

Abstract
Rotator cuff (RC) muscles undergo several detrimental changes following mechanical unloading resulting from RC tendon tear. In this review, we highlight the pathological causes and consequences of mechanical alterations at the whole muscle, muscle fiber, and muscle resident cell level as they relate to RC disease progression. In brief, the altered mechanical loads associated with RC tear lead to architectural, structural, and compositional changes at the whole-muscle and muscle fiber level. At the cellular level, these changes equate to direct disruption of mechanobiological signaling, which is exacerbated by mechanically-regulated biophysical and biochemical changes to the cellular and extra-cellular environment (also known as the stem cell ‘niche’). Together, these data have important implications for both pre-clinical models and clinical practice. In pre-clinical models, it is important to recapitulate both the atrophic and degenerative muscle loss found in humans using clinically relevant modes of injury. Clinically, understanding the mechanics and underlying biology of the muscle will impact both surgical decision-making and rehabilitation protocols, as interventions that may be good for atrophic muscle will have a detrimental effect on degenerating muscle, and vice versa.
Keywords: Rotator Cuff, Muscle Mechanics, Skeletal Muscle Biology, Muscle Mechanobiology, Muscle Atrophy, Muscle Degeneration
Introduction
Rotator cuff tears affect over 20% of the general population, with prevalence increasing with age1. Mechanical failure of the RC tendon, often preceded by pathological changes in the tendon microstucture2–6, is the primary cause of RC disease. Equally important from a clinical perspective are the resulting detrimental changes in RC muscle structure and composition, which play a prominent role in both the failure of RC tendon repairs as well as the persistent functional deficits observed in many patients7. Indeed, these muscular changes are a primary method by which RC disease is staged clinically8, based on the relative ease of noninvasive CT and MRI imaging of muscle compared to tendon and the negative relationship between intramuscular fat accumulation and clinical outcomes7; 9. Our understanding of the biological processes that govern the replacement of muscle by fat and fibrosis remains limited, but the role of altered mechanics in the progressive decline of RC muscle is central both to the existing data and prevailing hypotheses regarding RC muscle fate after tendon tear and/or repair. A summary of key factors involved in controlling and actuating skeletal muscle remodeling, which is arguably the most sensitive tissue to changes in mechanical loading in the RC, is outlined in Figure 1.
Figure 1.
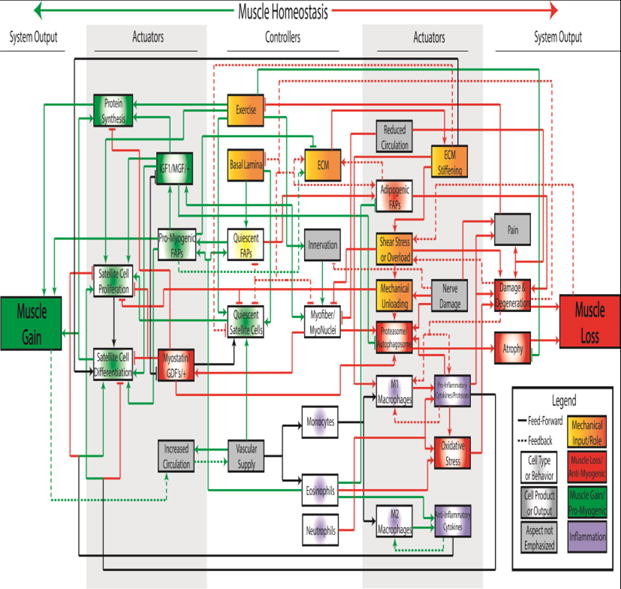
Conceptual control system diagram representing 33 key controllers and actuators of muscle gain (green) and loss (red) that may be affected in rotator cuff muscle pathology. Black lines represent processes that may contribute to muscle gain or loss, depending on context. Lines ending in arrowheads indicate a proliferative, intensifying or otherwise increasing effect on the indicated element, while lines ending in perpendicular lines indicate an inhibitory or otherwise diminishing effect. Dashed lines denote downstream processes that feed backward through the system to modulate upstream cell populations or processes. Additionally, system elements are color-coded by predominant functional category and shaded to indicate biological category, though again some elements may contribute to gain or loss in a context dependent manner. It is important to note that this is not a comprehensive map and does not show all known or possible interactions.
The phenotype of muscle disuse atrophy is primarily the result of mechanical unloading, which can occur via tendon failure, bed rest, casting, hindlimb suspension (in rodents), or otherwise decreased voluntary muscle activation10–14. Nerve injury or dysfunction may also cause atrophy, though the atrophy phenotype in denervation has a distinct molecular signature compared to disuse15. Regardless of the inciting event, atrophy is characterized by increased contractile protein turnover coupled with diminished protein synthesis15, leading to a reversible13; 16; 17 reduction in muscle force-producing capacity. In RC disease, disuse muscle atrophy, absent a nerve injury, appears to dominate the early phase of RC disease18–21 (Figure 2, Atrophic Stage).
Figure 2.
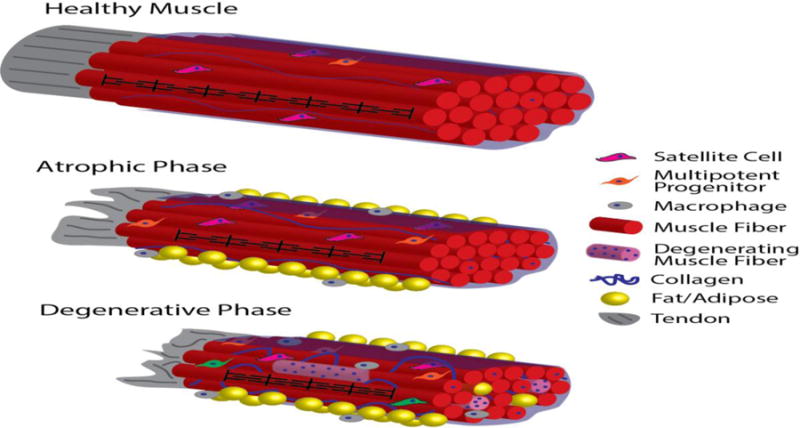
Schematic of progression of muscle pathology through different stages of RC disease. In healthy muscle with intact tendon (represented as a single fascicle connected to a tendon for simplicity), densely-packed muscle fibers are organized into fascicles by perimycium (translucent blue sheath), sarcomere length is maintained (black pattern on center-front fiber, not to scale), and muscle resident cells remain quiescent (note that cells are not to scale). With tendon disruption, RC muscle progresses to the atrophic disease stage, where muscle fibers become shorter, fiber cross-sectional area is reduced, and fat, fibrosis, and inflammation appear, while muscle architecture and overall muscle fiber and satellite cell numbers remain relatively constant. In the advanced, degenerative stage of RC disease, muscle fiber architecture is altered as sarcomeres remain chronically short, and damage and degeneration-regeneration becomes apparent (represented by the heterogeneous, hypercellular/myophagocytic light-pink fibers and centrally-nucleated, otherwise healthy fibers, respectively). The accumulation of inflammatory cells, collagen, and fat in both the inter-fascicular space (yellow spheres outside blue perimycium) and intra-fascicular space (yellow spheres between fibers) is more pronounced in the degenerative phase. Resident stem cell function is also disrupted in this stage of disease (represented in green in this schematic).
Contrary to muscle atrophy, which is self-limiting and not thought to alter overall muscle fiber structure, organization, or number22, muscle fibers can also accumulate structural damage, leading to altered sarcomere structure, degeneration and ultimately muscle fiber death (Figure 1, Damage/Degeneration). Altered mechanics are also implicated in this mode of muscle loss, though the mechanism is more complex than the unloading-induced muscle atrophy described above. For example, in other muscle pathologies including Duchenne muscular dystrophy23–25, abnormal shear stress is implicated in sarcolemmal disruption. Muscle overloading, particularly during eccentric contraction, is known to induce muscle damage and regeneration16; 17; 26–29 (Figure 1, Shear Stress or Overload). However, in these and other examples there are several mediators of muscle degeneration-regeneration that influence the rate and degree of muscle injury and recovery. These include inflammation and resident stem cell function, which are themselves influenced by passive changes in the micromechanical environment26; 30–33 (Figure 1, ECM Stiffening). In advanced RC disease, muscle degeneration and reduced muscle fiber numbers driven by these mechanobiological changes appear to be the prominent mode of muscle loss34 (Figure 2, Degenerative Stage).
Since RC disease progression can result in both unloading and overloading, the changing mechanobiological environment of the muscle can significantly impair the body’s attempt at regeneration, and this in turn may explain the irreversible loss of muscle and poor surgical outcomes that define RC disease. In this review, we summarize our current understanding of biomechanical environment changes and specific aspects of that environment that contribute to the clinically intractable deficits that result from chronic RC tear at the whole muscle, muscle fiber, and single cell levels. In this context, we will highlight differences between the atrophic and degenerative aspects of disease, focusing on the roles of mechanical unloading and matrix stiffening and their detrimental effects on muscle physiology at each length scale.
Whole Muscle Remodeling After RC Injury
As RC tendon tears progress over time, the RC muscles become mechanically unloaded and begin to retract. Mechanical unloading is potentially compounded by pain-induced diminution of voluntary muscle contraction. Suprascapular nerve dysfunction may also impact muscle remodeling, though the prevalence of neuropathy in RC tears appears to be limited35. As the muscle retracts, muscle fibers shorten via serial sarcomere subtraction36 and the muscle becomes fibrotic37 (discussed in further detail below). This causes the working range of the muscle to decrease, and it has been hypothesized to increase RC muscles’ susceptibility to injury during repair given the high muscle tension and strain required to repair the retracted tendon to its footprint38; 39.
A second consequence of chronic muscle retraction is increased muscle fiber-bundle stiffness37; 40. Because single fiber passive mechanics are not altered with RC tear, this stiffening is likely attributable to increased fibrosis, i.e. collagen content in the muscle40 (Figure 1, ECM Stiffening). While fibrosis predicts stiffness to a degree37; 40, the existing data suggests that muscle stiffness is dictated by additional variables. One possible contributor is reduced sarcomere number following tear, which would lead to higher average sarcomere strains for a given change in muscle length36; 41. Another possibility is that ECM modification, and not just content, dictate total ECM stiffness (and cell-ECM interactions). Specifically, increased collagen cross-linking (associated with increased stiffness42) has been demonstrated in degenerated RC tendon43, though it has yet to be measured in RC muscle. Whether or not increased matrix stiffness is protective (via increased load distribution in passive stretch) or detrimental (via increased active shear stress) to muscle fibers remains unknown.
In contrast to tissue-level stiffening, changes in whole muscle biomechanics are more complicated. Similar to findings in fiber bundles, a human case series44 and two separate animal models21; 45 found increased whole muscle stiffness over time following tendon transection. However, a cadaver study that involved samples with higher fat infiltration found that high fat accumulation correlated with softer muscle overall46. A possible explanation for these findings is that whole muscle stiffness tracks with fiber bundle stiffness until muscle tissue loss hits a critical threshold, at which point whole muscle mechanics are dictated by a combination of muscle- and non-muscle tissue mechanics. Indeed, one prevailing hypothesis is that accumulation of adipose tissue serves as a necessary mechanical buffer to counter changes in muscle volume and pennation angle, in order to preserve a maximum possible amount of muscle force producing capacity47. As RC disease progresses and stiff muscle40; 48 is replaced by more extensible fat49, overall organ extensibility increases, even though there is no evidence suggesting that remaining muscle tissue becomes more extensible itself. The resulting tissue is a complex and compartmentalized biomechanical system, which can in turn give rise to significant but localized changes in the micromechanical environment that impact both individual muscle fibers and resident stem cells.
Mechanical (Un)Loading Drives Muscle Fiber Pathology
The paradigm of mechanical unloading followed by muscle remodeling, progressive muscle atrophy, and fat accumulation has been central to RC muscle research for over two decades8. In the literature, this process is often interchangeably referred to as ‘fatty infiltration’, ‘fatty atrophy’, and ‘fatty degeneration’, with the latter two referencing specific modes of muscle loss. In this section, we discuss the effects of RC tear on individual muscle fibers, highlighting the biological importance of differentiating muscle atrophy from degeneration and focusing on the differential role that mechanical loads play in each of these distinct mechanisms of muscle loss.
In parallel with alterations at the whole muscle level, individual muscle fibers undergo several detrimental changes as a result of chronic unloading, with distinct stages across the disease spectrum. In terms of muscle architecture, muscle fibers in full-thickness tears have decreased sarcomeric order and increased accumulation of small (<1μm) intracellular lipid droplets50, but normal sarcomere length41 is maintained via serial sarcomere subtraction36; 51. However in large or massive (>5cm retraction) RC tears, normal sarcomere remodeling is not observed, and sarcomeres are significantly shorter than in either intact or full thickness tears36. The implications of these findings are two-fold: first, this suggest that muscle with moderate tears are more likely than larger tears to remodel and remain functional after repair, and second, that repair of massive tears may fail due to a combination of over-strained sarcomeres immediately after repair and failure to adapt sarcomere length over time36.
Beyond architectural changes, RC muscle undergoes continuous muscle loss throughout the course of disease. In the ‘early’ phase of disease, radial atrophy of the RC muscle occurs as mechanical load is reduced following smaller and/or less chronic RC tendon tear18; 19 (Figure 1, Mechanical Unloading, Atrophy; Figure 2, Atrophic Stage), possibly heightened by passive stress shielding resulting from the stiffer matrix discussed above, or inflammatory signals known to activate catabolic pathways within muscle discussed below10; 11; 15; 52. This atrophic muscle loss encompasses a cell-intrinsic, self-limiting process where protein turnover is tightly controlled by the interplay of anabolic52 and catabolic15 pathways; it is not generally considered a putative mechanism of muscle fiber deletion22. With decreased load, strain-dependent signaling proteins at the Z-disk53 and costamere54 become less active, leading to decreased anabolic signaling52; 55 and increased signaling and activity of the two principal mechanisms of protein degradation in the cell: the ubiquitin-proteasome55–57 and the autophagosome/lysosome15 (Figure 1, Protein Synthesis, Proteasome/ Autophagosome). In muscle atrophy, proteins are freed from the Z-disk via the combined action of calpains58; 59 and muscle-specific E3 ubiquitin ligases56. These freed contractile proteins are subsequently degraded, thus reducing the force-producing capacity of the muscle. Importantly, protein loss in muscle atrophy is highly specific; only ubiquitinated proteins are degraded, while proteins still anchored to the Z-disk are protected from the catabolic machinery and overall muscle architecture is preserved.
As RC disease progresses, the muscle is more likely to experience higher axial loads (due to diminished total cross-sectional area)60 and higher shear forces when activated (due to increased stiffness of the matrix)61; 62. Both axial and shear stress are known to cause direct mechanical injury to the sarcolemma23; 27 (Figure 1, Shear Stress or Overload). This, along with potential increases in permeability caused by local inflammatory cells (discussed below)61; 63, leads to increased calcium in the muscle fiber because of direct diffusion and sarcoplasmic reticulum disruption64; 65. Increased calcium may then increase calcium-sensitive calpain activity, which along with their role in contractile protein turnover in atrophy65; 66 are also implicated in cellular apoptosis67; 68 and necrosis69. Together with the direct overload/shear injury, these indirect mediators of protein turnover and cell death (as well as the hypothesized increase in oxidative stress and a protease-rich inflammatory environment discussed below) likely explain the recently described muscle damage and degeneration of muscle fibers found in advanced rotator cuff disease34 (Figure 1, Damage and Degeneration).
Can Satellite Cells Maintain Muscle Homeostasis Post-Injury?
Satellite cells (SCs), the primary myogenic stem cell of adult muscle, play an integral role in adult myogenesis and homeostasis, particularly in the context of muscle’s response to exercise/reloading26; 70 and recovery from damage or injury31; 71 (Figure 1, Quiescent Satellite Cells, Satellite Cell Proliferation/Differentiation). Notably, while SCs are required for muscle regeneration, they are dispensable in the context of fiber hypertrophy72; 73. Given the muscle degeneration present in advanced RC disease34, understanding the fate of SCs in RC disease is paramount to understanding why muscle loss is generally irreversible after RC tear.
Like many other adult stem cells, the SC is highly dependent on it’s physical environment, i.e. niche74. This niche consists of the region between the sarcolemma and the basal lamina of the muscle fiber75, with SC fate determined by dynamic interactions with each structure76 (Figure 1, Basal Lamina, ECM). The physical and biochemical composition of the niche is important in the maintenance of SC quiescence77, potency78, and possibly even myogenic fate79, with ECM proteins and particularly collagens IV and VI playing a notable role in the regulation of SCs by the basal lamina80; 81. The dependency of SC function on niche composition suggests a central role of force transmission in the maintenance of SC stemness. Changes in matrix composition directly alter the way SCs experience load based on local niche stiffness78, and may also affect SC focal adhesions based on altered integrin-binding substrate availability82. Alterations in the matrix may also alter SC function indirectly because the matrix serves as a local reservoir and co-modulator of growth factors critical to SC modulation78; 83; 84 (Figure 1, IGF1/MGF/+, Myostatin/GDF’s/+). As mechanical loads change, the matrix undergoes damage and remodeling which determine the rate of release and/or sequestration of these factors85; 86.
Indeed, SC function is highly dependent on niche stiffness (Figure 1, ECM Stiffening). During in vitro culture, SCs proliferate, self-renew, and differentiate best on substrates with normal muscle stiffness (~11kPa)48; 87. On stiff substrates (~106 kPa) differentiation into myotubes is favored87, while the maintenance of quiescence is favored on softer substrates (~2kPa)77. In RC disease, increased matrix stiffness may skew SCs toward differentiation and away from self-renewal, which may deplete the SC pool and diminish the muscle’s regenerative capacity in the long run88. However, evaluation of this hypothesis is difficult, because although RC muscles have reduced SC populations compared to other muscles89, once SCs are removed from the pathological environment of the torn RC, their proliferation, differentiation, and fusion characteristics are not different between no-tear and full-thickness tears89.
Beyond the static stiffness of the niche, mechanical loading is also important in the maintenance and activation of the SC pool (Figure 1, Exercise, Mechanical Unloading, Satellite Cell Proliferation/Differentiation). SCs can be activated by mechanical loading in vitro32 and exercise in vivo, though the effects of exercise are multifactorial and the relative contribution of mechanical load to overall exercise-induced activation is difficult to determine in vivo16; 26; 90 (Figure 1, Exercise). The complementary finding is also true; in most models, unloading reduces total SC number and potency of remaining SCs11. Therefore, reduced muscle activity levels following RC tear in the atrophic stage of disease may ultimately contribute to diminished regenerative capacity in the degenerative phase of disease.
Further complicating the evaluation of SCs in the context of RC disease are other potential environmental contributors to SC dysfunction known to occur in the torn RC. The torn RC environment demonstrates altered expression of soluble factors91–94 as well as increased inflammation34; 95; 96 and oxidative stress97–99, which are all potent mediators of SC function30; 100; 101 that may be exacerbated by muscle injury. Pro-inflammatory signaling and oxidative stress stimulate SC proliferation, but blocks differentiation100; 101 and self-renewal102 via the TNF/TWEAK/NF-κB and Notch signaling axes, while anti-inflammatory signals aid in differentiation103 (Figure 1, Monocytes, M1/M2 Macrophages, Eosinophils, Neutrophils, Pro-Inflammatory Cytokines/Proteases, Oxidative Stress, Anti-Inflammatory Cytokines). These effects may also be modulated by interplay of other myogenic regulatory factors that are likely altered in the torn RC, including members of the TGF-β superfamily (GDF and BMP subfamilies in particular)52; 104 and IGF/GH axis105–107, all of which influence SC dynamics (Figure 1, IGF1/MGF/+, Myostatin/GDF’s/+). While classically ‘pro-‘ and ‘anti-‘ inflammatory or myogenic signaling suggests a binary effect (i.e. either ‘good’ or ‘bad’), the effects of these factors on muscle homeostasis are dependent on the environment as a whole. For example, in acute inflammatory situations, these factors are coordinated to promote muscle homeostasis, as proliferation of SCs occurs prior to fusion, regeneration, and return to quiescence30; 76; 108; 109. However, with chronic inflammation, prolonged pro-inflammatory and anti-myogenic signaling may simultaneously inhibit muscle regeneration and SC pool maintenance, leading to both short- and long-term deficits in muscle regenerative capacity.
Multipotent Progenitors – Potential Sources of Fat and Fibrosis
Progressive fat accumulation and fibrosis in RC tears at the whole-muscle level is indisputable, but the cell population(s) responsible remain unresolved. Further complicating our understanding of fat accumulation mechanisms is the finding that lipid-filled cells accumulate in distinct locations (epimuscular, interfascicular, and intrafascicular) within the muscle structure34; 110, where lipid in different anatomical locations may be the result of disparate cellular processes or cell populations. SCs are theoretically capable of adopting an adipogenic fate111; 112, and the lipid content of mature muscle fibers is known to increase with tear50. However, there is no current consensus on whether SCs can trans-differentiate into adipocytes in vivo113; 114, and at a minimum the niche must be significantly manipulated in vitro to alter SC fate115; 116.
Multipotent adipose stem cells (ASCs) have also been implicated in fat accumulation, particularly at the epimuscular border110. In this region, adjacent to existing fat depots, there is evidence that RC tear leads to a whitening of the normally beige epimuscular fat depot, with implications for paracrine signaling that may affect muscle regeneration110. Yet fat accumulation does not strictly proceed from the muscle border, particularly in more advanced disease117. As such, two additional native muscle stem cell populations, fibro/adipogenic progenitors (FAPs) and perivascular stem cells/pericytes have been the focus of research into the source of fat in torn RC muscle. A comprehensive discussion of the role of these cell types in skeletal muscle is beyond the scope of this review and can be found elsewhere118; 119.
The bipotent FAP population, first described independently by Uezumi120 and Joe121 in 2010, is the most attractive candidate of the muscle resident cells implicated in RC disease progression (Figure 1, Quiescent FAPs). With an indispensible role in normal muscle regeneration (where they synthesize new, regenerative matrix)122; 123 and ability to differentiate into either adipocytes or fibroblasts120; 121 (Figure 1, Pro-Myogenic FAPs, Adipogenic FAPs), dysfunction of FAP’s could be implicated in all three detrimental changes in torn RC (muscle loss, fibrosis/stiffening, and fat accumulation). Indeed, the conditions under which FAPs take on an adipogenic fate exist in the inflammatory environment of the torn human RC123, and a small animal model has shown potential for adipogenic differentiation of FAPs in the context of RC tear124.
Unlike FAPs, pericytes are a heterogeneous population125, are not muscle specific, and are capable of differentiating into a wider array of mesenchymal cells, including skeletal muscle126, adipocytes127, fibroblasts128; 129, and others127; 130. In non-muscle tissues, matrix stiffening causes pericytes to adopt a myofibroblast/fibrotic phenotype128; 129, which leads to a positive fibrotic feedback loop131. In the same small animal RC tear model used to evaluate FAP fate, pericytes were found to contribute to both adipogenic and fibrogenic cell populations124, though more stringent cell classification and more clinically relevant models (discussed below) are needed to definitively characterize and study the source(s) of adipocytes in RC disease.
A caveat to the findings presented in this section is that, due to a lack of a definitive marker for either FAPs or pericytes, studies of these cell populations are inherently studies of mixed populations that share a small number of specific markers. Of particular note is the overlap between markers for pericytes, FAPs, and many other subpopulations that are generally considered mesenchymal stem cells132. One such marker, PW1, is a good example of this conundrum; PW1+ stem cells have been found in many tissue types133 and share many other markers and characteristics with pericytes, including myogenic potential134, yet PW1+ cells and classical pericytes are distinct populations135. As such, differentiating between these cell populations remains a challenge, and complicates efforts to identify or describe a unique cell population primarily responsible for fat accumulation in RC disease, if such a population exists.
Mechanobiology of Inflammatory Cells – Regulating the Regulators
Infiltration and persistence of inflammatory cells is a key feature of the RC disease process in both tendon5; 136 and muscle34; 95. Understanding inflammatory cell population dynamics is particularly important given the complex role of timing and specific inflammatory milieu in modulating the changes in muscle fibers and resident stem cells described in the preceding sections. Here, we will highlight findings in the broad field of inflammatory cell mechanobiology137; 138 as they relate to the pathology of RC tears.
Inflammatory cells are known to accumulate in the tendon in the earliest stages of disease, where they are thought to modulate the degeneration of the tendon that eventually leads to muscle unloading3; 96; 98; 139. Therefore, it is possible that the original inflammatory response observed in RC muscles is the result of local degeneration-related signaling from the tendon. While it is unclear if RC tendon and muscle share common degenerative mechanisms at the cellular or molecular level, based on the high degree of physical and biochemical cross-talk between the two tissues140; 141 it is possible that inflammatory or degenerative findings in muscle may inform research in tendon, and vice versa.
Of the inflammatory cell populations, the myeloid class generally and monocyte/macrophage subpopulation in particular is commonly implicated in modulating skeletal muscle responses to injury and disease142 (Figure 1, Monocytes, Eosinophils, Neutrophils, M1/M2 Macrophages). While the complexity of the inflammatory milieu is beyond the scope of this review and has been reviewed elsewhere143; 144, in general, when muscle fibers and surrounding matrix are damaged in healthy muscle, myeloid cells are recruited to the injury region by a combination of released cytokines/chemokines and subsequent increased matrix permeability145. This acute inflammation is marked by M1 macrophage polarization146 and release of pro-inflammatory cytokines142 and proteases147. Resolution, characterized by M2 macrophage polarization146 and expression of anti-inflammatory and often pro-fibrotic and pro-survival cytokine and growth factor release, quickly follows in non-pathological situations103. This normal regenerative cascade appears to be altered in RC disease.
Increased matrix stiffening and matrix remodeling in RC disease likely influence both inflammatory cell migration148 and cytokine release profile, skewing toward reduced migration speed149 and prolonged, enhanced release of pro-inflammatory cytokines150 on stiffer matrix (though these findings have not been evaluated specifically in RC muscle). While in some diseases abnormal inflammation alone is enough to drive muscle damage and degeneration151, it remains to be seen whether pro-inflammatory cells and signals are active participants in RC muscle degeneration, or if persistent inflammation is the result of continual muscle injury.
Clinical Considerations and Concerns of Pre-Clinical Models
These findings have important clinical significance, but many questions still remain. Muscle atrophy and degeneration are fundamentally different mechanisms of muscle loss, and must thus be treated with fundamentally different interventions. From a rehabilitation perspective, selecting the correct intervention is complicated by the existence of both modes of muscle loss along the spectrum of disease. Mechanical reloading (i.e. exercise and/or tendon repair) is generally sufficient to recover from atrophy13; 16; 17; 152, including limited evidence for this finding in RC153. But in a stiff, degenerating muscle, exercise and the damage that may result from mechanical overload may accelerate the pace of muscle loss23; 25; 34. In torn RC this is particularly concerning, as the normal inflammatory and regenerative processes that are associated with positive muscle changes following exercise are likely impaired30; 102; 150.
In terms of surgical intervention and repair, the altered muscle architecture and increased stiffness must be considered, particularly in chronic, massive tears154–156. These pathological changes suggest that repair of large tears may be futile from a functional standpoint, as repair may cause more damage38 and the muscle may not adapt sarcomere number and length normally36. Tendon repair alone is also unlikely to ameliorate the pro-degenerative/anti-regenerative environment of the chronically torn cuff, further diminishing the likelihood of successful functional restoration in large and massive tears. Given these findings, two issues still remain: our ability to reliably measure these important biological phenomena clinically, and a framework in which this type of biomechanical and biological information can be used for clinical and surgical decision-making.
Two final confounding issues in studying the biomechanical and cellular regulators of RC disease are discrepancies between animal models and human patients or patient-derived cells, and inconsistencies in findings between different laboratories. Briefly, while large animal models recapitulate the muscle stiffening and atrophy found in the early phase of RC disease18; 19; 21; 40; 45, small animal models require a nerve injury (denervation) to achieve a similar phenotype157–159. However, nerve injury (denervation in particular) is a relatively rare feature of clinical RC disease, and may confound results from such models as the mechanisms of denervation atrophy and disuse atrophy are not equivalent15. To date, no animal model has demonstrated the accumulation of muscle damage and cellular degeneration found in humans, and only the sheep model seems to accumulate fat to a degree that is similar to patients with advanced disease21. This is problematic when trying to translate animal research to the clinic, as preclinical models do not share the disease processes that govern the most intractable clinical cases. Complicating this issue are differences in cell markers and behavior between humans and animals, differences in patient demographics between human studies, and inconsistencies in isolation methods that are used to identify SCs, FAPs, and pericytes. This leads to potentially significant and confounding discrepancies between the cell populations evaluated from model to model and study to study, and likely contributes to the conflicting effects of various factors on resident cell dynamics reported in different publications.
Conclusions
Skeletal muscle is in a constant state of dynamic equilibrium between contractile protein gain and loss. In RC disease, the role of the macro- and micro-mechanical environment is paramount. The initial insult to the muscle is fundamentally an unloading event, where tendon tear and pain lead to direct mechanical unloading and indirect disuse, both of which lead to muscle atrophy. The secondary and tertiary effects of the initial unloading insult are also mechanically sensitive. With prolonged retraction of the muscle, the matrix becomes stiff, leading to potentially increased incidence of muscle damage while simultaneously creating an environment in which regeneration is likely inhibited, which may help explain the progressive, irreversible muscle loss due to degeneration observed in RC disease.
Acknowledgments
We would like to acknowledge funding from the National Institutes of Health (NIAMS T32-AR060712, M.C.G. and NICHD R01-HD073180 S.R.W.) and the Muscular Dystrophy Association (241665 A.J.E.).
Footnotes
We have no conflicts of interest to disclose.
Author Contributions:
M.C.G. performed the literature search, manuscript synthesis, and figure preparation
A.S. participated in manuscript preparation and provided clinical and translational expertise/perspective for all sections
A.J.E. participated in manuscript preparation and provided focused expertise in cellular and molecular mechanobiology
S.R.W participated in manuscript preparation and provided focused expertise in whole muscle and single fiber mechanobiology
References
- 1.Yamamoto A, Takagishi K, Osawa T, et al. Prevalence and risk factors of a rotator cuff tear in the general population. Journal of Shoulder and Elbow Surgery. 2010;19:116–120. doi: 10.1016/j.jse.2009.04.006. [DOI] [PubMed] [Google Scholar]
- 2.Brooks C, Revell W, Heatley F. A quantitative histological study of the vascularity of the rotator cuff tendon. Journal of Bone & Joint Surgery, British. 1992;74:151–153. doi: 10.1302/0301-620X.74B1.1732247. [DOI] [PubMed] [Google Scholar]
- 3.Longo UG, Franceschi F, Ruzzini L, et al. Histopathology of the supraspinatus tendon in rotator cuff tears. The American journal of sports medicine. 2008;36:533–538. doi: 10.1177/0363546507308549. [DOI] [PubMed] [Google Scholar]
- 4.Lundgreen K, Lian Ø, Scott A, et al. Increased levels of apoptosis and p53 in partial-thickness supraspinatus tendon tears. Knee Surgery, Sports Traumatology, Arthroscopy. 2013;21:1636–1641. doi: 10.1007/s00167-012-2226-9. [DOI] [PubMed] [Google Scholar]
- 5.Matthews T, Hand G, Rees J, et al. Pathology of the torn rotator cuff tendon. Bone & Joint Journal. 2006;88:489–495. doi: 10.1302/0301-620X.88B4.16845. [DOI] [PubMed] [Google Scholar]
- 6.Riley G, Goddard M, Hazleman B. Histopathological assessment and pathological significance of matrix degeneration in supraspinatus tendons. Rheumatology. 2001;40:229–230. doi: 10.1093/rheumatology/40.2.229. [DOI] [PubMed] [Google Scholar]
- 7.Gladstone JN, Bishop JY, Lo IK, et al. Fatty infiltration and atrophy of the rotator cuff do not improve after rotator cuff repair and correlate with poor functional outcome. The American journal of sports medicine. 2007;35:719–728. doi: 10.1177/0363546506297539. [DOI] [PubMed] [Google Scholar]
- 8.Goutallier D, Postel J-M, Bernageau J, et al. Fatty muscle degeneration in cuff ruptures: pre-and postoperative evaluation by CT scan. Clinical orthopaedics and related research. 1994;304:78–83. [PubMed] [Google Scholar]
- 9.Hebert-Davies J, Teefey SA, Steger-May K, et al. Progression of Fatty Muscle Degeneration in Atraumatic Rotator Cuff Tears. JBJS. 2017;99:832–839. doi: 10.2106/JBJS.16.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bialek P, Morris CA, Parkington J, et al. Distinct protein degradation profiles are induced by different disuse models of skeletal muscle atrophy. Physiological genomics. 2011 doi: 10.1152/physiolgenomics.00247.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brooks NE, Myburgh KH. Skeletal muscle wasting with disuse atrophy is multi-dimensional: the response and interaction of myonuclei, satellite cells and signaling pathways. Frontiers in physiology. 2014;5:99. doi: 10.3389/fphys.2014.00099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clark BC. In vivo alterations in skeletal muscle form and function after disuse atrophy. Medicine and science in sports and exercise. 2009;41:1869–1875. doi: 10.1249/MSS.0b013e3181a645a6. [DOI] [PubMed] [Google Scholar]
- 13.Jones SW, Hill RJ, Krasney PA, et al. Disuse atrophy and exercise rehabilitation in humans profoundly affects the expression of genes associated with the regulation of skeletal muscle mass. The FASEB journal. 2004;18:1025–1027. doi: 10.1096/fj.03-1228fje. [DOI] [PubMed] [Google Scholar]
- 14.Reardon KA, Davis J, Kapsa RM, et al. Myostatin, insulin‐like growth factor‐1, and leukemia inhibitory factor mRNAs are upregulated in chronic human disuse muscle atrophy. Muscle & nerve. 2001;24:893–899. doi: 10.1002/mus.1086. [DOI] [PubMed] [Google Scholar]
- 15.Bonaldo P, Sandri M. Cellular and molecular mechanisms of muscle atrophy. Disease models & mechanisms. 2013;6:25–39. doi: 10.1242/dmm.010389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Darr KC, Schultz E. Exercise-induced satellite cell activation in growing and mature skeletal muscle. Journal of Applied Physiology. 1987;63:1816–1821. doi: 10.1152/jappl.1987.63.5.1816. [DOI] [PubMed] [Google Scholar]
- 17.Schoenfeld BJ. Does exercise-induced muscle damage play a role in skeletal muscle hypertrophy? The Journal of Strength & Conditioning Research. 2012;26:1441–1453. doi: 10.1519/JSC.0b013e31824f207e. [DOI] [PubMed] [Google Scholar]
- 18.Lundgreen K, Lian ØB, Engebretsen L, et al. Lower muscle regenerative potential in full-thickness supraspinatus tears compared to partial-thickness tears. Acta orthopaedica. 2013;84:565–570. doi: 10.3109/17453674.2013.858289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Steinbacher P, Tauber M, Kogler S, et al. Effects of rotator cuff ruptures on the cellular and intracellular composition of the human supraspinatus muscle. Tissue and Cell. 2010;42:37–41. doi: 10.1016/j.tice.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 20.Zanotti RM, Carpenter JE, Blasier RB, et al. The low incidence of suprascapular nerve injury after primary repair of massive rotator cuff tears. Journal of Shoulder and Elbow Surgery. 1997;6:258–264. doi: 10.1016/s1058-2746(97)90014-8. [DOI] [PubMed] [Google Scholar]
- 21.Gerber C, Meyer D, Schneeberger A, et al. Effect of tendon release and delayed repair on the structure of the muscles of the rotator cuff: an experimental study in sheep. The Journal of Bone & Joint Surgery. 2004;86:1973–1982. doi: 10.2106/00004623-200409000-00016. [DOI] [PubMed] [Google Scholar]
- 22.Cardenas D, Stolov W, Hardy R. Muscle fiber number in immobilization atrophy. Archives of physical medicine and rehabilitation. 1977;58:423–426. [PubMed] [Google Scholar]
- 23.Petrof BJ. The molecular basis of activity-induced muscle injury in Duchenne muscular dystrophy. Molecular and cellular biochemistry. 1998;179:111–124. doi: 10.1023/a:1006812004945. [DOI] [PubMed] [Google Scholar]
- 24.McArdle A, Edwards R, Jackson M. How does dystrophin deficiency lead to muscle degeneration?—evidence from the mdx mouse. Neuromuscular Disorders. 1995;5:445–456. doi: 10.1016/0960-8966(95)00001-4. [DOI] [PubMed] [Google Scholar]
- 25.Wallace GQ, McNally EM. Mechanisms of muscle degeneration, regeneration, and repair in the muscular dystrophies. Annual review of physiology. 2009;71:37–57. doi: 10.1146/annurev.physiol.010908.163216. [DOI] [PubMed] [Google Scholar]
- 26.Crameri RM, Langberg H, Magnusson P, et al. Changes in satellite cells in human skeletal muscle after a single bout of high intensity exercise. The Journal of physiology. 2004;558:333–340. doi: 10.1113/jphysiol.2004.061846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Friden J, Lieber R. Eccentric exercise‐induced injuries to contractile and cytoskeletal muscle fibre components. Acta Physiologica Scandinavica. 2001;171:321–326. doi: 10.1046/j.1365-201x.2001.00834.x. [DOI] [PubMed] [Google Scholar]
- 28.Friden J, Lieber RL. Structural and mechanical basis of exercise-induced muscle injury. Medicine and science in sports and exercise. 1992;24:521–530. [PubMed] [Google Scholar]
- 29.Buford TW, MacNeil RG, Clough LG, et al. Active muscle regeneration following eccentric contraction-induced injury is similar between healthy young and older adults. Journal of Applied Physiology. 2014;116:1481–1490. doi: 10.1152/japplphysiol.01350.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Palacios D, Mozzetta C, Consalvi S, et al. TNF/p38α/polycomb signaling to Pax7 locus in satellite cells links inflammation to the epigenetic control of muscle regeneration. Cell stem cell. 2010;7:455–469. doi: 10.1016/j.stem.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sambasivan R, Yao R, Kissenpfennig A, et al. Pax7-expressing satellite cells are indispensable for adult skeletal muscle regeneration. Development. 2011;138:3647–3656. doi: 10.1242/dev.067587. [DOI] [PubMed] [Google Scholar]
- 32.Tatsumi R, Sheehan S, Iwasaki H, et al. Mechanical stretch induces activation of skeletal muscle satellite cells in vitro. Experimental cell research. 2001;267:107–114. doi: 10.1006/excr.2001.5252. [DOI] [PubMed] [Google Scholar]
- 33.Porter JD, Khanna S, Kaminski HJ, et al. A chronic inflammatory response dominates the skeletal muscle molecular signature in dystrophin-deficient mdx mice. Human Molecular Genetics. 2002;11:263–272. doi: 10.1093/hmg/11.3.263. [DOI] [PubMed] [Google Scholar]
- 34.Gibbons MC, Singh A, Anakwenze O, et al. Histological Evidence of Muscle Degeneration in Advanced Human Rotator Cuff Disease. The Journal of Bone & Joint Surgery. 2017;99:190–199. doi: 10.2106/JBJS.16.00335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bachasson D, Singh A, Shah SB, et al. The role of the peripheral and central nervous systems in rotator cuff disease. Journal of Shoulder and Elbow Surgery. 2015;24:1322–1335. doi: 10.1016/j.jse.2015.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gibbons MC, Sato EJ, Bachasson D, et al. Muscle architectural changes after massive human rotator cuff tear. Journal of Orthopaedic Research. 2016 doi: 10.1002/jor.23256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sato EJ, Killian ML, Choi AJ, et al. Skeletal muscle fibrosis and stiffness increase after rotator cuff tendon injury and neuromuscular compromise in a rat model. Journal of Orthopaedic Research. 2014;32:1111–1116. doi: 10.1002/jor.22646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Davis ME, Stafford PL, Jergenson MJ, et al. Muscle fibers are injured at the time of acute and chronic rotator cuff repair. Clinical Orthopaedics and Related Research®. 2015;473:226–232. doi: 10.1007/s11999-014-3860-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gerber C, Meyer DC, Frey E, et al. Neer Award 2007: Reversion of structural muscle changes caused by chronic rotator cuff tears using continuous musculotendinous traction. An experimental study in sheep. Journal of Shoulder and Elbow Surgery. 2009;18:163–171. doi: 10.1016/j.jse.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 40.Silldorff MD, Choo AD, Choi AJ, et al. Effect of Supraspinatus Tendon Injury on Supraspinatus and Infraspinatus Muscle Passive Tension and Associated Biochemistry. The Journal of Bone & Joint Surgery. 2014;96:e175. doi: 10.2106/JBJS.M.01315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ward SR, Hentzen ER, Smallwood LH, et al. Rotator cuff muscle architecture: implications for glenohumeral stability. Clinical orthopaedics and related research. 2006;448:157–163. doi: 10.1097/01.blo.0000194680.94882.d3. [DOI] [PubMed] [Google Scholar]
- 42.Reddy GK. Cross-linking in collagen by nonenzymatic glycation increases the matrix stiffness in rabbit achilles tendon. Experimental Diabetes Research. 2004;5:143–153. doi: 10.1080/15438600490277860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bank RA, TeKoppele JM, Oostingh G, et al. Lysylhydroxylation and non-reducible crosslinking of human supraspinatus tendon collagen: changes with age and in chronic rotator cuff tendinitis. Annals of the Rheumatic Diseases. 1999;58:35–41. doi: 10.1136/ard.58.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hersche O, Gerber C. Passive tension in the supraspinatus musculotendinous unit after long-standing rupture of its tendon: a preliminary report. Journal of Shoulder and Elbow Surgery. 1998;7:393–396. doi: 10.1016/s1058-2746(98)90030-1. [DOI] [PubMed] [Google Scholar]
- 45.Safran O, Derwin KA, Powell K, et al. Changes in rotator cuff muscle volume, fat content, and passive mechanics after chronic detachment in a canine model. J Bone Joint Surg Am. 2005;87:2662–2670. doi: 10.2106/JBJS.D.02421. [DOI] [PubMed] [Google Scholar]
- 46.Giambini H, Hatta T, Krzysztof GR, et al. Intramuscular Fat Infiltration Evaluated by Magnetic Resonance Imaging Predicts the Extensibility of the Supraspinatus Muscle. Muscle & Nerve. 2017 doi: 10.1002/mus.25673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Meyer DC, Hoppeler H, von Rechenberg B, et al. A pathomechanical concept explains muscle loss and fatty muscular changes following surgical tendon release. Journal of orthopaedic research. 2004;22:1004–1007. doi: 10.1016/j.orthres.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 48.Engler AJ, Griffin MA, Sen S, et al. Myotubes differentiate optimally on substrates with tissue-like stiffness. J Cell Biol. 2004;166:877–887. doi: 10.1083/jcb.200405004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Comley K, Fleck NA. A micromechanical model for the Young’s modulus of adipose tissue. International Journal of Solids and Structures. 2010;47:2982–2990. [Google Scholar]
- 50.Mendias CL, Roche SM, Harning JA, et al. Reduced muscle fiber force production and disrupted myofibril architecture in patients with chronic rotator cuff tears. Journal of Shoulder and Elbow Surgery. 2015;24:111–119. doi: 10.1016/j.jse.2014.06.037. [DOI] [PubMed] [Google Scholar]
- 51.Tomioka T, Minagawa H, Kijima H, et al. Sarcomere length of torn rotator cuff muscle. Journal of Shoulder and Elbow Surgery. 2009;18:955–959. doi: 10.1016/j.jse.2009.03.009. [DOI] [PubMed] [Google Scholar]
- 52.Glass DJ. Skeletal muscle hypertrophy and atrophy signaling pathways. Int J Biochem Cell Biol. 2005;37:1974–1984. doi: 10.1016/j.biocel.2005.04.018. [DOI] [PubMed] [Google Scholar]
- 53.Miller MK, Bang M-L, Witt CC, et al. The muscle ankyrin repeat proteins: CARP, ankrd2/Arpp and DARP as a family of titin filament-based stress response molecules. Journal of molecular biology. 2003;333:951–964. doi: 10.1016/j.jmb.2003.09.012. [DOI] [PubMed] [Google Scholar]
- 54.Peter AK, Cheng H, Ross RS, et al. The costamere bridges sarcomeres to the sarcolemma in striated muscle. Progress in pediatric cardiology. 2011;31:83–88. doi: 10.1016/j.ppedcard.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Glass DJ. Molecular mechanisms modulating muscle mass. Trends in molecular medicine. 2003;9:344–350. doi: 10.1016/s1471-4914(03)00138-2. [DOI] [PubMed] [Google Scholar]
- 56.Bodine SC, Latres E, Baumhueter S, et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science. 2001;294:1704–1708. doi: 10.1126/science.1065874. [DOI] [PubMed] [Google Scholar]
- 57.Mitch WE, Goldberg AL. Mechanisms of muscle wasting—the role of the ubiquitin–proteasome pathway. New England Journal of Medicine. 1996;335:1897–1905. doi: 10.1056/NEJM199612193352507. [DOI] [PubMed] [Google Scholar]
- 58.Bartoli M, Richard I. Calpains in muscle wasting. The international journal of biochemistry & cell biology. 2005;37:2115–2133. doi: 10.1016/j.biocel.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 59.Huang J, Forsberg NE. Role of calpain in skeletal-muscle protein degradation. Proceedings of the national academy of sciences. 1998;95:12100–12105. doi: 10.1073/pnas.95.21.12100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Demiray H. A note on the elasticity of soft biological tissues. Journal of biomechanics. 1972;5:309–311. doi: 10.1016/0021-9290(72)90047-4. [DOI] [PubMed] [Google Scholar]
- 61.McNeil PL, Steinhardt RA. Loss, restoration, and maintenance of plasma membrane integrity. The Journal of cell biology. 1997;137:1–4. doi: 10.1083/jcb.137.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Trotter J. Functional morphology of force transmission in skeletal muscle. Cells Tissues Organs. 1993;146:205–222. doi: 10.1159/000147459. [DOI] [PubMed] [Google Scholar]
- 63.Bradley W, Fulthorpe J. Studies of sarcolemmal integrity in myopathic muscle. Neurology. 1978;28:670–670. doi: 10.1212/wnl.28.7.670. [DOI] [PubMed] [Google Scholar]
- 64.Turner PR, Fong P, Denetclaw WF, et al. Increased calcium influx in dystrophic muscle. The Journal of cell biology. 1991;115:1701–1712. doi: 10.1083/jcb.115.6.1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Turner PR, Westwood T, Regen CM, et al. Increased protein degradation results from elevated free calcium levels found in muscle from mdx mice. Nature. 1988;335:735–738. doi: 10.1038/335735a0. [DOI] [PubMed] [Google Scholar]
- 66.Alderton JM, Steinhardt RA. Calcium influx through calcium leak channels is responsible for the elevated levels of calcium-dependent proteolysis in dystrophic myotubes. Journal of Biological Chemistry. 2000;275:9452–9460. doi: 10.1074/jbc.275.13.9452. [DOI] [PubMed] [Google Scholar]
- 67.Squier MK, Miller AC, Malkinson AM, et al. Calpain activation in apoptosis. Journal of cellular physiology. 1994;159:229–237. doi: 10.1002/jcp.1041590206. [DOI] [PubMed] [Google Scholar]
- 68.Orrenius S, Zhivotovsky B, Nicotera P. Regulation of cell death: the calcium–apoptosis link. Nature reviews Molecular cell biology. 2003;4:552–565. doi: 10.1038/nrm1150. [DOI] [PubMed] [Google Scholar]
- 69.Spencer MJ, Mellgren RL. Overexpression of a calpastatin transgene in mdx muscle reduces dystrophic pathology. Human Molecular Genetics. 2002;11:2645–2655. doi: 10.1093/hmg/11.21.2645. [DOI] [PubMed] [Google Scholar]
- 70.Wang X, Kawano F, Matsuoka Y, et al. Mechanical load-dependent regulation of satellite cell and fiber size in rat soleus muscle. American Journal of Physiology-Cell Physiology. 2006;290:C981–C989. doi: 10.1152/ajpcell.00298.2005. [DOI] [PubMed] [Google Scholar]
- 71.Lepper C, Partridge TA, Fan C-M. An absolute requirement for Pax7-positive satellite cells in acute injury-induced skeletal muscle regeneration. Development. 2011;138:3639–3646. doi: 10.1242/dev.067595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lee S-J, Huynh TV, Lee Y-S, et al. Role of satellite cells versus myofibers in muscle hypertrophy induced by inhibition of the myostatin/activin signaling pathway. Proceedings of the National Academy of Sciences. 2012;109:E2353–E2360. doi: 10.1073/pnas.1206410109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.McCarthy JJ, Mula J, Miyazaki M, et al. Effective fiber hypertrophy in satellite cell-depleted skeletal muscle. Development. 2011;138:3657–3666. doi: 10.1242/dev.068858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yin H, Price F, Rudnicki MA. Satellite cells and the muscle stem cell niche. Physiological reviews. 2013;93:23–67. doi: 10.1152/physrev.00043.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mauro A. Satellite cell of skeletal muscle fibers. The Journal of biophysical and biochemical cytology. 1961;9:493–495. doi: 10.1083/jcb.9.2.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kuang S, Kuroda K, Le Grand F, et al. Asymmetric self-renewal and commitment of satellite stem cells in muscle. Cell. 2007;129:999–1010. doi: 10.1016/j.cell.2007.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Quarta M, Brett JO, DiMarco R, et al. An artificial niche preserves the quiescence of muscle stem cells and enhances their therapeutic efficacy. Nature biotechnology. 2016 doi: 10.1038/nbt.3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Thomas K, Engler AJ, Meyer GA. Extracellular matrix regulation in the muscle satellite cell niche. Connective tissue research. 2015;56:1–8. doi: 10.3109/03008207.2014.947369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hosoyama T, Ishiguro N, Yamanouchi K, et al. Degenerative muscle fiber accelerates adipogenesis of intramuscular cells via RhoA signaling pathway. Differentiation. 2009;77:350–359. doi: 10.1016/j.diff.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 80.Urciuolo A, Quarta M, Morbidoni V, et al. Collagen VI regulates satellite cell self-renewal and muscle regeneration. Nature communications. 2013;4 doi: 10.1038/ncomms2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sanes JR. The basement membrane/basal lamina of skeletal muscle. Journal of Biological Chemistry. 2003;278:12601–12604. doi: 10.1074/jbc.R200027200. [DOI] [PubMed] [Google Scholar]
- 82.Carson JA, Wei L. Integrin signaling9s potential for mediating gene expression in hypertrophying skeletal muscle. Journal of applied physiology. 2000;88:337–343. doi: 10.1152/jappl.2000.88.1.337. [DOI] [PubMed] [Google Scholar]
- 83.Hynes RO. The extracellular matrix: not just pretty fibrils. Science. 2009;326:1216–1219. doi: 10.1126/science.1176009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bentzinger CF, Wang YX, von Maltzahn J, et al. Fibronectin regulates Wnt7a signaling and satellite cell expansion. Cell stem cell. 2013;12:75–87. doi: 10.1016/j.stem.2012.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Taipale J, Keski-Oja J. Growth factors in the extracellular matrix. The FASEB Journal. 1997;11:51–59. doi: 10.1096/fasebj.11.1.9034166. [DOI] [PubMed] [Google Scholar]
- 86.Hinz B. The extracellular matrix and transforming growth factor-β1: Tale of a strained relationship. Matrix Biology. 2015;47:54–65. doi: 10.1016/j.matbio.2015.05.006. [DOI] [PubMed] [Google Scholar]
- 87.Gilbert PM, Havenstrite KL, Magnusson KE, et al. Substrate elasticity regulates skeletal muscle stem cell self-renewal in culture. Science. 2010;329:1078–1081. doi: 10.1126/science.1191035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Shefer G, Van de Mark DP, Richardson JB, et al. Satellite-cell pool size does matter: defining the myogenic potency of aging skeletal muscle. Developmental biology. 2006;294:50–66. doi: 10.1016/j.ydbio.2006.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Meyer GA, Farris AL, Sato E, et al. Muscle progenitor cell regenerative capacity in the torn rotator cuff. Journal of Orthopaedic Research. 2015;33:421–429. doi: 10.1002/jor.22786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kadi F, Schjerling P, Andersen LL, et al. The effects of heavy resistance training and detraining on satellite cells in human skeletal muscles. The Journal of physiology. 2004;558:1005–1012. doi: 10.1113/jphysiol.2004.065904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Choo A, McCarthy M, Pichika R, et al. Muscle Gene Expression Patterns in Human Rotator Cuff Pathology. The Journal of Bone & Joint Surgery. 2014;96:1558–1565. doi: 10.2106/JBJS.M.01585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Davies MR, Liu X, Lee L, et al. TGF-β Small Molecule Inhibitor SB431542 Reduces Rotator Cuff Muscle Fibrosis and Fatty Infiltration By Promoting Fibro/Adipogenic Progenitor Apoptosis. PloS one. 2016;11:e0155486. doi: 10.1371/journal.pone.0155486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.De Giorgi S, Saracino M, Castagna A. Degenerative disease in rotator cuff tears: what are the biochemical and histological changes? Joints. 2013;2:26–28. [PMC free article] [PubMed] [Google Scholar]
- 94.Flück M, Ruoss S, Möhl CB, et al. Genomic and lipidomic actions of nandrolone on detached rotator cuff muscle in sheep. The Journal of Steroid Biochemistry and Molecular Biology. 2016 doi: 10.1016/j.jsbmb.2016.08.005. [DOI] [PubMed] [Google Scholar]
- 95.Gumucio JP, Davis ME, Bradley JR, et al. Rotator cuff tear reduces muscle fiber specific force production and induces macrophage accumulation and autophagy. Journal of Orthopaedic Research. 2012;30:1963–1970. doi: 10.1002/jor.22168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Perry SM, McIlhenny SE, Hoffman MC, et al. Inflammatory and angiogenic mRNA levels are altered in a supraspinatus tendon overuse animal model. Journal of Shoulder and Elbow Surgery. 2005;14:S79–S83. doi: 10.1016/j.jse.2004.09.020. [DOI] [PubMed] [Google Scholar]
- 97.Morikawa D, Itoigawa Y, Nojiri H, et al. Contribution of oxidative stress to the degeneration of rotator cuff entheses. Journal of Shoulder and Elbow Surgery. 2014;23:628–635. doi: 10.1016/j.jse.2014.01.041. [DOI] [PubMed] [Google Scholar]
- 98.Wang M-X, Wei A, Yuan J, et al. Antioxidant enzyme peroxiredoxin 5 is upregulated in degenerative human tendon. Biochemical and biophysical research communications. 2001;284:667–673. doi: 10.1006/bbrc.2001.4991. [DOI] [PubMed] [Google Scholar]
- 99.Gumucio J, Rittman D, McDonagh B, et al. Molecular mechanisms behind the accumulation of lipid that occurs after skeletal muscle injury. The FASEB Journal. 2016;30:1244.1248–1244.1248. [Google Scholar]
- 100.Kozakowska M, Pietraszek-Gremplewicz K, Jozkowicz A, et al. The role of oxidative stress in skeletal muscle injury and regeneration: focus on antioxidant enzymes. Journal of muscle research and cell motility. 2015;36:377–393. doi: 10.1007/s10974-015-9438-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chen S-E, Jin B, Li Y-P. TNF-α regulates myogenesis and muscle regeneration by activating p38 MAPK. American Journal of Physiology-Cell Physiology. 2007;292:C1660–C1671. doi: 10.1152/ajpcell.00486.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ogura Y, Mishra V, Hindi SM, et al. Proinflammatory cytokine TWEAK suppresses satellite cell self-renewal through inversely modulating Notch and NF-κB signaling pathways. Journal of Biological Chemistry. 2013 doi: 10.1074/jbc.M113.517300. jbc. M113.517300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Arnold L, Henry A, Poron F, et al. Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. The Journal of experimental medicine. 2007;204:1057–1069. doi: 10.1084/jem.20070075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Trendelenburg AU, Meyer A, Rohner D, et al. Myostatin reduces Akt/TORC1/p70S6K signaling, inhibiting myoblast differentiation and myotube size. American Journal of Physiology-Cell Physiology. 2009;296:C1258–C1270. doi: 10.1152/ajpcell.00105.2009. [DOI] [PubMed] [Google Scholar]
- 105.Stitt TN, Drujan D, Clarke BA, et al. The IGF-1/PI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Molecular cell. 2004;14:395–403. doi: 10.1016/s1097-2765(04)00211-4. [DOI] [PubMed] [Google Scholar]
- 106.Rabinovsky ED, Gelir E, Gelir S, et al. Targeted expression of IGF-1 transgene to skeletal muscle accelerates muscle and motor neuron regeneration. The FASEB Journal. 2003;17:53–55. doi: 10.1096/fj.02-0183fje. [DOI] [PubMed] [Google Scholar]
- 107.Tonkin J, Temmerman L, Sampson RD, et al. Monocyte/Macrophage-derived IGF-1 Orchestrates Murine Skeletal Muscle Regeneration and Modulates Autocrine Polarization. Molecular Therapy. 2015 doi: 10.1038/mt.2015.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bentzinger CF, Wang YX, Dumont NA, et al. Cellular dynamics in the muscle satellite cell niche. EMBO reports. 2013;14:1062–1072. doi: 10.1038/embor.2013.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Charge SB, Rudnicki MA. Cellular and molecular regulation of muscle regeneration. Physiological reviews. 2004;84:209–238. doi: 10.1152/physrev.00019.2003. [DOI] [PubMed] [Google Scholar]
- 110.Meyer GA, Gibbons MC, Sato E, et al. Epimuscular Fat in the Human Rotator Cuff Is a Novel Beige Depot. Stem cells translational medicine. 2015 doi: 10.5966/sctm.2014-0287. sctm.2014-0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Asakura A, Rudnicki MA, Komaki M. Muscle satellite cells are multipotential stem cells that exhibit myogenic, osteogenic, and adipogenic differentiation. Differentiation. 2001;68:245–253. doi: 10.1046/j.1432-0436.2001.680412.x. [DOI] [PubMed] [Google Scholar]
- 112.Delaigle AlM. Jonas J-C, Bauche IB, et al. Induction of adiponectin in skeletal muscle by inflammatory cytokines: in vivo and in vitro studies. Endocrinology. 2004;145:5589–5597. doi: 10.1210/en.2004-0503. [DOI] [PubMed] [Google Scholar]
- 113.Starkey JD, Yamamoto M, Yamamoto S, et al. Skeletal muscle satellite cells are committed to myogenesis and do not spontaneously adopt nonmyogenic fates. Journal of Histochemistry & Cytochemistry. 2011;59:33–46. doi: 10.1369/jhc.2010.956995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Shefer G, Wleklinski-Lee M, Yablonka-Reuveni Z. Skeletal muscle satellite cells can spontaneously enter an alternative mesenchymal pathway. Journal of cell science. 2004;117:5393–5404. doi: 10.1242/jcs.01419. [DOI] [PubMed] [Google Scholar]
- 115.Yin H, Pasut A, Soleimani VD, et al. MicroRNA-133 controls brown adipose determination in skeletal muscle satellite cells by targeting Prdm16. Cell metabolism. 2013;17:210–224. doi: 10.1016/j.cmet.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Itoigawa Y, Kishimoto KN, Sano H, et al. Molecular mechanism of fatty degeneration in rotator cuff muscle with tendon rupture. Journal of orthopaedic research. 2011;29:861–866. doi: 10.1002/jor.21317. [DOI] [PubMed] [Google Scholar]
- 117.Beeler S, Ek ET, Gerber C. A comparative analysis of fatty infiltration and muscle atrophy in patients with chronic rotator cuff tears and suprascapular neuropathy. Journal of Shoulder and Elbow Surgery. 2013;22:1537–1546. doi: 10.1016/j.jse.2013.01.028. [DOI] [PubMed] [Google Scholar]
- 118.Judson RN, Zhang RH, Rossi F. Tissue‐resident mesenchymal stem/progenitor cells in skeletal muscle: collaborators or saboteurs? FEBS Journal. 2013;280:4100–4108. doi: 10.1111/febs.12370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Birbrair A, Zhang T, Wang Z-M, et al. Pericytes: multitasking cells in the regeneration of injured, diseased, and aged skeletal muscle. Frontiers in aging neuroscience. 2014;6:245. doi: 10.3389/fnagi.2014.00245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Uezumi A, Fukada S-i, Yamamoto N, et al. Mesenchymal progenitors distinct from satellite cells contribute to ectopic fat cell formation in skeletal muscle. Nature cell biology. 2010;12:143–152. doi: 10.1038/ncb2014. [DOI] [PubMed] [Google Scholar]
- 121.Joe AW, Yi L, Natarajan A, et al. Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nature cell biology. 2010;12:153–163. doi: 10.1038/ncb2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Uezumi A, Ikemoto-Uezumi M, Tsuchida K. Roles of nonmyogenic mesenchymal progenitors in pathogenesis and regeneration of skeletal muscle. Frontiers in physiology. 2014;5 doi: 10.3389/fphys.2014.00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Heredia JE, Mukundan L, Chen FM, et al. Type 2 innate signals stimulate fibro/adipogenic progenitors to facilitate muscle regeneration. Cell. 2013;153:376–388. doi: 10.1016/j.cell.2013.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Liu X, Ning AY, Chang NC, et al. Investigating the cellular origin of rotator cuff muscle fatty infiltration and fibrosis after injury. Muscles, Ligaments and Tendons Journal. 2016;6:6. doi: 10.11138/mltj/2016.6.1.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Birbrair A, Zhang T, Wang Z-M, et al. Role of pericytes in skeletal muscle regeneration and fat accumulation. Stem cells and development. 2013;22:2298–2314. doi: 10.1089/scd.2012.0647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Dellavalle A, Sampaolesi M, Tonlorenzi R, et al. Pericytes of human skeletal muscle are myogenic precursors distinct from satellite cells. Nature cell biology. 2007;9:255–267. doi: 10.1038/ncb1542. [DOI] [PubMed] [Google Scholar]
- 127.Farrington-Rock C, Crofts N, Doherty M, et al. Chondrogenic and adipogenic potential of microvascular pericytes. Circulation. 2004;110:2226–2232. doi: 10.1161/01.CIR.0000144457.55518.E5. [DOI] [PubMed] [Google Scholar]
- 128.Lin S-L, Kisseleva T, Brenner DA, et al. Pericytes and perivascular fibroblasts are the primary source of collagen-producing cells in obstructive fibrosis of the kidney. The American journal of pathology. 2008;173:1617–1627. doi: 10.2353/ajpath.2008.080433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Rajkumar VS, Howell K, Csiszar K, et al. Shared expression of phenotypic markers in systemic sclerosis indicates a convergence of pericytes and fibroblasts to a myofibroblast lineage in fibrosis. Arthritis research & therapy. 2005;7:R1113. doi: 10.1186/ar1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Schor AM, Canfield A, Sutton A, et al. Pericyte differentiation. Clinical orthopaedics and related research. 1995;313:81–91. [PubMed] [Google Scholar]
- 131.Hinz B. Tissue stiffness, latent TGF-β1 activation, and mechanical signal transduction: implications for the pathogenesis and treatment of fibrosis. Current rheumatology reports. 2009;11:120–126. doi: 10.1007/s11926-009-0017-1. [DOI] [PubMed] [Google Scholar]
- 132.Covas DT, Panepucci RA, Fontes AM, et al. Multipotent mesenchymal stromal cells obtained from diverse human tissues share functional properties and gene-expression profile with CD146+ perivascular cells and fibroblasts. Experimental hematology. 2008;36:642–654. doi: 10.1016/j.exphem.2007.12.015. [DOI] [PubMed] [Google Scholar]
- 133.Besson V, Smeriglio P, Wegener A, et al. PW1 gene/paternally expressed gene 3 (PW1/Peg3) identifies multiple adult stem and progenitor cell populations. Proceedings of the National Academy of Sciences. 2011;108:11470–11475. doi: 10.1073/pnas.1103873108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Mitchell KJ, Pannérec A, Cadot B, et al. Identification and characterization of a non-satellite cell muscle resident progenitor during postnatal development. Nature cell biology. 2010;12:257–266. doi: 10.1038/ncb2025. [DOI] [PubMed] [Google Scholar]
- 135.Malecova B, Puri PL. “Mix of Mics”-phenotypic and biological heterogeneity of “Multipotent” muscle interstitial cells (MICs) Journal of stem cell research & therapy. 2012 [PMC free article] [PubMed] [Google Scholar]
- 136.Millar NL, Hueber AJ, Reilly JH, et al. Inflammation is present in early human tendinopathy. The American journal of sports medicine. 2010;38:2085–2091. doi: 10.1177/0363546510372613. [DOI] [PubMed] [Google Scholar]
- 137.McWhorter FY, Davis CT, Liu WF. Physical and mechanical regulation of macrophage phenotype and function. Cellular and Molecular Life Sciences. 2015;72:1303–1316. doi: 10.1007/s00018-014-1796-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lautenschläger F, Paschke S, Schinkinger S, et al. The regulatory role of cell mechanics for migration of differentiating myeloid cells. Proceedings of the National Academy of Sciences. 2009;106:15696–15701. doi: 10.1073/pnas.0811261106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Oak NR, Gumucio JP, Flood MD, et al. Inhibition of 5-LOX, COX-1, and COX-2 Increases Tendon Healing and Reduces Muscle Fibrosis and Lipid Accumulation After Rotator Cuff Repair. The American journal of sports medicine. 2014;42:2860–2868. doi: 10.1177/0363546514549943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Wang JH-C. Mechanobiology of tendon. Journal of biomechanics. 2006;39:1563–1582. doi: 10.1016/j.jbiomech.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 141.Kjaer M. Role of extracellular matrix in adaptation of tendon and skeletal muscle to mechanical loading. Physiological reviews. 2004;84:649–698. doi: 10.1152/physrev.00031.2003. [DOI] [PubMed] [Google Scholar]
- 142.Tidball JG, Dorshkind K, Wehling-Henricks M. Shared signaling systems in myeloid cell-mediated muscle regeneration. Development. 2014;141:1184–1196. doi: 10.1242/dev.098285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kharraz Y, Guerra J, Mann CJ, et al. Macrophage plasticity and the role of inflammation in skeletal muscle repair. Mediators of inflammation. 2013;2013 doi: 10.1155/2013/491497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Lawrence T, Natoli G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nature reviews immunology. 2011;11:750–761. doi: 10.1038/nri3088. [DOI] [PubMed] [Google Scholar]
- 145.Friedl P, Weigelin B. Interstitial leukocyte migration and immune function. Nature immunology. 2008;9:960–969. doi: 10.1038/ni.f.212. [DOI] [PubMed] [Google Scholar]
- 146.Mantovani A, Sica A, Sozzani S, et al. The chemokine system in diverse forms of macrophage activation and polarization. Trends in immunology. 2004;25:677–686. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 147.Sugihara R, Kumamoto T, Ito T, et al. Human muscle protein degradation in vitro by eosinophil cationic protein (ECP) Muscle & nerve. 2001;24:1627–1634. doi: 10.1002/mus.1198. [DOI] [PubMed] [Google Scholar]
- 148.Van Goethem E, Poincloux R, Gauffre F, et al. Matrix architecture dictates three-dimensional migration modes of human macrophages: differential involvement of proteases and podosome-like structures. The journal of immunology. 2010;184:1049–1061. doi: 10.4049/jimmunol.0902223. [DOI] [PubMed] [Google Scholar]
- 149.Wolf K, Friedl P. Extracellular matrix determinants of proteolytic and non-proteolytic cell migration. Trends in cell biology. 2011;21:736–744. doi: 10.1016/j.tcb.2011.09.006. [DOI] [PubMed] [Google Scholar]
- 150.Previtera ML, Sengupta A. Substrate stiffness regulates proinflammatory mediator production through TLR4 activity in macrophages. PloS one. 2015;10:e0145813. doi: 10.1371/journal.pone.0145813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Dorph C, Lundberg IE. Idiopathic inflammatory myopathies–myositis. Best Practice & Research Clinical Rheumatology. 2002;16:817–832. doi: 10.1053/berh.2002.0261. [DOI] [PubMed] [Google Scholar]
- 152.Frontera WR, Meredith CN, O’reilly K, et al. Strength conditioning in older men: skeletal muscle hypertrophy and improved function. Journal of applied physiology. 1988;64:1038–1044. doi: 10.1152/jappl.1988.64.3.1038. [DOI] [PubMed] [Google Scholar]
- 153.Butt U, Rashid M, Temperley D, et al. Muscle regeneration following repair of the rotator cuff. Bone Joint J. 2016;98:1389–1394. doi: 10.1302/0301-620X.98B10.37231. [DOI] [PubMed] [Google Scholar]
- 154.Galatz LM, Ball CM, Teefey SA, et al. The outcome and repair integrity of completely arthroscopically repaired large and massive rotator cuff tears. The Journal of Bone & Joint Surgery. 2004;86:219–224. doi: 10.2106/00004623-200402000-00002. [DOI] [PubMed] [Google Scholar]
- 155.Gerber C, Fuchs B, Hodler J. The Results of Repair of Massive Tears of the Rotator Cuff*†. The Journal of Bone & Joint Surgery. 2000;82:505–505. doi: 10.2106/00004623-200004000-00006. [DOI] [PubMed] [Google Scholar]
- 156.Zumstein MA, Jost B, Hempel J, et al. The clinical and structural long-term results of open repair of massive tears of the rotator cuff. J Bone Joint Surg Am. 2008;90:2423–2431. doi: 10.2106/JBJS.G.00677. [DOI] [PubMed] [Google Scholar]
- 157.Kim HM, Galatz LM, Lim C, et al. The effect of tear size and nerve injury on rotator cuff muscle fatty degeneration in a rodent animal model. Journal of shoulder and elbow surgery. 2012;21:847–858. doi: 10.1016/j.jse.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Liu X, Laron D, Natsuhara K, et al. A mouse model of massive rotator cuff tears. The Journal of Bone & Joint Surgery. 2012;94:e41. doi: 10.2106/JBJS.K.00620. [DOI] [PubMed] [Google Scholar]
- 159.Liu X, Manzano G, Kim HT, et al. A rat model of massive rotator cuff tears. Journal of orthopaedic research. 2011;29:588–595. doi: 10.1002/jor.21266. [DOI] [PubMed] [Google Scholar]