

Abstract
With multi-drug and pan-drug resistant bacteria becoming increasingly common in hospitals, antibiotic resistance has threatened to return us to a pre-antibiotic era that would completely undermine modern medicine. There is an urgent need to develop new antibiotics and strategies to combat resistance that are substantially different from earlier drug discovery efforts. One such strategy that would complement current and future antibiotics would be a class of co-drugs that target the evolution of resistance and thereby extend the efficacy of specific classes of antibiotics. A critical step in the development of such strategies lies in understanding the critical evolutionary trajectories responsible for resistance and which proteins or biochemical pathways within those trajectories would be good candidates for co-drug discovery. We identify the most important steps in the evolution of resistance for a specific pathogen and antibiotic combination by evolving highly polymorphic populations of pathogens to resistance in a novel bioreactor that favors biofilm development. As the populations evolve to increasing drug concentrations, we use deep sequencing to elucidate the network of genetic changes responsible for resistance and subsequent in vitro biochemistry and often structure determination to determine how the adaptive mutations produce resistance. Importantly, the identification of the molecular steps, their frequency within the populations and their chronology within the evolutionary trajectory toward resistance is critical to assessing their relative importance. In this work, we discuss findings from the evolution of the ESKAPE pathogen, Pseudomonas aeruginosa to the drug of last resort, colistin to illustrate the power of this approach.
Keywords: Pseudomonas aeuruginosa, colistin, antibiotic resistance, experimental evolution, bioreactor, polymorphic population, metagenomic deep sequencing
Introduction
Due to the misuse and overuse of antibiotics alongside the innate ability of bacteria to evolve, antibiotic resistance is on the rise and threatens to return us to a pre-antibiotic period. In September of 2016, the World Health Organization held a high global summit dedicated to addressing the need for novel antimicrobial development. Nearly every aspect of modern medicine relies on the availability of effective antibiotics to treat common infections. It is an unfortunate but well established fact that Hospital-Associated Infections (HAIs) are common complications of hospitalization. In 2013, the Centers for Disease Control and Prevention estimated that HAIs were the cause of over two million cases, resulting in at least 23,000 deaths in the United States.1 The organisms most strongly associated with US hospital infections and to which new antibiotics are urgently needed are frequently referred to as the no ‘ESKAPE’ pathogens (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter spp.).2,3 For these organisms, important therapeutic obstacles – resistance, toxicities, and bacteriostatic effects – limit treatment options. Most importantly, these strains and others will continue to become increasingly resistant. As we more broadly employ drugs of ‘last resort’, such as colistin for P. aeruginosa, the number of resistant strains will only increase. As multi-drug resistance spreads, the drugs of ‘last resort’ will join the list of marginally effective antibiotics, making it more difficult to ensure patient health. New drugs with novel mechanisms of action and staying power in the clinic will be essential to ensure patient health. An approach to solving this crisis is to increase the longevity of antibiotics currently in use by developing co-drugs that could be employed with antibiotics to inhibit the development of resistance and thereby increase the efficacy of current antibiotics while simultaneously reducing the spread of resistance. The development of such a co-drug strategy relies upon a clear molecular understanding of how specific pathogens evolve resistance during exposure to antibiotics.
Antibiotic resistance mechanisms are typically identified in clinical settings when an antibiotic treatment plan was unsuccessful, often leading to terrible consequences for the patient4. Even when genomic data is available from patient infections, there is little understanding of which genetic changes are the most critical to resistance since the original organism or ‘ancestor’ was not sequenced and thus there are only related genomes for comparative genomics. Even when such comparative genomics are successful and produce a list of mutations associated with resistance, there is often no clear way to classify which mutations are the most important for driving the overall evolutionary trajectory towards resistance. We set out to design an experimental approach that mimics clinical adaptation and can be used to delineate the importance of each acquired mutation in response to antibiotic therapy with the specific goal of identifying and ranking the molecular pathways and proteins responsible for resistance. Our work has benefited greatly from earlier experimental evolution studies and theory in model systems.5 The most common method traditionally uses serial flask transfers to evolve populations to resistance, but these works often focus on the final end-point strains rather than taking a metagenomic approach to elucidate and evaluate various evolutionary trajectories. Also, serial transfers select against the formation of complex biofilms, as the populations are transferred to new flasks each day, and while other approaches to study adaptation to antibiotic resistance have been used 6–8 they typically do not take into account all aspects of a growing bacterial community which includes biofilms and selection conditions that favor highly polymorphic populations.
To better understand the development of antibiotic resistance, we have designed a quantitative experimental evolution pipeline revolving around the evolution of an organism to resistance in a modified bioreactor. During adaptation to increasing antibiotic concentrations, samples of the entire heterogenous population are collected each day for metagenomic deep sequencing to quantitate the frequency of each mutation over time. At the end of the experiment, genetically diverse end-point isolates are sequenced to determine the genetic linkages between the acquired mutations. By combining the time dependent accumulation of mutations from deep metagenomic sequencing with the genotypes identified from resistant end-point isolates, we create a detailed timeline outlining how and when resistance is acquired within a given population. Using this timeline, we identify potential drug targets for the development of co-drugs that can be administered to increase the longevity of current antibiotics.
The strength of our pipeline lies in the detailed genomic analysis of our polymorphic populations. These populations give rise to multiple adaptive trajectories within a single experiment and our genomic analysis allows us to tease apart the sequence of events that result in the success of each trajectory9,10. Mutations which occur early in adaptation are likely to provide the organism with the highest fitness advantage; similarly, mutations that occur at a high frequency and are more successful across the population are most likely to contribute to resistance. Using our pipeline, we have successfully identified a variety of resistance mechanisms that recapitulate and predict resistance mechanisms found in the clinic.9–14 While resistance is more commonly conceived as a series of steps, our experiments have shown additional approaches that promote resistance: the emergence of hyperconjugation of transposable elements carrying genes needed for resistance10, or, early deletions in mutS that result in the development of a hypermutator phenotype which rapidly mutates thousands of alleles.9
Pseudomonas aeruginosa, classified by CDC as a serious threat1, is an opportunistic pathogen that can cause serious infections in immunocompromised individuals. The high level of intrinsic resistance to various antibiotics possessed by this organism15 makes it very difficult to treat infections. In this study, we describe the evolution of P. aeruginosa to colistin, a drug of last resort for Gram negative organisms. We show that in a hypermutator background, multiple mutations within the same operon lead to resistance. Furthermore, we identify mutant alleles that aid in the development of resistance but do not necessarily survive in the final resistant population.
Materials and methods
Bioreactor set up
A modified Sartorius Stedim Biostat Bplus bioreactor was used for evolution experiments. Long-term experimental evolution studies are critically dependent on absolute sterility. A single cell contaminant introduced at any point in an experiment can easily ruin the entire study and thus the utmost care must be taken in both establishing the initial population and the subsequent introduction of reagents, air and media. The empty glass vessel (1 liter volume) was sealed with all tubing and connections in place; aluminum foil was used to cover any open ends. All components were sterilized in an autoclave using a 45 minute sterilization time. The lid of the vessel has 4 inlet ports, of which two were used as media inlet ports, one as an inoculation port and one port was reserved for backup. After sterilization, the sparger delivering air to the vessel was connected to the air source via a sterile 0.2 μm air filter (Acro® 50 Vent Filters, Pall Laboratory). Spent air flowed through the condenser and was passed through a sterile 0.2 μm air filter before passing through a desiccating agent (W.A. Hammond Drierite Co. Ltd.) and entering a Tandem Pro Gas Analyzer (Magellan Biotech, Hertforshire, UK) that detected the exhaust CO2 concentration as percentage total air. The analyzer was pre-calibrated using gas tanks with known CO2 concentration. The empty media tanks that would serve as media reservoirs were pre-sterilized in an autoclave before the addition of media. The media was prepared and sterilized in an Integra Mediaclave 10 Media Sterilizer using the standard cycle. The choice of media depends on the organism under study. In general, it is a rich medium providing all the necessary resources for the bacteria to grow under conditions with no limiting resources. For P. aeruginosa, a mixture of 80% Luria Bertani broth (Becton Dickinson B214906) and 20% Brain Heart Infusion (Becton Dickinson B11060) supplemented with 2 mM magnesium sulfate was used. Antibiotics and supplements were added to the media through the addition port upon completion of the sterilization cycle. Media was then aseptically dispensed into the pre-sterilized tank. Filled tanks were set at room temperature for 16–24 hours prior to use to ensure sterility. The tubing from the media tank feeding the vessel was split into two arms. Each arm passed through a separate pump head on the Sartorius bioreactor control unit, was connected to a sterile media filter (Millipore opticap XL2 durapore) and was then connected to one of the media inlet ports on the vessel. The vessel lid had two outlet ports, one dedicated for sampling and the other dedicated to continuous waste removal. A pictorial representation of the set-up is shown in Fig. 1.
Figure 1. Evolution in a Bioreactor.

This illustration represents the workflow of an evolution experiment in a bioreactor. The culture in the vessel is maintained at exponential growth phase with fresh media and appropriate concentration of antibiotic being fed into it. The flow of media is regulated by peristaltic pumps that are programmed to be controlled by the carbon dioxide output from the vessel, which is used as a proxy for turbidity of the culture. With an increase in drug concentration, the rapidly dividing bacteria acquire mutations and form a polymorphic population of resistant bacteria.
The bioreactor was controlled using the Sartorius MFCSPID controller program. The controller was modified to use CO2 output from the gas analyzer as a signal to regulate pumps. One pump maintained a constant flow of media to the culture. The second pump was designated to respond to CO2 concentration and maintain mid-logarithmic growth. Turbidity measurements were made arbitrarily during the experiment using a sample of the effluent waste. As the effluent waste typically contains multi-drug resistant pathogens, the waste was collected and properly sterilized.
Experimental evolution in a bioreactor
The bioreactor was assembled as described above. The air inflow was set between 0.16 to 0.2 lpm and the water jacketed vessel was set to 37°C. Antibiotic-free, sterile media was pumped through the media filters and into the vessel to a predetermined volume based upon the organism being studied. This volume can be varied from 0.2 to 0.6 L. The rotor speed was set between 50 and 300 rpm (depending on the organism). P. aeruginosa was adapted as a 0.3 L culture with a rotor speed of 300 rpm. All pumps were turned off and this system was maintained for 24 to 48 hours prior to inoculation to ensure sterility. Care was taken to prevent the system from having any unnecessary openings. All inlet and outlet lines were pinched off with metal clamps when not in use. The positive pressure inside the vessel established by the sterile air flow prevented contaminants from seeping into the system. The entire system was kept in a biosafety cabinet to further maintain sterility.
The inoculum was prepared using a single colony from a freshly streaked plate of non-selective media. The single colony was suspended in 1 ml of the growth medium and added to the vessel through the inoculation port. For some fastidious organisms, 1 ml of an overnight culture started from a single colony was used as inoculum. Post inoculation, the cells grew to mid-exponential phase before pumps were turned on. Once the desired cell density was reached, the pump feeding media at a steady rate was turned on to a speed which was sufficient for maintaining culture density. As the population density rises, the CO2 produced by cellular metabolism also rises. If the CO2 rose above the set point established for mid-log, a second pump was activated at a set speed, diluting the culture to maintain it at the set point. Once the appropriate dilution was achieved, the CO2 concentration dropped, turning off the second pump.
As the culture grew for longer periods of time in the vessel, biofilms started forming. The presence of the biofilms made it difficult to determine absolute cell density in the vessel. Since biofilms did not contribute to planktonic culture, the turbidity sample did not reflect the actual number of cells in the vessel. However, the biofilms contributed to the CO2 generated in the vessel. So, the CO2 set point had to be re-set several times during the experiment based on the amount of accumulated biofilm. The culture was maintained in such a way that the planktonic population was always in mid-exponential phase. In some of the earlier experiments, samples of the culture were collected every 12 hours and frozen as 20% glycerol stocks. The sample was also streaked on the appropriate solid medium to observe colony morphology and check for contaminants. In later experiments, the sampling interval was changed to 24 hours. 15 ml of the sample was frozen as a glycerol stock and ~45 ml was pelleted down and divided into three tubes that were stored at -80°C. The supernatant was also frozen.
Selection conditions for the establishment of polymorphic populations
The goal of the experiment is to identify mutations linked to antibiotic resistance and to minimize adaptation conditions that might be the result of growth within the bioreactor environment. To avoid adaptive mutations unrelated to antibiotic selection we use non-limiting growth conditions such that the population is always experiencing excess nutrient and highly favorable growth conditions. Thus the bioreactors are not chemostats but instead allow for maximal exponential growth at all times during the experiment. After the culture established itself in the vessel, the first drug concentration was introduced in the vessel. This concentration was typically 0.25–0.5X MIC of the inoculated ancestor. After the cells were exposed to this drug concentration for 24 hours, a sample was taken and a broth MIC test was performed using the same growth medium present in the vessel. Throughout the experiment the drug concentration is maintained well below the MIC of the population and it is this sub-MIC selection that favors the evolution of multiple simultaneous evolutionary trajectories, resulting in a complex polymorphic population within each vessel. The highest drug concentration at which the cells grew to an equivalent density after 18 hours as the current drug concentration was chosen as the next increment. If there was no increase, cells in the vessel were maintained at the same concentration for an additional 24 hours before repeating the MIC test.
At the end of the experiment, the sampling process was performed as described above. The remainder of the liquid contents of the vessel was collected through the sampling port. Saving 1 ml for serial dilution, the rest was frozen away as 20% glycerol stocks in individual 50 ml tubes. The vessel was then opened and the biofilm was collected from the vessel walls and the metal portions of the lid using a sterile spatula and collected in separate tubes. The biofilm was then suspended in a small volume of growth media and used for serial dilution. The rest was frozen as a glycerol stock.
Serial dilutions of the planktonic and biofilm samples from the final day of the experiment were plated on appropriate non-selective, solid growth media. After incubation, 50 to 90 single colonies (end-point isolates) were picked from the plates for phenotypic analysis. If obvious differences in morphology were observed, a few representative colonies from each morphology were picked. If not, colonies were picked randomly. The colonies were either suspended in 20% glycerol or were used to inoculate non-selective liquid media before making 20% glycerol stocks. Various phenotypic screens were performed to characterize the end-point isolates which included but were not limited to MIC testing, testing for cross-sensitivity or cross-resistance to other antibiotics, growth rate studies, biochemical tests and biofilm assays. Based on the diversity observed in these assays, 10 to 20 end-point isolates were selected for whole genome sequencing.
Whole genome sequencing and analysis
The ancestor strain for each experiment, samples collected from each day of the experiment, as well as end-point isolates were sequenced. Genomic DNA was isolated using the MO BIO Ultraclean® Microbial DNA Isolation Kit following manufacturer’s instructions. End-point isolates were grown overnight and pelleted before DNA extraction. The frozen pellets from daily samples were thawed on ice and immediately extracted. These samples were not outgrown to prevent loss of diversity due to freeze/thaw. Genomic DNA obtained was used to make paired-end sequencing libraries using Illumina’s Nextera XT DNA library preparation kit. The Illumina HiSeq platform was used for whole genome deep sequencing of the daily population samples to obtain at least 300X coverage. The end-point isolates and ancestors were sequenced to obtain at least 100X coverage.
Comparison of the raw sequencing reads to the reference genome of the ancestor was done using Breseq13. The consensus mode was used for identifying mutations in end-point isolates and the polymorphism mode (-p) was used for the daily populations. In cases where a closed reference genome was not available, the Pacific BioSciences sequencing platform was used to sequence the ancestor and assemble a closed reference. The pile-ups for called mutations were manually examined to verify their accuracy.
Results
The quantitative experimental evolution pipeline is composed of 5 key components: 1) Evolve resistant populations in a bioreactor under polymorphic selection conditions; 2) Identify the frequency and order of mutations correlated with antibiotic resistance as a function of time; 3) Identify the genotypes of end-point isolates to establish genetic linkages; 4) Validate the effect of mutations by physicochemical characterization; 5) Rank candidates for potential drug development (Fig. 2).
Figure 2. Pipeline of quantitative experimental evolution to predict antibiotic resistance and identify targets for drug discovery.

Each stage produces essential data and approaches that when taken together predict resistance, identify the most important targets, suggests potential biochemical mechanisms, and leads to assay development. The stages are: 1) Evolve resistant populations in a bioreactor under polymorphic selection conditions; 2) Identify the frequency and order of mutations correlated with antibiotic resistance as a function of time; 3) Identify the genotypes of end-point isolates to establish genetic linkages; 4) Validate the effect of mutations by physicochemical characterization; 5) Rank candidates for potential drug development.
Identifying an adaptive trajectory to colistin resistance in P. aeruginosa
P. aeruginosa, PAO1 was evolved to colistin resistance using our quantitative experimental evolution approach in our modified bioreactor. After 26 days, we obtained a final population that was resistant to 16 mg/L colistin, 2-fold higher than the clinical breakpoint of colistin for P. aeruginosa as determined by 2014 MIC Interpretive standards set by the Clinical and Laboratory Standards Institute (CLSI). In our experiment, we observed a single point mutation in the gene encoding the DNA mismatch repair enzyme, MutS (L142P), which arose and rapidly swept through the population, eventually comprising 78% of the population on the last day. The mutation in MutS resulted in a strong hypermutator phenotype. Hypermutator phenotypes in P. aeruginosa have been observed at high frequency in clinical isolates of sputum from cystic fibrosis patients16,17. Hypermutators facilitate rapid adaptation albeit at the expense of the accumulation of many non-adaptive mutations that hitchhike together with adaptive mutations. In the presence of this rapid mutation rate, mutations specific to resistance were seen to accumulate in the two-component system, pmrAB, which has been clinically linked to colistin resistance18–20. As a hypermutator, several hundred additional mutations accumulated in the resistant population. The deconvolution of all the mutations within this hypermutator will be discussed in later work.
PmrAB is a two-component regulatory system that activates downstream lipopolysaccharide modification systems in response to cationic antimicrobial peptides21. Belonging to the polymyxin class of antibiotics, colistin is a cationic antimicrobial peptide that is believed to cause membrane damage in Gram negative bacteria22,23. Modification of the negative charges on the bacterial outer membrane by PmrAB regulated genes is postulated to allow the bacteria to protect themselves from colistin, which can bind negatively charged phosphate groups on outer membrane lipopolysaccharides24.
Quantitative analysis of the metagenomic deep sequencing data for each daily, heterogenous sample using Breseq25 allowed us to identify the frequency at which mutations in pmrAB rose and spread throughout the population (Fig. 3). Based on the predicted domain structure of the membrane bound sensor kinase PmrB18, single nucleotide polymorphisms (SNPs) leading to amino acid substitutions in the transmembrane domains (L17P, L18P, L167P, L170P), in the periplasmic domain (D47G, V136M), in the HAMP linker domain (present in Histidine kinases, Adenyl cyclases, Methyl-accepting proteins and Phosphatases) (V185A) and in the C-terminal ATP binding domain (A330P, F408L) were observed in our evolved population. Mutations in these domains have also been observed in colistin resistant clinical isolates18,26,27 as well as lab adapted colistin resistant strains of P. aeruginosa18,19,28. A SNP leading to a substitution G15S was observed in pmrA. Fig. 3 traces the frequencies of these mutations and the point at which they arose during the experiment. We observe that all these mutations start appearing only after the organism is exposed to a colistin concentration >2.25 mg/L. This suggests that there may be other mutations that occur prior to the pmrAB mutations which predispose PAO1 to colistin resistance. One unintentional benefit of hypermutators is that they can provide a very extensive survey of the entire adaptive landscape and thus provide a comprehensive catalog of mutations that may facilitate resistance.9
Figure 3. Frequencies of mutant alleles associated with colistin resistance in P. aeruginosa PAO1.
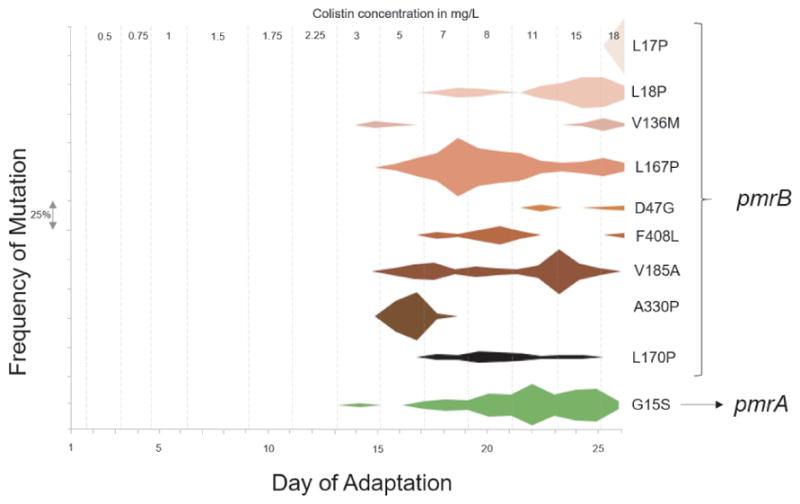
Mutations in the pmrAB operon were identified by analysis of whole genome sequencing data obtained from each daily population collected from the bioreactor. The frequency of each mutation is on Y-axis and each interval on the axis represents a frequency of 25%. The corresponding day on which the mutation is observed is on the X-axis. The grey dashed lines represent the distinct colistin concentrations the culture was exposed to during evolution. The mutations on the right are the amino acid changes in the protein caused by single nucleotide polymorphisms (SNPs) in the corresponding gene (pmrA or pmrB). Note that some mutations such as pmrB A330P have early success but then become extinct as other more successful pmrA alleles confer greater success to drug selection.
Although there are several positions on the pmrB gene that developed SNPs during the course of adaptation, not all of them persisted till the end. The final end-point isolates we sequenced had only one (L18P) out of the nine mutations observed in the daily populations. Our results suggest that our approach provides a fairly comprehensive survey of all the mutations appearing throughout the course of adaptation and limits the role of population bottlenecks in limiting the accessible evolutionary trajectories. We are currently working on deconvoluting the complex hypermutator genomic data which will not only help in identifying the weaker trajectories (<1% frequency within the population), but will also identify early mutations that will provide useful basis for providing data needed for early stage molecular diagnostics.
Discussion
The modified bioreactor we use for adaptation provides several advantages over traditional serial transfer evolution experiments. Many clinically significant bacteria form thick biofilms and bioreactor culturing can select for this formation. This long-term establishment of biofilm more accurately mimics the natural ecology that many of these organisms create. The organism studied in this work, P. aeruginosa, has a strong propensity to form biofilms, especially in the lungs of cystic fibrosis patients, where 65–80% of all infections can be attributed to biofilms 29,30. Fig. 4 shows the biofilm that developed in the bioreactor vessel during the evolution of P. aeruginosa to colistin. Additionally, it is clear that the evolutionary trajectories obtained from these studies do not address the molecular mechanisms of pathogenesis. Pathogenesis requires an appropriate host or host cell line. In vitro experimental evolution is very informative however in determining the molecular basis for antibiotic resistance. Adaptive mutations conferring antibiotic resistance have very strong effects on the fitness of the organism that typically far out-weigh those of adapting to the bioreactor growth conditions since we do not limit critical resources such as carbon and nitrogen.
Figure 4. Biofilm build up in the bioreactor vessel during evolution of P. aeruginosa to colistin.

The bioreactor design favors the development of strong biofilms. Since the evolution of the populations takes place over weeks in a single vessel, those adaptive alleles that favor biofilm formation have a selective advantage as they can adhere to surfaces and not be removed as new media is added to maintain a constant exponentially growing planktonic phase. Acinetobacter, enterococci, and Pseudomonas have all exhibited strong biofilm formation in this experimental system.
The bioreactor also maintains a continuous culture at its fastest growth rate while slowly increasing the antibiotic concentration in an empirically designed, stepwise manner. One of the major advantages of evolving resistance in a bioreactor is the evolution of a highly polymorphic population to study the subtle nuances of antibiotic resistance. This polymorphism arises from the large culture volume, continuous logarithmic growth, reduced bottleneck, and growth in sub-inhibitory concentrations of antibiotic. Bioreactor experiments are carried out with culture volumes ranging from 0.2–0.6 L, over 20 times larger than a typical flask transfer experiment, and are not passaged in 100-fold dilutions that can bottleneck populations. Flask transfer experiments also enter stationary phase each day, reducing the number of doublings. While P. aeruginosa can only achieve 6–8 generations before reaching stationary phase in batch culture when growing in a rich medium, it experiences roughly 20 generations every day in the bioreactor. This increase in replication allows for a more thorough survey of possible mutations across the genome12.
Quantitative analysis of the deep sequencing data obtained from the daily populations provides us with a comprehensive list of all mutations occurring in the population during the process of evolution and their relative frequencies in the population. It also allows us to look at the rise and fall of genotypes that help in the early adaptation of the population but may not persist at higher antibiotic concentrations due to a more favorable mutation arising and taking over the population. This is clear in Fig. 3 where early mutations like A330P and L167P within PmrB are seen at high frequencies during early adaptation but are replaced by other mutations at higher drug concentration. The appearance and persistence of a mutation relies on the fine balance between the resistance conferred by that mutation and the fitness cost associated with it. From our analysis, we can capture these unsuccessful mutations, which serve as progenitors for the more successful lineages. Having knowledge of these early mutations is useful in the clinic. With the decreasing cost of whole genome sequencing, clinicians are moving towards the sequencing approach to characterize pathogenic isolates from patients. Knowing which mutations predispose cells to becoming resistant to a particular drug is important information when deciding what antibiotics to administer as treatment.
An essential component of our analysis is the establishment of the order of mutations as well as their frequency (Fig 3). Targets for potential drug development are those identified in these critical first steps towards resistance. Work performed by C. Miller et al. on daptomycin resistance in E. faecalis shows that mutations specific to the liaFSR operon serve as an essential opening step to all the successful evolutionary trajectories leading to resistance.12 In another study by K. Beabout et al. showing the evolution of tigecycline (TGC) resistance in E. faecalis, metagenomic deep sequencing helped identify an increase in transconjugation that lead to the widespread presence of transposon Tn916, containing the TGC resistance gene, tetM..10 Another example is the evolution of the ESKAPE pathogen A. baumannii to TGC resistance.9 Several organisms have been observed to evolve a hypermutator phenotype in the clinic which suggests that their adaptive strategy involves increasing their mutation rate.31 Hypermutator phenotypes are frequently seen in TGC resistant strains of A. baumannii in the clinic, as well as in our bioreactor evolved populations of P. aeruginosa. The sheer number of mutations acquired in hypermutator populations pose a serious challenge for analysis. Thousands of non-adaptive “hitchhiker” mutations were acquired across the genome yet only a few, key mutations were responsible for the increase in TGC resistance. Metagenomic data from the daily populations as well as frequency data and statistical analysis were essential to identifying the key mutations associated with resistance in this complex genomic background.
A holistic approach that uses experimental evolution, metagenomic deep sequencing and in vitro biochemistry is also very useful for deconstructing complex strategies of antibiotic resistance. The next step in the quantitative experimental evolution pipeline is the validation of targets identified from genome sequence analysis. Ideally, gene deletions and allelic replacement are used to validate the mutations’ ability to confer antibiotic resistance. However, many bacteria do not possess the genetic tools necessary for gene validation. Also, the epistatic relations between multiple adaptive alleles can prove to be nearly impossible as even five mutations will generate 120 combinations of potential pair-wise interactions. The order of mutations from our time frequency metagenomics can help to establish the epistatic relationship of complex evolutionary trajectories. However, a combination of in vitro biochemistry, biophysics and modeling can be used to link physicochemical measurements to predictions of phenotypes such as drug resistance.32 An important goal of our work is to move beyond qualitative explanations for antibiotic resistance and towards predictive modeling to determine how specific mutations associated with resistance change protein function or amount to predict changes in drug sensitivity. Measured physicochemical data also provides vital information for the drug design process. From previous studies in our lab on daptomycin resistance in E. faecalis12, we showed that the LiaFSR three component system was crucial in conferring resistance. By studying the LiaR protein, we determined that the adaptive mutation in LiaR causing resistance (D191N), constitutively activated the protein and hence, its regulon.33 We also identified a downstream target of LiaR, a previously unidentified protein, LiaX, whose upregulation was also necessary for loss of susceptibility. These two proteins form putative targets for drug design.34
Whenever a mutation confers antibiotic resistance, this gene and the protein it encodes for can serve as a potential target for adjuvant co-drug development to limit resistance. For example, after identifying the upregulation of the liaFSR system as a major player in daptomycin resistance in E. faecalis12, we adapted strains of E. faecium with liaR deletions to daptomycin resistance. These strains evolve resistance 2 times slower than the original, susceptible ancestor (unpublished), suggesting that administering a drug to inhibit liaR during daptomycin treatment would increase the likelihood of successful treatment. While bacteria will continue to evolve resistance mechanisms, even to this two-pronged attack, the efficacy of current antibiotics would be extended while novel antimicrobials are developed. Additionally, the physicochemical assays that are used to assess how a protein promotes resistance can often be used as high-throughput assays for drug discovery. Quantitative experimental evolution also determines important adaptive mutations that arise early in treatment. With the push for personalized medicine, identifying these mutations in a patient’s bacterial infection may help clinicians administer more effective treatment plans. By a more strategic administration of antibacterials, we can slow the emergence and subsequent spread of resistance in pathogens. The wealth of data derived from our adaptive pipeline provides multiple approaches to extending the efficacy of current antibiotics and potentially slowing the rate at which resistance is observed in the clinic.
Conclusions
Using quantitative experimental evolution as a drug development pipeline, we are able to successfully recapitulate resistance mutations observed in the clinic as well as predict resistance mutations before they are observed. The CDC has classified pathogens based on their threat level and some of the pathogens categorized as serious threats include Acinetobacter, enterococci and Pseudomonas.1 We have used our quantitative evolution pipeline on these organisms and have shown that mutations observed in the clinic in drug resistant isolates of these pathogens are also observed in our bioreactor adapted strains.9,12 Moving forward, we are using quantitative experimental evolution to predict how emerging pathogens will acquire resistance to antibiotics currently in use and move us away from a reactive understanding of resistance towards a predictive one, where new drugs can be developed in anticipation of resistance rather than in response. This method also allows us to identify how likely a therapy will fail, which in turn, will lead to the design of new treatment strategies, such as adjuvant molecules that may prevent or postpone development of resistance, or combination therapies that can restore susceptibility to current drugs.
Acknowledgments
Funding
This work was supported by the Defense Threat Reduction Agency [HDTRA1-15-1-0069] and National Institutes of Health Grant [R01AI080714] to Y.S. The content of the information does not necessarily reflect the position or the policy of the federal government, and no official endorsement should be inferred.
Footnotes
Dedication
This article is dedicated to K.C. Nicolaou. His passion and dedication as a scientist are unmatched. A great colleague and an outstanding leader in the field of chemistry.
References
- 1.Centers for Disease Control and Prevention. [Accessed 3 August 2017];Antibiotic resistance threats in the United States, 2013. 2013 https://www.cdc.gov/drugresistance/pdf/ar-threats-2013-508.pdf.
- 2.Boucher HW, et al. Bad bugs, no drugs: No ESKAPE! An update from the Infectious Diseases Society of America. Clin Infect Dis. 2009;48:1–12. doi: 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- 3.Rice LB. Federal funding for the study of antimicrobial resistance in nosocomial pathogens: No ESKAPE. J Infect Dis. 2008;197:1079–1081. doi: 10.1086/533452. [DOI] [PubMed] [Google Scholar]
- 4.Munoz-price LS, Lolans K, Quinn JP. Emergence of resistance to daptomycin during treatment of vancomycin-resistant Enterococcus faecalis infection. Clin Infect Dis. 2005;41:565–566. doi: 10.1086/432121. [DOI] [PubMed] [Google Scholar]
- 5.Barrick JE, Lenski RE. Genome dynamics during experimental evolution. Nat Rev Genet. 2013;14:827–839. doi: 10.1038/nrg3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baym M, et al. Spatiotemporal microbial evolution on antibiotic landscapes. Science. 2016;353:1147–1151. doi: 10.1126/science.aag0822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Toprak E, et al. Evolutionary paths to antibiotic resistance under dynamically sustained drug selection. Nat Genet. 2012;44:101–105. doi: 10.1038/ng.1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang Q, et al. Acceleration of emergence of bacterial antibiotic resistance in connected microenvironments. Science. 2011;333:1764–1767. doi: 10.1126/science.1208747. [DOI] [PubMed] [Google Scholar]
- 9.Hammerstrom TG, Beabout K, Clements TP, Saxer G, Shamoo Y. Acinetobacter baumannii repeatedly evolves a hypermutator phenotype in response to tigecycline that effectively surveys evolutionary trajectories to resistance. PLoS One. 2015;10:e0140489. doi: 10.1371/journal.pone.0140489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beabout K, et al. Rampant parasexuality evolves in a hospital pathogen during antibiotic selection. Mol Biol Evol. 2015;32:2585–2597. doi: 10.1093/molbev/msv133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arias CA, et al. Genetic basis for in vivo daptomycin resistance in enterococci. N Engl J Med. 2011;365:892–900. doi: 10.1056/NEJMoa1011138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miller C, et al. Adaptation of Enterococcus faecalis to daptomycin reveals an ordered progression to resistance. Antimicrob Agents Chemother. 2013;57:5373–5383. doi: 10.1128/AAC.01473-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li X, et al. Rapid emergence of high-level tigecycline resistance in Escherichia coli strains harbouring blaNDM-5 in vivo. Int J Antimicrob Agents. 2016;47:324–327. doi: 10.1016/j.ijantimicag.2016.01.005. [DOI] [PubMed] [Google Scholar]
- 14.Niebel M, et al. Deletions in a ribosomal protein-coding gene are associated with tigecycline resistance in Enterococcus faecium. Int J Antimicrob Agents. 2015;46:572–575. doi: 10.1016/j.ijantimicag.2015.07.009. [DOI] [PubMed] [Google Scholar]
- 15.Murray JL, Kwon T, Marcotte EM, Whiteley M. Intrinsic antimicrobial resistance determinants in the superbug Pseudomonas aeruginosa. mBio. 2015;6:e01603–15. doi: 10.1128/mBio.01603-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oliver A, Cantón R, Campo P, Baquero F, Blázquez J. High frequency of hypermutable Pseudomonas aeruginosa in cystic fibrosis lung infection. Science. 2000;288:1251–1253. doi: 10.1126/science.288.5469.1251. [DOI] [PubMed] [Google Scholar]
- 17.Waine DJ, Honeybourne D, Smith EG, Whitehouse JL, Dowson CG. Association between hypermutator phenotype, clinical variables, mucoid phenotype, and antimicrobial resistance in Pseudomonas aeruginosa. J Clin Microbiol. 2008;46:3491–3493. doi: 10.1128/JCM.00357-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moskowitz SM, et al. PmrB mutations promote polymyxin resistance of Pseudomonas aeruginosa isolated from colistin-treated cystic fibrosis patients. Antimicrob Agents Chemother. 2012;56:1019–1030. doi: 10.1128/AAC.05829-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee J-Y, Park YK, Chung ES, Na IY, Ko KS. Evolved resistance to colistin and its loss due to genetic reversion in Pseudomonas aeruginosa. Sci Rep. 2016;6:25543. doi: 10.1038/srep25543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee JY, Ko KS. Mutations and expression of PmrAB and PhoPQ related with colistin resistance in Pseudomonas aeruginosa clinical isolates. Diagn Microbiol Infect Dis. 2014;78:271–276. doi: 10.1016/j.diagmicrobio.2013.11.027. [DOI] [PubMed] [Google Scholar]
- 21.McPhee JB, Lewenza S, Hancock REW. Cationic antimicrobial peptides activate a two-component regulatory system, PmrA-PmrB, that regulates resistance to polymyxin B and cationic antimicrobial peptides in Pseudomonas aeruginosa. Mol Microbiol. 2003;50:205–217. doi: 10.1046/j.1365-2958.2003.03673.x. [DOI] [PubMed] [Google Scholar]
- 22.Olaitan AO, Morand S, Rolain JM. Mechanisms of polymyxin resistance: acquired and intrinsic resistance in bacteria. Front Microbiol. 2014;5:643. doi: 10.3389/fmicb.2014.00643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu Z, Qin W, Lin J, Fang S, Qiu J. Antibacterial mechanisms of polymyxin and bacterial resistance. Biomed Res Int. 2015;2015:679109. doi: 10.1155/2015/679109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Joo H, Fu C, Otto M. Bacterial strategies of resistance to antimicrobial peptides. Phil Trans R Soc B. 2016;371:20150292. doi: 10.1098/rstb.2015.0292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deatherage DE, Barrick JE. Identification of mutations in laboratory evolved microbes from next-generation sequencing data using breseq. Methods Mol Biol. 2014;1151:165–188. doi: 10.1007/978-1-4939-0554-6_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barrow K, Kwon DH. Alterations in two-component regulatory systems of phoPQ and pmrAB are associated with polymyxin B resistance in clinical isolates of Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2009;53:5150–5154. doi: 10.1128/AAC.00893-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abraham N, Kwon DH. A single amino acid substitution in PmrB is associated with polymyxin B resistance in clinical isolate of Pseudomonas aeruginosa. FEMS Microbiol Lett. 2009;298:249–254. doi: 10.1111/j.1574-6968.2009.01720.x. [DOI] [PubMed] [Google Scholar]
- 28.Jochumsen N, et al. The evolution of antimicrobial peptide resistance in Pseudomonas aeruginosa is shaped by strong epistatic interactions. Nat Commun. 2016;7:13002. doi: 10.1038/ncomms13002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Flynn KM, Dowell G, Johnson TM, Koestler BJ, Waters CM. Evolution of ecological diversity in biofilms of Pseudomonas aeruginosa by altered cyclic diguanylate signaling. J Bacteriol. 2016;198:2608–2618. doi: 10.1128/JB.00048-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mcdaniel CT, Panmanee W, Hassett DJ. In: Cystic Fibrosis in the Light of New Research. Wat D, editor. InTech; 2015. [DOI] [Google Scholar]
- 31.Weigand MR, Sundin GW. General and inducible hypermutation facilitate parallel adaptation in Pseudomonas aeruginosa despite divergent mutation spectra. Proc Natl Acad Sci USA. 2012;109:13680–13685. doi: 10.1073/pnas.1205357109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Walkiewicz K, et al. Small changes in enzyme function can lead to surprisingly large fitness effects during adaptive evolution of antibiotic resistance. Proc Natl Acad Sci USA. 2012;109:21408–21413. doi: 10.1073/pnas.1209335110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Davlieva M, et al. A variable DNA recognition site organization establishes the LiaR-mediated cell envelope stress response of enterococci to daptomycin. Nucleic Acids Res. 2015;43:4758–4773. doi: 10.1093/nar/gkv321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tran TT, Miller WR, Shamoo Y, Arias CA. Targeting cell membrane adaptation as a novel antimicrobial strategy. Curr Opin Microbiol. 2016;33:91–96. doi: 10.1016/j.mib.2016.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]