

ABSTRACT
The core promoters of protein-encoding genes play a central role in regulating transcription. M1BP is a transcriptional activator that associates with a core promoter element known as Motif 1 that resides at thousands of genes in Drosophila. To gain insight into how M1BP functions, we identified an interacting protein called GFZF. GFZF had been previously identified in genetic screens for factors involved in maintenance of hybrid inviability, the G2-M DNA damage checkpoint, and RAS/mitogen-activated protein kinase (MAPK) signaling, but its contribution to these processes was unknown. Here, we show that GFZF resides in the nucleus and functions as a transcriptional coactivator. In addition, we show that GFZF is a glutathione S-transferase (GST). Thus, GFZF is the first transcriptional coactivator with intrinsic GST activity, and its identification as a transcriptional coactivator provides an explanation for its role in numerous biological processes.
KEYWORDS: glutathione S-transferase, cell cycle, coactivator, transcription
INTRODUCTION
Regulation of RNA polymerase II (Pol II)-transcribed genes is one of the primary mechanisms by which cells coordinate the myriad of processes required for survival, proliferation, and development. The core promoter, defined as the 80- to 100-bp region centered on the transcription start site (TSS), is the hub of transcription regulation (1). Transcription initiates when general transcription factors (GTFs) bind elements within the core promoter region, forming a complex consisting of Pol II and other highly conserved Pol II-associated transcription factors (2). In recent years, our understanding has advanced from a model where the core promoter and the GTFs act as static integrators of signals from sequence-specific transcription factors that bind enhancer regions and modulate transcription levels to one where the core promoter and its machinery constitute a more dynamic assembly with different enhancer specificities (3, 4) and intrinsic regulatory properties (1).
One particular core promoter element that provides a clear contrast to the models arising from canonical promoters has emerged. The element, named Motif 1 (5, 6), is present in the promoter regions of thousands of genes in Drosophila. We identified and characterized a factor that binds this conserved element and named it M1BP (7). M1BP is enriched at housekeeping gene promoters, and M1BP-bound genes tend to have moderate to high levels of paused Pol II, are constitutively expressed, and show little spatiotemporal fluctuation in transcription levels (7). Additionally, Motif 1 and, by extension, M1BP-bound promoters tend to lack many of the elements once thought to be essential for initiation, such as the TATA box and initiator (6), so how initiation occurs at these promoters remains a mystery. Thus, the study of M1BP promoters might provide insights into previously unknown mechanisms of transcription initiation and activation.
Here we characterized a factor called GFZF that M1BP recruits to promoters. GFZF turns out to be a novel transcriptional coactivator that has glutathione S-transferase (GST) activity. GFZF has been identified in many genetic screens since its initial characterization (8). These screens have implicated GFZF in a wide variety of processes, including regulation of the cell cycle (9), DNA damage checkpoints during the transition from G2 to M phase (10), transcriptional and splicing control of RAS/mitogen-activated protein kinase (MAPK) signaling (11), response to oxidative stress (12), three-dimensional organization of polycomb complexes (13), and speciation (14), among other processes (15–17). Despite its involvement in these critical cellular processes, little is known about the mechanism by which it carries out these seemingly disparate functions. Early work reported that GFZF resides in the cytoplasm (8). Here, we present data supporting a parsimonious conclusion that GFZF is a transcription factor required for expression of the many factors that carry out the functions described in the above-mentioned screens.
Historically, GSTs have been studied for their role in cellular detoxification (18). However, there are notable examples of GSTs performing additional cellular functions, which include the regulation of signal transduction (19), inhibition of apoptosis (20), and the response to oxidative stress (21). Thus, it seems that GSTs play a critical, and perhaps underappreciated, role in cellular function and homeostasis. Our unprecedented finding of a transcription factor with GST activity raises the possibility of additional layers of complexity to the already-complex process of metazoan transcriptional regulation.
RESULTS AND DISCUSSION
M1BP associates with GFZF.
In order to understand the function of the core promoter, it is essential to know what factors associate with it. To identify factors that associate with Motif 1-containing core promoters, we immobilized promoter DNA from a mitochondrial ribosomal protein subunit gene (mRpS30) with a strong consensus Motif 1 and incubated this with Drosophila embryo nuclear extract. As a negative control, we also incubated extract with a mutant version of the promoter DNA that no longer binds M1BP. Bound proteins were then detected by SDS-PAGE and identified by mass spectrometry. Comparison of the factors bound to these two promoters identified several factors, including Putzig and GFZF (Fig. 1A). The identification of Putzig is consistent with previous findings that Putzig exists in a complex with TRF2 (22) and that TRF2 interacts with M1BP (23).
FIG 1.

GFZF is a nuclear protein recruited to promoter DNA by M1BP. (A) Silver-stained SDS-PAGE analysis of eluates from immobilized template pulldowns using the mRpS30 promoter DNA with either the wild-type Motif 1 sequence (wt Motif 1) or the Motif 1 sequence with 3 mutated nucleotides (Δ Motif 1). wt and Δ Motif 1 sequences for the mRpS30 promoter are shown below the gel. Nuclear extracts were incubated with the immobilized templates, and bound proteins were recovered and analyzed by 8% SDS-PAGE. (B) Coomassie blue-stained SDS-PAGE analysis of immobilized template pulldown experiments using the RpLP1 promoter sequence performed with recombinant, purified His-M1BP and His-GFZF. wt and Δ Motif 1 sequences for the RpLP1 promoter are shown below the gel. (C) Coomassie blue-stained SDS-PAGE analysis of Mal fusion pulldown experiments performed with recombinant, purified Mal-lacZα or Mal-M1BP fusion proteins immobilized on amylose beads and His-GFZF.
To determine if GFZF interacts directly with M1BP, we performed the immobilized template pulldown analysis with recombinant proteins expressed in and purified from Escherichia coli. The data are consistent with our results from extracts and show, as expected, that neither of the two factors associated with the mutated Motif 1 template (Fig. 1B, lanes 3, 5, and 7). In contrast, M1BP was able to associate with the wild-type (wt) motif 1 template regardless of whether GFZF was present in the reaction (Fig. 1B, cf. lanes 4 and 8). Conversely, GFZF was able to associate with the wild-type Motif 1 template only in the presence of M1BP (Fig. 1B, cf. lanes 6 and 8). Thus, direct interaction between M1BP and GFZF is likely to be involved in recruiting GFZF to promoter DNA.
To determine if M1BP and GFZF interact in the absence of a DNA template, we performed pulldowns with purified maltose binding protein (Mal) fusions. Using either the alpha fragment of LacZ as a control or full-length M1BP fused to Mal, we determined that GFZF interacts specifically with M1BP (Fig. 1C, cf. lanes 3 and 5). Notably, the immobilized template pulldown (Fig. 1B) showed a roughly stoichiometric recovery of both GFZF and M1BP, whereas in the case of the Mal fusion pulldowns, GFZF is recovered substoichiometrically. This suggests that GFZF may have a greater propensity to bind M1BP in a DNA-templated context.
GFZF associates with chromosomes.
While GFZF was originally reported to be primarily a cytoplasmic protein (8), the results of our immobilized template pulldown experiments indicated that GFZF might associate with chromosomes. To test this, we used immunofluorescence microscopy with GFZF antibody to detect GFZF on polytene chromosomes. Antibody against GFZF localized it to distinct bands broadly distributed across each chromosome (Fig. 2A). Since our pulldown analysis indicated that M1BP and GFZF associate with each other, we compared their distributions on chromosomes. A comparison of M1BP and GFZF staining patterns on different polytene chromosome spreads revealed very similar staining patterns (Fig. 2B). However, since both M1BP and GFZF antisera were prepared in rabbits, we could not detect both proteins at the same time on the same specimens. To circumvent this problem, we constructed a transgenic fly line that expresses FLAG-tagged M1BP (Fig. 2C and D) and localized the two proteins with a mouse monoclonal antibody targeting the FLAG epitope on M1BP and rabbit antibody targeting GFZF. This revealed significant overlap in staining for the two factors (Fig. 2E), suggesting that M1BP and GFZF bind the same genomic regions.
FIG 2.
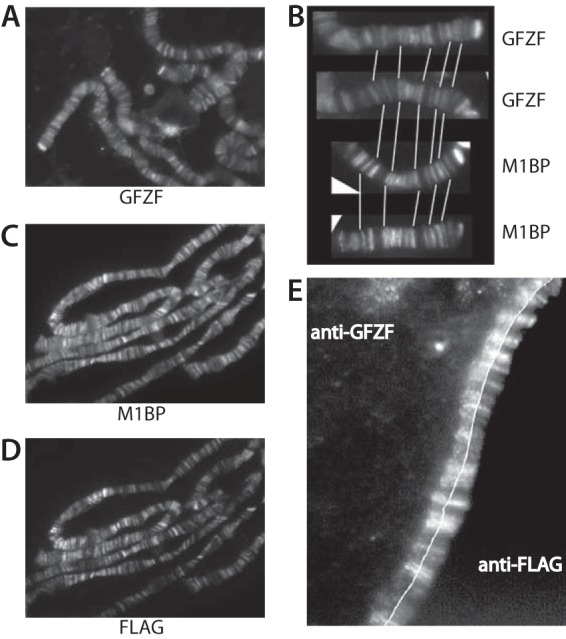
GFZF is associated with polytene chromosomes. (A) Polytene chromosomes from third-instar larvae were spread and stained with antibody against GFZF. (B) Comparison of M1BP and GFZF staining patterns at the end of chromosome 2R on separately stained samples. White lines guide an alignment of the patterns. (C and D) Polytene chromosomes from third-instar larvae expressing M1BP with N-terminal double FLAG and single HA tags under the control of the Hsp83 promoter were spread and stained with rabbit antibody against M1BP (C) and mouse monoclonal antibody against the FLAG epitope (D). (E) Polytene chromosomes from flies expressing 2×FLAG-HA-M1BP were spread and stained with mouse antibody against the FLAG epitope and rabbit antibody against GFZF. For ease of comparison, the images were split and aligned so that GFZF is to the left of the thin white line and FLAG-M1BP is to the right.
GFZF colocalizes with M1BP at many promoters.
To gain further insights into GFZF's role, we mapped the distribution of GFZF on the genome using chromatin immunoprecipitation with exonuclease (ChIP-exo). In ChIP-exo, chromatin is isolated from cross-linked cells, an immunoprecipitation is performed, and libraries are generated. In the course of library preparation, lambda exonuclease is applied to the immunoprecipitates and digests DNA in the 5′-to-3′ direction until its progression is stopped, typically when it encounters a cross-link point that prevents its continued digestion. This provides a snapshot of the 5′ cross-link borders of factors present at the time of cross-linking (24). We applied this to both M1BP and GFZF and found that GFZF was present on over 1,000 promoters in proliferating Drosophila S2R+ cells (Fig. 3A). A composite plot shows that the ChIP-exo signal for GFZF largely overlaps with M1BP and is concentrated in a 100-bp region just upstream from the transcription start site (Fig. 3B). Additionally, after calling peaks for both factors, we identified genes that have an M1BP or GFZF peak within 100 bp of the TSS. A total of 3,013 genes are bound by M1BP, while 1,885 are bound by GFZF. Furthermore, both factors are bound almost exclusively to the promoter regions of active genes (Fig. 3C). Gene ontology analysis of genes with a GFZF peak within 100 bp of the TSS revealed that, like M1BP (7), GFZF is highly enriched at the promoters of genes that perform housekeeping functions (i.e., metabolism, organization, and cellular physiology) (Fig. 3D). Thus, we conclude that GFZF and M1BP show remarkable overlap throughout the genome.
FIG 3.

GFZF and M1BP colocalize at approximately 1,800 active promoters. (A) Heat maps showing M1BP (left panel) or GFZF (center panel) ChIP-exo reads mapped in a 1,000-bp window centered on the transcription start sites (TSS) of 5,339 active genes lacking a neighboring gene TSS within 500 bp. ChIP-exo reads for the forward and reverse strands are separated and displayed in blue and red, respectively. The presence of Motif 1 within 100 bp of the TSS is indicated by blue lines in the right panel. (B) Composite plot of the heat maps displayed in a 600-bp window centered on the TSS. ChIP-exo reads for the forward and reverse strands are displayed above and below the x axis, respectively. (C) Venn diagram showing the overlap between active genes with M1BP and GFZF peak calls within 100 bp of a TSS. Genes have not been filtered for neighboring gene TSSs. (D) Enriched gene ontology terms for GFZF-bound promoters.
GFZF is a transcriptional coactivator.
The extensive colocalization of GFZF with M1BP, a known transcription factor (7), raises the possibility that GFZF is a transcription factor. To test whether GFZF activates transcription, we performed a dual-luciferase reporter assay following GFZF depletion in S2R+ cells (Illustrated in Fig. 4A). We used the GFZF-associated promoters for the ribosomal protein gene RpLP1, sex-lethal (Sxl) gene, roX2 gene, or abnormal wing disc gene (awd) to drive transcription of a firefly luciferase reporter. These promoters were chosen because previous studies had linked GFZF to processes and pathways in which their genes or gene products are involved (14, 15). As an internal control for the transfection efficiency, we used the RpIII128 promoter, which lacks M1BP and GFZF, to drive expression of a sequence coding Renilla luciferase. Both firefly and Renilla luciferase-encoding plasmids were transfected with either an empty expression vector or one that expressed a FLAG-tagged version of GFZF. Cells were treated for 1 day with double-stranded RNA (dsRNA) targeting either lacZ as a control, exon 2 of GFZF, or the 5′ untranslated region (5′ UTR) of GFZF and subsequently transfected with reporter plasmids. Two days later, cells were lysed and assayed for firefly and Renilla luciferase activity. Ectopically expressed FLAG-GFZF activated each of the promoters in the presence of the lacZ RNA interference (RNAi) control (Fig. 4B to E, cf. bars 1 and 2). This suggests that GFZF levels in the cell are limiting. RNAi targeting exon 2 of both the endogenous and ectopic GFZF inhibited GFZF-dependent activation (Fig. 4B to E, cf. bars 2 and 4). In contrast, RNAi targeting the 5′ UTR of endogenous GFZF, which is different from the 5′ UTR of ectopic GFZF, did not inhibit activation by FLAG-GFZF (Fig. 4B to E, cf. bars 2 and 6). Instead, the level of expression mediated by endogenous GFZF was diminished (Fig. 4B to E, cf. bars 1 and 5).
FIG 4.

GFZF is a transcriptional coactivator. (A) Overview of the luciferase assay used to test GFZF's role in transcription activation. S2R+ cells were treated with dsRNA targeting either lacZ (RNAi control), exon 2 of GFZF, or the 5′ UTR of endogenous GFZF. The exon 2 RNAi targets both the endogenous and transfected versions of GFZF, and the 5′ UTR RNAi targets only the endogenous GFZF. After 24 h of incubation with dsRNA, luciferase reporter plasmids and a vector that expresses either empty or wild-type GFZF were transfected into the cells. Cells were incubated for an additional 48 h and then assayed in tandem for firefly and Renilla luciferase activity. Firefly luciferase expression was driven by a GFZF-bound promoter, while Renilla luciferase expression was driven by the GFZF- and M1BP-less RpIII128 promoter. (B to E) Firefly/Renilla luciferase ratios are displayed for RpLP1, Sxl, roX2, and awd. All values are normalized to the lacZ dsRNA, empty-vector sample at each promoter.
To determine if GFZF is involved in activation of endogenous genes, we knocked down the level of GFZF and used ChIP to monitor the association of GFZF, M1BP, and Pol II with the same promoters that were tested in our transient-expression assay. RNAi targeting GFZF caused significant decreases in the level of GFZF associated with the RpLP1, Sxl, roX2, and awd promoters (Fig. 5A). These results confirm that our ChIP-exo analysis with the GFZF antibody indeed monitors GFZF. Furthermore, the ChIP analysis also confirmed our ChIP-exo data showing that little if any GFZF associates with the hsp70 promoter. Knockdown of GFZF also caused decreases in the level of M1BP associating with the promoters (Fig. 5B). This was unexpected since our biochemical analysis showed that M1BP bound a promoter fragment independently of GFZF (Fig. 1B). Western blot analysis showed that the knockdown of GFZF does not affect the level of M1BP (Fig. 5D). Thus, the contribution GFZF to M1BP promoter occupancy must reflect some role for GFZF contributing to M1BP in the cellular context. GFZF might be stabilizing the binding of M1BP or inducing some conformational change that affects the cross-linking efficiency. In accordance with the transient-expression data, the knockdown of GFZF caused a marked decrease in the level of a Pol II subunit, Rpb3, detected at GFZF-associated promoters but had an insignificant impact on Rpb3 associated with the hsp70 promoter (Fig. 5C). Taken together, the transient expression data and the ChIP analysis establish that GFZF is a transcriptional coactivator.
FIG 5.

GFZF knockdown results in Pol II and M1BP loss at GFZF-bound promoters. (A to C) qPCR quantification of GFZF (A), M1BP (B), and Rpb3 (C) ChIP samples following 3 days of RNAi treatment with dsRNA targeting either lacZ (lacZi) as a control or GFZF (GFZFi). Error bars show standard deviation (n = 4). Two-tailed t tests assuming equal variance were used to generate P values (reported for each locus tested). (D) Anti-GFZF-, anti-M1BP-, and anti-NELF E-probed Western blots of S2R+ cell lysates following 3 days of RNAi. Western blots are from experiments with RNAi-treated cells performed as biological replicates and are annotated as 1 and 2. MWM, molecular weight marker.
GFZF has GST activity.
An intriguing feature of GFZF is its glutathione S-transferase (GST) homology region, which is unprecedented for a transcription factor. A previous study demonstrated that GFZF binds a glutathione (GSH) column and can be eluted with GSH in a dose-dependent fashion (8). To test whether GFZF functions as a glutathione S-transferase and to measure its affinity for GSH, we expressed the GST domain of GFZF with a His tag in E. coli and purified it using metal affinity and ion-exchange chromatography (Fig. 6A). At the same time, we designed and expressed a catalytic mutant (S876A) of GFZF using the structure of a related GST in silkworm for reference (25). We next assayed GST activity by monitoring the increase in absorbance at 340 nm that results when GSH is conjugated to 1-chloro-2,4-dinitrobenzene (CDNB). Based on initial reaction velocities, the Kms for glutathione for the wild-type (wt) and the S876A mutant were determined to be 0.07 mM and 3.28 mM, respectively (Fig. 6B). The Km of wt GFZF falls well below the physiological range of GSH concentrations, which has been reported to be between 1 and 10 mM (26), though it has been reported that GSH concentrations are lower in the nucleus (27). Thus, GFZF's high affinity for GSH suggests that it is probably almost always bound in a cellular context.
FIG 6.

GFZF has GST activity with high affinity for glutathione. (A) Coomassie blue-stained SDS-PAGE analysis of the His-tagged GST homology domain of GFZF (wt and S876A mutant). (B) Kinetic analysis of GFZF's GST domain. The Km was obtained by altering the GSH concentration while keeping CDNB constant.
To determine if the GST activity was involved in transcriptional activation, we measured activation of the luciferase reporter genes in the presence of a wild-type GFZF, a mutant GFZF (S876A), or a truncated GFZF which has the GST domain deleted (Fig. 7A). The wt and S876A mutant activated transcription to similar extents, while the truncated GST-less mutant had approximately half as much activity (Fig. 7B). While wt GFZF activates transcription more robustly than the GST-less mutant in the luciferase assay, it was critical to assess whether differences in protein expression could account for the differences between those samples. To that end, we performed Western blotting against the FLAG epitope to quantify ectopic GFZF expression in cells (representative Western blots are shown in Fig. 7C). Upon comparing the fold increase in luciferase activity with the fold increase in ectopic protein expression, we conclude that the GST portion of GFZF does not contribute to its ability to activate transcription in this assay (Fig. 7D) (P = 0.78 [two-tailed t test, assuming equal variance, comparing difference in fold activation for the wt and ΔGST mutant for the luciferase assay and ectopic GFZF expression]; n = 12).
FIG 7.
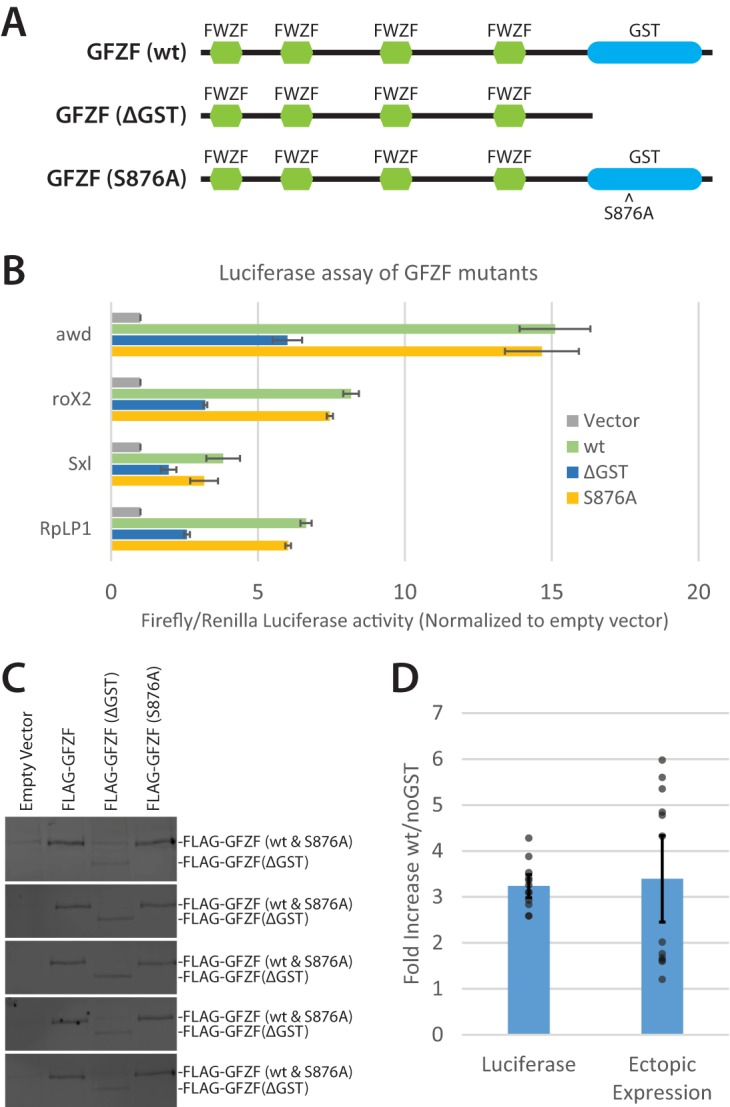
GFZF's GST domain is not required for transcription activation of transfected DNA. (A) Schematic of GFZF and mutations used to test the function of GFZF's GST domain in transcription activation. FWZF stands for FLYWCH zinc finger domain. (B) Normalized firefly/Renilla luciferase activities are displayed for four different reporter promoters with wt, ΔGST, and S876A mutant versions of GFZF ectopically expressed (n = 3). (C) Representative Western blots of luciferase lysates following ectopic expression of wt, ΔGST, or S876A GFZF. Western blots from 5 of 12 independent experiments are shown. The upper and lower band in each blot correspond to full-length (wt or S876A) and truncated (ΔGST) GFZF, respectively. The wt and ΔGST bands were quantified with ImageJ to determine the fold difference in ectopic expression. (D) Fold differences in luciferase activity and ectopic GFZF expression are compared for the wt and ΔGST GFZF samples after combining the data for all four of the tested promoters. Calculation of the ratio of activation by wild-type and ΔGST GZF was made by subtracting the endogenous (empty vector) luciferase values from the values for ectopically expressed GFZF samples and then taking the ratio of the wt to ΔGST adjusted values. Calculation of the ratio of levels of ectopically expressed wild-type and ΔGST GFZF was made by measuring the intensities of anti-FLAG Western blot signals. There is no significant difference between the fold increase in luciferase activity and the fold increase in ectopic expression for the wt versus ΔGST samples (P = 0.78). Error bars represent standard deviation; n = 12 (3 replicates from 4 promoters).
Identification of GFZF as a transcription factor provides insights into its roles in a broad spectrum of biological processes.
Since its initial discovery, GFZF has appeared as a “hit” in numerous screens (10, 11, 13–15). While possible explanations for GFZF's appearance in these screens have been put forth, they have lacked a unifying cellular function that could explain GFZF's seemingly disparate roles. Here we show that GFZF binds approximately 1,800 genes and functions as a transcriptional coactivator. This new information can explain the broad functionality of GFZF. The GFZF gene was first identified in Drosophila as a suppressor of a gene called killer of prune (also known as awd). Mutations in awd alone cause no phenotype, but these mutations cause lethality in flies that are homozygous for nonlethal mutations in another gene, called prune. It was proposed that mutations in the GFZF gene suppressed the lethality caused by the combination of mutations in awd and prune because wild-type GFZF was generating something toxic by conjugating glutathione to a metabolic product derived from the activities of mutant prune (encoding a cyclic AMP phosphodiesterase) and mutant awd (encoding a nucleoside diphosphate kinase) (15). However, our data provide a simpler explanation: GFZF associates with the awd promoter and activates transcription (Fig. 4E, 5A to C, and 8A). Hence, mutations in the GFZF gene would reduce the level of expression of mutant awd so there would no longer be sufficient mutant Awd protein to cause lethality with mutant Prune protein. In another case, GFZF's appearance in a screen for RAS-mediated MAPK activation can be explained by GFZF's binding to the core promoter region of mek (Fig. 8B, Dsor1). In accordance with GFZF's function as a transcriptional coactivator, the authors demonstrated that knockdown of GFZF results in reduced levels of mek transcripts (11). Likewise, GFZF's appearance in the G2-M DNA damage checkpoint screen could be simply explained by GFZF being required for the transcription of other factors involved in this DNA damage checkpoint. Our ChIP-exo analysis indicates that GFZF associates with 22 of the 64 genes that were identified in this screen, including the promoters of factors known to have roles in this DNA damage checkpoint, including myt1, 14-3-3ε, and tefu (10) (Fig. 9).
FIG 8.
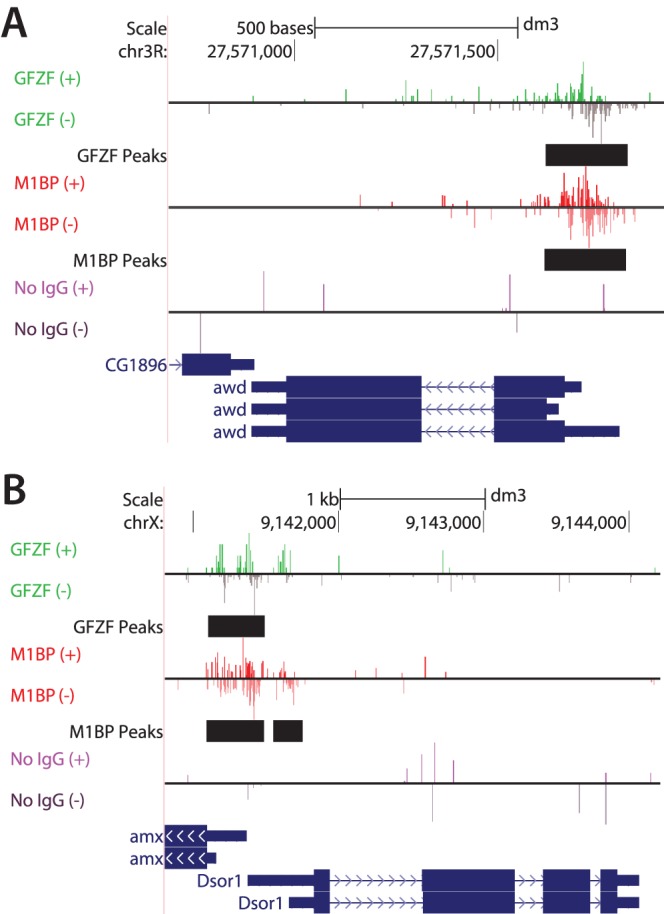
GFZF binds the promoters of genes integral to genetic screens in which GFZF was a “hit.” (A) UCSC genome browser shot showing the association of GFZF and M1BP with the promoter of the gene awd. awd is also known as killer of prune. Mutations in awd alone cause no phenotype, but such mutations cause lethality in flies that are homozygous for nonlethal mutations in another gene, called prune (15). The reads above and below the horizontal line correspond to ChIP-exo reads from the forward (+) and reverse (−) strands, respectively. (B) UCSC genome browser shot showing GFZF's association with the promoter of Dsor1, which is critical for the RAS/MAPK signaling pathway (11). Dsor1 encodes the MEK kinase. Shown are forward (+)- and reverse (−)-strand ChIP-exo reads for M1BP, GFZF, or a no-IgG ChIP control. Black bars show the genomic location of MultiGPS-generated peaks for either GFZF or M1BP.
FIG 9.
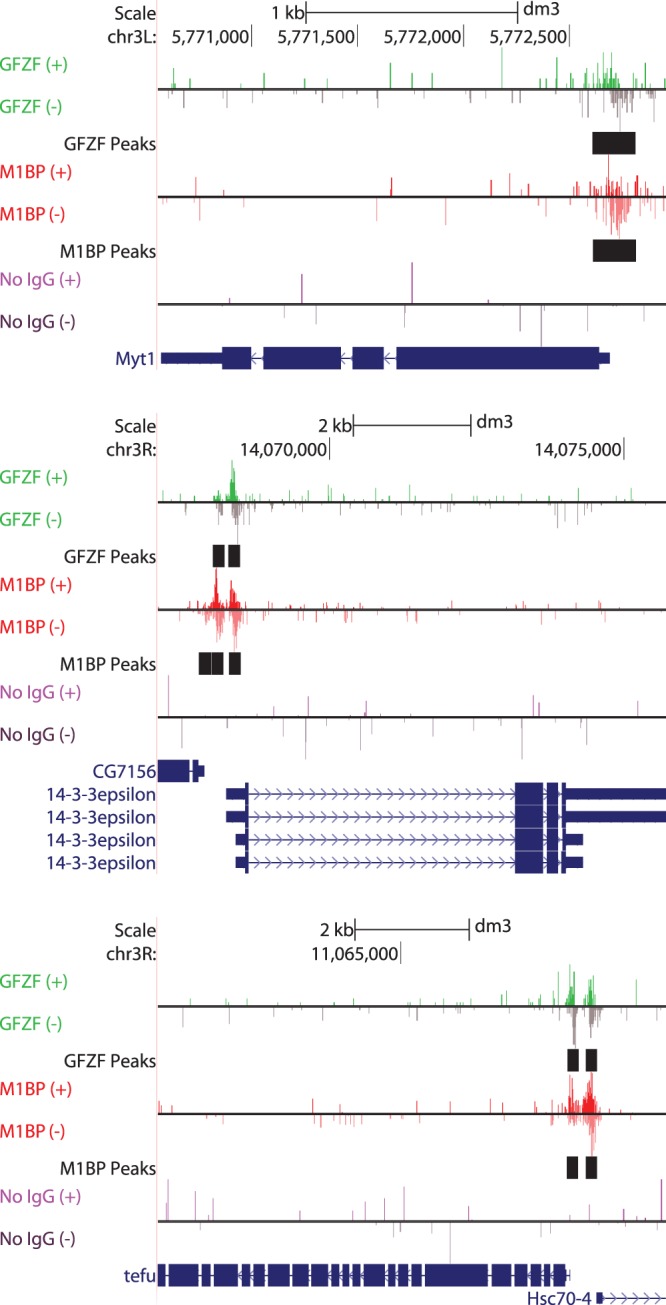
GFZF binds the promoters of genes involved in the G-to-M DNA damage checkpoint. Based on an RNAi screen, Myt1, 14-3-3ε, and tefu were among the top genes to affect the G2-to-M DNA damage checkpoint (10). UCSC Genome browser shots of ChIP-exo data show that GFZF associates with the promoters of all three genes. Shown are forward (+)- and reverse (−)-strand ChIP-exo reads for M1BP, GFZF, or a no-IgG ChIP control. Bars in black show the genomic location of MultiGPS-generated peaks for either GFZF or M1BP.
GFZF was also identified in a screen for mutations that affect hybrid inviability (14). When female Drosophila melanogaster organisms are mated to male Drosophila simulans organisms, no male progeny are produced. Mutations in GFZF in male D. simulans allowed production of male progeny in this interspecies mating. GFZF binds to the promoters of three (msl-1, msl-2, and mle) out of five subunits that comprise the male-specific lethal (MSL) complex in flies. Additionally, it binds to the promoter region of roX2, one of the noncoding RNAs (ncRNAs) that is part of the MSL complex (Fig. 10). The MSL complex functions in dosage compensation in male flies by doubling the amount of transcription arising from genes on the X chromosome; disrupting the function of the MSL complex causes male lethality. Since GFZF is a transcriptional coactivator and binds the promoters of several genes encoding the MSL complex, we speculate that hybrid-specific GFZF-mediated misregulation of MSL components might be contributing to male lethality. This would be consistent with the work of others who have provided evidence that defects in dosage compensation contribute to hybrid inviability (28–30). However, a follow-up study which tested the hypothesis that defects in the MSL complex contribute to hybrid inviability concluded that defects in MSL function cannot fully explain hybrid inviability (31). It could be that GFZF's role in hybrid inviability is more nuanced than misregulation of MSL complex components and might involve misexpression of other factors involved in maintaining incompatibility. Whatever the case, it is reasonable to speculate that GFZF's role will involve misregulation of genes required for maintenance of hybrid inviability.
FIG 10.

GFZF binds the promoter region of male-specific lethal (MSL) complex genes. UCSC Genome browser shots of genes encoding components of the MSL complex are shown. GFZF's appearance in a screen for hybrid inviability, which leads to male lethality, could pertain to its role in expression of components of the MSL complex. Shown are forward (+)- and reverse (−)-strand ChIP-exo reads for M1BP, GFZF, or a no-IgG ChIP control. Bars in black show the genomic location of MultiGPS-generated peaks for either GFZF or M1BP.
The GST activity of GFZF.
GFZF is unusual because of its unique combination of zinc fingers and a functional GST domain. Our search for homologous genes in other organisms indicates that genes sharing homology to the entirety of GFZF are limited to Schizophora, the section of true flies which includes the common housefly. Since other neopterans, including mosquitoes, have GST proteins that share homology with GFZF's GST domain but lack GFZF's zinc fingers, it is likely that GFZF evolved recently as a result of a gene fusion (see the Clustal alignments in Fig. S1 in the supplemental material and the cladogram in Fig. S2 in the supplemental material). In accordance with this hypothesis, mRNA expression data show that there is a second promoter in the intron that immediately precedes the GST domain of the full-length GFZF gene, and the resulting transcript is predicted to encode a functional GST. This transcript is detected from 14-h-old embryos to adults, whereas the full-length GFZF is detected throughout development beginning with 0- to 2-h-old embryos (32).
At this point, the function of the GST domain is unclear. We observed that deletion of this domain reduced the level of expression of the remainder of the protein and that the remaining part still activated transcription. Since we assayed for function only on transiently transfected DNA, it remains possible that the GST activity is important in a natural chromatin context, which is not formed on transiently transfected DNA. Mutations in the GST domain of GFZF that cause larval lethality have been identified, so the domain appears to be essential (15).
It is possible that the gene fusion resulting in GFZF is fortuitous and that the GST domain's function is not linked to gene regulation. On the other hand, this fusion raises the intriguing possibility that GST activity is important for gene expression and that other organisms bring GST activity to a gene's promoter through protein-protein interactions. GST proteins are best known for their roles in protecting cells from toxic endogenous and xenobiotic compounds, so GST might function at promoters to inhibit DNA damage (21). Another possibility is that GFZF serves as a sensor of the redox potential of the cell. Having a GST transcription factor act as a nuclear sensor of the redox state of the cell could ensure that cells can quickly alter their transcriptional program in response to stress and chemical insult. There is precedent for redox regulation of transcription factors, both directly (33) and through signal transduction (19). Brf2, a Pol III core transcription factor, has a single oxidation-prone cysteine residue that when oxidized inhibits Brf2's ability to form a complex with TATA binding protein (TBP) at some Pol III-dependent promoters. In cells, oxidative stress caused a sharp decline in Brf2-dependent gene transcripts (34). In an example of redox regulation through signal transduction, a GST protein acts to inhibit c-Jun N-terminal kinase (JNK) activity under normal physiological conditions. However, when cells are treated with hydrogen peroxide or UV irradiation, the GST dimerizes and no longer inhibits JNK, thus allowing the signaling cascade to commence (19). As further evidence of redox-driven transcriptional regulation, sublethal levels of hydrogen peroxide globally reduce the turnover rate of Pol II paused in the promoter-proximal regions of genes (35). Finally, PrfA, a protein in the intracellular pathogenic bacterium Listeria monocytogenes, appears to be allosterically regulated by glutathione (36). If, as in the above examples, such a molecular switch regulates GFZF function in response to redox perturbations, it would represent an elegant means of quickly altering the expression of a multitude of genes in response to stress.
MATERIALS AND METHODS
Nuclear extracts.
Nuclear extracts were prepared from 0- to 12-h Oregon R embryos as previously described (37).
Immobilized-template pulldowns.
Immobilized-template pulldowns were performed by annealing oligonucleotides (sequences are listed in the supplemental material) corresponding to the core promoter sequence of mRpS30 (−32 to +18) or RpLP1 (−37 to +13) and gel purifying the annealed templates from a polyacrylamide gel. One strand of the annealed template was biotinylated at the 5′ end. Oligonucleotides used for pulldowns from nuclear extracts have an inverted 3′ dT to inhibit degradation by 3′ exonuclease present in the nuclear extracts. For immobilized-template pulldowns from nuclear extracts, 3.5 μg of wt or mutant Motif 1 mRpS30 template was immobilized on 100-μl streptavidin Dynabeads (Thermo Fisher, 11205D) according to the manufacturer's instructions. Template-bound beads were equilibrated in 0.18 M HEMGN (180 mM KCl, 25 mM HEPES [pH 7.6], 12.5 mM MgCl2, 0.1 mM EDTA [pH 7.9], 10% glycerol, 0.1% NP-40, and 1 mM dithiothreitol [DTT]). The beads were then incubated at room temperature for 45 min with 250 μl of nuclear extract and 40 μg of HaeIII-digested E. coli DNA. Beads were washed 5 times at room temperature for 10 min each with 0.18 M HEMGN. Finally, beads were transferred to a new tube and boiled in gel loading buffer, and the resulting proteins were analyzed on an 8% SDS-polyacrylamide gel.
For immobilized-template pulldowns using purified factors, 800 ng wt or mutant Motif 1 RpLP1 template was immobilized on 20-μl streptavidin Dynabeads. Binding reaction mixtures (150 μl) had the following composition: 25 mM HEPES (pH 7.6), 200 mM NaCl, 40 mM KCl, 10% glycerol, 10 μM ZnCl2, 0.1% NP-40, 1 mM DTT, 3.5 μg His-GFZF (or dialysis buffer), 3.5 μg His-M1BP (purification previously described [23]), and 25 μg sonicated salmon sperm DNA. Samples were incubated at room temperature for 1 h, washed 3 times with wash buffer consisting of 25 mM HEPES (pH 7.6), 200 mM NaCl, 40 mM KCl, 10% glycerol, 10 μM ZnCl2, 0.1% NP-40, and 1 mM DTT, and then transferred to a new tube and eluted with 15 μl gel loading buffer for 10 min at 75°C. Samples were loaded on an 8% SDS-polyacrylamide gel and stained with Coomassie brilliant blue. Ten percent (15 μl) of the unbound fractions was also analyzed by SDS-PAGE as described above.
Purification of Mal fusions.
One liter of BL21(DE3) E. coli culture transformed with plasmids encoding N-terminal maltose binding protein (Mal) fused with a rigid linker to the lacZα fragment or M1BP was grown at 37°C to an optical density at 600 nm (OD600) of 0.8. Expression of the Mal fusion protein was induced by addition of IPTG (isopropyl-β-d-thiogalactopyranoside) to a final concentration of 300 μM. Cultures were incubated at 37°C for an additional 2 h and then placed on ice and harvested by centrifugation for 10 min at 7,500 × g. The pellet was frozen in liquid nitrogen and then resuspended in 125 ml Mal lysis buffer (25 mM HEPES [pH 7.6], 500 mM NaCl, 10% glycerol, 0.1% NP-40, 1 mM DTT, 0.4 mM phenylmethylsulfonyl fluoride [PMSF], and a protease inhibitor cocktail). All subsequent steps were performed at 4°C. Cells were lysed by passing the cell suspension 3 times through a Microfluidizer. Lysates were cleared by ultracentrifugation for 1 h in a Beckman type 70 Ti rotor at 35,000 rpm. Fifty milliliters of supernatant was passed through 0.5 ml of amylose resin (NEB E8021) packed in a Poly-Prep gravity column (Bio-Rad, 7311550). The resin was washed with 12 column volumes of Mal lysis buffer and finally with 4 column volumes of Mal lysis buffer with 180 mM NaCl. Protein-bound resin was stored at 4°C.
Mal protein fusion pulldowns with His-GFZF.
Binding reaction mixtures totaling 500 μl consisted of 25 mM HEPES (pH 7.6), 180 mM NaCl, 10% glycerol, 0.1% NP-40, 1 mM DTT, 0.4 mM PMSF, 2 μl Mal-bound amylose resin (∼9 μg Mal fusion protein), and 3.5 μg His-GFZF. Reaction mixtures were incubated at 4°C for 2 h with end-over-end rotation and then washed with 25 mM HEPES (pH 7.6), 180 mM NaCl, 10% glycerol, 0.1% NP-40, 1 mM DTT, and 0.4 mM PMSF at 4°C for 2 h with end-over-end rotation. The resin was transferred to a new tube, and material was eluted in 20 μl gel loading buffer by heating to 75°C for 10 min. The material was analyzed by 10% SDS-PAGE and stained with Coomassie brilliant blue.
Polytene chromosome squashes and immunofluorescence.
Salivary glands were dissected from third-instar larvae and stained with antibody against GFZF (8) or M1BP (7) or with anti-FLAG M2 antibody (Sigma, F1804) as previously described (38).
Purification of full-length His-GFZF.
The coding sequence of GFZF was cloned from S2R+ cDNA into the NheI and EcoRI restriction sites of the pET28 expression vector. Rosetta (DE3)pLysS cells were grown in 1 liter of LB medium at 37°C to an OD600 of 0.4. IPTG was added to a final concentration of 1 mM, and cells were incubated at 18°C for 24 h. Cells were collected, lysed in lysis buffer (50 mM HEPES [pH 7.6], 500 mM NaCl, 10 mM imidazole, 10% glycerol, 0.1% Triton X-100, 2 mM PMSF, and 20 mM 2-mercaptoethanol), sonicated, and centrifuged at 20,000 × g for 20 min. Lysates were applied to 300 μl Ni-nitrilotriacetic acid (NTA) resin (Qiagen, 30210) and bound in batch at 4°C for 1 h. Resin was collected and washed in batch at 4°C for 15 min with 30 ml lysis buffer. Resin was packed in a column, washed with an additional 5 ml lysis buffer, and eluted with 50 mM HEPES (pH 7.6), 150 mM NaCl, 10% glycerol, 200 mM imidazole, 0.1% Triton X-100, 2 mM PMSF, and 20 mM 2-mercaptoethanol. The eluates were further purified through Mono Q using a buffer consisting of 50 mM HEPES (pH 7.6), 10% glycerol, 0.1 mM ZnCl2, 0.1% Triton X-100, and 1 mM DTT with a NaCl gradient from 150 mM to 550 mM.
Generation of FLAG- and HA-tagged M1BP-transgenic flies.
The M1BP-coding sequence was amplified from the plasmid described previously (7) and inserted downstream of the hsp83 promoter in pCaSpeR-hs83 (39). The resulting plasmid encoded M1BP with two consecutive FLAG tags and a hemagglutinin (HA) tag at the N terminus. Drosophila transformation was performed by Rainbow Transgenic Flies, Inc.
ChIP-exo.
ChIP-exo was performed with antibodies against GFZF (8) and M1BP (7) essentially as described in reference 23 with minor modifications. Libraries were quantified by quantitative PCR (qPCR) and sequenced on an Illumina NextSeq 500. Base calls were performed using Bcl2FastQ version 2.16.0. Sequenced reads were masked for low-quality sequence and then mapped to the D. melanogaster dm3 whole genome using bwa mem (versions 0.7.9a, 0.7.12) with the default parameters. Heat maps were generated with HOMER bioinformatics software (47) and java Treeview (48). Tables for composite plots were generated with HOMER, and plots were visualized using R (49). Based on the knowledge that M1BP mediates GFZF's association with chromatin, we reasoned that we might be able to boost GFZF's ChIP-exo signal intensity by using additional cross-linking reagents which would increase the likelihood that GFZF and M1BP form protein-protein cross-links. To test this, we cross-linked cells first with dimethyl adipimidate and ethylene glycol bis(succinimidyl succinate) for 10 min and then with formaldehyde for an additional 10 min. We performed ChIP-exo with this material and compared this additionally cross-linked material with our formaldehyde-only material and found a good correlation (Pearson r2 = 0.87 for summed reads surrounding gene promoters) between the data sets and also noticed increased signal intensities using the chromatin with extra cross-linkers. Thus, we used the data from this chromatin preparation for our GFZF analysis. Genome browser images were generated with the UCSC genome browser (40).
Peak calling.
The 5′ ends of reads were obtained, and MultiGPS (41) was used to call peaks using the default settings with Preimmune ChIP-exo libraries serving as control samples. A list of genes with a GFZF peak within 100 bp of its TSS was used for gene ontology (GO) analysis. GO analysis was performed using DAVID with the GOTERM_BP_1 list (42, 43). The Venn diagram was generated with BioVenn (44). The active gene list was derived from reference 45 as described in reference 7.
Luciferase reporter assays.
S2R+ cells were grown in M3+BPYE containing 10% FBS and at 25°C. For luciferase assays without dsRNA treatment (Fig. 7), 1.7 million S2R+ cells in 600 μl of medium were seeded per well in 24-well plates (Corning, 353047) and incubated overnight. The following day, cells in a well were transfected with a mixture of 25 ng pGL3-(RpLP1 [−500 to +50], Sxl [−500 to +53], roX2 [−258 to +60], or awd [−500 to +25]), 25 ng pRL-polIII-Renilla (46), 50 ng pAc5.1 (empty or 2XFLAG-GFZF wt, ΔGST, or S876A mutant), 20 μl serum-free medium, and 0.2 μl FuGENE HD (Promega, E2311). The plasmids and medium were premixed prior to the addition of FuGENE HD as indicated in the manufacturer's protocol. Two days after transfection, cells were lysed and assayed for luciferase activity using the dual-luciferase reporter assay system (Promega, E1910) according to the manufacturer's protocol. To monitor relative levels of transiently expressed derivatives of GFZF, a portion of cell lysate from the dual-luciferase assay containing 20 μg of total protein (as determined with the Bio-Rad Bradford assay 5000006) was subjected to Western blot analysis with M2 anti-FLAG antibody (Sigma, F1804). For luciferase assays with dsRNA treatment (Fig. 4), dsRNA was generated by in vitro transcription with T7 polymerase on PCR-generated templates flanked by T7 promoter sequences (the primer sequences used to generate each PCR template are included in Table S1). A total of 1.7 million cells in 300 μl serum-free medium were seeded per well in 24-well plates. The cells were then treated with 3 μg of dsRNA for 1 h, after which 300 μl of medium supplemented with 20% fetal bovine serum (FBS) and 2× antibiotic plus antifungal (Corning, 30-004-CI) was added. dsRNA-treated cells were then incubated overnight and transfected with DNA on the following day as described above.
RNAi depletion of GFZF followed by ChIP.
S2R+ cells were maintained in M3+BPYE containing 10% FBS and grown at 25°C. Ten milliliters of cells at 2.8 million cells per ml was plated in a 10-cm dish and incubated overnight. The following day, the medium was removed, the cells were rinsed with phosphate-buffered saline (PBS), and 6 ml of serum-free medium was added. Sixty micrograms of dsRNA targeting either lacZ or exon 2 of GFZF was added to the culture and incubated for 1 h. Six milliliters of medium containing 20% FBS and 2× antibiotic plus antifungal (Corning, 30-004-CI) was added, and cells were incubated for an additional 3 days. Chromatin immunoprecipitation (ChIP) experiments were performed as described previously (7) using rabbit polyclonal antisera against GFZF, M1BP, or Rpb3. Primers used to quantify percent recovery are listed in Table S1. For Western blots, cells were lysed in lithium dodecyl sulfate (LDS) sample buffer, and material equivalent to 2 million lysed cells was subjected to SDS-PAGE, transferred to nitrocellulose, and probed with antibodies against GFZF, M1BP, or NELF-E.
Expression and purification of the GFZF GST domain.
For the purification of the His-tagged GST domain of GFZF, the DNA sequence encoding residues 800 to 1045 of GFZF was cloned into pET28 using the NheI and EcoRI restriction sites. The S876A mutation was introduced using the In-Fusion cloning kit (Clontech, 638910) with primers bearing the desired mutations. Rosetta (DE3)pLysS cells were transformed, and cells were grown at 37°C in 4 liters of LB medium to an OD600 of 0.8 to 0.9 and induced with 0.5 mM IPTG. Following induction, cells were incubated at 15°C for 24 h. Cell were harvested and resuspended in 150 ml of lysis buffer (25 mM HEPES [pH 7.6], 500 mM NaCl, 5 mM imidazole, 10% glycerol, 0.1% Nonidet P-40, 1 μM ZnCl2, 0.1 mM PMSF, 20 mM 2-mercaptoethanol, and a protease inhibitor cocktail). The cells were lysed by passing the cell suspension through a Microfluidizer three times and then ultracentrifuged at 125,000 × g for 30 min. The cleared lysate was bound in batch with 1.5 ml of Talon resin at 4°C for 1 h and washed in batch with 45 ml of lysis buffer for 30 min at 4°C. The Talon resin was packed into a column, washed with an additional 5 column volumes, and eluted with buffer consisting of 50 mM HEPES (pH 7.6), 100 mM NaCl, 200 mM imidazole, 10% glycerol, 0.1% Nonidet P-40, 1 μM ZnCl2, 0.1 mM PMSF, and 20 mM 2-mercaptoethanol. The eluates were dialyzed overnight at 4°C in a buffer consisting of 25 mM HEPES (pH 7.6), 100 mM NaCl, 10% glycerol, 0.1% Nonidet P-40, 0.1 mM PMSF, and 2 mM DTT. Purified proteins were flash frozen in liquid nitrogen and stored at −80°C.
GST activity assay.
One-milliliter reaction mixtures consisting of 100 mM potassium phosphate (pH 6.5), 1 mM 1-chloro-2,4-dinitrobenzene (CDNB), 130 nM (8 μg) purified His-GFZF truncations, and various amounts of glutathione were assayed for absorbance at 340 nm every 20 s for 10 min. Automated readings were taken on a Pharmacia Biotech Ultrospec 3000. The slope for the linear part of the curve (typically 0 to 240 s) was taken as the initial velocity. R was used to generate the graph and determine the Vmax and Km using nonlinear least-squares regression.
Accession number(s).
Sequence data are in the Gene Expression Omnibus (GEO) database under accession number GSE105009.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health (NIH) (grant R01GM047477 to D.S.G.).
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/MCB.00476-17.
REFERENCES
- 1.Danino YM, Even D, Ideses D, Juven-Gershon T. 2015. The core promoter: at the heart of gene expression. Biochim Biophys Acta 1849:1116–1131. doi: 10.1016/j.bbagrm.2015.04.003. [DOI] [PubMed] [Google Scholar]
- 2.Thomas MC, Chiang C-M. 2008. The general transcription machinery and general cofactors. Crit Rev Biochem Mol Biol 41:105–178. doi: 10.1080/10409230600648736. [DOI] [PubMed] [Google Scholar]
- 3.Zabidi MA, Stark A. 2016. Regulatory enhancer–core-promoter communication via transcription factors and cofactors. Trends Genet 32:801–814. doi: 10.1016/j.tig.2016.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ohler U, Wassarman DA. 2010. Promoting developmental transcription. Development 137:15–26. doi: 10.1242/dev.035493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.FitzGerald PC, Sturgill D, Shyakhtenko A, Oliver B, Vinson C. 2006. Comparative genomics of Drosophila and human core promoters. Genome Biol 7:R53. doi: 10.1186/gb-2006-7-7-r53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ohler U, Liao G-C, Niemann H, Rubin GM. 2002. Computational analysis of core promoters in the Drosophila genome. Genome Biol 3:RESEARCH0087. doi: 10.1186/gb-2002-3-12-research0087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li J, Gilmour DS. 2013. Distinct mechanisms of transcriptional pausing orchestrated by GAGA factor and M1BP, a novel transcription factor. EMBO J 32:1829–1841. doi: 10.1038/emboj.2013.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dai M-S, Sun X-X, Qin J, Smolik SM, Lu H. 2004. Identification and characterization of a novel Drosophila melanogaster glutathione S-transferase-containing FLYWCH zinc finger protein. Gene 342:49–56. doi: 10.1016/j.gene.2004.07.043. [DOI] [PubMed] [Google Scholar]
- 9.Ambrus AM, Rasheva VI, Nicolay BN, Frolov MV. 2009. Mosaic genetic screen for suppressors of the de2f1 mutant phenotype in Drosophila. Genetics 183:79–92. doi: 10.1534/genetics.109.104661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kondo S, Perrimon N. 2011. A genome-wide RNAi screen identifies core components of the G2-M DNA damage checkpoint. Sci Signal 4:rs1. doi: 10.1126/scisignal.2001350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ashton-Beaucage D, Udell CM, Gendron P, Sahmi M, Lefrançois M, Baril C, Guenier A-S, Duchaine J, Lamarre D, Lemieux S, Therrien M. 2014. A functional screen reveals an extensive layer of transcriptional and splicing control underlying RAS/MAPK signaling in Drosophila. PLoS Biol 12:e1001809. doi: 10.1371/journal.pbio.1001809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li H-M, Buczkowski G, Mittapalli O, Xie J, Wu J, Westerman R, Schemerhorn BJ, Murdock LL, Pittendrigh BR. 2008. Transcriptomic profiles of Drosophila melanogaster third instar larval midgut and responses to oxidative stress. Insect Mol Biol 17:325–339. doi: 10.1111/j.1365-2583.2008.00808.x. [DOI] [PubMed] [Google Scholar]
- 13.Gonzalez I, Mateos-Langerak J, Thomas A, Cheutin T, Cavalli G. 2014. Identification of regulators of the three-dimensional polycomb organization by a microscopy-based genome-wide RNAi screen. Mol Cell 54:485–499. doi: 10.1016/j.molcel.2014.03.004. [DOI] [PubMed] [Google Scholar]
- 14.Phadnis N, Baker EP, Cooper JC, Frizzell KA, Hsieh E, de la Cruz AFA, Shendure J, Kitzman JO, Malik HS. 2015. An essential cell cycle regulation gene causes hybrid inviability in Drosophila. Science 350:1552–1555. doi: 10.1126/science.aac7504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Provost E, Hersperger G, Timmons L, Ho WQ, Hersperger E, Alcazar R, Shearn A. 2006. Loss-of-function mutations in a glutathione S-transferase suppress the prune-killer of prune lethal interaction. Genetics 172:207–219. doi: 10.1534/genetics.105.044669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barth TK, Schade GOM, Schmidt A, Vetter I, Wirth M, Heun P, Thomae AW, Imhof A. 2014. Identification of novel Drosophila centromere-associated proteins. Proteomics 14:2167–2178. doi: 10.1002/pmic.201400052. [DOI] [PubMed] [Google Scholar]
- 17.Ranson H, Rossiter L, Ortelli F, Jensen B, Wang X, Roth CW, Collins FH, Hemingway J. 2001. Identification of a novel class of insect glutathione S-transferases involved in resistance to DDT in the malaria vector Anopheles gambiae. Biochem J 359:295–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Salinas AE, Wong MG. 1999. Glutathione S-transferases—a review. Curr Med Chem 6:279–309. [PubMed] [Google Scholar]
- 19.Adler V, Yin Z, Fuchs SY, Benezra M, Rosario L, Tew KD, Pincus MR, Sardana M, Henderson CJ, Wolf CR, Davis RJ, Ronai Z. 1999. Regulation of JNK signaling by GSTp. EMBO J 18:1321–1334. doi: 10.1093/emboj/18.5.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kamada K, Goto S, Okunaga T, Ihara Y, Tsuji K, Kawai Y, Uchida K, Osawa T, Matsuo T, Nagata I, Kondo T. 2004. Nuclear glutathione S-transferase π prevents apoptosis by reducing the oxidative stress-induced formation of exocyclic dna products. Free Rad Biol Med 37:1875–1884. doi: 10.1016/j.freeradbiomed.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 21.Hayes JD, Flanagan JU, Jowsey IR. 2005. Glutathione transferases. Annu Rev Pharmacol Toxicol 45:51–88. doi: 10.1146/annurev.pharmtox.45.120403.095857. [DOI] [PubMed] [Google Scholar]
- 22.Hochheimer A, Zhou S, Zheng S, Holmes MC, Tjian R. 2002. TRF2 associates with DREF and directs promoter-selective gene expression in Drosophila. Nature 420:439. doi: 10.1038/nature01167. [DOI] [PubMed] [Google Scholar]
- 23.Baumann DG, Gilmour DS. 2017. A sequence-specific core promoter-binding transcription factor recruits TRF2 to coordinately transcribe ribosomal protein genes. Nucleic Acids Res 45:10481–10491. doi: 10.1093/nar/gkx676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rhee HS, Pugh BF. 2012. ChIP-exo method for identifying genomic location of DNA-binding proteins with near-single-nucleotide accuracy. Curr Protoc Mol Biol Chapter 21:Unit 21.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kakuta Y, Usuda K, Nakashima T, Kimura M, Aso Y, Yamamoto K. 2011. Crystallographic survey of active sites of an unclassified glutathione transferase from Bombyx mori. Biochim Biophys Acta 1810:1355–1360. doi: 10.1016/j.bbagen.2011.06.022. [DOI] [PubMed] [Google Scholar]
- 26.Montero D, Tachibana C, Rahr Winther J, Appenzeller-Herzog C. 2013. Intracellular glutathione pools are heterogeneously concentrated. Redox Biol 1:508–513. doi: 10.1016/j.redox.2013.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Söderdahl T, Enoksson M, Lundberg M, Holmgren A, Ottersen OP, Orrenius S, Bolcsfoldi G, Cotgreave IA. 2003. Visualization of the compartmentalization of glutathione and protein-glutathione mixed disulfides in cultured cells. FASEB J 17:124–126. [DOI] [PubMed] [Google Scholar]
- 28.Rodriguez MA, Vermaak D, Bayes JJ, Malik HS. 2007. Species-specific positive selection of the male-specific lethal complex that participates in dosage compensation in Drosophila. Proc Natl Acad Sci U S A 104:15412–15417. doi: 10.1073/pnas.0707445104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chatterjee RN, Chatterjee P, Pal A, Pal-Bhadra M. 2007. Drosophila simulans lethal hybrid rescue mutation (Lhr) rescues inviable hybrids by restoring X chromosomal dosage compensation and causes fluctuating asymmetry of development. J Genet 86:203–215. doi: 10.1007/s12041-007-0028-5. [DOI] [PubMed] [Google Scholar]
- 30.Bachtrog D. 2008. Positive selection at the binding sites of the male-specific lethal complex involved in dosage compensation in Drosophila. Genetics 180:1123–1129. doi: 10.1534/genetics.107.084244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barbash DA. 2010. Genetic testing of the hypothesis that hybrid male lethality results from a failure in dosage compensation. Genetics 184:313–316. doi: 10.1534/genetics.109.108100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gramates LS, Marygold SJ, Santos GD, Urbano J-M, Antonazzo G, Matthews BB, Rey AJ, Tabone CJ, Crosby MA, Emmert DB, Falls K, Goodman JL, Hu Y, Ponting L, Schroeder AJ, Strelets VB, Thurmond J, Zhou P, the FlyBase Consortium. 2017. FlyBase at 25: looking to the future. Nucleic Acids Res 45:D663–D671. doi: 10.1093/nar/gkw1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brigelius-Flohé R, Flohé L. 2011. Basic principles and emerging concepts in the redox control of transcription factors. Antioxid Redox Signal 15:2335–2381. doi: 10.1089/ars.2010.3534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gouge J, Satia K, Guthertz N, Widya M, Thompson AJ, Cousin P, Dergai O, Hernandez N, Vannini A. 2015. Redox signaling by the RNA polymerase III TFIIB-related factor Brf2. Cell 163:1375–1387. doi: 10.1016/j.cell.2015.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nilson KA, Lawson CK, Mullen NJ, Ball CB, Spector BM, Meier JL, Price DH. 2017. Oxidative stress rapidly stabilizes promoter-proximal paused Pol II across the human genome. Nucleic Acids Res 45:11088–11105. doi: 10.1093/nar/gkx724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reniere ML, Whiteley AT, Hamilton KL, John SM, Lauer P, Brennan RG, Portnoy DA. 2015. Glutathione activates virulence gene expression of an intracellular pathogen. Nature 517:170–173. doi: 10.1038/nature14029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Biggin MD, Tjian R. 1988. Transcription factors that activate the Ultrabithorax promoter in developmentally staged extracts. Cell 53:699–711. doi: 10.1016/0092-8674(88)90088-8. [DOI] [PubMed] [Google Scholar]
- 38.Ghosh SKB, Missra A, Gilmour DS. 2011. Negative elongation factor accelerates the rate at which heat shock genes are shut off by facilitating dissociation of heat shock factor. Mol Cell Biol 31:4232–4243. doi: 10.1128/MCB.05930-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Missra A, Gilmour DS. 2010. Interactions between DSIF (DRB sensitivity inducing factor), NELF (negative elongation factor), and the Drosophila RNA polymerase II transcription elongation complex. Proc Natl Acad Sci U S A 107:11301–11306. doi: 10.1073/pnas.1000681107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. 2002. The human genome browser at UCSC. Genome Res 12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mahony S, Edwards MD, Mazzoni EO, Sherwood RI, Kakumanu A, Morrison CA, Wichterle H, Gifford DK. 2014. An integrated model of multiple-condition ChIP-Seq data reveals predeterminants of Cdx2 binding. PLoS Comput Biol 10:e1003501. doi: 10.1371/journal.pcbi.1003501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huang DW, Sherman BT, Lempicki RA. 2009. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang DW, Sherman BT, Lempicki RA. 2009. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 44.Hulsen T, de Vlieg J, Alkema W. 2008. BioVenn—a web application for the comparison and visualization of biological lists using area-proportional Venn diagrams. BMC Genomics 9:488. doi: 10.1186/1471-2164-9-488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nechaev S, Fargo DC, dos Santos G, Liu L, Gao Y, Adelman K. 2010. Global analysis of short RNAs reveals widespread promoter-proximal stalling and arrest of Pol II in Drosophila. Science 327:335–338. doi: 10.1126/science.1181421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gilchrist DA, Nechaev S, Lee C, Ghosh SKB, Collins JB, Li L, Gilmour DS, Adelman K. 2008. NELF-mediated stalling of Pol II can enhance gene expression by blocking promoter-proximal nucleosome assembly. Genes Dev 22:1921–1933. doi: 10.1101/gad.1643208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. 2010. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38:576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Saldanha AJ. 2004. Java Treeview–extensible visualization of microarray data. Bioinformatics 20:3246–3248. [DOI] [PubMed] [Google Scholar]
- 49.R Core Team 2014. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.