

ABSTRACT
The E3 ubiquitin ligase NEDD4 has been intensively studied in processes involved in viral infections, such as virus budding. However, little is known about its functions in bacterial infections. Our investigations into the role of NEDD4 in intracellular bacterial infections demonstrate that Mycobacterium tuberculosis and Listeria monocytogenes, but not Mycobacterium bovis BCG, replicate more efficiently in NEDD4 knockdown macrophages. In parallel, NEDD4 knockdown or knockout impaired basal macroautophagy/autophagy, as well as infection-induced autophagy. Conversely, NEDD4 expression promoted autophagy in an E3 catalytic activity-dependent manner, thereby restricting intracellular Listeria replication. Mechanistic studies uncovered that endogenous NEDD4 interacted with BECN1/Beclin 1 and this interaction increased during Listeria infection. Deficiency of NEDD4 resulted in elevated K48-linkage ubiquitination of endogenous BECN1. Further, NEDD4 mediated K6- and K27- linkage ubiquitination of BECN1, leading to elevated stability of BECN1 and increased autophagy. Thus, NEDD4 participates in killing of intracellular bacterial pathogens via autophagy by sustaining the stability of BECN1.
KEYWORDS: ATG8, autophagy, BECN1, E3 ubiquitin ligase, Listeria monocytogenes, Mycobacterium tuberculosis, NEDD4
Introduction
Autophagy is an evolutionarily conserved process by which cytosolic components are sequestered into double-membrane vesicles and consequently fused with lysosomes.1 Under physiological conditions autophagy plays a critical role in cellular pathways, as diverse as growth, development and differentiation, aging and cell death.1-3 Autophagy has also been described as an innate defense mechanism against certain intracellular bacteria, including Mycobacterium tuberculosis (Mtb) and Listeria monocytogenes (Lm).4-6 Dysregulated autophagy has also been implicated in pathological development, emphasizing its critical involvement in maintaining homeostasis at the cellular and organismic level.3,7
The ULK1 and BECN1 complexes are 2 key players in the formation of autophagosomes. The ULK1 complex, which plays pivotal roles in autophagy initiation, consists of the serine/threonine kinase ULK1, ATG13, RB1CC1 and ATG101.8,9 ULK1 is activated by AMPK-mediated phosphorylation or loss of MTOR-mediated phosphorylation.10-12 After activation, ULK1 phosphorylates downstream targets to initiate autophagy. One such target is the BECN1 complex, comprising BECN1, PIK3R4/VPS15, PIK3C3/VPS34 and ATG14.13,14
Both abundances and activities of ULK1 and BECN1 are extensively regulated by ubiquitination. TRAF6 mediates K63 ubiquitination of ULK1 through AMBRA1, which increases stability and function of ULK1.15 TRAF6 also mediates K63 ubiquitination of BECN1, facilitating TLR4-triggered autophagy.15 In contrast, the deubiquitinase TNFAIP3/A20 reduces TLR4-triggered autophagy by decreasing K63 ubiquitination of BECN1.16 RNF216 also regulates K48 ubiquitination of BECN1, resulting in proteasomal degradation of BECN1 and decreased autophagy.17 Recently CUL3-KLHL20 was shown to ubiquitinate ULK1, leading to the proteasomal degradation of ULK1.18
NEDD4 (neuronal precursor cell expressed, developmentally downregulated 4) is a HECT (homologous E6-AP carboxyl terminus) type E3 ubiquitin ligase, and is comprised of one N-terminal C2 domain, 3 or 4 WW domains and 1 C-terminal HECT domain.19,20 The C2 domain mediates its intracellular localization by interacting with phospholipids and proteins. The WW domain, named for the presence of 2 conserved tryptophan residues, is responsible for substrate recognition by binding to a PPxY motif. The HECT domain confers E3 catalytic activity.19,20 Numerous studies have demonstrated that NEDD4 facilitates budding of various viruses by ubiquitinating viral matrix proteins via PPxY motifs. These include vesicular stomatitis virus,21 Marburg virus,22 Ebola virus,23,24 Rous sarcoma virus,25 and human immunodeficiency virus (HIV). 26 NEDD4 also ubiquitinates capsid protein VI of adenovirus in a PPxY-dependent manner and facilitates its intracellular targeting and effective infection.27 Moreover, NEDD4 increases influenza virus infection by decreasing the abundance of the antiviral protein IFITM3 via ubiquitination.28 While the role of NEDD4 in viral infections is well documented, its function in bacterial infections remains elusive.
As intracellular pathogens, Lm and Mtb have evolved specific strategies to subvert defenses inside host cells.29,30 The common mechanism underlying intracellular survival shared by Lm and Mtb is the egression into the cytosol.30 For Lm, listeriolysin O (LLO) is essential for its rapid escape into the cytosol and for subsequent autophagy activation.31,32 Unlike Lm, Mtb induces phagosomal escape at later time points of infection via the ESX-1 secretion system,33,34 which is encoded in region of difference 1 (RD1). ESX-1 is also required for sequestering Mtb within autophagosomes.35 M. bovis BCG (BCG) lacks the RD1 region and hence remains in the phagosomes within macrophages.34,36 The capacity of LLO to facilitate egression has been harnessed for the generation of a recombinant BCG vaccine strain, BCG ΔureC::hly (rBCG).37 Although it perturbs the phagosomal membrane, this vaccine candidate remains in the phagosomes; however, bacterial components including proteins, glycolipids and double-stranded DNA are released into the cytosol, thereby inducing autophagy and inflammasome activation.38
Here we investigated the role of NEDD4 in bacterial infections with Mtb, BCG, rBCG and Lm. We show that NEDD4 contributes to killing of intracellular Mtb, rBCG and Lm in vitro. NEDD4 deficiency impaired basal autophagy, starvation-induced autophagy, and carbonyl cyanide m-chlorophenyl hydrazone (CCCP)-induced mitophagy, as well as autophagy upon Mtb and rBCG infection. Reciprocally, NEDD4 expression increased autophagy in an E3 catalytic activity-dependent manner. Moreover, we uncovered that NEDD4 interacts with Atg8-family proteins, forms a complex with ULK1 and BECN1, and mediates ubiquitination of BECN1 under basal conditions as well as Lm infection. Specifically, NEDD4 knockdown (KD) induced elevated K48-linkage ubiquitination of endogenous BECN1. BECN1 stability was increased by NEDD4 via K6 and K27 ubiquitination during autophagy induction, thereby promoting autophagy activation. Collectively, our data reveal a novel regulation of autophagy in which NEDD4 increases BECN1 stability via ubiquitination and hence promotes autophagy, thereby contributing to killing of membrane-perturbing intracellular bacterial pathogens.
Results
NEDD4 contributes to efficient control of bacteria perturbing the phagosomal membrane
To investigate the role of NEDD4 in bacterial infection, NEDD4 stable KD THP-1 and HeLa cell lines were established with lentivirus-delivered shRNAs (Fig. S1A-D). NEDD4 shRNA #2 (sh2) was selected for further experiments due to its robust KD efficiency. THP-1 cells expressing scrambled shRNA or sh2 were employed for in vitro infection with different intracellular bacteria. For Mtb infection, the intracellular bacterial load in NEDD4 KD cells was significantly higher than in control cells expressing scrambled shRNA at 48 h post-infection (p.i.) (Fig. 1A). However, the intracellular growth of attenuated strain Mtb ΔRD1, which lacks the ability of escaping into the cytosol, was not significantly different in NEDD4 KD and control cells (Fig. 1B). For both Mtb and Mtb ΔRD1, bacterial uptake at 4 h p.i. was not affected by NEDD4 KD (Fig. 1A and B).
Figure 1.
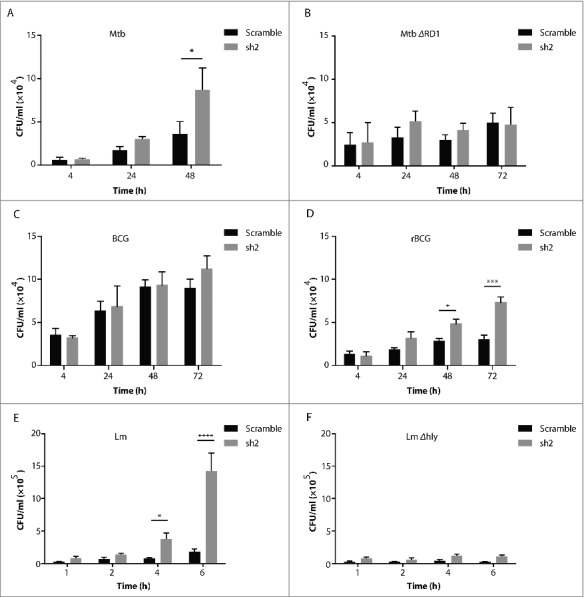
NEDD4 contributes to efficient control of intracellular bacteria perturbing the phagosomal membrane. (A-D) Colony-forming unit (CFU) assay of intracellular growth of Mycobacterium tuberculosis (Mtb) (A), Mtb ΔRD1 (B), M. bovis BCG (BCG) (C) and M. bovis BCG ΔureC::hly (rBCG) (D) in scramble (Scr) or NEDD4 knockdown (KD) THP-1 cells at 4, 24, 48 and h post-infection (p.i.). CFU assay of Listeria monocytogenes (Lm) (E) and Lm Δhly (F) in scramble and NEDD4 KD THP-1 cells at 1, 2, 4 and 6 h p.i. Data are shown as mean ± SD of 3 independent experiments. P values were calculated using two-way ANOVA with Bonferroni's multiple comparisons test. (*) p ≤ 0.05, (***) p ≤ 0.001, (****) p ≤ 0.0001.
To further investigate the role of NEDD4 in intracellular mycobacterial infection, BCG and rBCG were employed. rBCG impairs the integrity of the phagosomal membrane, leading to increased inflammasome activation and autophagy.38 We found that compared to BCG, rBCG replicated more efficiently in NEDD4 KD cells (Fig. 1C and D). Lm rapidly escapes from phagosomes into the cytosol by secreting the pore-forming LLO.32 Accordingly, Lm wild type (WT) and LLO-deficient mutant-Lm Δhly were employed to infect NEDD4 KD and control THP-1 cells. Lm WT replicated significantly faster in NEDD4 KD cells than in control cells. For Lm Δhly, there was no significant difference in NEDD4 KD and control cells (Fig. 1E and F). Lactate dehydrogenase (LDH) cytotoxicity assay verified that NEDD4 KD did not affect cell death upon Lm infection, suggesting that the increased replication in NEDD4 KD cells is not due to cell death (Fig. S1E). Taken together, these results demonstrate that in macrophages, NEDD4 is required for efficient control of intracellular bacteria perturbing the phagosomal membrane.
NEDD4 contributes to autophagy
Autophagy is an innate defense mechanism by which replication of certain intracellular bacteria is restrained.6 We interrogated whether NEDD4 participates in efficient control of bacterial growth via autophagy. To this end, the levels of LC3-II, a marker of autophagy induction, were examined by western blot in NEDD4 stable KD HeLa and THP-1 cells. Cells were treated with choloroquine (CQ) to block autophagy flux. We found that LC3-II abundance was significantly lower in NEDD4 KD HeLa and THP-1 cells with or without CQ treatment (Fig. 2A and B and Fig. S2A and B), suggesting that autophagy was impaired in these NEDD4 KD cells. For further validation, LC3-II abundance was monitored in Nedd4+/+ and nedd4−/− mouse embryonic fibroblasts (MEFs). LC3-II abundance in nedd4−/− MEFs was also significantly lower than in Nedd4+/+ cells with or without CQ treatment (Fig. 2C and Fig. S2C). Because SQSTM1/p62 is a selective substrate for autophagy, protein levels of SQSTM1 were examined by western blot to monitor autophagy activation. Levels of SQSTM1 in nedd4−/− cells were significantly higher than in WT cells, further verifying compromised autophagy in nedd4−/− cells (Fig. 2D and Fig. S2D). To monitor autophagosome formation, Nedd4+/+ and nedd4−/− MEFs stably expressing YFP-LC3B were generated. Immunofluorescence (IF) analysis revealed that the numbers of LC3B puncta in Nedd4+/+ cells were significantly higher than in nedd4−/− cells (Fig. 2E and F). Taken together, these data indicate that NEDD4 deficiency impairs basal autophagy.
Figure 2.
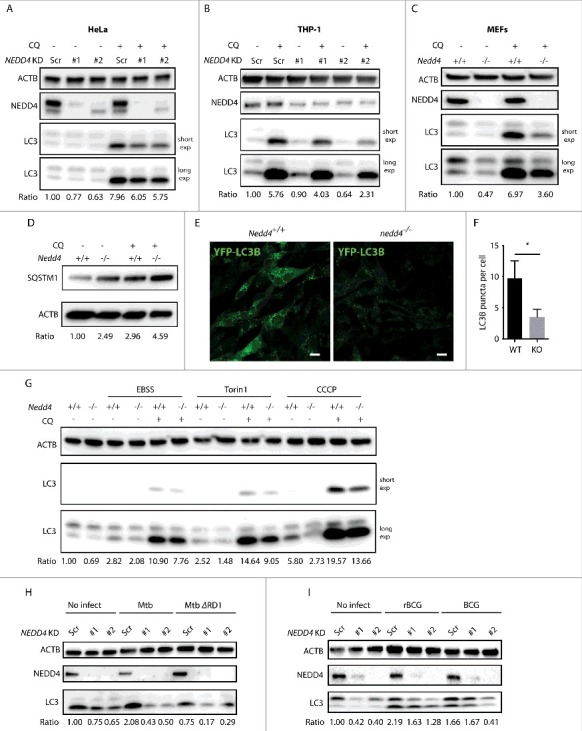
NEDD4 contributes to autophagy. Western blot analysis of LC3 levels in NEDD4 Scr and KD HeLa cells (A), NEDD4 Scr and KD THP-1 cells (B) and Nedd4+/+ or nedd4−/− MEFs (C) with or without choloroquine (CQ) treatment. (D) Western blot analysis of SQSTM1 levels in Nedd4+/+ or nedd4−/− MEFs with or without CQ treatment. (E) Immunofluorescence analysis of YFP-LC3B puncta in Nedd4+/+ or nedd4−/− MEFs stably expressing YFP-LC3B. (F) Quantification of YFP-LC3B puncta per cell. Data are shown as mean ± SD of 3 independent experiments. A total of 50 cells per group for each experiment were quantified. P values were calculated using the Student 2-tailed unpaired t test. (*) p ≤0.05. (G) Western blot analysis of LC3 levels in Nedd4+/+ or nedd4−/− MEFs after starvation, Torin1 or CCCP treatment with or without CQ. (H) Western blot analysis of LC3 levels in Scr and NEDD4 KD THP-1 cells upon Mtb or Mtb ΔRD1 infection. (I) Western blot analysis of LC3 levels in Scr and NEDD4 KD THP-1 cells upon BCG or rBCG infection. LC3-II:ACTB ratio was determined using ImageJ software and normalized to control. Results are representative of 3 independent experiments.
To determine whether NEDD4 also functions in other types of autophagy, Nedd4+/+ and nedd4−/− MEFs were treated with Earle's balanced salt solution (EBSS), MTOR inhibitor Torin1 or CCCP to stimulate starvation-induced autophagy or mitophagy. Western blots revealed significantly lower LC3-II abundance in nedd4−/− than in Nedd4+/+ MEFs for all stimuli (Fig. 2G and Fig. S2E). Furthermore, we investigated whether autophagy upon bacterial infection was affected by NEDD4 deficiency. Indeed, LC3-II abundance in NEDD4 KD THP-1 cells upon Mtb infection was decreased compared to controls. Moreover, Mtb ΔRD1 infection induced significantly lower LC3-II abundance compared to Mtb (Fig. 2H and Fig. S2F and H). Similarly, rBCG infection induced higher LC3-II levels than BCG infection and LC3-II levels in NEDD4 KD cells upon rBCG infection were lower than controls (Fig. 2I and Fig. S2G). To evaluate whether NEDD4 is involved in xenophagy, the colocalization of Lm with endogenous LC3 was examined in THP-1 control and NEDD4 KD cells. Indeed, LC3 recruitment to Lm was impaired by NEDD4 KD (Fig. S3A). Further, the colocalization of Lm with LAMP2 in NEDD4 KD cells was also lower than in control cells, demonstrating defective trafficking of Lm into LAMP2-positive organelles by NEDD4 KD (Fig. S3B). Overall, our results demonstrate a general role of NEDD4 in autophagy.
NEDD4 involvement in autophagy depends on its ubiquitin ligase activity
We questioned whether the enzymatic activity of NEDD4 was involved in autophagy. To this end, HEK293T cells were transfected with control plasmid, catalytically inactive mutant (NEDD4C867A) or NEDD4WT and then LC3-II levels were evaluated by western blot. We found that in cells expressing NEDD4WT, LC3-II abundance was significantly higher than in controls, demonstrating that NEDD4WT expression increased autophagy. Moreover, in cells expressing NEDD4C867A, LC3-II abundance was significantly lower compared to cells expressing NEDD4WT, establishing that the E3 ubiquitin ligase activity of NEDD4 was required for its biological function in autophagy (Fig. 3A and Fig. S4A). Additionally, complementation with NEDD4WT or NEDD4C867A in nedd4−/− MEFs further demonstrated that NEDD4WT expression induced significantly higher LC3-II levels, confirming that the defect of autophagy in nedd4−/− MEFs can be rescued by expression of NEDD4WT but not NEDD4C867A (Fig. 3B and Fig. S4B). Furthermore, LC3-II abundances were evaluated in cells with different expression levels of NEDD4WT or NEDD4C867A. LC3-II abundance was induced by NEDD4WT expression in a dose-dependent manner, whereas overexpression of NEDD4C867A had a marginal or no effect on LC3-II levels (Fig. 3C and Fig. S4C). Furthermore, complementation of NEDD4 in THP-1 KD cells induced significantly higher autophagy under Lm infection and basal conditions (Fig. 3D and Fig. S4D). Consistently, intracellular Lm replication was restricted by NEDD4 expression (Fig. 3E). Altogether, our results show that NEDD4 promotes autophagy and that its function critically depends on its E3 catalytic activity.
Figure 3.
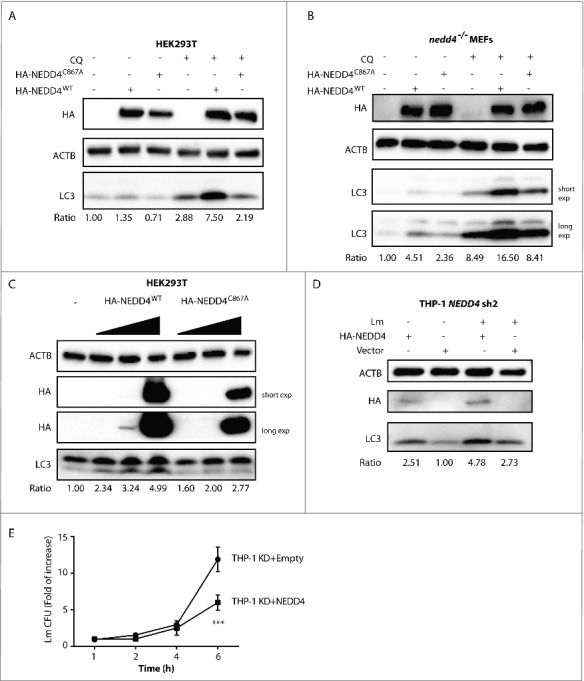
NEDD4 involvement in autophagy depends on its ubiquitin ligase activity. Western blot analysis of LC3 levels in HEK293T cells (A) or nedd4−/− MEFs (B) expressing empty vector, HA-NEDD4WT or HA-NEDD4C867A with or without CQ treatment. HEK293T cells or nedd4−/− MEFs were transfected with empty vector, or plasmids expressing HA-NEDD4WT or HA-NEDD4C867A and cells were treated with or without CQ for 4 h at 48 h after transfection. (C) Western blot analysis of LC3 levels in HEK293T cells expressing different levels of HA-NEDD4WT or HA-NEDD4C867A. HEK293T cells were transfected with 0.1, 0.3, or 1.0 µg of HA-NEDD4WT or HA-NEDD4C867A and lysates were collected after 48 h of transfection. (D) Western blot analysis of LC3 levels in THP-1 NEDD4 KD cells expressing empty vector or HA-NEDD4WT. LC3-II:ACTB ratio was determined using ImageJ software and normalized to control. Results are representative of 3 independent experiments. (E) CFU assay of Lm in THP-1 NEDD4 KD cells expressing empty vector or HA-NEDD4WT at 1, 2, 4 and 6 h p.i. Data are shown as mean ± SD of 3 independent experiments. P values were calculated using two-way ANOVA with Bonferroni's multiple comparisons test. (***) p ≤ 0.001.
NEDD4 interacts with Atg8-family proteins
NEDD4 has been shown to be part of the Atg8-family protein interaction network.39 To interrogate whether NEDD4 interacts with MAP1LC3B/LC3B, HA-NEDD4 and YFP-LC3B were co-expressed in HEK293T cells and then immunoprecipitation (IP) was performed with anti-HA agarose. Western blot revealed that exogenous HA-NEDD4 interacted with YFP-LC3B (Fig. 4A). Further IP with HEK293T cells expressing HA-NEDD4 demonstrated that HA-NEDD4 interacted with endogenous LC3 (Fig. 4B). In addition, treatment with Torin1 increased the interaction between NEDD4 and LC3 (Fig. 4B). Immunofluorescence confirmed colocalization of NEDD4 with YFP-LC3B (Fig. 4C). Electron microscopy with immunogold labeling revealed that endogenous NEDD4 was associated with double-membrane autophagosomes (Fig. 4D). IP with HEK293T cells expressing HA-NEDD4 and Atg8-family homologs showed that HA-NEDD4 interacted with all Atg8-family homologs (Fig. 4E). Pulldown assay using agarose conjugated with different Atg8-family proteins demonstrated direct interaction of NEDD4 with all the Atg8-family homologs (Fig. S5A). To identify the LC3-interacting region of NEDD4, different truncation mutants were generated and IP with anti-HA agarose was performed. The mutant NEDD4Δ381–900 still interacted with LC3B. In contrast, this interaction was completely abrogated in cells expressing mutants NEDD4Δ255–900, demonstrating that the region of 256–380 within NEDD4 was indispensable for interaction with LC3B (Fig. 4F). Immunofluorescence also confirmed that the 255–380 region of NEDD4 was required for its colocalization with LC3B (Fig. S5B). In silico analysis with the iLIR web server has identified a putative LC3-interacting region (264-269, ENWEII) within the sequence 256–380 of NEDD4.40 Indeed, the interaction between NEDD4 with LC3B was abolished by deleting the motif of 264–269, demonstrating that this motif served as an interacting domain of NEDD4 with Atg8-family proteins (Fig. 4F). To determine whether this NEDD4 motif was required for autophagy, HEK293T cells were transfected with a plasmid encoding NEDD4WT or NEDD4Δ264–269, and LC3-II abundances were monitored by western blot. In cells expressing NEDD4Δ264–269, LC3-II abundance was significantly lower than in NEDD4WT, confirming that the LC3-interacting region 264–269 of NEDD4 is critical for its function in autophagy (Fig. 4G and Fig. S5C). In sum, NEDD4 directly interacts with Atg8-family proteins, and this interaction contributes to its functional role in autophagy.
Figure 4.
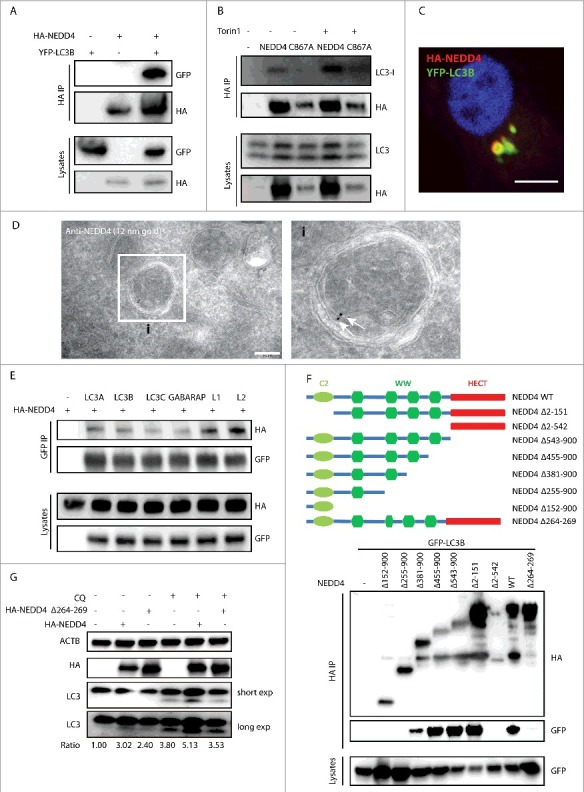
NEDD4 interacts with Atg8-family proteins. (A) Interaction between YFP-LC3B and HA-NEDD4. HEK293T cells were transfected with YFP-LC3B and HA-NEDD4 as indicated. Afterwards, lysates were harvested for immunoprecipitation (IP) with anti-HA agarose followed by anti-GFP and anti-HA immunoblotting. (B) Interaction between exogenously expressed HA-NEDD4WT and HA-NEDD4C867A with endogenous LC3 in the absence or presence of Torin1 treatment. HEK293T cells were transfected with HA-NEDD4WT or HA-NEDD4C867A, and after 48 h cells were treated with or without 1 µM Torin1 for 2 h. Lysates were harvested for IP with anti-HA agarose. (C) Colocalization of HA-NEDD4 and YFP-LC3B in HeLa cells. (D) Electron microscopy analysis of NEDD4 immunogold labeling on autophagosomes. NEDD4 was detected using a mouse anti-NEDD4 antibody followed by protein A-gold 12 nm labeling. Arrows indicate the association of NEDD4 with autophagosomes. (E) Interactions between NEDD4 and Atg8-family proteins. HEK293T cells were transfected with HA-NEDD4 and YFP-ATG8s as indicated. After 48 h, lysates were harvested for IP with anti-GFP agarose followed by anti-HA immunoblotting. (F) The interaction of NEDD4 mutants with LC3. HEK293T cells were transfected with plasmids encoding YFP-LC3B and HA-NEDD4 mutants as indicated. After 48 h, lysates were harvested for IP with anti-HA agarose followed by anti-GFP immunoblotting. (G) Western blot analysis of LC3 levels in HEK293T cells expressing HA-NEDD4WT or HA-NEDD4Δ264-269 with or without CQ treatment. LC3-II:ACTB ratio was determined using ImageJ software and normalized to control. Results are representative of 3 independent experiments.
NEDD4 forms a complex with ULK1 and BECN1
Next, we investigated whether NEDD4 interacts with ULK1 or BECN1. To this end, HA-NEDD4 and MYC-ULK1 were co-expressed in HEK293T cells and IP was performed with anti-HA agarose hereafter. Western blot revealed that HA-NEDD4 interacted with MYC-ULK1 and this interaction increased after Torin1 treatment, demonstrating that autophagy activation induced the interaction between HA-NEDD4 and MYC-ULK1 (Fig. 5A). IP with cells expressing the kinase-inactive mutant MYC-ULK1K46I or the phosphorylation-deficient mutant MYC-ULK1S4A demonstrated that HA-NEDD4 also interacted with MYC-ULK1K46I or MYC-ULK1S4A. Hence, interactions between NEDD4 and ULK1 were independent of kinase activity or phosphorylation of ULK1 (Fig. S6A).
Figure 5.
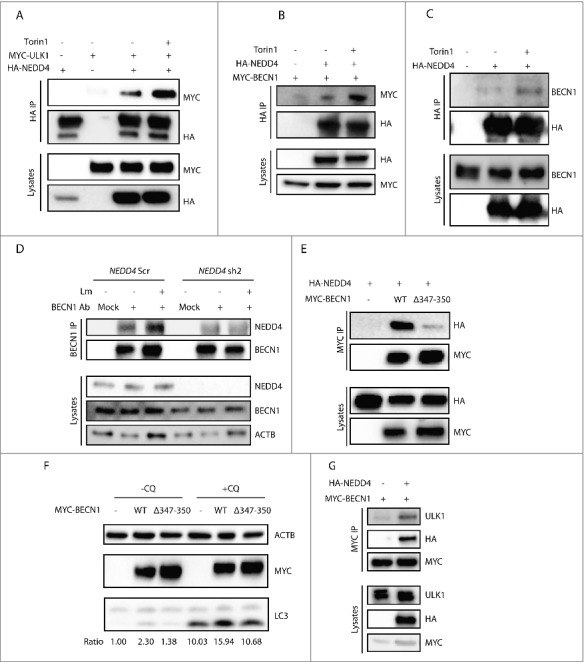
NEDD4 forms a complex with ULK1 and BECN1. (A) Interaction between MYC-ULK1 mutants with HA-NEDD4, with or without Torin1 treatment. HEK293T cells were transfected with plasmids encoding HA-NEDD4 and MYC-ULK1 as indicated. After 48 h, cells were treated with or without 1 µM Torin1 for 2 h. Lysates were harvested for IP with anti-HA agarose followed by anti-MYC immunoblotting. (B) Interaction between MYC-BECN1 and HA-NEDD4 in HEK293T cells with or without Torin1 treatment. HEK293T cells were transfected with MYC-BECN1 and HA-NEDD4WT or HA-NEDD4C867A as indicated. After 48 h, cells were treated with or without 1 µM Torin1 for 2 h. Lysates were harvested for IP with anti-HA agarose and followed by anti-MYC immunoblotting. (C) Interaction between exogenously expressed HA-NEDD4 with endogenous BECN1 with or without Torin1 treatment. HEK293T cells were transfected with HA-NEDD4 as indicated. After 48 h, cells were treated with or without 1 µM Torin1 for 2 h. Lysates were harvested for IP with anti-HA agarose. (D) Interaction between endogenous BECN1 and NEDD4 with or without Lm infection. THP-1 NEDD4 Scr and KD cells were infected with or without Lm at MOI = 10 for 2 h and cell lysates were collected for IP with isotype control or anti-BECN1 antibody. (E) Interaction between HA-NEDD4 and MYC-BECN1WT or MYC-BECN1Δ347-350. HEK293T cells were transfected with plasmids encoding HA-NEDD4 and MYC-BECN1WT or MYC-BECN1Δ347–350 as indicated. After 48 h, lysates were harvested for IP with anti-HA agarose and followed by anti-MYC immunoblotting. (F) Western blot analysis of LC3 levels in HEK293T cells expressing empty vector, MYC-BECN1WT or MYC-BECN1Δ347–350. HEK293T cells were transfected with the same amount of empty vector, or plasmids encoding MYC-BECN1WT or MYC-BECN1Δ347–350 and lysates were collected after 48 h of transfection. (G) NEDD4 forms a complex with ULK1 and BECN1. HEK293T cells were co-transfected with plasmids encoding MYC-BECN1 and HA-NEDD4WT or HA-NEDD4C867A as indicated. After 48 h, lysates were harvested and subjected to IP with anti-MYC agarose followed by anti-ULK1 immunoblotting.
To determine whether NEDD4 interacted with BECN1, HA-NEDD4 and MYC-BECN1 were co-expressed in HEK293T cells and IP was performed with anti-HA agarose. HA-NEDD4 interacted with MYC-BECN1 and this interaction was increased by Torin1 treatment, proving the elevated interaction between NEDD4 and BECN1 during autophagy (Fig. 5B). IP confirmed increased interactions between HA-NEDD4 and endogenous BECN1 during autophagy (Fig. 5C). However, the serine 15 phosphorylation of BECN1 was not affected by its interaction with NEDD4 (Fig. S6B). To investigate whether endogenous NEDD4 interacts with BECN1, IP was performed in THP-1 control and NEDD4 KD cells. We found that endogenous BECN1 indeed interacted with NEDD4 and the interaction was induced by Lm as well as Torin1 stimulation; however, this interaction was impaired by NEDD4 KD (Fig. 5D and Fig. S6C). The substrates of NEDD4 have common L/PPxY motifs, which are recognized by its WW domains.19,20 By sequence alignment, we identified the LPxY motif in BECN1 (corresponding to amino acids 347–350 of mouse BECN1), which is evolutionarily conserved from yeast to human (Fig. S6D). IP with cells expressing BECN1Δ347–350 demonstrated loss of interaction with NEDD4 by deletion of this motif, proving that the LPxY motif of BECN1 is critical for its interaction with NEDD4 (Fig. 5E). Moreover, to investigate the functional role of this motif in autophagy, HEK293T cells were transfected with BECN1WT or BECN1Δ347–350, and LC3-II abundance was examined by western blot. LC3-II abundance was impaired by deletion of the LPxY motif of BECN1, confirming that this NEDD4-interacting motif of BECN1 is required for autophagy (Fig. 5F and Fig. S6E). To further investigate whether HA-NEDD4 forms a complex with ULK1 and BECN1, MYC-BECN1 and HA-NEDD4 were co-expressed in HEK293T cells and IP was performed thereafter. Both endogenous ULK1 and HA-NEDD4 were co-precipitated with MYC-BECN1. Thus, NEDD4 forms a complex with ULK1 and BECN1 (Fig. 5G).
NEDD4 mediates ubiquitination of BECN1
Because NEDD4 is an E3 ubiquitin ligase, we investigated whether it mediates ubiquitination of BECN1. MYC-BECN1 was co-expressed together with NEDD4WT or NEDD4C867A in HEK293T cells and IP was performed with anti-MYC agarose. Western blot using anti-ubiquitin antibody FK2 demonstrated that ubiquitination of BECN1 was enhanced in cells expressing NEDD4WT but not in cells expressing NEDD4C867A, suggesting that NEDD4 mediated ubiquitination of BECN1 (Fig. 6A). Consistent with the increased interaction of NEDD4 with BECN1 upon Torin1 treatment, the ubiquitination of BECN1 by NEDD4 was also increased by Torin1 (Fig. 6B). Moreover, the ubiquitination of BECN1Δ347–350 was diminished compared with BECN1WT, confirming that the interaction motif of BECN1 is critical for its ubiquitination mediated by NEDD4 (Fig. 6C). To characterize the ubiquitination type of BECN1 in more detail, MYC-BECN1 was co-expressed with various FLAG-ubiquitin mutants in HEK293T cells and IP was performed. Western blot using anti-FLAG antibody revealed that BECN1 was mainly K6- and K27-type ubiquitinated (Fig. 6D). To further investigate the ubiquitination pattern of endogenous BECN1, IP was performed with anti-BECN1 antibody using THP-1 control and NEDD4 KD cells upon Lm infection. Western blot revealed higher K48 linkage ubiquitination of BECN1 in NEDD4 KD cells than in controls without or with Lm infection. However, there was no distinct difference of K63-linkage ubiquitination of BECN1 in control and NEDD4 KD cells (Fig. 6E). In sum, these data emphasize that NEDD4 mediates ubiquitination of BECN1 and that this ubiquitination increases during autophagy.
Figure 6.
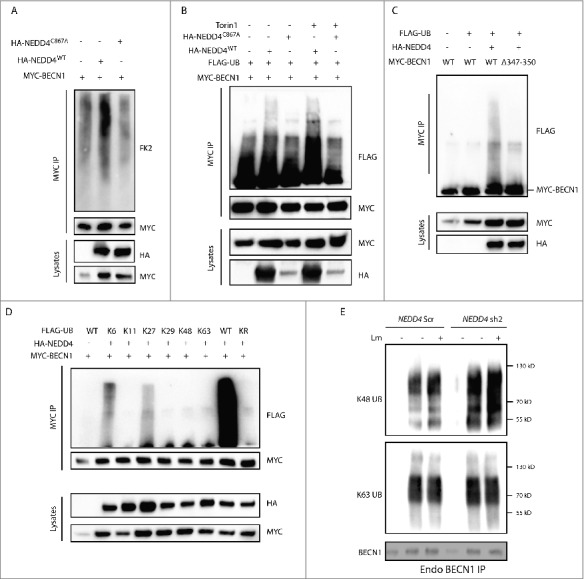
NEDD4 mediates ubiquitination of BECN1. (A) MYC-BECN1 ubiquitination in HEK293T cells. HEK293T cells were transfected with plasmids encoding MYC-BECN1 and HA-NEDD4WT or HA-NEDD4C867A as indicated. After 48 h, lysates were harvested for IP with anti-MYC agarose followed by immunoblotting with endogenous ubiquitin antibody FK2 (Enzo Life Sciences, BML-PW8810). (B) MYC-BECN1 ubiquitination mediated by NEDD4 in HEK293T cells with or without Torin1 treatment. HEK293T cells were co-transfected with plasmids encoding MYC-BECN1, FLAG-UB and HA-NEDD4WT or HA-NEDD4C867A as indicated. After 48 h, cells were treated with or without 1 µM Torin1 for 2 h. Lysates were harvested for IP with anti-MYC agarose followed by anti-FLAG immunoblotting. (C) MYC-BECN1WT and MYC-BECN1Δ347–350 ubiquitination is mediated by NEDD4 in HEK293T cells. HEK293T cells were co-transfected with plasmids encoding FLAG-UB, HA-NEDD4 and MYC-BECN1WT and MYC-BECN1Δ347–350 as indicated. After 48 h, lysates were harvested for IP with anti-MYC agarose followed by anti-FLAG immunoblotting. (D) Ubiquitination type of BECN1 mediated by NEDD4. HEK293T cells were co-transfected with MYC-BECN1, HA-NEDD4 and FLAG-ubiquitin variants as indicated. After 48 h, lysates were harvested and IP with anti-MYC agarose was followed by anti-FLAG immunoblotting. (E) Ubiquitination pattern of endogenous BECN1 in THP-1 NEDD4 Scr and KD cells with or without Lm infection. THP-1 control cells and NEDD4 KD cells were infected with or without Lm at MOI = 10 for 2 h and cell lysates were collected for IP with isotype control or anti-BECN1 antibody. The amount of BECN1 in IP samples of THP-1 control and NEDD4 KD cells was adjusted to the same level and afterwards samples were blotted with specific anti-K48 or anti-K63 linkage ubiquitin antibodies.
NEDD4 sustains BECN1 stability and autophagy
We next hypothesized that BECN1 stability was affected by NEDD4. Indeed, BECN1 protein abundance in nedd4−/− MEFs was lower than in Nedd4+/+ cells (Fig. 7A). Consistently, NEDD4 KD in HeLa cells reduced protein levels of BECN1 (Fig. 7B), which could be rescued by proteasome inhibitor MG132 treatment in nedd4−/− MEFs (Fig. 7C). To determine the stability of BECN1, Nedd4+/+ and nedd4−/− MEFs were treated with the translation inhibitor cyclohexamide (CHX) at different time points. Western blot against BECN1 demonstrated that the abundance of BECN1 in nedd4−/− MEFs at 4, 6, and 8 h post-CHX treatment significantly declined compared to Nedd4+/+ MEFs, establishing that BECN1 stability was compromised by NEDD4 deficiency (Fig. 7D). Because the interaction between NEDD4 and BECN1 was increased during autophagy activation, we hypothesized that BECN1 stability was sustained by NEDD4 during autophagy. In support of this notion, western blot against BECN1 revealed significantly diminished levels of BECN1 in nedd4−/− MEFs at 6 and 8 h post-Torin1 treatment as compared to Nedd4+/+ MEFs. Thus, NEDD4 enhanced BECN1 stability during basal autophagy (Fig. 7E).
Figure 7.
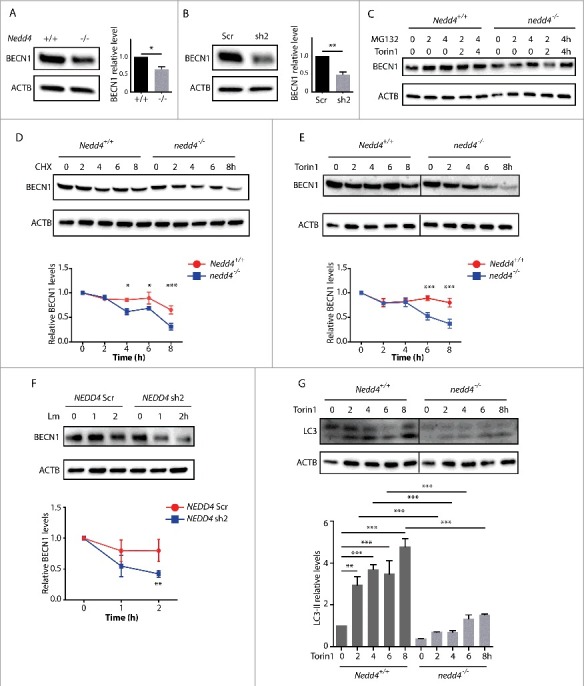
NEDD4 sustains BECN1 stability and autophagy. (A) Western blot analysis of BECN1 levels in Nedd4+/+ or nedd4−/− MEFs. Right panel, quantification of BECN1 levels in Nedd4+/+ or nedd4−/− MEFs. (B) Western blot analysis of BECN1 levels in NEDD4 Scr and KD HeLa cells. Right panel, quantification of BECN1 levels in NEDD4 Scr and KD HeLa cells. Relative levels were calculated against ACTB as a loading control and separately normalized to the level of control cells. Data are shown as mean ± SD of 3 independent experiments. P values were calculated using the Student 2-tailed unpaired t test. (*) p ≤0.05, (**) p ≤0.01. (C) Western blot analysis of BECN1 levels in Nedd4+/+ or nedd4−/− MEFs with or without MG132 treatment. (D) Western blot analysis of BECN1 stability with cycloheximide (CHX) treatment in Nedd4+/+ or nedd4−/− MEFs. Nedd4+/+ or nedd4−/− MEFs were treated with 50 µg/ml CHX for 0, 2, 4, 6, and 8 h and lysates were collected for SDS-PAGE. Lower panel, quantification of BECN1 levels presented in the upper panel. Relative levels were calculated against ACTB as a loading control and separately normalized to Nedd4+/+ or nedd4−/− MEFs with 0 h CHX treatment. (E) Western blot analysis of BECN1 levels with Torin1 treatment in Nedd4+/+ or nedd4−/− MEFs. Nedd4+/+ or nedd4−/− MEFs were treated with 1 µM Torin1 for 0, 2, 4, 6, and 8 h and lysates were collected for SDS-PAGE. Lower panel, quantification of BECN1 levels presented in the upper panel. The relative levels were calculated against ACTB as a loading control and separately normalized to Nedd4+/+ or nedd4−/− MEFs with 0 h Torin1 treatment. (F) Western blot analysis of BECN1 stability in THP-1 Scr and NEDD4 sh2 KD cells upon Lm infection. THP-1 control and NEDD4 KD cells were infected with Lm at MOI = 10 for 0, 1, 2 h and lysates were collected for SDS-PAGE. Lower panel, quantification of BECN1 levels presented in the upper panel. Relative levels were calculated against ACTB as a loading control and separately normalized to THP-1 Scr or THP-1 NEDD4 KD cells without Lm infection. For (D-F), data are shown as mean ± SEM of 3 independent experiments. P values were calculated using two-way ANOVA with Bonferroni's multiple comparisons test. (*) p ≤0.05, (**) p ≤0.01, (***) p ≤0.001. (G) Western blot analysis of LC3 levels with Torin1 treatment in Nedd4+/+ or nedd4−/− MEFs. Nedd4+/+ or nedd4−/− MEFs were treated with 1 µM Torin1 for 0, 2, 4, 6, and 8 h and lysates were collected for SDS-PAGE. LC3-II:ACTB ratio was determined using ImageJ software and normalized to Nedd4+/+ without Torin1 treatment. Results are representative of 3 independent experiments. Data are shown as mean ± SEM of 3 independent experiments. P values were calculated using one-way ANOVA with Bonferroni's multiple comparisons test. (**) p ≤ 0.01, (***) p ≤ 0.001.
Furthermore, to investigate whether BECN1 stability is increased by NEDD4 during Lm infection, THP-1 control and NEDD4 KD cells were infected with Lm for different time periods and BECN1 abundance was examined by western blot. Indeed, BECN1 abundance was significantly higher in control cells than in NEDD4 KD cells at 2 h of infection, confirming that NEDD4 also enhanced BECN1 stability during Lm infection (Fig. 7F). To investigate the effect of NEDD4 on autophagy induction, Nedd4+/+ and nedd4−/− MEFs were treated with Torin1, and LC3-II abundance was evaluated. Compared with nedd4−/− MEFs, Nedd4+/+ MEFs displayed more profoundly elevated LC3-II levels during the entire period of Torin1 treatment, suggesting that NEDD4 enhances autophagy activation (Fig. 7G). Thus, by sustaining BECN1 stability during autophagy activation, NEDD4 enhances the amplitude of autophagy.
Discussion
Here, we demonstrate a critical role of NEDD4 in the control of intracellular bacteria that perturb the phagosomal membrane to differing degrees and by distinct mechanisms. Lm has been defined as paragon “cytosolic pathogen” because it rapidly perforates the phagosomal membranes in an LLO-dependent manner.32 Mtb also translocates into the cytosol by means of the ESX-1 secretion system, albeit at later time points postinfection.33,34,41 In contrast to Mtb, the vaccine strain BCG and the Mtb ΔRD1 mutant, which both lack the RD1 region, fail to egress into the cytosol.34,36 The vaccine candidate rBCG takes advantage of LLO, thereby perturbing the phagosomal membrane without escaping into the cytosol.37,42 In our experiments, Mtb, rBCG and Lm replicated faster in NEDD4 KD macrophages compared to controls, suggesting that NEDD4 functions via a conserved mechanism by which it efficiently controls not only cytosolic bacteria, but also bacteria in impaired phagosomes. The expression of LLO and RD1 gene products is necessary for autophagy induction during Lm and Mtb infection, respectively.31,35 Consistent with these reports, our data reveal an important role for NEDD4 in autophagy induction upon Mtb and rBCG infection. There are some discrepancies in the literature concerning the role of autophagy in intracellular replication of Lm. Py et al. showed that Lm replicates more efficiently in atg5−/− MEFs.5 However, another study demonstrated that Lm replicates normally in atg5−/− bone marrow-derived macrophages.31 A major difference between our study and others is that we employed THP-1 human macrophage-like cells. We found increased growth of intracellular Lm in ATG5 KD THP-1 cells (Fig. S6F), confirming the restrictive role of autophagy on intracellular replication of Lm in THP-1 cells. In sum, we conclude that NEDD4 is important for the efficient control via autophagy of intracellular bacteria that impair the integrity of phagosomal membranes.
The vaccine strain rBCG, termed VPM1002, is rapidly advancing through the clinical pipeline. It has successfully completed phase I and phase IIa safety and immunogenicity trials (NCT01479972, NCT01113281, and NCT00749034) and is currently undergoing a phase II safety and immunogenicity trial in HIV-exposed newborns (NCT02391415).43,44 In preclinical studies, this vaccine induces improved protection accompanied by better safety as compared to parental BCG.42,45 In addition to enhanced cross-presentation following apoptosis,46,47 autophagy facilitates antigen presentation to T cells by delivering cytosolic antigens to products of the major histocompatibility complex (MHC).48 Therefore, it is tempting to speculate that NEDD4 contributes to the improved efficacy and safety of rBCG over BCG. In addition, NEDD4 can also be harnessed for host-directed therapy against multidrug-resistant tuberculosis (MDR-TB).49
Although our study focused on bacterial infections, it also sheds light on the role of NEDD4 in general autophagy induced by various stimuli. A previous report has demonstrated that NEDD4 expression increases basal macroautophagy.50 Consistent with this finding, we demonstrated that not only basal autophagy, but also starvation-induced autophagy and CCCP-induced mitophagy were all compromised by nedd4 KO, suggesting a general role of NEDD4 in autophagy induction. Moreover, upon Mtb and rBCG infection, autophagy was impaired by NEDD4 KD in terms of LC3-II abundance. NEDD4 also contributes to Lm targeting into phagophores and subsequently lysosomes, demonstrating its role in xenophagy. For xenophagy, cytosolic bacteria are initially coated by polyubiquitin and then recognized by autophagic receptors for delivery into phagophores.51 It is still controversial whether the polyubiquitin coat is composed of ubiquitinated host proteins or of ubiquitinated bacterial components.51,52 Considering the different location of cytosolic bacteria and bacteria perforating the phagosomal membrane but remaining in the phagosomes, it is possible that both mechanisms are involved in xenophagy. E3 ubiquitin ligases participating in this process are incompletely characterized. LRSAM1, an E3 ubiquitin ligase, has been found to contribute to autophagy by mediating direct ubiquitination of Salmonella Typhimurium.53 Various studies have demonstrated that NEDD4 ubiquitinates viral matrix proteins.21-26 Future investigations will determine whether NEDD4 contributes to xenophagy by ubiquitinating membrane perturbing bacteria.
Finally, we uncovered a novel regulation by NEDD4 during autophagy involving complex formation with ULK1 and BECN1. In addition, NEDD4 directly interacted with Atg8-family proteins, and deletion of the LC3-interacting region of NEDD4 lowered autophagy. Thus, we hypothesize that NEDD4 was recruited to phagophores by means of Atg8-family proteins and, once on phagophores, ubiquitinated BECN1. In contrast to a previous report,54 we found that NEDD4 modulated novel K6- and K27-type ubiquitination of BECN1. Moreover, we demonstrated that K48 linkage ubiquitination of BECN1 was increased by NEDD4 KD during basal autophagy or Lm-induced autophagy. We also revealed that NEDD4 stabilized BECN1 during autophagy. Therefore, our study provides compelling evidence that NEDD4 enhances autophagy by increasing BECN1 stability via novel types of ubiquitination. Hence the E3 ubiquitin ligase NEDD4 represents an important player in autophagy regulation at the post-translational level. Several other E3 ubiquitin ligases have been reported to positively or negatively regulate ULK1 and BECN1 by K63- or K48-type ubiquitination during induction of autophagy.15-18 It is thus reasonable to postulate that the NEDD4-mediated ubiquitination of BECN1 decreases its proteasomal degradation via K48 ubiquitination. The detailed mechanisms by which E3 ubiquitin ligases are coordinated during autophagy remain to be elucidated.
Overall, a novel role of NEDD4 in control of intracellular bacterial infections by autophagy has been revealed and insights into the maintenance of autophagy by specific ubiquitination of key autophagic machineries have been provided. By employing distinct membrane-perturbing microbes, our study not only unraveled critical host defense mechanisms against the deadliest pathogen on earth,55 but also deepened our understanding of the mechanisms underlying better protective and safety profiles of the clinically advanced rBCG vaccine candidate against TB-VPM1002.
Material and methods
Cell culture
The human monocytic cell line THP-1 was obtained from the American Type Culture Collection (ATCC, TIB-202) and maintained in RPMI 1640 (Gibco, 31870) with 10% (v:v) heat-inactivated fetal bovine serum (Sigma-Aldrich, F0804), 1 mM sodium pyruvate (Gibco, 11360070), 2 mM L-glutamine (Gibco, 25030081), 10 mM HEPES buffer (Gibco, 15630080), pH 7.2-7.5, 50 µM 2-mercaptoethanol (Gibco, 31350010). To differentiate THP-1 into macrophage-like cells, THP-1 cells were stimulated with 50 ng/ml phorbol 12-myristate 13-acetate (Sigma-Aldrich, P8139) for 24 h and then incubated with fresh medium for another 48 h. HeLa cells (ATCC, CCL-2) and HEK293T cells (ATCC, CRL-11268) were maintained in complete Dulbecco's modified Eagle's medium (DMEM) 4.5 g/l glucose (Gibco, 10938) with 10% (v:v) heat-inactivated fetal bovine serum, 1 mM sodium pyruvate, 2 mM L-glutamine. Nedd4+/+ and nedd4−/− immortalized MEFs were generated and maintained as previously described.56 All cells were incubated at 37°C, 5% CO2 in a humidified incubator.
Bacteria culture and CFU (colony-forming unit) assay
Mtb H37Rv, Mtb ΔRD1,57 M. bovis BCG and M. bovis BCG ΔureC::hly were grown in 7H9 medium (Sigma-Aldrich, M0178) containing 0.05% Tween-80 (Sigma-Aldrich, P8074) to OD600 less than 0.6. Single bacteria were prepared by passing through syringes. Then cells were infected with bacteria at a multiplicity of infection (MOI) of 10. After bacteria had been added, cells were centrifuged at 311 × g for 5 min and then incubated for 2 h. Afterwards, cells were washed with phosphate-buffered saline (PBS; Gibco, 14190144) to remove extracellular bacteria and incubated for another 4, 24, 48, and 72 h. At each time point, cells were lysed with sterile water, serially diluted with PBS supplemented with 0.05% Tween-80 and then plated on 7H11 agar plates (Sigma-Aldrich, M0428).
Lm EGD WT and Lm EGD Δhly were kindly provided by Dr. Marc Lecuit (Institute of Pasteur, France). Cells were grown in brain heart infusion (BHI; Sigma-Aldrich, 53286) medium overnight to stationary phase and then subcultured 1:10 in BHI medium for 2 h at 37°C. Bacteria were washed 3 times in DMEM medium. Cells were infected with bacteria at MOI of 5. After addition of bacteria, cells were centrifuged at 311 × g for 5 min and incubated at 37°C for 1 h. After washing 3 times with warm PBS, infected cells were placed in growth medium supplemented with 50 µg/ml gentamicin (Sigma-Aldrich, G1264) for 1 h and 20 µg/ml for another 1, 3, or 5 h. At each time point, cells were lysed with sterile water, serially diluted with PBS and then plated on BHI agar plates.
Plasmids, antibodies and reagents
HA-NEDD4WT (Addgene, 27002), and HA-NEDD4C867A (Addgene, 26999) were gifts from Dr. Joan Massague.58 MYC-ULK1WT (Addgene, 27629), MYC-ULK1K46I (Addgene, 27630), and MYC-ULK1S4A (Addgene, 27631) were gifts from Dr. Reuben Shaw.59 FLAG-ubiquitin WT and related mutants (K6WT, K11WT K27WT, K29WT, K33WT, K48WT, K63WT and KallR) were provided by the MRC Protein Phosphorylation and Ubiquitylation Unit, UK. YFP-Atg8-family proteins encoding plasmids were a kind gift from Dr. Felix Randow (MRC Laboratory of Molecular Biology, UK). MYC-BECN1 plasmid (Biocat GmbH, MR207162-OR), MG132 (Merck Chemicals GmbH, 474791), Torin1 (Tocris Bioscience, 4247), cyclohexamide (Sigma-Aldrich, C1988) and choloroquine (Sigma-Aldrich, C6628) were purchased as indicated. Antibodies used in this study include: anti-ACTB (Sigma-Aldrich, A2228), anti-LC3 (MBL International, PM036), anti-human NEDD4 (Cell Signaling Technology, 3607), anti-mouse NEDD4 (BD Biosciences, 611481), anti-K48 ubiquitin (Merck Millipore, 05–1037), anti-K63 ubiquitin (Enzo Life Sciences, HWA4C4), anti-ubiquitin (FK2) (Enzo Life Sciences, BML-PW8810), rabbit anti-BECN1 (Cell Signaling Technology, 3495), mouse anti-BECN1 (Cell Signaling Technology, 4122), anti-ULK1 (Cell Signaling Technology, 8054), anti-LC3B (Sigma-Aldrich, L7543), anti-LAMP2 (Santa Cruz Biotechnology, SC-18822).
Electron microscopy
Nedd4+/+ and nedd4−/− MEFs were fixed with 2% paraformaldehyde (Electron Microscopy Sciences, 15714) plus 0.05% glutaraldehyde (Sigma-Aldrich, G5882) for 15 min at 37°C. The cell pellet was embedded in 10% bovine gelatin (Sigma-Aldrich, G1890), cut into small cubes and infiltrated with 2.3 M sucrose overnight. Samples were frozen in liquid nitrogen and 100-nm Tokuyasu sections were cut with a RMC MTX & CRX cryo-ultramicrotome (RMC Boeckeler, USA).60 Sections were transferred on pioloform (Plano GmbH, R1275)-carbon-coated TEM grids (Science Services, G200HEX-G3). 5% cold-water fish skin gelatin (Sigma-Aldrich, G7765) and 0.5% bovine serum albumin (BSA; Sigma-Aldrich, A2058) in PBS was used for blocking of nonspecific binding and dilution of antibody and goat-anti-mouse IgG-gold (Jackson ImmunoResearch, 115-205-068). Mouse antibody against NEDD4 (BD Biosciences, 611481) was used at 1:10 dilution overnight. 12 nm goat-anti-mouse IgG-gold was used at 1:40 for 30 min. Sections were embedded in 2% methyl cellulose with 0.2% uranyl acetate (Electron Microscopy Sciences, 22400) and analyzed with a TEM Zeiss LEO906 (Zeiss, Germany). Images were recorded digitally with a Morada camera (Olympus Soft Imaging Solutions, Germany) and iTEM software.
Lentivirus-based shRNA knockdown
pLK0.1 shRNA vectors against human NEDD4 were purchased from Sigma-Aldrich. The targeting sequences against human NEDD4 are: 5′CCGGCGGTTGGAGAATGTAGCAATACTCGAGTATTGCTACATTCTCCAACCGTTTTTG-3′ (sh1) and 5′-CCGGTACGTGAGAGTGACGTTATATCTCGAGATATAACGTCACTCTCACGTATTTTTG-3′ (sh2). Lentivirus production was performed according to the manufacturer's instructions. After transduction, THP-1 or HeLa cells were selected with 5 µg/ml puromycin (Sigma-Aldrich, P8833) to obtain stable KD cell lines.
Complementary expression of shRNA-resistant HA-NEDD4 in THP-1 NEDD4 KD cells
To express HA-NEDD4WT in stable THP-1 NEDD4 KD cells, silent mutations (TATGTTAGGGTAACA) were introduced into the target region of NEDD4 sh2 (TACGTGAGAGTGACG) by using a Q5 mutagenesis kit (New England Biolabs, E0554S). The mutated plasmid was subcloned into a Gateway entry vector pENTR 4 (ThermoFisher Scientific, A10465) and afterwards transferred into a lentiviral expression vector pLenti6/V5-DEST (ThermoFisher Scientific, V49610). THP-1 NEDD4 sh2 cells were transduced with virus containing HA-NEDD4 for 48 h and selected with 10 µg/ml blasticidin (Invivogen, ant-bl-05) for stable clones.
Immunofluorescence
Cells were fixed with 4% (v:v) paraformaldehyde in PBS, pH 7.4 for 10 min at room temperature (RT). After washing twice with PBS, cells were incubated with 50 mM glycine in PBS, pH 7.4 for 10 min and afterwards incubated with 0.05% saponin (Sigma-Aldrich, 47036), 1% BSA in PBS for 10 min. Primary and secondary antibodies were diluted in PBS at the indicated dilutions and incubated for 1 h at RT. DAPI (Sigma-Aldrich, D9542) was used for nuclear staining. Cells were mounted with DAKO mounting medium (Dako Cytomation, S3023).
Immunoprecipitation
HEK293T cells were transfected with the indicated plasmids and incubated for 48 h. Cells of one 10-cm dish were lysed with 500 µl of lysis buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.5% NP-40 (Sigma-Aldrich, 74385) supplemented with protease inhibitors (Roche, 05892791001) on ice for 20 min. Lysates were centrifuged at 16,000 × g at 4°C for 10 min and supernatants were mixed with anti-HA (Sigma-Aldrich, A2095), anti-MYC (Sigma-Aldrich, A7470) or anti-FLAG agarose (Sigma-Aldrich, A2220) overnight at 4°C. For endogenous protein IP, supernatants were mixed with the indicated antibodies and protein G Dynabeads (Life Technologies, 10003D) overnight at 4°C. Afterwards, agaroses were washed twice with PBS and boiled with sample buffer for SDS-PAGE.
Ubiquitination assay
For analysis of the ubiquitination of overexpressed BECN1, HEK293T cells were transfected with MYC-BECN1, FLAG-ubiquitin wild type, or HA-ubiquitin mutants and HA-NEDD4. The cell extracts were immunoprecipitated with anti-MYC agarose and analyzed by immunoblotting with anti-FLAG antibody.
SDS-PAGE and western blot
Cells were lysed in RIPA buffer supplemented with protease inhibitor cocktail and PhosSTOP (Roche, 4906837001) on ice for 10 min. After centrifugation at 16,000 × g for 10 min, supernatants were heated with SDS sample buffer at 95°C for 10 min. Proteins were separated on 4–15% SDS-PAGE gels (Bio-Rad, 4561086) and transferred onto nitrocellulose membranes. The membranes were incubated with primary antibodies at 4°C overnight and secondary antibodies at RT for 1 h, respectively. All antibodies were diluted in PBS supplemented with 0.1% Tween-20 (Sigma-Aldrich, P1379) and 5% BSA. Membranes were developed with ECL detection Kit (ThermoFisher Scientific, 34094), exposed with ChemiDoc (Bio-Rad Laboratories, Germany) and evaluated using ImageJ software. The antibody against ACTB/β-actin was used as the loading control.
Statistical analysis
Statistical analysis was performed with GraphPad Prism v6.04 (GraphPad software Inc., USA). P-values were calculated using Student t test, one-way or 2-way ANOVA (Bonferroni's multiple comparisons test) and 95% confidence interval.
Supplementary Material
Abbreviations
- BCG
Mycobacterium bovis bacille Calmette-Guérin (BCG)
- BHI
brain heart infusion
- BSA
bovine serum albumin
- CCCP
carbonyl cyanide m-chlorophenyl hydrazone
- CFU
colony-forming unit
- CHX
cyclohexamide
- CQ
chloroquine
- EBSS
Earle's balanced salt solution
- FCS
fetal calf serum
- HECT
homologous E6-AP carboxyl terminus
- HIV
human immunodeficiency virus
- IF
immunofluorescence
- IP
immunoprecipitation
- KD
knockdown
- KO
knockout
- LDH
lactate dehydrogenase
- LLO
listeriolysin O
- Lm
Listeria monocytogenes
- MDR-TB
multi-drug resistant tuberculosis
- MEFs
mouse embryonic fibroblasts
- MHC
major histocompatibility complex
- MOI
multiplicity of infection
- Mtb
Mycobacterium tuberculosis
- NEDD4
neuronal precursor cell expressed, developmentally down-regulated 4
- PBS
phosphate-buffered saline
- p.i.
post-infection
- rBCG
Mycobacterium bovis BCG ΔureC::hly
- RT
room temperature
- WT
wild type
Disclosure of potential conflicts of interest
All other authors declare no conflicts of interest.
Acknowledgments
We thank Dr. Marc Lecuit (Institute of Pasteur, France) for kindly providing us with Listeria monocytogenes EGD WT and Δhly mutant. We thank Dr. Felix Randow (MRC Laboratory of Molecular Biology, UK) for providing us with YFP-ATG8s plasmids. We thank Anne Koehler and Mary Louise Grossman for technical and editorial assistance, respectively.
Funding
This work was partially supported by the European Union's Seventh Framework Program TBVAC2020 grant no. 643381), “ADITEC” (HEALTH-F4-2011-280873) as well as by the German Federal Ministry of Education and Research (Bundesministerium für Bildung und Forschung [BMBF]) “inVAC” (grant no. 03ZZ0806A to S.H.E.K.).
References
- 1.He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67–93. doi: 10.1146/annurev-genet-102808-114910. PMID:19653858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469:323–35. doi: 10.1038/nature09782. PMID:21248839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368:1845–6. doi: 10.1056/NEJMra1205406. PMID:23656658. [DOI] [PubMed] [Google Scholar]
- 4.Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. 2004;119:753–66. doi: 10.1016/j.cell.2004.11.038. PMID:15607973. [DOI] [PubMed] [Google Scholar]
- 5.Py BF, Lipinski MM, Yuan J. Autophagy limits Listeria monocytogenes intracellular growth in the early phase of primary infection. Autophagy. 2007;3:117–25. doi: 10.4161/auto.3618. PMID:17204850. [DOI] [PubMed] [Google Scholar]
- 6.Deretic V. Autophagy as an immune defense mechanism. Curr Opin Immunol. 2006; 18:375–82. doi: 10.1016/j.coi.2006.05.019. PMID:16782319. [DOI] [PubMed] [Google Scholar]
- 7.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. PMID:18191218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mizushima N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr Opin Cell Biol. 2010;22:132–9. doi: 10.1016/j.ceb.2009.12.004. PMID:20056399. [DOI] [PubMed] [Google Scholar]
- 9.Hosokawa N, Sasaki T, Iemura S, Natsume T, Hara T, Mizushima N. Atg101, a novel mammalian autophagy protein interacting with Atg13. Autophagy. 2009;5:973–9. doi: 10.4161/auto.5.7.9296. PMID:19597335. [DOI] [PubMed] [Google Scholar]
- 10.Ganley IG, Lam du H, Wang J, Ding X, Chen S, Jiang X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem. 2009;284:12297–305. doi: 10.1074/jbc.M900573200. PMID:19258318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, Kundu M, Kim DH. ULK–Atg13–FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell. 2009;20:1992–2003. doi: 10.1091/mbc.E08-12-1249. PMID:19225151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–41. doi: 10.1038/ncb2152. PMID:21258367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Itakura E, Mizushima N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy. 2010;6:764–76. doi: 10.4161/auto.6.6.12709. PMID:20639694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Russell RC, Tian Y, Yuan H, Park HW, Chang YY, Kim J, Kim H, Neufeld TP, Dillin A, Guan KL. ULK1 induces autophagy by phosphorylating Beclin–1 and activating VPS34 lipid kinase. Nat Cell Biol. 2013;15:741–50. doi: 10.1038/ncb2757. PMID:23685627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nazio F, Strappazzon F, Antonioli M, Bielli P, Cianfanelli V, Bordi M, Gretzmeier C, Dengjel J, Piacentini M, Fimia GM, et al. . mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self–association and function through AMBRA1 and TRAF6. Nat Cell Biol. 2013;15:406–16. doi: 10.1038/ncb2708. PMID:23524951. [DOI] [PubMed] [Google Scholar]
- 16.Shi CS, Kehrl JH. TRAF6 and A20 regulate lysine 63–linked ubiquitination of Beclin–1 to control TLR4–induced autophagy. Sci Signal. 2010;3:ra42. doi: 10.1126/scisignal.2000751. PMID:20501938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu C, Feng K, Zhao X, Huang S, Cheng Y, Qian L, Wang Y, Sun H, Jin M, Chuang TH, et al. . Regulation of autophagy by E3 ubiquitin ligase RNF216 through BECN1 ubiquitination. Autophagy. 2014;10:2239–50. doi: 10.4161/15548627.2014.981792. PMID:25484083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu CC, Lin YC, Chen YH, Chen CM, Pang LY, Chen HA, Wu PR, Lin MY, Jiang ST, Tsai TF, et al. . Cul3–KLHL20 Ubiquitin Ligase governs the turnover of ULK1 and VPS34 complexes to control autophagy termination. Mol Cell. 2016;61:84–97. doi: 10.1016/j.molcel.2015.11.001. PMID:26687681. [DOI] [PubMed] [Google Scholar]
- 19.Ingham RJ, Gish G, Pawson T. The Nedd4 family of E3 ubiquitin ligases: functional diversity within a common modular architecture. Oncogene. 2004;23:1972–84. doi: 10.1038/sj.onc.1207436. PMID:15021885. [DOI] [PubMed] [Google Scholar]
- 20.Boase NA, Kumar S. NEDD4: The founding member of a family of ubiquitin–protein ligases. Gene. 2015;557:113–22. doi: 10.1016/j.gene.2014.12.020. PMID:25527121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harty RN, Paragas J, Sudol M, Palese P. A proline–rich motif within the matrix protein of vesicular stomatitis virus and rabies virus interacts with WW domains of cellular proteins: implications for viral budding. J Virol. 1999;73:2921–9. PMID:10074141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Urata S, Yasuda J. Regulation of Marburg virus (MARV) budding by Nedd4.1: a different WW domain of Nedd4.1 is critical for binding to MARV and Ebola virus VP40. J Gen Virol. 2010;91:228–34. doi: 10.1099/vir.0.015495-0. PMID:19812267. [DOI] [PubMed] [Google Scholar]
- 23.Harty RN, Brown ME, Wang G, Huibregtse J, Hayes FP. A PPxY motif within the VP40 protein of Ebola virus interacts physically and functionally with a ubiquitin ligase: implications for filovirus budding. Proc Natl Acad Sci USA. 2000;97:13871–6. doi: 10.1073/pnas.250277297. PMID:11095724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yasuda J, Nakao M, Kawaoka Y, Shida H. Nedd4 regulates egress of Ebola virus–like particles from host cells. J Virol. 2003;77:9987–92. doi: 10.1128/JVI.77.18.9987-9992.2003. PMID:12941909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kikonyogo A, Bouamr F, Vana ML, Xiang Y, Aiyar A, Carter C, Leis J. Proteins related to the Nedd4 family of ubiquitin protein ligases interact with the L domain of Rous sarcoma virus and are required for gag budding from cells. Proc Natl Acad Sci USA. 2001;98:11199–204. doi: 10.1073/pnas.201268998. PMID:11562473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sette P, Jadwin JA, Dussupt V, Bello NF, Bouamr F. The ESCRT–associated protein Alix recruits the ubiquitin ligase Nedd4–1 to facilitate HIV–1 release through the LYPXnL L domain motif. J Virol. 2010;84:8181–92. doi: 10.1128/JVI.00634-10. PMID:20519395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wodrich H, Henaff D, Jammart B, Segura–Morales C, Seelmeir S, Coux O, Ruzsics Z, Wiethoff CM, Kremer EJ. A capsid–encoded PPxY–motif facilitates adenovirus entry. PLoS Pathog. 2010;6:e1000808. doi: 10.1371/journal.ppat.1000808. PMID:20333243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chesarino NM, McMichael TM, Yount JS. E3 Ubiquitin Ligase NEDD4 promotes influenza virus infection by decreasing levels of the antiviral protein IFITM3. PLoS Pathog. 2015;11:e1005095. doi: 10.1371/journal.ppat.1005095. PMID:26263374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kaufmann SH, Dorhoi A. Molecular Determinants in Phagocyte–Bacteria Interactions. Immunity. 2016;44:476–91. doi: 10.1016/j.immuni.2016.02.014. PMID:26982355. [DOI] [PubMed] [Google Scholar]
- 30.Ray K, Marteyn B, Sansonetti PJ, Tang CM. Life on the inside: the intracellular lifestyle of cytosolic bacteria. Nat Rev Microbiol. 2009;7:333–40. doi: 10.1038/nrmicro2112. PMID:19369949. [DOI] [PubMed] [Google Scholar]
- 31.Meyer–Morse N, Robbins JR, Rae CS, Mochegova SN, Swanson MS, Zhao Z, Virgin HW, Portnoy D. Listeriolysin O is necessary and sufficient to induce autophagy during Listeria monocytogenes infection. PLoS One. 2010;5:e8610. doi: 10.1371/journal.pone.0008610. PMID:20062534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Portnoy DA, Auerbuch V, Glomski IJ. The cell biology of Listeria monocytogenes infection: the intersection of bacterial pathogenesis and cell–mediated immunity. J Cell Biol. 2002;158:409–14. doi: 10.1083/jcb.200205009. PMID:12163465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van der Wel N, Hava D, Houben D, Fluitsma D, van Zon M, Pierson J, Brenner M, Peters PJ. M. tuberculosis and M. leprae translocate from the phagolysosome to the cytosol in myeloid cells. Cell. 2007;129:1287–98. doi: 10.1016/j.cell.2007.05.059. PMID:17604718. [DOI] [PubMed] [Google Scholar]
- 34.Houben D, Demangel C, van Ingen J, Perez J, Baldeon L, Abdallah AM, Caleechurn L, Bottai D, van Zon M, de Punder K, et al. . ESX–1–mediated translocation to the cytosol controls virulence of mycobacteria. Cell Microbiol. 2012;14:1287–98. doi: 10.1111/j.1462-5822.2012.01799.x. PMID:22524898. [DOI] [PubMed] [Google Scholar]
- 35.Watson RO, Manzanillo PS, Cox JS. Extracellular M. tuberculosis DNA targets bacteria for autophagy by activating the host DNA–sensing pathway. Cell. 2012;150:803–15. doi: 10.1016/j.cell.2012.06.040. PMID:22901810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lewis KN, Liao R, Guinn KM, Hickey MJ, Smith S, Behr MA, Sherman DR. Deletion of RD1 from Mycobacterium tuberculosis mimics bacille Calmette–Guerin attenuation. J Infect Dis. 2003;187:117–23. doi: 10.1086/345862. PMID:12508154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hess J, Miko D, Catic A, Lehmensiek V, Russell DG, Kaufmann SH. Mycobacterium bovis Bacille Calmette–Guerin strains secreting listeriolysin of Listeria monocytogenes. Proc Natl Acad Sci USA. 1998;95:5299–304. doi: 10.1073/pnas.95.9.5299. PMID:9560270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Saiga H, Nieuwenhuizen N, Gengenbacher M, Koehler AB, Schuerer S, Moura–Alves P, Wagner I, Mollenkopf HJ, Dorhoi A, Kaufmann SH. The recombinant BCG DeltaureC::hly vaccine targets the AIM2 inflammasome to induce autophagy and inflammation. J Infect Dis. 2015;211:1831–41. doi: 10.1093/infdis/jiu675. PMID:25505299. [DOI] [PubMed] [Google Scholar]
- 39.Behrends C, Sowa ME, Gygi SP, Harper JW. Network organization of the human autophagy system. Nature. 2010;466:68–76. doi: 10.1038/nature09204. PMID:20562859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kalvari I, Tsompanis S, Mulakkal NC, Osgood R, Johansen T, Nezis IP, Promponas VJ. iLIR: A web resource for prediction of Atg8–family interacting proteins. Autophagy. 2014;10:913–25. doi: 10.4161/auto.28260. PMID:24589857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Simeone R, Sayes F, Song O, Groschel MI, Brodin P, Brosch R, Majlessi L. Cytosolic access of Mycobacterium tuberculosis: critical impact of phagosomal acidification control and demonstration of occurrence in vivo. PLoS Pathog. 2015;11:e1004650. doi: 10.1371/journal.ppat.1004650. PMID:25658322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Grode L, Seiler P, Baumann S, Hess J, Brinkmann V, Nasser Eddine A, Mann P, Goosmann C, Bandermann S, Smith D, et al. . Increased vaccine efficacy against tuberculosis of recombinant Mycobacterium bovis bacille Calmette–Guerin mutants that secrete listeriolysin. J Clin Invest. 2005;115:2472–9. doi: 10.1172/JCI24617. PMID:16110326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kaufmann SH, Cotton MF, Eisele B, Gengenbacher M, Grode L, Hesseling AC, Walzl G. The BCG replacement vaccine VPM1002: from drawing board to clinical trial. Expert Rev Vaccines. 2014;13:619–30. doi: 10.1586/14760584.2014.905746. PMID:24702486. [DOI] [PubMed] [Google Scholar]
- 44.Kaufmann SH, Lange C, Rao M, Balaji KN, Lotze M, Schito M, Zumla AI, Maeurer M. Progress in tuberculosis vaccine development and host–directed therapies––a state of the art review. Lancet Respir Med. 2014;4:301–20. doi: 10.1016/S2213-2600(14)70033-5.. [DOI] [PubMed] [Google Scholar]
- 45.Desel C, Dorhoi A, Bandermann S, Grode L, Eisele B, Kaufmann SH. Recombinant BCG DeltaureC hly+ induces superior protection over parental BCG by stimulating a balanced combination of type 1 and type 17 cytokine responses. J Infect Dis. 2011;204:1573–84. doi: 10.1093/infdis/jir592. PMID:21933877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schaible UE, Winau F, Sieling PA, Fischer K, Collins HL, Hagens K, Modlin RL, Brinkmann V, Kaufmann SH. Apoptosis facilitates antigen presentation to T lymphocytes through MHC–I and CD1 in tuberculosis. Nat Med. 2003;9:1039–46. doi: 10.1038/nm906. PMID:12872166. [DOI] [PubMed] [Google Scholar]
- 47.Winau F, Weber S, Sad S, de Diego J, Hoops SL, Breiden B, Sandhoff K, Brinkmann V, Kaufmann SH, Schaible UE. Apoptotic vesicles crossprime CD8 T cells and protect against tuberculosis. Immunity. 2006;24:105–17. doi: 10.1016/j.immuni.2005.12.001. PMID:16413927. [DOI] [PubMed] [Google Scholar]
- 48.Jagannath C, Lindsey DR, Dhandayuthapani S, Xu Y, Hunter RL Jr, Eissa NT. Autophagy enhances the efficacy of BCG vaccine by increasing peptide presentation in mouse dendritic cells. Nat Med. 2009;15:267–76. doi: 10.1038/nm.1928. PMID:19252503. [DOI] [PubMed] [Google Scholar]
- 49.Wallis RS, Hafner R. Adancing host–directed therapy for tuberculosis. Nat Rev Immunol. 2015;15:255–63. doi: 10.1038/nri3813. PMID:25765201. [DOI] [PubMed] [Google Scholar]
- 50.Li Y, Zhang L, Zhou J, Luo S, Huang R, Zhao C, Diao A. Nedd4 E3 ubiquitin ligase promotes cell proliferation and autophagy. Cell Prolif. 2015;48:338–47. doi: 10.1111/cpr.12184. PMID:25809873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Randow F, Youle RJ. Self and nonself: how autophagy targets mitochondria and bacteria. Cell Host Microbe. 2014;15:403–11. doi: 10.1016/j.chom.2014.03.012. PMID:24721569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fujita N, Morita E, Itoh T, Tanaka A, Nakaoka M, Osada Y, Umemoto T, Saitoh T, Nakatogawa H, Kobayashi S, et al. . Recruitment of the autophagic machinery to endosomes during infection is mediated by ubiquitin. J Cell Biol. 2013;203:115–28. doi: 10.1083/jcb.201304188. PMID:24100292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huett A, Heath RJ, Begun J, Sassi SO, Baxt LA, Vyas JM, Goldberg MB, Xavier RJ. The LRR and RING domain protein LRSAM1 is an E3 ligase crucial for ubiquitin–dependent autophagy of intracellular Salmonella Typhimurium. Cell Host Microbe. 2012;12:778–90. doi: 10.1016/j.chom.2012.10.019. PMID:23245322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Platta HW, Abrahamsen H, Thoresen SB, Stenmark H. Nedd4–dependent lysine–11–linked polyubiquitination of the tumour suppressor Beclin 1. Biochem J. 2012;441:399–406. doi: 10.1042/BJ20111424. PMID:21936852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.WHO Global tuberculosis report 2015. Geneva: World Health Organization , 2015. http://www.who.int/tb/publications/global_report/en [Google Scholar]
- 56.Hsia HE, Kumar R, Luca R, Takeda M, Courchet J, Nakashima J, Wu S, Goebbels S, An W, Eickholt BJ, et al. . Ubiquitin E3 ligase Nedd4–1 acts as a downstream target of PI3K/PTEN–mTORC1 signaling to promote neurite growth. Proc Natl Acad Sci USA. 2014;111:13205–10. doi: 10.1073/pnas.1400737111. PMID:25157163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hsai T, Hingley–Wilson SM, Chen B, Chen M, Dai AZ, Morin PM, Marks CB, Padiyar J, Goulding C, Gingery M, et al. . The primary mechanism of attenuation of bacillus Calmette–Guerin is a loss of secreted lytic function required for invasion of lung interstitial tissue. Proc Natl Acad Sci USA. 2003;100:12420–5. doi: 10.1073/pnas.1635213100. PMID:14557547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gao S, Alarcon C, Sapkota G, Rahman S, Chen PY, Goerner N, Macias MJ, Erdjument–Bromage H, Tempst P, Massagué J. Ubiquitin ligase Nedd4L targets activated Smad2/3 to limit TGF–beta signaling. Mol Cell. 2009;36:457–68. doi: 10.1016/j.molcel.2009.09.043. PMID:19917253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Egan DF, Shackelford DB, Mihaylova MM, Gelino SR, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, et al. . Phosphorylation of ULK1 (hATG1) by AMP–activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456–61. doi: 10.1126/science.1196371. PMID:21205641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tokuyasu KT. A technique for ultracryotomy of cell suspensions and tissues. J Cell Biol. 1973;57:551–65. doi: 10.1083/jcb.57.2.551. PMID:4121290. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.