

ABSTRACT
Alzheimer disease (AD) is the most common neurodegenerative disease characterized by the deposition of amyloid plaque in the brain. The autophagy-associated PIK3C3-containing phosphatidylinositol 3-kinase (PtdIns3K) complex has been shown to interfere with APP metabolism and amyloid beta peptide (Aβ) homeostasis via poorly understood mechanisms. Here we report that NRBF2 (nuclear receptor binding factor 2), a key component and regulator of the PtdIns3K, is involved in APP-CTFs homeostasis in AD cell models. We found that NRBF2 interacts with APP in vivo and its expression levels are reduced in hippocampus of 5XFAD AD mice; we further demonstrated that NRBF2 overexpression promotes degradation of APP C-terminal fragments (APP-CTFs), and reduces Aβ1–40 and Aβ1-42 levels in human mutant APP-overexpressing cells. Conversely, APP-CTFs, Aβ1–40 and Aβ1-42 levels were increased in Nrbf2 knockdown or nrbf2 knockout cells. Furthermore, NRBF2 positively regulates autophagy in neuronal cells and NRBF2-mediated reduction of APP-CTFs levels is autophagy dependent. Importantly, nrbf2 knockout attenuates the recruitment of APP and APP-CTFs into phagophores and the sorting of APP and APP-CTFs into endosomal intralumenal vesicles, which is accompanied by the accumulation of the APP and APP-CTFs into RAB5-positive early endosomes. Collectively, our results reveal the potential connection between NRBF2 and the AD-associated protein APP by showing that NRBF2 plays an important role in regulating degradation of APP-CTFs through modulating autophagy.
KEYWORDS: Aβ, Alzheimer's disease, APP, autophagy, class III phosphatidylinositol 3-kinase (PtdIns3K), NRBF2
Introduction
Alzheimer disease (AD) is the most common age-related neurodegenerative disease characterized by the intracellular aggregation of amyloid beta (Aβ) peptide and hyperphosphorylated MAPT/tau, as well as extracellular Aβ plaque deposition in the brain.1,2 Aβ generation has been regarded as the critical event during the progression and pathogenesis of AD.3,4 Aβ is produced from APP (amyloid beta precursor protein) by the amyloidogenic processing pathway.4–6 APP is a type I transmembrane protein that can be processed by either non-amyloidogenic processing or amyloidogenic processing.4–6 Non-amyloidogenic APP processing occurs at the cell surface, where APP is cleaved by α-secretase to produce a soluble APP ectodomain and the membrane-associated C83 APP C-terminal fragment alpha (APP-CTFα), which is further cleaved by γ-secretase to produce nontoxic P3 peptides.4–7 Amyloidogenic APP processing takes places in intracellular compartments, including the trans-Golgi network, endosomes, and autophagosomes, where APP is first cleaved by BACE1/β-secretase, which generates the soluble sAPPβ and the membrane-bound C99 APP-CTFβ. Subsequently APP-CTFβ is cleaved into Aβ1–40 or Aβ1-42 by γ-secretase.4–7 Thus, APP-CTFs (hereafter referred to as APP-CTFα and APP-CTFβ) levels and their subcellular locations are essential for Aβ production. In addition, the production of Aβ is also tightly regulated by autophagy-mediated degradation of APP-CTFs.8–10
Autophagy is a major pathway involved in the delivery of long-lived proteins, protein aggregates, and organelles to lysosomes for degradation.10,11 Autophagy can be generally divided into several steps, including autophagosome formation (initiation), fusion of the autophagosome with a lysosome (maturation), and breakdown of the cargo followed by efflux of the resulting macromolecules.10,11 Autophagy dysfunction has been implicated in the pathogenesis of AD. In AD patients, MTOR (mechanistic target of rapamycin) signaling, an important signaling pathway controlling autophagy, is enhanced, which may reflect lowered levels of basal autophagy.12 In addition, accumulated autophagosomes and high levels of lysosomal hydrolases have been found in AD brain, which may indicate impaired autophagosomal-lysosomal clearance.13,14 In AD cells and animal models, deletion of BECN1/Becn1, the gene encoding a key protein for the formation of autophagosomes, increases both the intracellular and extracellular Aβ loads.15–17 Moreover, knockout of Atg7 (autophagy-related 7) affects both the production and secretion of Aβ.18,19 In line with these findings, increasing evidence indicates that induction of autophagy may serve as a viable therapeutic strategy that can ameliorate the pathological features associated with AD. For instance, neuronal-specific overexpression of Tfeb (transcription factor EB), a master regulator of the autophagy-lysosome pathway, induces APP degradation and reduces Aβ generation.20 Pharmacological induction of autophagy by rapamycin decreases intracellular Aβ levels, reduces Aβ plaque load and improves cognition in AD mice.12,21
Autophagy initiation can be regulated by the PIK3C3/VPS34-containing PtdIns3K complex, which also includes PIK3R4/VPS15, BECN1/Beclin 1 and ATG14.22–24 Our and others’ recent studies have shown that NRBF2 is a novel component of the PtdIns3K complex, and associates with ATG14 in both mammalian cells25–28 and in yeast (where it is named Atg38).29 In addition, NRBF2 has been demonstrated to positively regulate autophagy.25,28,30 However, there is also a controversial report,26 suggesting that the role of NRBF2 in regulating autophagy may be cell type dependent. Whether NRBF2 is a positive or negative regulator in neuronal cells remains unclear. In addition, though autophagy has been implicated in Aβ metabolism, the roles and mechanisms of the PtdIns3K complex components involved in autophagy initiation and regulating APP metabolism are unclear. Here, we investigated the functions of NRBF2 in regulating APP metabolism in cell models. Our results suggest that NRBF2, via activation of autophagy, facilitates APP-CTFs degradation. The mechanisms for NRBF2-mediated APP metabolism may be related to its interaction with APP and the sorting of APP and APP-CTFs from early endosomes into autophagosomes and endosomal intralumenal vesicles (ILVs) for lysosomal degradation. Our findings highlight the importance of NRBF2, a PtdIns3K component, in regulating APP metabolism.
Results
NRBF2 is reduced in brains of 5XFAD AD mice
Autophagy has been linked to AD pathogenesis.33 To identify whether NRBF2, a key component of the PtdIns3K complex, is involved in AD, we measured NRBF2 protein levels in brains of 5XFAD transgenic AD mice. 5XFAD mice co-express 5 mutations of familial AD, which recapitulate major features of AD amyloid pathology.31 As expected, both full-length APP (FL-APP) and APP-CTFs levels were increased in the hippocampus of 5XFAD mice (Fig. 1A). Interestingly, as shown in Fig. 1 A and B, NRBF2 protein levels were significantly reduced in the hippocampus of 12-mo-old 5XFAD mice (n = 6) compared with age-controlled wild-type C57BL/6 mice. In contrast, ATG12–ATG5 levels were not changed in 5XFAD mice (Fig. 1 A and B), suggesting that NRBF2 may play a specific role in this AD animal model. Previous results show that BECN1 is reduced in the brains of AD patients and APPswePS1dE9 mice.15,32 Consistent with this finding, BECN1 proteins levels were also reduced in 5XFAD mice (Fig. 1 A and B). Interestingly, our results show that both LC3B-II and SQSTM1/p62 levels were increased in the hippocampus of 5XFAD mice (Fig. 1 A and B), which is consistent with previous reports that autophagic degradation defects are found in AD patients and several other AD animal models.33,34 These results reveal the potential role of NRBF2 in the pathogenesis AD.
Figure 1.
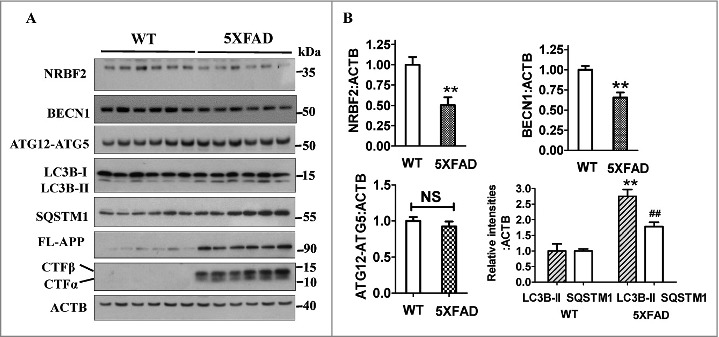
NRBF2 protein levels are reduced in the hippocampus region of 5XFAD AD mice. (A) Western blotting was used to detect the expression of NRBF2, BECN1, ATG12–ATG5, LC3B, SQSTM1, FL-APP, and APP-CTFs levels in the hippocampus region of 12-mo-old C57BL/6 and 5XFAD AD mice; ACTB was used as an internal control. (B) Quantitative data in panel 1A (n = 6) showed that NRBF2 and BECN1 but not ATG12–ATG5 levels are significantly decreased, while LC3B-II and SQSTM1/p62 levels are significantly increased in 5XFAD mice. NS, not significant; *, P < 0.05, **, P < 0.01 vs. the control.
Overexpression of Nrbf2 decreases APP-CTFs and Aβ levels
As we found that the NRBF2 level is reduced in 5XFAD mice, we wondered whether NRBF2 is important in regulating APP-CTFs (APP-CTFα and APP-CTFβ) degradation and Aβ production. We first overexpressed NRBF2 in N2a mouse neuroblastoma cells stably expressing the human Swedish mutant APP695 (N2S cells), which is commonly used as an AD cell model.35 We found that Nrbf2 overexpression significantly reduced the expression of APP-CTFα and APP-CTFβ levels but did not affect the levels of FL-APP (Fig. 2A and B). However, the amounts of sAPPα and sAPPβ were not different in Nrbf2-overexpressed cells compared with control cells (Fig. 2C and D). In addition, Nrbf2 overexpression did not affect PSEN1 levels, a subunit of γ-secretase, which is responsible for the production of Aβ by cleaving of APP-CTFβ. These results suggest that NRBF2 may affect APP-CTFs degradation rather than proteolytic processing of APP.
Figure 2.
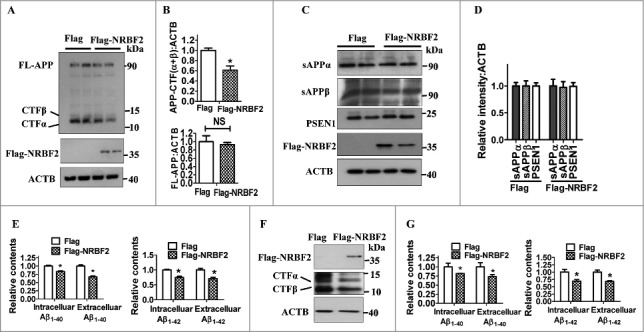
Nrbf2 overexpression reduces APP-CTFs, and intracellular and extracellular Aβ1–40 and Aβ1-42 levels. (A) After N2a mouse neuroblastoma cells stably expressing human Swedish mutant APP695 (N2S cells) were transfected with Flag-NRBF2 plasmid or vehicle plasmid, the expression of NRBF2, full-length APP (FL-APP), APP-CTFα, and APP-CTFβ were examined by western blotting; ACTB was used as an internal control. (B) Quantitative data in Fig. 2A showed that NRBF2 overexpression significantly reduced APP-CTFs levels but not FL-APP levels. (C and D) Western blotting and quantification show that Nrbf2 overexpression does not affect sAPPα, sAPPβ, and PSEN1 levels. (E) ELISA analysis results showed that Nrbf2 overexpression significantly reduces intracellular and extracellular Aβ1–40 and Aβ1-42 levels in N2S cells. (F) Overexpression of Nrbf2 in Chinese hamster ovary cells stably transfected with a plasmid encoding human APP751 bearing the V717F mutation (7PA2 cells) reduced the expression of APP-CTFs. (G) ELISA analysis results demonstrated that Nrbf2 overexpression significantly reduces intracellular and extracellular Aβ1–40 and Aβ1-42 levels in N2S cells. Quantification data are presented as the mean ± SEM, n = 3 from independent experiments. NS, not significant; *, P < 0.05 vs. the control.
After cleavage by γ-secretase, APP-CTFβ generated Aβ1–40 or Aβ1-42,4 and Aβ1–40 accounts for about 90% of all Aβ fragments generated from APP-CTFβ in amyloid plaques.36 To further determine whether the decreased APP-CTFs in NRBF2 overexpressing cells was accompanied by lower levels of Aβ, we examined Aβ1–40 and Aβ1-42 levels by ELISA assay. As shown in Fig. 2E, overexpression of Nrbf2 lowered both the intracellular and extracellular Aβ40 levels. To further confirm these results, we used a non-neuronal cell, Chinese hamster ovary (CHO) cells, stably transfected with human APP751 bearing the V717F mutation (7PA2 cells), as another AD cell model, and we obtained similar results (Fig. 2F and G). These results suggest that Nrbf2 overexpression decreases APP-CTFα and APP-CTFβ, as well as intracellular and extracellular Aβ1–40 and Aβ1-42 levels in different cells.
Depletion of Nrbf2 increases APP-CTFs and Aβ levels
To further confirm the functions of NRBF2 in APP-CTFs degradation and Aβ production, we generated Nrbf2 knockdown (KD) and nrbf2 knockout (KO) N2S cells. As shown in Fig. 3 A and B, transient transfection of Nrbf2 siRNA resulted in about a 60% reduction in the protein levels of NRBF2. Meanwhile, the cellular APP-CTFα and APP-CTFβ levels were increased about 50% in Nrbf2 KD cells (Fig. 3C and D). In addition, CRISPR-Cas9 technology was used to generate nrbf2 KO cells, and the most potent clone (clone 2) among several nrbf2 KO stable clones generated was chosen for investigating the roles of NRBF2 in APP metabolism (Fig. S1 and 3E). Consistently, KO of nrbf2 dramatically increased APP-CTFα and APP-CTFβ levels but not FL-APP levels (Fig. 3F and G). Moreover, Nrbf2 overexpression could rescue nrbf2 KO-mediated increase of APP-CTFα and APP-CTFβ levels (Fig. 3H). In line with the findings in Nrbf2-overexpression cells, nrbf2 KO did not affect sAPPα, sAPPβ and PSEN1 levels (Fig. 3I and J). Moreover, the deficiency of Nrbf2 also increased the amount of the intracellular and secreted Aβ1–40 and Aβ1-42 levels when compared to vehicle control cells (Fig. 3K). Finally, we measured γ-secretase activity by using a luciferase assay as described previously.37 The result shows that nrbf2 KO did not affect γ-secretase activity (Fig. S2). Altogether, these results indicate that NRBF2 is an important factor for regulating APP-CTFα, APP-CTFβ levels and Aβ contents, with minimal effects on the secretase cleavage of APP.
Figure 3.
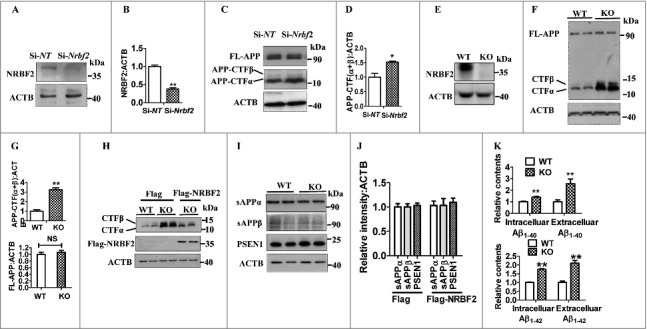
Silencing Nrbf2 increases APP-CTFs, and intracellular and extracellular Aβ1–40 and Aβ1-42 levels. (A and B) After transfecting N2S cells with Nrbf2 siRNA or nontargeting (NT) siRNA, the expression of NRBF2 was detected and quantified; ACTB was used as an internal control. (C) After Nrbf2 knockdown, the expression of FL-APP, APP-CTFα, and APP-CTFβ were detected by western blotting; ACTB was used as an internal control. (D) Quantitative data in Fig. 3C showed that Nrbf2 knockdown significantly increased APP-CTFα, and APP-CTFβ levels. (E) The expression of NRBF2 in WT and KO cells was examined by western blotting. (F) Western blotting was used to detect the expression of FL-APP, APP-CTFα, and APP-CTFβ levels after KO of nrbf2; ACTB was used as an internal control. (G) Quantitative data in Fig. 3F showed that nrbf2 KO significantly increased APP-CTFα and APP-CTFβ levels, but not FL-APP levels. (H) Overexpression of Nrbf2 rescues nrbf2 KO-mediated increase of APP-CTFs levels. (I and J) Western blotting and quantification shows that Nrbf2 overexpression does not affect sAPPα, sAPPβ, and PSEN1 levels. (K) ELISA analysis results demonstrated that nrbf2 KO significantly increases intracellular and extracellular Aβ1–40 and Aβ1-42 levels. Quantification data were presented as the mean ± SEM, n = 3 from independent experiments. NS, not significant; *, P < 0.05, **, P < 0.01 vs. the control.
NRBF2 positively regulates autophagy in neuronal N2S cells
NRBF2 was initially described as a regulator of nuclear receptors including PPARA/PPARα (peroxisome proliferator activated receptor alpha), RAR (retinoic acid receptor), and RXRA/RXRα (retinoid X receptor alpha).38,39 Our recent study and others' reports show that NRBF2 is an important component of the PtdIns3K complex and binds ATG14. However, the role of NRBF2 in regulating autophagy is still under debate.26 To test the function of NRBF2 in regulating autophagy in neuronal cells, we examined the levels of the autophagy substrate SQSTM1/p62 in Nrbf2 KD cells. We found that Nrbf2 KD significantly increased SQSTM1 levels (Fig. 4A and B). In addition, Nrbf2 KD attenuated the starvation (Earle's balanced salt solution)-induced reduction of SQSTM1 and increase of MAP1LC3B/LC3B (microtubule-associated protein 1 light chain 3 beta)-II levels (Fig. 4C and D), indicating that Nrbf2 KD attenuated starvation-induced autophagy. Similarly, nrbf2 KO also dramatically increased SQSTM1 protein levels (Fig. 4E and F) rather than mRNA levels (Fig. S3), suggesting that nrbf2 KO attenuates the degradation of SQSTM1 levels. Conversely, Nrbf2 overexpression significantly lowered cellular SQSTM1 (Fig. 4G and H). Moreover, there was also a significant increase in the number of LC3 puncta after Nrbf2 overexpression (Fig. 4 I and J). Taken together, these results demonstrate that Nrbf2 positively regulates autophagy in neuronal cells.
Figure 4.
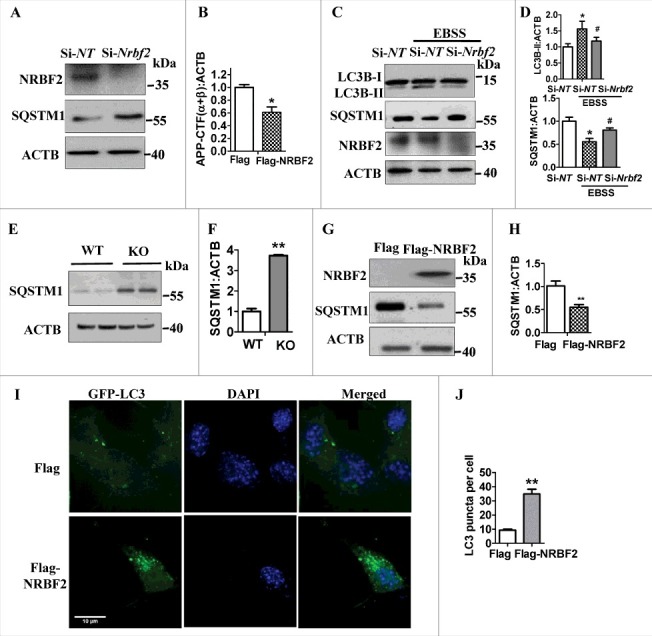
NRBF2 positively regulates autophagy. (A) After Nrbf2 knockdown, the expression of SQSTM1 was examined; ACTB was used as an internal control. (B) Quantitative data from panel A showed that Nrbf2 knockdown significantly increases SQSTM1 levels. (C) Western blotting detected the effects of starvation on the expression of LC3B-II and SQSTM1 levels after Nrbf2 knockdown; ACTB was used as an internal control. (D) Quantitative data demonstrated that Nrbf2 knockdown significantly attenuates the starvation induced increase of LC3B-II and reduction of SQSTM1/p62; ACTB was used as an internal control. (E and F) Western blotting and quantification results showed that nrbf2 KO significantly increases SQSTM1 levels. (G and H) Western blotting and quantification results showed that Nrbf2 overexpression significantly decreases SQSTM1 levels; ACTB was used as an internal control. (I and J) Effect of Nrbf2 overexpression on the formation of LC3 puncta was detected and quantified. Quantification data were presented as the mean ± SEM, n = 3 from independent experiments. *, #, P < 0.05, **, P< 0.01 vs. the relative control.
NRBF2 facilitates APP-CTFs degradation via autophagy
Autophagy has been implicated in the metabolism of APP by degradation of APP-CTFs,16,40 resulting in reduced Aβ via γ-secretase-mediated cleavage of APP-CTFβ. Because we found that NRBF2 is a positive regulator of autophagy (Fig. 4), to determine whether NRBF2-mediated autophagy is correlated with its role in regulating APP metabolism, we first tested the half-life of APP-CTFα, APP-CTFβ and FL-APP by treating cells with cycloheximide (CHX), which inhibits protein synthesis. As shown in Fig. 5A and B, the half-lives of APP-CTFs and FL-APP in nrbf2 KO cells were significantly increased compared with vehicle control cells. Importantly, the APP-CTFs:FL-APP ratio was increased in KO cells. These results suggest that the degradation of APP-CTFs in nrbf2 KO cells is impaired.
Figure 5.
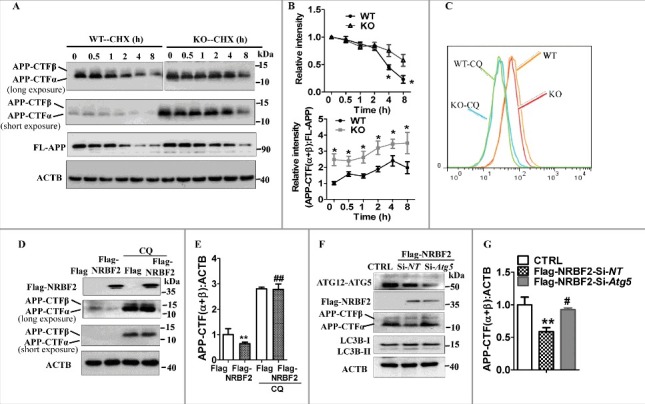
Involvement of autophagy for NRBF2-mediated the regulation of APP-CTFs levels. (A) After the protein synthesis inhibitor cycloheximide (CHX, 25 μg/ml) was added to WT and KO cells, the expression of APP-CTFα, APP-CTFβ, and FL-APP was examined; ACTB was used as an internal control. (B) Quantitative data from panel A showed that nrbf2 KO significantly attenuated the degradation of APP-CTFs, and increase the ratio between APP-CTFs to FL-APP in the presence of CHX. (C) Flow cytometry detected the effects of nrbf2 KO on the lysosomal numbers (LysoTracker Red staining) in the presence or absence of the lysosome inhibitor CQ. (D) Effect of CQ on the Nrbf2 overexpression-mediated degradation of APP-CTFs was examined; ACTB was used as an internal control. (E) Quantitative results from panel C showed that CQ blocked the Nrbf2 overexpression-mediated reduction of APP-CTF levels. (F) Effects of Atg5 knockdown on the Nrbf2 overexpression-mediated changes in the expression of APP-CTFs and LC3 levels were examined; ACTB was used as an internal control. (G) Quantitative data from panel F showed that Atg5 knockdown significantly attenuates the Nrbf2 overexpression-mediated degradation of APP-CTFs levels. Quantification data were presented as the mean ± SEM, n = 3. #, P< 0.05, **, ##, P < 0.01 vs. the relative control.
In addition, nrbf2 KO did not affect lysosomal numbers as reflected by LysoTracker Red staining (Fig. 5C), suggesting that nrbf2 KO may attenuate the delivery of APP-CTFs to lysosomes. Next, we treated Nrbf2 overexpressing cells with chloroquine (CQ) to inhibit lysosomal acidification41 and evaluated APP-CTFs abundance, we found that the Nrbf2 overexpression-mediated reduction of APP-CTFs was attenuated by CQ treatment in N2S cells (Fig. 5D and E) and in 7PA2 cells (Fig. S4). Third, we knocked down the expression of Atg5, an important factor for controlling autophagy initiation, and examined the effects of ATG5 on Nrbf2 overexpression-mediated degradation of APP-CTFs. We found that Atg5 knockdown prevented Nrbf2 overexpression-induced LC3B-II levels (Fig. 5F) as well as reduction of APP-CTFs (Fig. 5F and G). Atg5 was also important for regulating FL-APP levels (Fig. S5), which is consistent with a previous report.42 Taken together, these results indicate that autophagy is involved in NRBF2-mediated degradation of APP-CTFα and APP-CTFβ.
NRBF2 interacts with APP and NRBF2 is required for APP and APP-CTFs recruitment into autophagic structures
Using co-immunoprecipitation to examine how NRBF2 affects APP metabolism, we found that NRBF2 interacted with APP in the brains of 5XFAD AD mice (Fig. 6A) as well as in HEK293 cells (Fig. 6B). As a positive control, the interaction between NRBF2 and BECN1 was also confirmed (Fig. 6A). The interaction of NRBF2 with APP was further supported by immunostaining analysis showing the colocalization of NRBF2 with APP in N2S cells (Fig. 6C). Probably due to the fast turnover of APP-CTFs, we could not detect the interaction of NRBF2 with APP-CTFs by co-immunoprecipitation (data not shown). These results suggest the physiological association between NRBF2 and APP.
Figure 6.
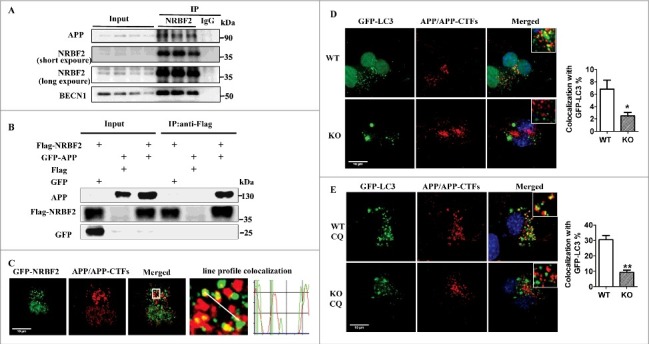
NRBF2 interacts with APP, and NRBF2 is required for APP and APP-CTFs recruitment into autophagic structures. (A) NRBF2 interacts with APP. Brain samples (n = 4) from 5XFAD mice were lysed and immunoprecipitated (IP) with NRBF2 antibody; eluates were resolved by SDS-PAGE and analyzed by immunoblotting with the corresponding antibodies. (B) After transient expression of Flag-NRBF2 or/and GFP-APP, coimmunoprecipitation of GFP-APP with Flag antibody was performed in HEK293 cells. (C) After transient expression of GFP-NRBF2 in N2S cells, the colocalization of NRBF2 with APP and APP-CTFs was analyzed by immunostaining and representative images are shown. (D-E) After transfection of WT and nrbf2 KO N2S cells with a plasmid encoding GFP-LC3, (D) followed by staining with APP C-terminal antibody, the colocalizaiton of LC3 with APP and APP-CTFs was recorded and representative pictures are shown. Quantitative data show the percentage of APP and APP-CTFs dots that colocalize with LC3 dots. (E) Colocalizaiton of LC3 with APP and APP-CTFs in WT and KO cells was recorded in the presence of the lysosome inhibitor chloroquine (CQ) and representative images are shown. Quantitative data show the percentage of APP and APP-CTFs dots that colocalize with LC3 dots. Quantification data were presented as the mean ± SEM, n = 20-25 cells from 3 independent experiments. *, P < 0.01 vs. the relative control.
Autophagy substrates are first recruited into phagophores, the precursors to autophagosomes, which then fuse with lysosomes for degradation.9 Our previous study suggested that NRBF2 is important for controlling autophagy initiation by modulating PtdIns3K activity28 and now we found that NRBF2 interacts with APP. Thus, to better understand how NRBF2 mediates autophagosome formation to affect APP metabolism, we examined the effects of NRBF2 on the recruitment of APP and APP-CTFs into autophagic structures. As shown in Fig. 6D and E, after transfecting cells with a plasmid encoding GFP-LC3, we found that the colocalization of LC3 puncta with APP and APP-CTFs was significantly reduced in nrbf2 KO cells, suggesting that nrbf2 KO attenuates the recruitment of APP and APP-CTFs into phagophores. Importantly, the sequestration of APP-CTFs within matured autophagosomes was also compromised in nrbf2 KO cells as reflected by the reduced colocalization of APP and APP-CTFs with LC3 in the presence of the lysosomal inhibitor CQ (Fig. 6E). Taken together, these results suggest that nrbf2 KO attenuates the delivery of APP and APP-CTFs into autophagic structures.
nrbf2 KO attenuates the sorting of APP and APP-CTFs into endosomal intralumenal vesicles (ILVs)
APP-CTFs can be sorted into ILVs of multivesicular endosomes, followed by fusion with lysosomes for degradation.4,43 PtdIns3K and its main product phosphatidylinositol-3-phosphate (PtdIns3P) enhance the sorting of APP and APP-CTFs into ILVs.44 Previous findings, including our study, found that NRBF2 positively regulates the production of PtdIns3P.27,28 Thus, we examined whether nrbf2 KO affects the sorting of APP and APP-CTFs into ILVs. Cells expressing the active (GTP-bound) form of RAB5, RAB5Q79L, show an enlargement of endosomes, which is commonly used to facilitate the discrimination between the limiting membrane and the lumen of these organelles.44,45 The lumen of these endosomes contains the ILVs.46 Consistent with a previous report,44 under normal conditions, APP and APP-CTFs were predominantly observed within the endosomal lumen (Fig. 7A) after overexpression of GFP-RAB5Q79L. In contrast, nrbf2 KO significantly increased the percentage of APP and APP-CTFs present in the periphery of endosomes (Fig. 7A), which means that nrbf2 KO attenuated the sorting of APP and APP-CTFs into ILVs. This notion was further confirmed by the results that nrbf2 KO impaired the distribution of APP and APP-CTFs in the RAB7- and LAMP1-positive late endosomes/lysosomes (Fig. 7C and D). Importantly, nrbf2 KO resulted in the accumulation of APP and APP-CTFs in RAB5-positive early endosomes (Fig. 7B). Taken together, these results suggest that nrbf2 KO results in the accumulation of APP and APP-CTFs in early endosomes and attenuates the sorting of APP and APP-CTFs into late endosomes/lysosomes for degradation.
Figure 7.
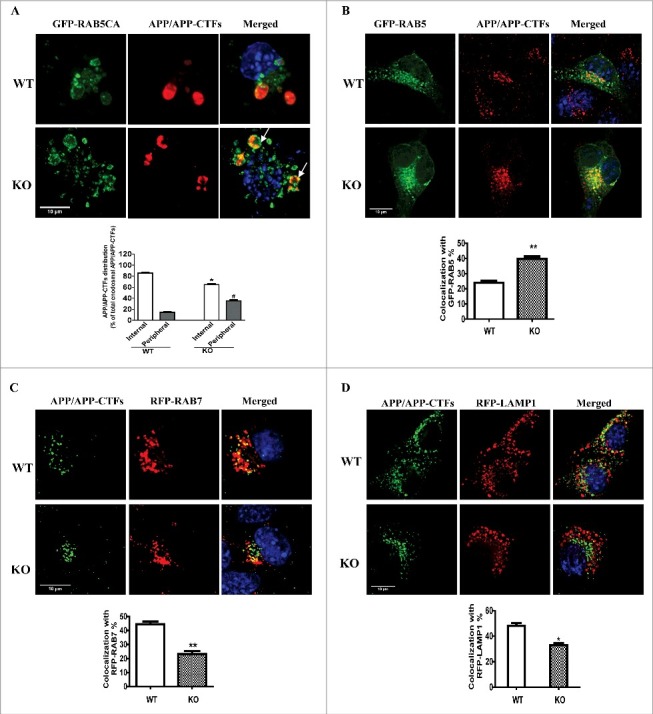
nrbf2 KO attenuates the sorting of APP and APP-CTFs into endosomal intralumenal vesicles (ILVs). (A) After WT or nrbf2 KO cells were transfected with a plasmid encoding the active form of RAB5, GFP-RAB5Q79L, followed by staining with APP C-terminal antibody and DAPI, the localization of APP and APP-CTFs in the periphery or lumen of enlarged endosomes were recorded and representative pictures were shown. Arrows indicate the colocalization of APP/APP-CTFs in the periphery of endosomes. Quantitative data showed that nrbf2 KO reduces the sorting of APP-CTFs into lumen and increases APP-CTFs in the periphery of giant endosomes. (B) After WT or nrbf2 KO cells were transfected with a plasmid encoding GFP-RAB5, followed by staining with APP C-terminal antibody and DAPI, the localization of APP and APP-CTFs with RAB5 was recorded and representative pictures are shown. Quantification results show that nrbf2 KO increases the distribution of APP and APP-CTFs in the RAB5-postive early endosomes. (C) After WT or nrbf2 KO cells were transfected with a plasmid encoding RFP-RAB7, followed by staining with APP C-terminal antibody and DAPI, the localization of APP and APP-CTFs with RAB7 was recorded and representative pictures are shown. Quantification results show that nrbf2 KO reduces the distribution of APP and APP-CTFs in the RAB7-postive late endosomes/lysosomes. (D) After WT or nrbf2 KO cells were transfected with a plasmid encoding RFP-LAMP1, followed by staining with APP C-terminal antibody and DAPI, the localization of APP and APP-CTFs with LAMP1 was recorded and representative pictures are shown. Quantification results show that nrbf2 KO decreases the distribution of APP and APP-CTFs in the LAMP1-postive late endosomes/lysosomes. The colocalization of APP and APP-CTFs with different organelle markers was presented as the percentage of APP and APP-CTFs dots which costained with differential organelle markers. Quantification data were presented as the mean ± SEM, n = 20-30 cells from 3 independent experiments. *, #, P < 0.05, **, P < 0.01 vs. the relative control.
Discussion
Autophagy dysregulation has been implicated in several neurodegenerative diseases including AD.9,47 There is an accumulation of autophagosomes in the brains of AD patients and animal models of AD.48 In addition, familial AD-associated mutations or loss of Psen1 or Psen2 impair autophagy-lysosomal functions.49–51 Consistent with these findings, autophagy dysfunction is also found in 5XFAD AD mice as reflected by the increase of both LC3B-II and SQSTM1 levels (Fig. 1). Moreover, autophagy-related proteins such as BECN1,15,17 ATG5,42 and ATG7 are associated with APP processing and Aβ metabolism. Importantly, BECN1 is reduced in brains of AD patients and APPswePS1dE9 AD mice;15,32 our results in 5XFAD AD mice also confirmed this result. Remarkably, pharmacological and genetic activation of autophagy induces APP and APP-CTFs degradation and reduces Aβ generation.12,20,21 These results highlight the crucial role of autophagy in APP metabolism. In the present study, we found that NRBF2, a component of the PtdIns3K, is reduced in the hippocampus region of 5XFAD mice, whereas ATG12–ATG5 contents remain unchanged, suggesting that NRBF2 may be involved in the AD process.
By using cellular AD models, we further demonstrated that NRBF2 plays an important role in regulating APP-CTFs homeostasis. Specifically, Nrbf2 overexpression reduces, whereas Nrbf2 depletion increases, both APP-CTFα, APP-CTFβ levels and Aβ contents (Fig. 2 and Fig. 3), whereas NRBF2 does not affect FL-APP levels. Further studies show that NRBF2 does not affect Swedish sAPPα, and sAPPβ contents (Fig. 2C and D; Fig. 3I and J). In addition, nrbf2 KO does not affect γ-secretase activity (Fig. S2). These results may indicate that proteolytic processing of APP is not the main pathway by which NRBF2-mediates the changes of APP-CTFα and APP-CTFβ levels.
Other studies as well as ours show that NRBF2 is a component of the PtdIns3K;25–28,30 however, its roles in the regulation of autophagy are controversial.25–28 In the current study, our results from Nrbf2 KD (Fig. 4A), nrbf2 KO (Fig. 4E), and Nrbf2 overexpression (Fig. 4G and I) in N2S cells support the notion that NRBF2 is a positive regulator of autophagy. These results are consistent with our previous findings in MEF cells and nrbf2 KO mice28 and others studies25,27,30 in mammalian cells as well as in yeast.29 The mechanisms by which NRBF2 induces autophagy may be related to its roles in regulating PtdIns3K activities.27,28
Next, we investigated the links between autophagy and NRBF2-mediated APP metabolism. We found that nrbf2 KO significantly increases the half-life for APP-CTFs degradation. In addition, inhibition of lysosomal activity with CQ and knockdown of Atg5 reverse NRBF2 overexpression-induced degradation of APP-CTFs. Notably, nrbf2 KO did not affect lysosomal numbers, suggesting that the delivery of APP and APP-CTFs into the lysosome for degradation may be compromised without Nrbf2 (Fig. 5). These lines of evidence support the hypothesis that autophagy is involved in NRBF2-mediated degradation of APP-CTFs. These results are also consistent with previous reports that autophagy is critical for modulating APP-CTFs degradation.52 Though FL-APP has been reported to be degraded by autophagy,20 our results showed that NRBF2 overexpression or silencing did not affect FL-APP levels (Fig. 2A, Fig. 3C and F), implying that the levels of FL-APP may not be affected by modulating NRBF2. As only slight changes in the amount of FL-APP may lead to an increase or decrease of APP-CTFs levels, we do not fully exclude the possibility that NRBF2 may affect the degradation of FL-APP to regulate APP-CTFs levels (NRBF2 overexpression or KO causes about 5% percent changes in FL-APP level but this is not statistically significant). It can be concluded that the autophagy pathway plays a major role for NRBF2-mediated changes of APP-CTFs levels.
Lysosomes mediate the clearance of cellular substrates originating from both autophagic and endocytic pathways.53 NRBF2 plays an important role in regulating autophagosome formation by modulation of PtdIns3K activities.28 Our results show that nrbf2 KO impairs the recruitment of APP and APP-CTFs into autophagic structures and ILVs (Fig. 6D and E, and Fig. 7); this impairment may finally attenuate the degradation of APP-CTFs by lysosomes. These effects may be responsible for the increased amyloidogenic processing of APP after silencing of Nrbf2. In addition to delivery into the autophagy pathway, APP and APP-CTFs can also be delivered into lysosomes for degradation by fusion of late endosomes with lysosomes.4 Thus, the sorting of APP and APP-CTFs into late endosomes also affects APP-CTFs levels and Aβ homeostasis.
One important factor responsible for APP and APP-CTFs sorting into late endosomes is PtdIns3P.44 PtdIns3P levels in the brains of AD patients and animal models are reduced. Silencing of the lipid kinase PIK3C3,44 a key enzyme responsible for PtdIns3P production, reduces APP sorting into intralumenal vesicles and enhances the amyloidogenic processing of APP. Our results suggest that nrbf2 KO reduces the sorting of APP and APP-CTFs into ILVs (Fig. 7A), this notion was further supported by the result that nrbf2 KO reduces the distribution of APP and APP-CTFs in the late endosomes/lysosomes (Fig. 7C and D). This effect may be due to the reduced production or activity of PtdIns3K because previous reports have shown that NRBF2 positively regulates the production of PtdIns3P by modulating PIK3C3 activities.27,28
Interestingly, nrbf2 KO results in the accumulation of APP and APP-CTFs in RAB5-positive early endosomes (Fig. 7B). We found that NRBF2 interacts with APP in both 5XFAD mice and in neuronal cells, and the interaction may contribute to the redistribution of APP and APP-CTFs in nrbf2 KO cells. Collectively, nrbf2 KO attenuates the degradation of APP-CTFs via reducing the recruitment of APP and APP-CTFs into both phagophores and ILVs, and increasing the distribution of APP and APP-CTFs into RAB5-positive early endosomes, which may further affect the degradation of APP-CTFs by lysosomes and the processing of APP in different organelles. These results may explain nrbf2 KO-increased amyloidogenic processing of APP. However, whether other PtdIns3K components such as BECN1 regulate APP metabolism in a similar way needs to be further explored.
Our results provide strong evidence showing that NRBF2 levels in the hippocampus of 5XFAD AD mice are reduced. Thus, it would be interesting in the future to examine whether NRBF2 levels in brains of AD patient are also reduced. We found that autophagy is important for NRBF2-mediated APP-CTFα and APP-CTFβ degradation, and the effects of NRBF2 on APP processing and trafficking are required to be further elucidated in order to fully understand the links between NRBF2 and AD. In addition, we need to understand in the near future the mechanisms by which NRBF2 specifically affect APP-CTFα and APP-CTFβ levels rather than FL-APP levels. Though we found that NRBF2 interacts with APP, and nrbf2 KO attenuates the delivery of APP and APP-CTFs into phagophores and subsequently lysosomes, other methods are required to further confirm the roles of NRBF2 in regulating APP and APP-CTFs redistribution and whether NRBF2 specifically affects the redistribution of APP and APP-CTFs, but not other endocytic proteins. In addition, how NRBF2 mediates the redistribution of APP and APP-CTFs into different organelles is largely unclear. Importantly, the connection between NRBF2 and AD needs to be confirmed by using nrbf2 KO AD animal models.
In summary, we found that NRBF2 protein levels are reduced in the hippocampus of 5XFAD AD mice. We showed that Nrbf2 overexpression reduces amyloidogenic processing of APP and that silencing of Nrbf2 increases amyloidogenic processing of APP in AD cell models. In addition, we demonstrated that NRBF2 is a positive regulator of autophagy in neuronal cells and NRBF2-mediated autophagy is critical for regulating APP-CTFs homeostasis (Fig. 8). Remarkably, we found that NRBF2 interacts with APP, and nrbf2 KO caused the accumulation of APP and APP-CTFs within RAB5-positive early endosomes and attenuated the recruitment of APP and APP-CTFs into phagophores and their delivery into ILVs for further degradation by lysosomes. Our work thus establishes an important link between NRBF2 and APP metabolism.
Figure 8.
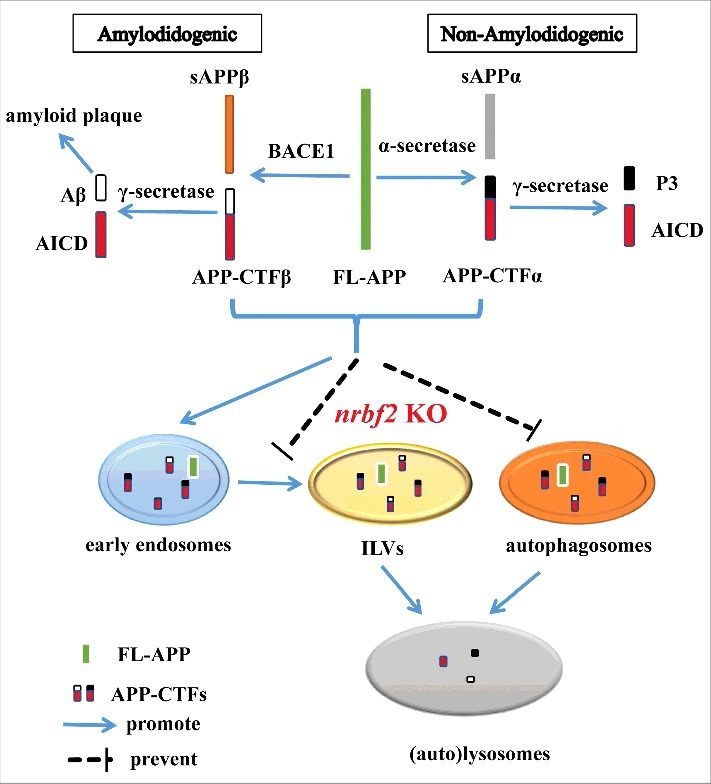
Schematic model for NRBF2 function in the autophagic turnover of APP-CTFs. Full-length amyloid precursor protein (FL-APP) can be processed by either α-secretase (non-amyloidogenic processing) or BACE1/β-secretase (amyloidogenic processing) to generate sAPPα/APP-CTFα and sAPPβ/APP-CTFβ, respectively. APP-CTFβ can be further cleaved to generate Aβ by γ-secretase. Aβ accumulation has been regarded as the critical event during the progression and pathogenesis of AD. In addition to proteolytic processing of APP to produce Aβ, autophagy regulators (e.g., NRBF2) also play a role for regulating Aβ production by degradation of APP-CTFs. By regulating autophagy and interaction with APP, nrbf2 KO attenuates the degradation of APP-CTFs via reducing the recruitment of APP and APP-CTFs into phagophores and sorting into ILVs, and increasing the distribution of APP and APP-CTFs into RAB5-positive early endosomes. AICD, APP intracellular domain; P3, P3 peptide.
Materials and methods
Reagents and antibodies
APP C-terminal antibody (51-2700) was obtained from Thermo Fisher Scientific. Anti-LC3B (NB100-2220) antibody was purchased from Novus Biologicals. Anti-ACTB/β-actin (sc-47778) was purchased from Santa Cruz Biotechnology. Anti-NRBF2 antibody was from Cell Signaling Technology (8633) and Bethyl Laboratories (A301-851A). Mouse anti-rabbit IgG (conformation specific; L27A9) monoclonal antibody (3678) were obtained from Cell Signaling Technology. Anti-SQSTM1/p62 (P0067), and anti-Flag (F3165) antibodies were purchased from Sigma-Aldrich. Anti-ATG5 (9980), and anti-PSEN1 antibodies were obtained from Cell Signaling Technology. Anti-sAPPα (2B3 clone, 11088), and anti-sAPPβ (6A1 clone, 10321) antibodies were purchased from Immuno-Biological Laboratories. Anti-Aβ1-16 monoclonal antibody (6E10, 803001) was purchased from BioLegend. Biotinylated monoclonal βA4(40)-5C3 (0060-100BIOTIN/βA4(40)-5C3, specific to a peptide corresponding to Aβ1–40) antibody was purchased from Nanotools. Flag-NRBF2 and GFP-RAB5-Q79L plasmids were used as previously described.28,54 Chloroquine (C6628) was purchased from Sigma-Aldrich. Mouse Nrbf2 siRNA and nontarget siRNA were purchased from Dharmacon. DMEM (11965-126), LysoTracker Red DND-99 (L7528), Dynabeads® protein G for immunoprecipitation (10003D), fetal bovine serum (FBS; 10270-106), Opti-MEM I (31985-070), G418 (10131-035), Alexa Fluor® 488 goat anti-mouse IgG (A-11001) and Alexa Fluor® 594 goat anti-rabbit IgG (A-11012) were purchased from Thermo Fisher Scientific.
Animal experiments
All animal experiments were approved by the Committee on the Use of Human & Animal Subjects in Teaching and Research (HASC) of Hong Kong Baptist University. 5XFAD mice overexpress the K670N, M671L (Swedish), I716V (Florida), and V717I (London) mutations in human APP (695), as well as M146L and L286V mutations in human PSEN1. 5XFAD AD and C57BL/6J mice were obtained from the Jackson Laboratory (Bar Harbor, ME, USA). Mice were housed in a pathogen-free facility under 12-h light, 12-h dark cycles with food and water provided.
Cell culture
Mouse neuroblastoma N2a cells stably transfected with human Swedish mutant were a gift from Dr. Gopal Thinakaran (University of Chicago, Chicago, IL, USA). N2S and nrbf2 KO N2S cells were maintained in 1:1 Dulbecco's modified Eagle's medium (DMEM):Opti-MEM supplemented with 5% FBS, penicillin and streptomycin and 200 μg/mL G418.35,55 Chinese hamster ovary cells stably transfected with human APP751 bearing the V717F mutation (7PA2 cells) were a gift from Prof. Dennis J. Selkoe (Harvard Medical School, Boston, MA).56 The cells were maintained in DMEM with 10% FBS, penicillin and streptomycin and 200 μg/mL G418. Cells were incubated at 37°C in a humid 5% CO2:95% air environment.56
Establishment of nrbf2 KO N2S cells
nrbf2 KO N2S cells were established using the lentivirus-mediated CRISPR/Cas9 system transduction in N2S cells. Briefly, the Nrbf2 sgRNA/Cas9 plasmid, psPAX2 (Addgene, 12260; deposited by Didier Trono) and pCMV-VSV-G (Addgene, 8454; deposited by Bob Weinberg) were co-transfected into HEK293T cell for 48 h to generate virus particles. Virus-containing medium was collected and filtered through a 0.45-μm filter (Pall, KA2DBLP6G). N2S cells in 24-well plates at 70–80% confluence, were infected with virus-containing medium for another 24 h, followed by puromycin treatment for 3 days. Surviving cells were reseeded to a 96-well plate for isolation of single cell clones. nrbf2 knockout in the expanded colonies was confirmed by western blotting.
Cell transfection
N2S cells were transfected with mouse Nrbf2 siRNA and the nontarget siRNA by using Lipofectamine RNAiMAX (Invitrogen, 13778030). The transfection with plasmids encoding Flag-NRBF2, GFP-NRBF2, GFP-APP, GFP-LC3, GFP-RAB5, RFP-RAB7, RFP-LAMP1, and GFP-RAB5-Q79L was done with Lipofectamine® LTX with Plus™ Reagent (15338030) or Lipofectamine® 3000 Transfection Reagent (L300008) from Invitrogen. After 24–72 h post-transfection, cells were used for analysis.
Determination of Aβ1–40 and Aβ1-42 contents by ELISA
The levels of both secreted and intracellular Aβ contents were determined by using a sandwich ELISA as previously described.55 Briefly, cell culture medium was collected and cleared by centrifugation for use in detecting extracellular Aβ1–40 levels. For intracellular Aβ determination, after the cell surface Aβ was removed by treating harvested cells with a solution containing 0.25% trypsin (Invitrogen, 15090046) and 0.02% EDTA, the cells were then lysed in RIPA buffer (Cell Signaling Technology, 9803) containing proteinase inhibitor (Roche Applied Science, 04693124001). For ELISA processing, the monoclonal antibody 6E-10 (Immuno-Biological Laboratories, 803001) was used as the capture antibody and was added to ELISA plates (0.2 μg per well diluted in 0.1M Na2CO3, pH 9.6) and incubated overnight at 4°C. The plates were then blocked with Block ACE (Bio-Rad, BUF029) for 2 h at room temperature. Media samples and equilibrated protein extracts from each treatment (a 100-μl volume was adjusted in all treatment groups) were applied in duplicate and incubated at room temperature for 2 h. Biotinylated monoclonal anti-Aβ1–40 5C3 (50 ng per well), which recognizes the sequence 3–6 of Aβ, was used for detection of Aβ40. Following the addition of streptavidin-conjugated horseradish peroxidase (DAKO, P0397) for 2 h at room temperature, the substrate tetramethylbenzidine (Thermo Fisher Scientific, 34028) was added to the plates. Finally, absorbance values at 450 nm were measured in duplicate wells after addition of 2 M H2SO4. Aβ1-42 contents were determined by using a commercially available kit (Thermo Fisher Scientific, KHB3441) according to the manufacturer's instructions. All ELISA experimental data were from 3 different independent experiments.
Western blotting and immunoprecipitation analysis
For cell experiments, proteins were extracted from cells by using ice-cold RIPA buffer with complete protease inhibitor mixture (Roche Applied Science, 04693124001). For animal experiments, animal tissues were homogenized in 9 volumes of ice-cold TBS (50 mM Tris-Cl, pH 7.5 150 mM NaCl) containing 0.5% SDS (Sigma-Aldrich, L3771) with complete protease inhibitor mixture. For immunoprecipitation, cells and animal brains were lysed in cell lysis buffer (25 mM Tris, pH 7.6, 100 mM NaCl, 0.5% NP40 [Sigma, 74385], 1 mM EDTA, 10% glycerol [Affymetrix/USB – J16374] with protease inhibitors). After immunoprecipitation with the indicated antibodies, Dynabeads® Protein G (Life Technologies, 10003D) were used for immunoprecipitation. Proteins were resolved by gel electrophoresis in 10–15% SDS-polyacrylamide gels, and subsequently transferred onto PVDF membranes. Following blocking with TBS-T (Tris-buffered saline with 0.1% Tween-20 [Sigma, P1379]) buffer containing 5% nonfat milk powder, the blots were probed with specific primary antibodies and secondary antibody, and finally visualized using the Pierce ECL kit (Pierce, 32106) detection reagent. ImageJ software (NIH) was used for quantification of the western blotting data.
Endogenous γ-secretase luciferase assay
The endogenaous γ-secretase activity was evaluated using cell-based γ-secretase as described previously37 with some modification. For each well of the 24-well cell culture dish, 200 ng of UAS-responsive reporter gene construct MH100, 50 ng of pRL-TK plasmid (Renilla luciferase, control reporter), and 100 ng of C99-Gal4 DNA-binding/VP16 transactivation (GVP) plasmids were transfected into wild-type (WT) or nrbf2 KO N2S cells using Lipofectamine® 3000 Transfection Reagent. After 48 h, the cells were harvested and luciferase activity was determined with the Dual-GLo™ Luciferase assay system (Promega, E2920) according to the manufacturer's instructions. DAPT (γ-secretase inhibitor; Sigma, D5942) was added at a concentration of 4 µM for 18 h as a positive control. The signal was measured according to the manufacturer's instructions. The MH-100, C99-GVP, and pRL-TK plasmids were gifts from Dr. Karlström (Department of Cell and Molecular Biology, Medical Nobel Institute, Karolinska Institute, Sweden).
Determination of lysosomal numbers using LysoTracker Red staining
The number of lysosomes was estimated using LysoTracker Red (Invitrogen, L7528) following the manufacturer's instructions. After WT and nrbf2 KO cells were treated with CQ (50 µM; Sigma, C6628) or vehicle control for overnight, lysosomes were stained by incubating cells with 50 nM LysoTracker Red for 30 min. Fluorescence intensities of 10,000 cells per sample were measured by flow cytometry (BD Biosciences).
Immunocytochemistry
Cells were seeded on coverslips placed in 24-well plates. For GFP-LC3 puncta formation assays, cells were transfected with GFP-LC3 plasmid and Flag-NRBF2 plasmid or vehicle plasmid for 48 h and then fixed with 3.7% paraformaldehyde, followed by nuclear staining with 4′,6-diamidino-2-phenylindole (DAPI; Roche, 10236276001). For LC3, RAB5, RAB7, LAMP1 and RAB5Q79L, APP and APP-CTFs colocalization assays, cells were transfected with plasmids encoding GFP-LC3, GFP-RAB5, RFP-RAB7, RFP-LAMP1 or GFP-RAB5Q79L for 48 h and then fixed with 3.7% paraformaldehyde, permeabilized in 0.2% Triton X-100 (Sigma, T8787) and blocked with 3% BSA (Sigma, B2046), followed by staining with anti-APP C-terminal antibody (1:500; Thermo Fisher Scientific, 51–2700) overnight at 4°C and then Alexa Fluor® 594 (red) or Alexa Fluor® 488 (green) secondary antibodies (1:500) for 1 h at room temperature. After nuclear staining with DAPI, the slices were mounted with FluorSave reagent (Calbiochem, 345789). Cells were visualized using a DeltaVision Deconvolution Microscope (GE Healthcare). The colocalization of APP and APP-CTFs with different organelle markers was presented as the percentage of APP and APP-CTFs dots which costained with differential organelle markers from 20–30 selected cells from 3 independent experiments with cells expressing GFP-LC3, GFP-RAB5, RFP-RAB7, or RFP-LAMP1 in each group (manually counted for the number of APP and APP-CTFs dots and costaining dots).
The method for determination of sorting of APP and APP-CTFs into ILVs was used as described previously.57 Briefly, individual large endosomes were imaged from distinct cells (total 50–70 endosomes scored per experiment per condition), whereas more than half of APP and APP-CTFs colocalization with “ring-like” RAB5Q79L (RAB5CA) was defined as “peripheral.” Data were presented as percentage of endosomes where APP and APP-CTFs were localized to the lumen and the periphery.
Statistical analysis
Each experiment was performed at least 3 times, and the results were presented as mean ± SEM. One-way analysis of variance (ANOVA) was followed by the Student-Newman-Keuls test using the SigmaPlot 11.0 software package. A probability value of P< 0.05 was considered to be statistically significant.
Abbreviations
- 5XFAD
5 familial AD mutations
- Aβ
amyloid beta peptide
- Aβ40
amyploid beta peptide 1–40
- Aβ42
amyploid beta peptide 1–42
- AD
Alzheimer disease
- APP
amyloid beta (A4) precursor protein
- APP-CTFs
APP C-terminal fragments
- ATG5
autophagy related 5
- CHX
cycloheximide
- CQ
chloroquine
- DAPI
4′,6-diamidino-2-phenylindole
- ILVs
endosomal intralumenal vesicles
- KD
knockdown
- KO
knockout
- LAMP1
lysosomal-associated membrane protein 1
- MAP1LC3B/LC3B
microtubule-associated protein 1 light chain 3 beta
- NRBF2
nuclear receptor binding factor 2
- PIK3C3
phosphatidylinositol 3-kinase catalytic subunit type 3
- PtdIns3K
class III phosphatidylinositol 3-kinase
- PtdIns3P
phosphatidylinositol-3-phosphate
- SQSTM1/p62
sequestosome 1
- WT
wild type
Competing financial interests
The authors declare that they have no competing financial interests.
Acknowledgments
This study was supported by the grants of NSFC-31500831, EF001/ICMS-LJH/2015/HKBU, SKL-QRCM-2014-2016, FDCT-022/2015/A1, FDCT-092-2015-A3 and MYRG2016-00119-ICMS-QRCM (to Jiahong Lu) and the grants of RGC/HKBU-121009/14, HMRF12132091, HMRF14150811, RC-IRMS/15-16/04, FRG II/15-16/034 and FRG II/16-17/018 (to Min Li). The authors would like to thank Dr. King-Ho Cheung and Dr. Benjamin Tong for their kind technique support and Dr. Martha Dahlen for her English editing of this manuscript.
References
- [1].O'Brien RJ, Wong PC. Amyloid precursor protein processing and Alzheimer's disease. Ann Rev Neurosci. 2011;34:185. doi: 10.1146/annurev-neuro-061010-113613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Krstic D, Knuesel I. Deciphering the mechanism underlying late-onset Alzheimer disease. Nat Rev Neurol. 2013;9:25–34. doi: 10.1038/nrneurol.2012.236. [DOI] [PubMed] [Google Scholar]
- [3].LaFerla FM, Green KN, Oddo S. Intracellular amyloid-β in Alzheimer's disease. Nat Rev Neurosci. 2007;8:499–509. doi: 10.1038/nrn2168. [DOI] [PubMed] [Google Scholar]
- [4].Haass C, Kaether C, Thinakaran G, Sisodia S. Trafficking and proteolytic processing of APP. Cold Spring Harbor Perspect Med. 2012;2:a006270. doi: 10.1101/cshperspect.a006270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R. APP processing and synaptic function. Neuron. 2003;37:925–37. doi: 10.1016/S0896-6273(03)00124-7. [DOI] [PubMed] [Google Scholar]
- [6].Hardy J, DJ S. The amyloid hypothesis of Alzheimer's disease: Progress and problems on the road to therapeutics. Science. 2002;297:353–6. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- [7].van der Kant R, Goldstein LS. Cellular functions of the amyloid precursor protein from development to dementia. Dev Cell. 2015;32:502–15. doi: 10.1016/j.devcel.2015.01.022. [DOI] [PubMed] [Google Scholar]
- [8].Ułamek-Kozioł M, Furmaga-Jabłońska W, Januszewski S, Brzozowska J, Ściślewska M, Jabłoński M, Pluta R. Neuronal autophagy: Self-eating or self-cannibalism in Alzheimer's disease. Neurochem Res. 2013;38:1769–73. doi: 10.1007/s11064-013-1082-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med. 2013;19:983–97. doi: 10.1038/nm.3232. [DOI] [PubMed] [Google Scholar]
- [10].Menzies FM, Fleming A, Rubinsztein DC. Compromised autophagy and neurodegenerative diseases. Nat Rev Neurosci. 2015;16:345–57. doi: 10.1038/nrn3961. [DOI] [PubMed] [Google Scholar]
- [11].Bento CF, Renna M, Ghislat G, Puri C, Ashkenazi A, Vicinanza M, Menzies FM, Rubinsztein DC. Mammalian Autophagy: How Does It Work? Ann Rev Biochem. 2016;85:685–713. doi: 10.1146/annurev-biochem-060815-014556. [DOI] [PubMed] [Google Scholar]
- [12].Caccamo A, Majumder S, Richardson A, Strong R, Oddo S. Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-β, and tau effects on cognitive impairments. J Biol Chem. 2010;285:13107–20. doi: 10.1074/jbc.M110.100420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Nixon RA, Yang D-S. Autophagy failure in Alzheimer's disease—locating the primary defect. Neurobiol Dis. 2011;43:38–45. doi: 10.1016/j.nbd.2011.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Boland B, Kumar A, Lee S, Platt FM, Wegiel J, Yu WH, Nixon RA. Autophagy induction and autophagosome clearance in neurons: Relationship to autophagic pathology in Alzheimer's disease. J Neurosci. 2008;28:6926–37. doi: 10.1523/JNEUROSCI.0800-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Pickford F, Masliah E, Britschgi M, Lucin K, Narasimhan R, Jaeger PA, Small S, Spencer B, Rockenstein E, Levine B, et al.. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid β accumulation in mice. J Clin Invest. 2008;118:2190–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Jaeger PA, Pickford F, Sun C-H, Lucin KM, Masliah E, Wyss-Coray T. Regulation of amyloid precursor protein processing by the Beclin 1 complex. PloS One. 2010;5:e11102. doi: 10.1371/journal.pone.0011102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Swaminathan G, Zhu W, Plowey ED. BECN1/Beclin 1 sorts cell-surface APP/amyloid β precursor protein for lysosomal degradation. Autophagy. 2016;12:2404–19. doi: 10.1080/15548627.2016.1234561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Nilsson P, Loganathan K, Sekiguchi M, Matsuba Y, Hui K, Tsubuki S, Tanaka M, Iwata N, Saito T, Saido TC. Aβ secretion and plaque formation depend on autophagy. Cell Rep. 2013;5:61–9. doi: 10.1016/j.celrep.2013.08.042. [DOI] [PubMed] [Google Scholar]
- [19].Nilsson P, Sekiguchi M, Akagi T, Izumi S, Komori T, Hui K, Sörgjerd K, Tanaka M, Saito T, Iwata N, et al.. Autophagy-related protein 7 deficiency in amyloid β (aβ) precursor protein transgenic mice decreases aβ in the multivesicular bodies and induces aβ accumulation in the golgi. Am J Pathol. 2015;185:305–13. doi: 10.1016/j.ajpath.2014.10.011. [DOI] [PubMed] [Google Scholar]
- [20].Xiao Q, Yan P, Ma X, Liu H, Perez R, Zhu A, Gonzales E, Tripoli DL, Czerniewski L, Ballabio A, et al.. Neuronal-targeted TFEB accelerates lysosomal degradation of APP, reducing Aβ generation and amyloid plaque pathogenesis. J Neurosci. 2015;35:12137–51. doi: 10.1523/JNEUROSCI.0705-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Spilman P, Podlutskaya N, Hart MJ, Debnath J, Gorostiza O, Bredesen D, Richardson A, Strong R, Galvan V. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-β levels in a mouse model of Alzheimer's disease. PloS One. 2010;5:e9979. doi: 10.1371/journal.pone.0009979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Li X, He L, Che KH, Funderburk SF, Pan L, Pan N, Zhang M, Yue Z, Zhao Y. Imperfect interface of Beclin1 coiled-coil domain regulates homodimer and heterodimer formation with Atg14L and UVRAG. Nat Commun. 2012;3:662. doi: 10.1038/ncomms1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Baskaran S, Carlson L-A, Stjepanovic G, Young LN, Grob P, Kim DJ, Stanley RE, Nogales E, Hurley JH. Architecture and dynamics of the autophagic phosphatidylinositol 3-kinase complex. Elife. 2014;3:e05115. doi: 10.7554/eLife.05115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Funderburk SF, Wang QJ, Yue Z. The Beclin 1–VPS34 complex–at the crossroads of autophagy and beyond. Trends Cell Biol. 2010;20:355–62. doi: 10.1016/j.tcb.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Cao Y, Wang Y, Saab WFA, Yang F, Pessin JE, Backer JM. NRBF2 regulates macroautophagy as a component of Vps34 Complex I. Biochem J. 2014;461:315–22. doi: 10.1042/BJ20140515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Zhong Y, Morris DH, Jin L, Patel MS, Karunakaran SK, Fu Y-J, Matuszak EA, Weiss HL, Chait BT, Wang QJ. Nrbf2 protein suppresses autophagy by modulating Atg14L protein-containing Beclin 1-Vps34 complex architecture and reducing intracellular phosphatidylinositol-3 phosphate levels. J Biol Chem. 2014;289:26021–37. doi: 10.1074/jbc.M114.561134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Young LN, Cho K, Lawrence R, Zoncu R, Hurley JH. Dynamics and architecture of the NRBF2-containing phosphatidylinositol 3-kinase complex I of autophagy. Proc Natl Acad Sci U S A. 2016;113(29):8224–9. doi: 10.1073/pnas.1603650113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Lu J, He L, Behrends C, Araki M, Araki K, Wang QJ, Catanzaro JM, Friedman SL, Zong WX, Fiel MI, et al.. NRBF2 regulates autophagy and prevents liver injury by modulating Atg14L-linked phosphatidylinositol-3 kinase III activity. Nat Commun. 2014;5:3920. doi: 10.1038/ncomms4920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Araki Y, Ku W-C, Akioka M, May AI, Hayashi Y, Arisaka F, Ishihama Y, Ohsumi Y. Atg38 is required for autophagy-specific phosphatidylinositol 3-kinase complex integrity. J Cell Biol. 2013;203:299–313. doi: 10.1083/jcb.201304123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Ma X, Zhang S, He L, Rong Y, Brier LW, Sun Q, Liu R, Fan W, Chen S, Yue Z, et al.. MTORC1-mediated NRBF2 phosphorylation functions as a switch for the class III PtdIns3K and autophagy. Autophagy. 2017;13:592–607. doi: 10.1080/15548627.2016.1269988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Jawhar S, Trawicka A, Jenneckens C, Bayer TA, Wirths O. Motor deficits, neuron loss, and reduced anxiety coinciding with axonal degeneration and intraneuronal Aβ aggregation in the 5XFAD mouse model of Alzheimer's disease. Neurobiol Aging. 2012;33:196 e29-. e40. doi: 10.1016/j.neurobiolaging.2010.05.027. [DOI] [PubMed] [Google Scholar]
- [32].François A, Bilan AR, Quellard N, Fernandez B, Janet T, Chassaing D, Paccalin M, Terro F, Page G. Longitudinal follow-up of autophagy and inflammation in brain of APPswePS1dE9 transgenic mice. J Neuroinflammation. 2014;11:139. doi: 10.1186/s12974-014-0139-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Boland B, Kumar A, Lee S, Platt FM, Wegiel J, Yu WH, Nixon RA. Autophagy induction and autophagosome clearance in neurons: Relationship to autophagic pathology in Alzheimer's disease. J Neurosci. 2008;28:6926–37. doi: 10.1523/JNEUROSCI.0800-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Vidal RL, Matus S, Bargsted L, Hetz C. Targeting autophagy in neurodegenerative diseases. Trends Pharmacol Sci. 2014;35:583–91. doi: 10.1016/j.tips.2014.09.002. [DOI] [PubMed] [Google Scholar]
- [35].Thinakaran G, Teplow DB, Siman R, Greenberg B, Sisodia SS. Metabolism of the Swedish Amyloid Precursor Protein Variant in Neuro2a (N2a) Cells Evidence That Cleavage at the “β-secretase” site occurs in the golgi apparatus. J Biol Chem. 1996;271:9390–7. doi: 10.1074/jbc.271.16.9390. [DOI] [PubMed] [Google Scholar]
- [36].Sisodia SS, St George-Hyslop PH. γ-Secretase, Notch, Aβ and Alzheimer's disease: Where do the presenilins fit in? Nat Rev Neurosci. 2002;3:281–90. doi: 10.1038/nrn785. [DOI] [PubMed] [Google Scholar]
- [37].Karlström H, Bergman A, Lendahl U, Näslund J, Lundkvist J. A sensitive and quantitative assay for measuring cleavage of presenilin substrates. J Biol Chem. 2002;277:6763–6. doi: 10.1074/jbc.C100649200. [DOI] [PubMed] [Google Scholar]
- [38].Yasumo H, Masuda N, Furusawa T, Tsukamoto T, Sadano H, Osumi T. Nuclear receptor binding factor-2 (NRBF-2), a possible gene activator protein interacting with nuclear hormone receptors. Biochim Biophys Acta. 2000;1490:189–97. doi: 10.1016/S0167-4781(99)00244-4. [DOI] [PubMed] [Google Scholar]
- [39].Flores AM, Li L, Aneskievich BJ. Isolation and functional analysis of a keratinocyte-derived, ligand-regulated nuclear receptor comodulator. J Invest Dermatol. 2004;123:1092–101. doi: 10.1111/j.0022-202X.2004.23424.x. [DOI] [PubMed] [Google Scholar]
- [40].Parr C, Carzaniga R, Gentleman SM, Van Leuven F, Walter J, Sastre M. Glycogen synthase kinase 3 inhibition promotes lysosomal biogenesis and autophagic degradation of the amyloid-β precursor protein. Mol Cell Biol. 2012;32:4410–8. doi: 10.1128/MCB.00930-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Homewood C, Warhurst D, Peters W, Baggaley V. Lysosomes, pH and the anti-malarial action of chloroquine. Nature. 1972;23:50–2. doi: 10.1038/235050a0. [DOI] [PubMed] [Google Scholar]
- [42].Tian Y, Bustos V, Flajolet M, Greengard P. A small-molecule enhancer of autophagy decreases levels of Aβ and APP-CTF via Atg5-dependent autophagy pathway. FASEB J. 2011;25:1934–42. doi: 10.1096/fj.10-175158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Thinakaran G, Koo EH. Amyloid precursor protein trafficking, processing, and function. J Biol Chem. 2008;283:29615–9. doi: 10.1074/jbc.R800019200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Morel E, Chamoun Z, Lasiecka ZM, Chan RB, Williamson RL, Vetanovetz C, Dall'Armi C, Simoes S, Point Du Jour KS, McCabe BD, et al.. Phosphatidylinositol-3-phosphate regulates sorting and processing of amyloid precursor protein through the endosomal system. Nat Commun. 2013;4:2250. doi: 10.1038/ncomms3250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Lawe DC, Chawla A, Merithew E, Dumas J, Carrington W, Fogarty K, Lifshitz L, Tuft R, Lambright D, Corvera S. Sequential roles for phosphatidylinositol 3-phosphate and Rab5 in tethering and fusion of early endosomes via their interaction with EEA1. J Biol Chem. 2002;277:8611–7. doi: 10.1074/jbc.M109239200. [DOI] [PubMed] [Google Scholar]
- [46].Stenmark H, Parton RG, Steele-Mortimer O, Lütcke A, Gruenberg J, Zerial M. Inhibition of rab5 GTPase activity stimulates membrane fusion in endocytosis. EMBO J. 1994;13:1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Kroemer G. Autophagy: A druggable process that is deregulated in aging and human disease. J Clin Invest. 2015;125:1–4. doi: 10.1172/JCI78652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Yu WH, Cuervo AM, Kumar A, Peterhoff CM, Schmidt SD, Lee J-H, Mohan PS, Mercken M, Farmery MR, Tjernberg LO, et al.. Macroautophagy—a novel β-amyloid peptide-generating pathway activated in Alzheimer's disease. J Cell Biol. 2005;171:87–98. doi: 10.1083/jcb.200505082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Shen H-M, Mizushima N. At the end of the autophagic road: An emerging understanding of lysosomal functions in autophagy. Trends Biochem Sci. 2014;39:61–71. doi: 10.1016/j.tibs.2013.12.001. [DOI] [PubMed] [Google Scholar]
- [50].Zhang X, Garbett K, Veeraraghavalu K, Wilburn B, Gilmore R, Mirnics K, Sisodia SS. A role for presenilins in autophagy revisited: Normal acidification of lysosomes in cells lacking PSEN1 and PSEN2. J Neurosci. 2012;32:8633–48. doi: 10.1523/JNEUROSCI.0556-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Reddy K, Cusack CL, Nnah IC, Khayati K, Saqcena C, Huynh TB, Noggle SA, Ballabio A, Dobrowolski R. Dysregulation of nutrient sensing and CLEARance in presenilin deficiency. Cell Rep. 2016;14:2166–79. doi: 10.1016/j.celrep.2016.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Salminen A, Kaarniranta K, Kauppinen A, Ojala J, Haapasalo A, Soininen H, Hiltunen M. Impaired autophagy and APP processing in Alzheimer's disease: The potential role of Beclin 1 interactome. Prog Neurobiol. 2013;106:33–54. doi: 10.1016/j.pneurobio.2013.06.002. [DOI] [PubMed] [Google Scholar]
- [53].Fader C, Colombo M. Autophagy and multivesicular bodies: Two closely related partners. Cell Death Differ. 2009;16:70–8. doi: 10.1038/cdd.2008.168. [DOI] [PubMed] [Google Scholar]
- [54].McKnight NC, Zhong Y, Wold MS, Gong S, Phillips GR, Dou Z, Zhao Y, Heintz N, Zong WX, Yue Z. Beclin 1 is required for neuron viability and regulates endosome pathways via the UVRAG-VPS34 complex. PLoS Genet. 2014;10:e1004626. doi: 10.1371/journal.pgen.1004626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Durairajan SSK, Liu L-F, Lu J-H, Koo I, Maruyama K, Chung SK, Huang JD, Li M. Stimulation of non-amyloidogenic processing of amyloid-β protein precursor by cryptotanshinone involves activation and translocation of ADAM10 and PKC-α. J Alzheimer's Dis. 2011;25:245–62. [DOI] [PubMed] [Google Scholar]
- [56].Podlisny MB, Ostaszewski BL, Squazzo SL, Koo EH, Rydell RE, Teplow DB, Selkoe DJ. Aggregation of secreted amyloid-protein into sodium dodecyl sulfate-stable oligomers in cell culture. J Biol Chem. 1995;270:9564–70. doi: 10.1074/jbc.270.16.9564. [DOI] [PubMed] [Google Scholar]
- [57].Gómez-Suaga P, Rivero-Ríos P, Fdez E, Ramírez MB, Ferrer I, Aiastui A, López De Munain A, Hilfiker S. LRRK2 delays degradative receptor trafficking by impeding late endosomal budding through decreasing Rab7 activity. Hum Mol Genet. 2014;23:6779–96. doi: 10.1093/hmg/ddu395. [DOI] [PubMed] [Google Scholar]