals were randomized to receive either vehicle or AdipoRon (50 mg/kg, IV). This dose was selected from a recently published study demonstrating significant skeletal and liver PRKAA phosphorylation occurs after a single bolus injection.16 After 30 min of MI, the slipknot was released. The myocardium was reperfused for either 3 h (for all assays excluding cardiac function and infarct size measurements) or 24 h. All assays were performed using tissue from the ischemic-reperfused area identified by Evans blue negative staining. Sham-operated control mice (sham MI-R) underwent the same surgical procedure, except the suture placed under the left coronary artery was not tied. To determine autophagic flux, chloroquine (CQ; Sigma-Aldrich, C6628) was administered (dose 10 mg/kg, intraperitoneal) 5 min before coronary artery occlusion.10
Determination of autophagic flux in adult mice
RFP-GFP-Map1lc3b transgenic mice were subjected to 30 min of ischemia and 3 h of reperfusion.39 At the end of the experiment, the heart was removed and the nonischemic-reperfused section was discarded. The ischemic-reperfused cardiac section was frozen in liquid nitrogen, and 3 slides/heart were prepared. Slides were visualized using a FV1000 confocal microscope with x60 oil-immersion objective lenses (Olympus, Tokyo, Japan). Red and green fluorescence images were taken from 5 fields/slide. Images were merged. The yellow puncta (green/red overlay, indicators of autophagosomes) and red-only puncta (RFP only, indicators of autolysosomes as GFP fluorescence was quenched by the acidic pH in lysosomes) in each field were counted by IP Lab Imaging Analysis Software (Version 4.2, Scanalytics) via a custom-made script (by Mr. Ken Anderson, Bio Vision Technologies). The numbers of yellow and red-only puncta in the merged image were automatically calculated and exported to Microsoft Excel for further analysis. Results from the same animal (3 slides/heart x 5 fields/slide) were averaged and counted as 1 sample. Assays were performed in a blinded manner.
Echocardiography and myocardial infarct size
Twenty-four h after reperfusion, mice were re-anesthetized and transthoracic 2-dimensional echocardiography was performed (Vevo 2100 system). M-mode recordings were obtained in the short axis view of the left ventricle (LV) at the level of the papillary muscles. Left ventricle ejection fraction (LVEF) was calculated as done previously.18 After echocardiographic measurement, the chest was re-opened, and the ligature around the coronary artery was retied. Myocardial infarct size was determined by Evan's blue-TTC (Sigma-Aldrich, E2129; 697079) double-staining protocol as previously reported.18
Quantification of superoxide production
Superoxide production in the ischemic-reperfused heart tissue was measured by lucigenin-enhanced chemiluminescence as described previously.18 Superoxide production was expressed as relative light units (RLU) per second per milligram heart. Reactive oxygen species (ROS) production in heart tissue was assessed by dihydroethidine staining (Thermo Fisher Scientific, D1168) as described previously.18
Cardiomyocyte culture and simulated ischemia reperfusion
Adult mouse cardiomyocytes were isolated from WT or Prkaa2 DN mice as previously reported and plated at 1 × 105 cells per well in a 6-well dish pre-coated with mouse LAM/laminin.40 After 1 h culture ina 5% CO2 incubator at 37oC, cardiomyocytes were subjected to simulated ischemia-reperfusion (SI-R) as originally described by Isner and colleagues and modified in our recently published study.40,41 The left ventricle from 1–2-d-old Sprague-Dawley rats were collected and digested with collagenase. The resulting cell suspension was pre-plated to clear fibroblasts. The cells were then plated at a density of 1250 cells per 1 mm2 in medium (containing 10% fetal bovine serum and 100 μmol/L bromodeoxyuridine [Sigma-Aldrich, B5002]). Twenty-four h after plating, cells were infected with lentivirus coding mCherry-GFP-Map1lc3b (multiplicity of infection 10) and subjected to SI-R. At the end of experiments, cells were fixed with 4% paraformaldehyde and examined by confocal fluorescence microscopy.
Western blot
Cardiac tissue from ischemic-reperfused area and cultured cardiomyocytes were homogenized with lysis buffer. The homogenates were centrifuged at 1000 g for 10 min at 4°C. The protein concentration of the supernatant was measured via the Bradford method.[5000001, Bio-Rad, Hercules,CA] Equal amounts of proteins derived from each sample were separated on 10% SDS-PAGE gels (Thermo Fisher Scientific, NPO335BOX), and electroblotted onto polyvinylidene fluoride membranes (EMD Millipore, IPVH00010). Blocked membranes were incubated overnight at 4°C with antibodies against target proteins as specified in the Results section. The membranes were probed with HRP-conjugated secondary antibodies at room temperature for 1 h. Immunoreactive protein bands were visualized by use of an enhanced chemiluminescence detection system (Kodak Molecular 4000R Pro) per the manufacturer's protocol. Densitometry data were analyzed by Carestream MI SE (Carestream Health).
PtdIns3K kinase assay
PtdIns3K was immnoprecipitated using anti-PIK3C3 antibody. Its activity was determined by a PtdIns3K kinase activity assay kit as recently described.42 Briefly, 20 µl of kinase reaction buffer (10 mM Tris, pH 8, 100 mM NaCl, 1 mM EDTA, 10 mM MnCl2), 4 µl of 500 µM phosphatidylinositol substrate, and 1 µl of 1.25 mM ATP (provided in kit) were added to the beads immunoprecipitated with anti-PIK3C3, and incubated at 37°C for 2 h. The reaction mixture was quenched with 5 µl of 100 mM EDTA, diluted with 130 µl H2O and 40 µl PtdIns3P detection buffer (provided in kit). Finally, the quenched reaction mixture and PtdIns3P detector protein (provided in the kit) were added together to the PtdIns3P-coated microplate for competitive binding to the PtdIns3P detector protein. The amount of PtdIns3P detector protein bound to the plate was determined through colorimetric detection of absorbance at 450 nm via a Spectra Max M5 (Molecular Devices).
Cathepsin activity
Activity of CTSB and CTSL was measured via the CTSB (Millipore, CBA001) and CTSL (Abcam, ab65306) activity kits as recently described.43 In brief, 100 µg protein lysates were added to a total of 200 µl reaction buffer (50 mmol/L sodium acetate, 8 mmol/L EDTA, 8 mmol/L dithiothreitol, pH 5.0), and incubated with the substrate for 1 h at 37°C. CTSB and CTSL activity was measured using the Spectra Max M5 microplate reader (Molecular Devices; excitation = 400 nm, emission = 505 nm for CTSL; excitation = 380 nm, emission = 450 nm for CTSB).
In vitro kinase assay and protein-protein interaction assay
An in vitro kinase assay was performed as recently described.44 In brief, 2 μg of BECN1 recombinant protein (EMD Millipore, 03–231) was incubated with a kinase assay buffer (50 μl reaction volume) containing 5 mM MOPS (pH 7.2), 5 mM MgCl2, 0.5 mM DTT, 2.5 mM β-glycerophosphate, 1 mM EGTA, 0.4 mM EDTA, 0.2 mM ATP. BECN1 phosphorylation was initiated by adding inactive AMPK recombinant protein (EMD Millipore, 171536, 200 ng) in the presence or absence of AMP (0.1 mM; SignalChem A46–09–500,). Reactions commenced for 30 min at 30°C, and were terminated by adding 5x sample loading buffer. Samples were heated for 5 min at 95°C. The reaction mixture was loaded onto a 4–20% Tris-glycine gel. After electrophoresis, all proteins were transferred to PVDF membrane for western blot.
To determine the effect of BECN1 (Thr119) phosphorylation upon its interaction with BCL2L1, 2 µg WT (EMD Millipore, 03–231) or mutated (T119A, S93 S96 T119A) recombinant BECN1 proteins were incubated with 50-µl kinase assay buffer as described above. BECN1 phosphorylation was initiated by adding 200 ng activated AMPK (SignalChem, P48–10H). Thirty min after AMPK addition, 1 µg recombinant BCL2L1 (R&D systems, 894-BX) was added and reactions were performed for an additional 30 min at 30°C. Samples were immunoprecipitated with antibody against BECN1, followed by immunoblotting with antibody against BCL2L1.
Statistical analysis
All data are reported as mean ± SEM. Data were analyzed by one-way or 2-way analysis of variance (ANOVA) followed by the Tukey post hoc test for pairwise comparisons (details in Figure legends). For all statistical tests, p values less than 0.05 were considered statistically significant. All statistical analyses were performed via GraphPad Prism 6.0.
Supplementary Material
Funding Statement
This research was supported by the following grants: NIH HL-096686, HL-123404, American Diabetes Association 1–15-BS-122 (XLM), and American Diabetes Association 1–14-BS-228 (YJW).
Abbreviation List
- ACACA
acetyl-coenzyme A carboxylase α
- ADIPOR1
adiponectin receptor 1
- ADIPOR2
adiponectin receptor 2
- ACTC1
actin, α, cardiac muscle 1
- ADIPOQ
adiponectin, C1Q and collagen domain containing
- Adipoq KO
adiponectin knockout
- AKT2
thymoma viral proto-oncogene 2
- AMPK
AMP-activated protein kinase
- ANOVA
analysis of variance
- BCL2L1
BCL2-like 1
- BECN1
Beclin 1, autophagy related
- CTSB
cathepsin B
- CTSL
cathepsin L
- CQ
chloroquine
- DB
diabetes
- DN
dominant negative
- GFP
green fluorescent protein
- HRP
horseradish peroxidase
- LAMP2
lysosomal-associated membrane protein 2
- MAP1LC3B
microtubule-associated protein 1, light chain 3 β
- LV
left ventricle
- LVEF
left ventricle ejection fraction
- MI-R
myocardial ischemia-reperfusion
- MTOR
mechanistic target of rapamycin (serine/threonine kinase)
- MTORC1/2
mechanistic target of rapamycin (serine/threonine kinase) complex 1/2
- NC
normal control
- PRKAA2/AMPKα2
protein kinase, AMP-activated, α 2 catalytic subunit
- PtdIns3K
class III phosphatidylinositol 3-kinase
- PtdIns3P
phosphatidylinositol 3-phosphate
- RFP
red fluorescent protein
- RLU
relative light units
- ROS
reactive oxygen species
- SDS-PAGE
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- SI-R
simulated ischemia-reperfusion
- SQSTM1
sequestosome 1
- TSC1
tuberous sclerosis 1
- TSC2
tuberous sclerosis 2
- TTC
2,3,5-triphenyltetrazolium chloride
- ULK1
unc-51 like kinase 1
- WT
wild type
Disclosures of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgment
We are greatly appreciative of Mr. Nadan Wang in the Center for Translational Medicine, Thomas Jefferson University, for his expertise in evaluation of cardiac function by echocardiography.
References
- [1].Lavandero S, Chiong M, Rothermel BA, Hill JA. Autophagy in cardiovascular biology. J Clin Invest. 2015;125:55-64. doi: 10.1172/JCI73943. PMID:25654551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Gatica D, Chiong M, Lavandero S, Klionsky DJ. Molecular mechanisms of autophagy in the cardiovascular system. Circ Res. 2015;116:456-67. doi: 10.1161/CIRCRESAHA.114.303788. PMID:25634969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Gottlieb RA, Mentzer RM Jr. Autophagy: An affair of the heart. Heart Fail Rev. 2013;18:575-84. doi: 10.1007/s10741-012-9367-2. PMID:23188163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Sciarretta S, Boppana VS, Umapathi M, Frati G, Sadoshima J. Boosting autophagy in the diabetic heart: A translational perspective. Cardiovasc Diagn Ther. 2015;5:394-402. doi: 10.3978/j.issn.2223-3652.2015.07.02. PMID:26543826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Kubli DA, Gustafsson AB. Unbreak my heart: Targeting mitochondrial autophagy in diabetic cardiomyopathy. Antioxid Redox Signal. 2015;22:1527-44. doi: 10.1089/ars.2015.6322. PMID:25808102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Munasinghe PE, Riu F, Dixit P, Edamatsu M, Saxena P, Hamer NS, Galvin IF, Bunton RW, Lequeux S, Jones G, et al.. Type-2 diabetes increases autophagy in the human heart through promotion of Beclin-1 mediated pathway. Int J Cardiol. 2016;202:13-20. doi: 10.1016/j.ijcard.2015.08.111. PMID:26386349 [DOI] [PubMed] [Google Scholar]
- [7].Kubli DA, Gustafsson AB. Cardiomyocyte health: Adapting to metabolic changes through autophagy. Trends Endocrinol Metab. 2014;25:156-64. doi: 10.1016/j.tem.2013.11.004. PMID:24370004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Gottlieb RA, Andres AM, Sin J, Taylor DP. Untangling autophagy measurements: All fluxed up. Circ Res. 2015;116:504-14. doi: 10.1161/CIRCRESAHA.116.303787. PMID:25634973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, Levine B, Sadoshima J. Distinct roles of autophagy in the heart during ischemia and reperfusion: Roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res. 2007;100:914-22. doi: 10.1161/01.RES.0000261924.76669.36. PMID:17332429 [DOI] [PubMed] [Google Scholar]
- [10].Ma X, Liu H, Foyil SR, Godar RJ, Weinheimer CJ, Hill JA, Diwan A. Impaired autophagosome clearance contributes to cardiomyocyte death in ischemia/reperfusion injury. Circulation. 2012;125:3170-81. doi: 10.1161/CIRCULATIONAHA.111.041814. PMID:22592897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Buse JB, Ginsberg HN, Bakris GL, Clark NG, Costa F, Eckel R, Fonseca V, Gerstein HC, Grundy S, Nesto RW, et al.. Primary prevention of cardiovascular diseases in people with diabetes mellitus: A scientific statement from the American heart association and the American diabetes Association. Circulation. 2007;115:114-26. doi: 10.1161/CIRCULATIONAHA.106.179294. PMID:17192512 [DOI] [PubMed] [Google Scholar]
- [12].Berg AH, Combs TP, Scherer PE. ACRP30/adiponectin: An adipokine regulating glucose and lipid metabolism. Trends Endocrinol Metab. 2002;13:84-9. doi: 10.1016/S1043-2760(01)00524-0. PMID:11854024 [DOI] [PubMed] [Google Scholar]
- [13].Chandran M, Phillips SA, Ciaraldi T, Henry RR. Adiponectin: More than just another fat cell hormone? Diabetes Care. 2003;26:2442-50. PMID:12882876 [DOI] [PubMed] [Google Scholar]
- [14].Ouchi N, Shibata R, Walsh K. Targeting adiponectin for cardioprotection. Expert Opin Ther Targets. 2006;10:573-81. doi: 10.1517/14728222.10.4.573. PMID:16848693 [DOI] [PubMed] [Google Scholar]
- [15].Goldstein BJ, Scalia RG, Ma XL. Protective vascular and myocardial effects of adiponectin. Nat. Clin Pract Cardiovasc Med. 2009;6:27-35. doi: 10.1038/ncpcardio1398. PMID:19029992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Okada-Iwabu M, Yamauchi T, Iwabu M, Honma T, Hamagami K, Matsuda K, Yamaguchi M, Tanabe H, Kimura-Someya T, Shirouzu M, et al.. A small-molecule AdipoR agonist for type 2 diabetes and short life in obesity. Nature. 2013;503:493-9. doi: 10.1038/nature12656. PMID:24172895 [DOI] [PubMed] [Google Scholar]
- [17].Zou MH, Xie Z. Regulation of interplay between autophagy and apoptosis in the diabetic heart: New role of AMPK. Autophagy. 2013;9:624-5. doi: 10.4161/auto.23577. PMID:23380689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Wang Y, Gao E, Tao L, Lau WB, Yuan Y, Goldstein BJ, Lopez BL, Christopher TA, Tian R, Koch W, et al.. AMP-activated protein kinase deficiency enhances myocardial ischemia/reperfusion injury but has minimal effect on the antioxidant/antinitrative protection of adiponectin. Circulation. 2009;119:835-44. doi: 10.1161/CIRCULATIONAHA.108.815043. PMID:19188503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Murase H, Kuno A, Miki T, Tanno M, Yano T, Kouzu H, Ishikawa S, Tobisawa T, Ogasawara M, Nishizawa K, et al.. Inhibition of DPP-4 reduces acute mortality after myocardial infarction with restoration of autophagic response in type 2 diabetic rats. Cardiovasc Diabetol. 2015;14:103. doi: 10.1186/s12933-015-0264-6. PMID:26259714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kobayashi S, Liang Q. Autophagy and mitophagy in diabetic cardiomyopathy. Biochim Biophys Acta. 2015;1852:252-61. doi: 10.1016/j.bbadis.2014.05.020. PMID:24882754 [DOI] [PubMed] [Google Scholar]
- [21].Eguchi M, Kim YH, Kang KW, Shim CY, Jang Y, Dorval T, Kim KJ, Sweeney G. Ischemia-reperfusion injury leads to distinct temporal cardiac remodeling in normal versus diabetic mice. PLoS One. 2012;7:e30450. doi: 10.1371/journal.pone.0030450. PMID:22347376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Chou IP, Chiu YP, Ding ST, Liu BH, Lin YY, Chen CY. Adiponectin receptor 1 overexpression reduces lipid accumulation and hypertrophy in the heart of diet-induced obese mice–possible involvement of oxidative stress and autophagy. Endocr Res. 2014;39:173-9. doi: 10.3109/07435800.2013.879165. PMID:24679155 [DOI] [PubMed] [Google Scholar]
- [23].Essick EE, Wilson RM, Pimentel DR, Shimano M, Baid S, Ouchi N, Sam F. Adiponectin modulates oxidative stress-induced autophagy in cardiomyocytes. PLoS One. 2013;8:e68697. doi: 10.1371/journal.pone.0068697. PMID:23894332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Jahng JW, Turdi S, Kovacevic V, Dadson K, Li RK, Sweeney G. Pressure overload-induced cardiac dysfunction in aged male adiponectin knockout mice is associated with autophagy deficiency. Endocrinology. 2015;156:2667-77. doi: 10.1210/en.2015-1162. PMID:25961840 [DOI] [PubMed] [Google Scholar]
- [25].Guo R, Zhang Y, Turdi S, Ren J. Adiponectin knockout accentuates high fat diet-induced obesity and cardiac dysfunction: Role of autophagy. Biochim Biophys Acta. 2013;1832:1136-48. doi: 10.1016/j.bbadis.2013.03.013. PMID:23524376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Maeda N, Shimomura I, Kishida K, Nishizawa H, Matsuda M, Nagaretani H, Furuyama N, Kondo H, Takahashi M, Arita Y, et al.. Diet-induced insulin resistance in mice lacking adiponectin/ACRP30. Nat Med. 2002;8:731-7. doi: 10.1038/nm724. PMID:12068289 [DOI] [PubMed] [Google Scholar]
- [27].Wang Y, Wang X, Jasmin JF, Lau WB, Li R, Yuan Y, Yi W, Chuprun K, Lisanti MP, Koch WJ, et al.. Essential role of caveolin-3 in adiponectin signalsome formation and adiponectin cardioprotection. Arterioscler Thromb Vasc Biol. 2012;32:934-42. doi: 10.1161/ATVBAHA.111.242164. PMID:22328772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Cai L, Wang J, Li Y, Sun X, Wang L, Zhou Z, Kang YJ. Inhibition of superoxide generation and associated nitrosative damage is involved in metallothionein prevention of diabetic cardiomyopathy. Diabetes. 2005;54:1829-37. doi: 10.2337/diabetes.54.6.1829. PMID:15919806 [DOI] [PubMed] [Google Scholar]
- [29].Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577-90. doi: 10.1016/S0092-8674(03)00929-2. PMID:14651849 [DOI] [PubMed] [Google Scholar]
- [30].Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. 2011;13:1016-23. doi: 10.1038/ncb2329. PMID:21892142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Xie Z, He C, Zou MH. AMP-activated protein kinase modulates cardiac autophagy in diabetic cardiomyopathy. Autophagy. 2011;7:1254-5. doi: 10.4161/auto.7.10.16740. PMID:21685727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Kim J, Kim YC, Fang C, Russell RC, Kim JH, Fan W, Liu R, Zhong Q, Guan KL. Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell. 2013;152:290-303. doi: 10.1016/j.cell.2012.12.016. PMID:23332761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Wen Y, Graybill WS, Previs RA, Hu W, Ivan C, Mangala LS, Zand B, Nick AM, Jennings NB, Dalton HJ, et al.. Immunotherapy targeting folate receptor induces cell death associated with autophagy in ovarian cancer. Clin Cancer Res. 2015;21:448-59. doi: 10.1158/1078-0432.CCR-14-1578. PMID:25416196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Xu X, Hua Y, Nair S, Zhang Y, Ren J. Akt2 knockout preserves cardiac function in high-fat diet-induced obesity by rescuing cardiac autophagosome maturation. J Mol Cell Biol. 2013;5:61-3. doi: 10.1093/jmcb/mjs055. PMID:23258696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Xu X, Ren J. Macrophage migration inhibitory factor (MIF) knockout preserves cardiac homeostasis through alleviating Akt-mediated myocardial autophagy suppression in high-fat diet-induced obesity. Int J Obes (Lond). 2015;39:387-96. doi: 10.1038/ijo.2014.174. PMID:25248618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lamming DW, Ye L, Katajisto P, Goncalves MD, Saitoh M, Stevens DM, Davis JG, Salmon AB, Richardson A, Ahima RS, et al.. Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science. 2012;335:1638-43. doi: 10.1126/science.1215135. PMID:22461615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Orogo AM, Gustafsson AB. Therapeutic targeting of autophagy: Potential and concerns in treating cardiovascular disease. Circ Res. 2015;116:489-503. doi: 10.1161/CIRCRESAHA.116.303791. PMID:25634972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Xing Y, Musi N, Fujii N, Zou L, Luptak I, Hirshman MF, Goodyear LJ, Tian R. Glucose metabolism and energy homeostasis in mouse hearts overexpressing dominant negative alpha2 subunit of AMP-activated protein kinase. J Biol Chem. 2003;278:28372-7. doi: 10.1074/jbc.M303521200. PMID:12766162 [DOI] [PubMed] [Google Scholar]
- [39].Xie M, Kong Y, Tan W, May H, Battiprolu PK, Pedrozo Z, Wang ZV, Morales C, Luo X, Cho G, et al.. Histone deacetylase inhibition blunts ischemia/reperfusion injury by inducing cardiomyocyte autophagy. Circulation. 2014;129:1139-51. doi: 10.1161/CIRCULATIONAHA.113.002416. PMID:24396039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Wang Y, Tao L, Yuan Y, Lau WB, Li R, Lopez BL, Christopher TA, Tian R, Ma XL. Cardioprotective effect of adiponectin is partially mediated by its AMPK-independent antinitrative action. Am J Physiol Endocrinol Metab. 2009;297:E384-91. doi: 10.1152/ajpendo.90975.2008. PMID:19470831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Namiki A, Brogi E, Kearney M, Kim EA, Wu T, Couffinhal T, Varticovski L, Isner JM. Hypoxia induces vascular endothelial growth factor in cultured human endothelial cells. J Biol Chem. 1995;270:31189-95. doi: 10.1074/jbc.270.52.31189. PMID:8537383 [DOI] [PubMed] [Google Scholar]
- [42].Ma B, Cao W, Li W, Gao C, Qi Z, Zhao Y, Du J, Xue H, Peng J, Wen J, et al.. Dapper1 promotes autophagy by enhancing the Beclin1-Vps34-Atg14L complex formation. Cell Res. 2014;24:912-24. doi: 10.1038/cr.2014.84. PMID:24980960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Li DL, Wang ZV, Ding G, Tan W, Luo X, Criollo A, Xie M, Jiang N, May H, Kyrychenko V, et al.. Doxorubicin blocks cardiomyocyte autophagic flux by inhibiting lysosome acidification. Circulation. 2016;133:1668-87. doi: 10.1161/CIRCULATIONAHA.115.017443. PMID:26984939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Wang Y, Gao E, Lau WB, Wang Y, Liu G, Li JJ, Wang X, Yuan Y, Koch WJ, Ma XL. G-protein-coupled receptor kinase 2-mediated desensitization of adiponectin receptor 1 in failing heart. Circulation. 2015;131:1392-404. doi: 10.1161/CIRCULATIONAHA.114.015248. PMID:25696921 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.