

Abstract
Null mutations in the human IQCB1/NPHP5 (nephrocystin-5) gene that encodes NPHP5 are the most frequent cause of Senior-Løken syndrome, a ciliopathy that is characterized by Leber congenital amaurosis and nephronophthisis. We generated germline Nphp5-knockout mice by placing a β-Geo gene trap in intron 4, thereby truncating NPHP5 at Leu87 and removing all known functional domains. At eye opening, Nphp5−/− mice exhibited absence of scotopic and photopic electroretinogram responses, a phenotype that resembles Leber congenital amaurosis. Outer segment transmembrane protein accumulation in Nphp5−/− endoplasmic reticulum was evident as early as postnatal day (P)6. EGFP-CETN2, a centrosome and transition zone marker, identified basal bodies in Nphp5−/− photoreceptors, but without fully developed transition zones. Ultrastructure of P6 and 10 Nphp5−/− photoreceptors revealed aberrant transition zones of reduced diameter. Nphp5−/− photoreceptor degeneration was complete at 1 mo of age but was delayed significantly in Nphp5−/−;Nrl−/− (cone only) retina. Nphp5−/− mouse embryonic fibroblast developed normal cilia, and Nphp5−/− kidney histology at 1 yr of age showed no significant pathology. Results establish that nephrocystin-5 is essential for photoreceptor outer segment formation but is dispensable for kidney and mouse embryonic fibroblast ciliary formation.—Ronquillo, C. C., Hanke-Gogokhia, C., Revelo, M. P., Frederick, J. M., Jiang, L., Baehr, W. Ciliopathy-associated IQCB1/NPHP5 protein is required for mouse photoreceptor outer segment formation.
Keywords: Senior-Løken syndrome, nephronophthisis, Leber congenital amaurosis, nephrocystins
Resident proteins of primary cilia, nephrocystins (NPHPs) are distinct in sequence and structure (1) and do not form a large complex as observed for the BBSome (2). Nephrocystins are expressed ubiquitously among ciliated cells and localize to the basal body and cilium where they may mediate a variety of sensory functions, including mechanosensation, chemosensation, olfaction, and vision (3). In renal epithelia, NPHP1, -4, -5, -6, and -8 are present in the transition zone, whereas NPHP2, -3, and -9 localize to the adjacent Inversin compartment. Null mutations in NPHP genes are associated with nonsyndromic and syndromic ciliopathies, for example, Senior-Løken syndrome (SLS), Meckel syndrome, and Joubert syndrome. SLS is an autosomal-recessive, retina-renal ciliopathy that is characterized by progressive retinitis pigmentosa (RP) (4) or Leber congenital amaurosis (LCA) (5) with nephronophthisis (6, 7). LCA and RP are characterized by presence of nonfunctional rods and cones at birth and progressive loss of rod photoreceptors, respectively (4). Nephronophthisis presents with diminished kidney size, corticomedullary cysts, and tubulointerstitial fibrosis (7).
Mutations in the gene that encodes the protein, termed IQ calmodulin-binding motif containing B1 (IQCB1 alias NPHP5), are the most common cause of SLS (6, 8–10). Numerous NPHP5-null mutations clustered in IQ and coiled-coil (CC) domains have been identified in patients with SLS, most of which are deletions, stop codons, or frameshifts (Fig. 1A, B). NPHP5 mutations have also been identified in patients with nonsyndromic LCA (11, 12). Retina phenotype of NPHP5-LCA is very severe, as the outer nuclear layer (ONL) is barely detectable in young patients and it is unclear whether outer segments (OSs) even develop (13). Foveal cone nuclei seem to be retained much longer, but gradual thinning was observed with eccentricity (13). Cone rod dystrophy 2 (crd2) in the American Pit Bull Terrier carries a frameshift mutation in exon 12 of the canine Nphp5 gene and is associated with LCA (14).
Figure 1.

Generation of Nphp5−/− mice. A) Diagram of the 15 exon Nphp5 gene. Translation of NPHP5 initiates in exon 3 and the β-Geo gene trap is located in intron 4, truncating NPHP5 at Leu87. A loxP site was placed in intron 5. B) IQCB1/NPHP5 functional domains. NPHP5 has BBSome interaction sites (BBS; yellow), 3 CC domains (green), 2 IQCB motifs in exons 10 and 12 (IQ; blue), respectively, and a CEP290/NPHP6-binding site (Cep). Position of a frameshift mutation in exon 12 of the Crd2 dog associated with SLS is noted (14). Mutations associated with SLS (red) and nonsyndromic LCA (black) are indicated. C) X-gal staining of embryonic d 13.5 heterozygous and homozygous embryos shows ubiquitous expression of NPHP5. D) Presence of β-Geo trap was determined by PCR with LAR3/R (294 bp), whereas WT Nphp5 gene was identified with F/R (452 bp). E) Immunohistochemistry of Nphp5+/− and Nphp5−/− retina cryosections at postnatal day (P)6, P10, and P15. Sections were probed with anti–NPHP5-KK antibody (red) and contrasted with DAPI (blue) to reveal ONL nuclei; absence of NPHP5 immunoreactivity in Nphp5−/− retina confirms germline knockout. IS, inner segment.
Among vertebrates, photoreceptor OS is considered a specialized primary cilium that houses phototransduction machinery (15) and is responsible for initial encoding of visual information. NPHP5 protein was shown to distribute in the photoreceptor proximal OS (16) as well as to the basal body/transition zone in primary cilia of renal epithelial cells (8, 17), but without a known physiologic function. Human and murine NPHP5 are proteins that consist of 598 aa with BBSome interaction sites, IQCB motifs, and CC domains that encompass the centrosomal localization domain and a C-terminal CEP290 binding site (Fig. 1) (6, 8, 17). NPHP5, calmodulin, and RP GTPase regulator coprecipitate, probably as a multiprotein complex (6, 18). NPHP5 interacts with RP GTPase regulator indirectly (19, 20) and with CEP290/NPHP6 directly (21, 22). In vitro studies showed that CEP290-NPHP5 interaction is required for ciliogenesis (8). Pathogenic NPHP5 mutations, which truncate the protein, delete the CEP290 binding site, thus impairing ciliogenesis (8). NPHP5 knockdown studies in vitro show decreased numbers of primary cilia in hTert-RPE1 cells (8, 22). Morpholino knockdown of NPHP5 in zebrafish shows development of pronephric cysts and blocks trafficking of GFP that is tagged with a rhodopsin targeting signal but not GFP tagged with peripherin-2 targeting signal (21, 23). These results suggest that NPHP5, at least in immortalized cell lines, acts as an early positive regulator of ciliogenesis and its absence inhibits migration and/or anchoring of the basal body to the cell cortex (8).
We generated Nphp5−/− mice and show that they recapitulate the human pathology of rapid retinal degeneration but without obvious renal disease. Absence of NPHP5 does not prevent proper anchoring of the basal body to the cell cortex, but formation of the transition zone is impaired and OSs fail to develop. Rod and cone photoreceptor degeneration is complete at 1 mo of age. By contrast, cone photoreceptor degeneration is delayed in Nphp5−/−;Nrl−/− double-knockout mice. Nphp5−/− mouse will be important for development of therapeutic strategies to inhibit progression of LCA.
MATERIALS AND METHODS
Mice
Wild-type (WT; C57BL/6J), Nrl−/− (021152), and transgenic mice that expressed EGFP-CETN2 (008234) were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). All animal experiments were approved by the Institutional Animal Care and Use Committee of the University of Utah.
Generation and genotyping of Nphp5−/− mice
An embryonic stell cell line that carried the Nphp5 gene into which a β-Geo gene trap was inserted (International Knockout Mouse Consortium, European Conditional Mouse Mutagenesis Program) was used to generate chimeras (Fig. 1) as described previously (24–26). Proper arrangement of the trap in intron 4, presence of 5′ and 3′ arms, and location of loxP in intron 5 were confirmed by PCR and sequencing. PCR was performed with an initial melting temperature of 94°C for 3 min, then 35 cycles of 94°C (30 s), 64/61°C (WT/mutant) (30 s), and 72°C (30 s), followed by a final 3-min extension at 72°C. Animals were genotyped by using primers specific for Nphp5 5′-CCTTTAGGGTGATAGTAGCCAATTCC-3′ (forward), 5′-AGGAACTAAGCTGTGAAATGGACC-3′ (reverse) and specific for the gene trap 5′-CAACGGGTTCTTCTGTTAGTCC-3′ (forward). WT allele yields an amplicon of 452 bp, and mutant allele, 294 bp. Chimeric mice were mated to WT C57BL/6J mice to generate heterozygous (Nphp5+/−) mice. Heterozygous mice are indistinguishable from WT littermates and breed, thrive, and survive normally. Breeding Nphp5+/− mice produced Nphp5-knockout animals at mendelian ratios.
Nphp5−/−;Nrl−/− mice
Nphp5−/− males were bred with Nrl−/− females to generate Nphp5+/−;Nrl+/− double heterozygotes. Male and female Nphp5+/−;Nrl+/− mice were bred to generate double knockouts. Nrl−/− mice were genotyped as described (27).
Multiple tissue RT-PCR
RNA was isolated (RNeasy Mini kit; Qiagen, Valencia CA, USA) from various tissues (eye, brain, heart, kidney, intestines, testis, ovary, lung, muscle, and thymus), and cDNA was synthesized by using SuperScriptIII First-Strand Synthesis System (Thermo Fisher Scientific Life Sciences, Waltham, MA, USA). PCR was performed by using primers specific for Nphp5 5′-GGTCCTCAGTCAAGATTCTTCTCG-3′ (forward) and 5′-ACCTTCTCCCCAGGATAAGAAA-3′ (reverse) to yield a 172 bp amplicon. Glyceraldehyde 3-phosphate dehydrogenase was used as a control.
Whole-mount embryo staining
Nphp5+/− breeding pairs were mated for 1–2 d, then separated. Weight of each female mouse was tracked daily starting 10 d after mating. A ∼1 g/d increase in weight suggested pregnancy. At embryonic d 13.5, females were euthanized, and embryos were dissected and measured. Yolk sac was collected for genotyping. Embryos were then fixed for 25–30 min in fixative [2% paraformaldehyde (PFA), 0.1 M PIPES pH 6.9, 2 mM MgCl2, 1.25 mM EGTA pH 8.0, in water] and washed 3× 5–15 min with PBS/1 mM MgCl2. Subsequently, embryos were incubated in X-gal staining solution (20 mM K3Fe(CN)6, 20 mM K4Fe(CN)6, 1 mM MgCl2, 0.01% sodium deoxycholate, 0.02% NP-40 in PBS, pH 7.2, with addition of 1 mg/ml X-gal before incubation) for 1–2 nights at room temperature, postfixed with 4% PFA in PBS for 3 h at room temperature, rinsed in PBS, imaged, and stored at 4°C.
Antibodies
Rhodopsin, ML-opsin, S-opsin, rod Tα, rod PDE6, cone arrestin, cone PDE6, cone Tγ, GRK1, prominin 1, and Ac-tubulin antibodies have been described (24, 26, 28–31). Anti–NPHP5-KK antibody was provided by Dr. Edgar Otto (University of Michigan, Ann Arbor, MI, USA) (6). Anti-CEP290 antibody was provided by Dr. Rob Mullins (University of Iowa, Iowa City, IA, USA).
Confocal immunolocalization of photoreceptor proteins
Littermate mouse eyes of various ages were dark-adapted, harvested under dim red light, fixed, and processed as described (24, 26, 28). Cryosections were blocked with 2% bovine serum albumin in 0.1 M phosphate buffer pH 7.4 with 0.1% Triton X-100 (blocking buffer) for 1 h at room temperature and incubated with primary antibody in blocking buffer overnight in a humidified chamber at 4°C. Sections were washed 3 times with 0.1 M phosphate buffer and incubated with a fluorescent (i.e., Alexa Fluor 555, Cy3) goat secondary antibody and DAPI (1:1000) in blocking buffer for 1 h in a humidified chamber at room temperature. Sections were washed 3 times with 0.1 M phosphate buffer, cover-slipped, and imaged using an Olympus Fluoview (FV1000; Olympus, Tokyo, Japan) or Zeiss LSM800 confocal microscope (Zeiss, Jena, Germany).
Immunohistochemistry of kidney sections
Mice of various ages were euthanized and their kidneys were isolated and fixed in 4% PFA for 4–6 h. Fixed kidneys were washed in 0.1 M phosphate buffer, pH 7.4, transferred to 15% sucrose, and incubated overnight at 4°C, then transferred to 30% sucrose and incubated a second night at 4°C. Kidneys were embedded in optimal cutting temperature compound and stored at −80°C. Cryosections were blocked with 2% bovine serum albumin in 0.1 M phosphate buffer, pH 7.4, that contained 0.1% Triton X-100 for 1 h, then incubated with primary antibody directed against Ac-tubulin (1:1000) in blocking buffer overnight in a humidified chamber at 4°C. After washing with 0.1 M phosphate buffer (10 min × 3), sections were incubated with a fluorescent goat anti-mouse secondary antibody (1:1000) in blocking buffer for 1 h in a humidified chamber at room temperature, washed again with 0.1 M phosphate buffer (10 min × 3), and cover-slipped using antifade medium. Section images were acquired with an Olympus Fluoview (FV1000) inverted confocal microscope.
Light microscopy
Mouse kidneys were collected immediately after euthanasia, sectioned on anterior/posterior plane, and fixed in 10% formaldehyde. Samples were paraffin-embedded, sectioned (3 μm) and stained with hematoxylin and eosin/periodic acid-Schiff per manufacturer guidelines. All histologic sections were imaged using an Olympus bright field microscope (BX51).
Electron microscopy
Isolated mouse eyecups were fixed by immersion in fixative solution (1% glutaraldehyde and 1% paraformaldehyde in 0.1 M cacodylate buffer, pH 7.4) at 4°C overnight as previously described (28, 32, 33). Transmission electron microscopy was performed at 75 kV by using a Jeol electron microscope (Jeol Ltd., Tokyo, Japan).
Fundoscopy
Mice were anesthetized by initial inhalation of a mixture of 3% issoflurane/O2 in a closed canister at a flow rate of 1.0 L/min. Mice were transferred on a heated water pad (37°C) while anesthesia was maintained at 1.5% isoflurane/O2 at the same flow rate. Pupils were dilated with a 1% tropicamide solution. Pupils were hydrated periodically with saline solution to prevent corneal desiccation. Imaging was accomplished by using Heidelberg Retina Angiography–Optical Coherence Tomography (Heidelberg, Germany) with a 488-nm argon blue laser with standard 500-nm long-pass filter. Images were acquired from both eyes with a 30° lens. After acquiring images, animals were transferred to a recovery heat pad that was maintained at 37°C and were monitored until they regained full consciousness.
Electroretinography
Scotopic and photopic electroretinogram (ERG) responses were recorded from Nphp5+/− and Nphp5−/− mouse eyes at P14 and P18, and from Nrl−/− single and double knockouts at P15 and P30 by using a UTAS BigShot Ganzfeld system (LKC Technologies, Gaithersburg, MD, USA). Before recording, mice were allowed to dark-adapt overnight. Under dim red illumination, dark-adapted mice were anesthetized, and their eyes were dilated with tropicamide and anesthetized topically. A reference electrode was punctured into the skin of the mouse forehead, and a coiled gold recording electrode was placed in contact with the cornea of the eye via a drop of 1% carboxymethylcellulose (Refresh Liquigel; Allergan, Dublin, Ireland). For scotopic ERG, dark-adapted mice were tested for rod function with an LED light source at intensities that ranged from −40 dB (log −3.52 cd·s/m2) to 10 dB (2.38 cd·s/m2), and with a xenon lamp at intensity of 20 dB (2.38 cd·s/m2). For photopic ERGs to record cone function, a rod-saturating background light of 10 dB (1.39 cd·s/m2) was used for 5-min light adaptation and during recording. Single-flash responses were recorded at stimulus intensities from 0 dB (0.48 cd·s/m2) to 20 dB (2.38 cd·s/m2). During the procedure, mouse was kept comfortable on a heating pad until waking. Triple antibiotic ointment was placed on the eye after ERG testing to prevent infection.
Mouse embryonic stem cells
Mouse embryonic fibroblasts (MEFs) were derived from embryonic d 13.5 littermate embryos, EGFP-Cetn2+;Nphp5+/− and EGFP-Cetn2+;Nphp5−/−, using standard procedures (34). MEFs were grown in DMEM that was supplemented with 15% fetal bovine serum, and serum-starved cells were fixed in 4% PFA, permeabilized in 0.1% Triton X-100, and incubated with anti-acetylated α-tubulin antibody (T7451; 1:2000 dilution; Sigma-Aldrich, St. Louis, MO, USA) in the presence of goat serum blocking solution. Cells were washed 3× in PBS, incubated with goat anti-mouse Cy3 secondary antibody (1:1000; Thermo Fisher Scientific Life Sciences) and DAPI (1:3000), and imaged as previously described.
Statistics
Retina degeneration was evaluated by ONL thickness, which was measured in μm ± sd (n = 3), by using Zeiss imaging software. Statistical significance was tested with Student’s t test (P < 0.05) using Sigma Plot (Sigma-Aldrich) (24).
RESULTS
Generation of Nphp5 germline knockout mice
The mouse Nphp5 gene consists of 15 exons, in which exons 1 and 2 are noncoding (Fig. 1A). Numerous null mutations centered on the N-terminal BBS and central CC domains of NPHP5 have been identified in human patients with SLS (Fig. 1B). Embryonic stem cells that contain a β-geo gene trap (35) in intron 4 of mouse Nphp5 (Fig. 1A) were used to generate chimeras. The trap is predicted to truncate NPHP5 after exon 4 at residue Leu87, removing all known NPHP5 functional domains, for example, the IQ and CC domains and the BBSome and CEP290 binding sites (Fig. 1B). Trap contains a splice-acceptor sequence upstream of the promoterless β-geo reporter gene essentially converting the gene trap into an exon. The reporter gene consists of a fusion of β-galactosidase (β-gal) and neomycin phosphotransferase II, which allows determination of β-gal expression under control of the Nphp5 promoter. Embryonic day 13.5 Nphp5−/− embryos show that β-gal is expressed throughout the body, confirming ubiquitous expression of NPHP5 (Fig. 1C). Independently, RT-PCR revealed ubiquitous expression of NPHP5 in different mouse organs, with exception of the lungs, and reduced expressions in muscle, ovaries, and intestines (Supplemental Fig. S1).
Chimeric mice were outbred to C57BL/6J WT mice and heterozygotes with verified germline transmission were bred to generate homozygous knockouts (Nphp5−/−). rd8 mutation of the Crb1 gene (36) was detected in the knockout mouse line but was subsequently eliminated by successive mating with rd8-free C57BL/6J mice. Genotyping results confirm germline knockout (Fig. 1D) and immunohistochemistry (Fig. 1E, right panels) shows absence of NPHP5 in mutant retina. In control animals at P6, when ciliogenesis is complete and OSs begin to form, NPHP5 expression is undetectable by immunohistochemistry, but expression becomes stronger at P10 and P15 (Fig. 1E). Despite widespread expression of NPHP5, germline knockout animals were viable, fertile, and developed normally.
Scotopic (Fig. 2A, B, left) and photopic (Fig. 2A, B, right) ERG responses (rod and cone vision, respectively) of P14 and P18 Nphp5−/− animals (red traces) reveal absence of a- and b-waves at any light intensity, which indicates absence of phototransduction. At P28, fundoscopy demonstrates widespread degeneration of the central and peripheral Nphp5−/− retina (Fig. 2C) In human patients with NPHP5-LCA and NPHP6-LCA, ERG responses were also undetectable (13).
Figure 2.
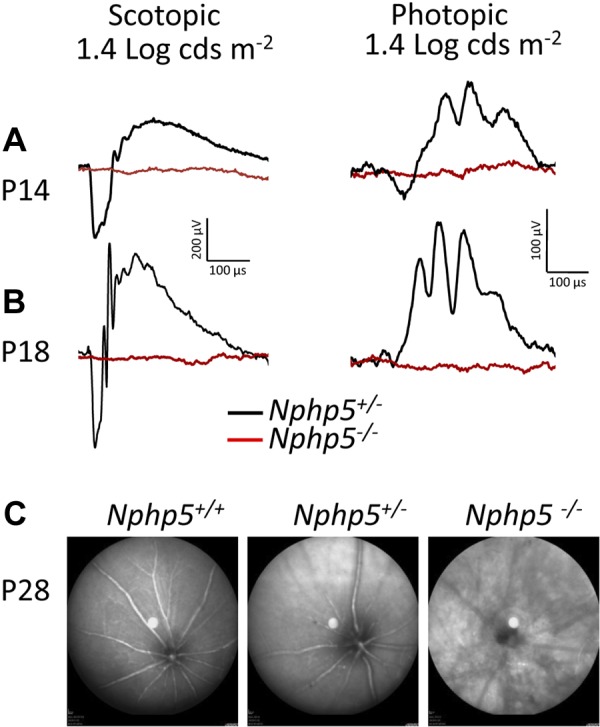
Phenotype of Nphp5−/− retinas. A, B) Scotopic (left) and photopic (right) electroretinography of Nphp5+/− and Nphp5−/− mice at P14 (A) and P18 (B). Lack of response (red) indicates that Nphp5−/− mice are blind early. C) Fundoscopy of WT, Nphp5+/−, and Nphp5−/− retinas at P28. Knockout mouse fundus shows peripheral and central mottling of the retina.
Rhodopsin and visual pigment trafficking
Rhodopsin is expressed as early as P3 in mouse photoreceptors (37). In P6 Nphp5+/− controls, rhodopsin begins to accumulate at the apical inner segment (Fig. 3A, left) and traffics to the OS at P10 and P15 (Fig. 3B, C, left). By contrast, rhodopsin accumulates in Nphp5−/− inner segments, the cell body, including the endoplasmic reticulum (ER), and in the outer plexiform layer (Fig. 3A–C, right). Rds/peripherin (peripherin-2), a disk structural component, and CNG channel subunits, present normally in the photoreceptor cell membrane, accumulate in perinuclear regions and axons of the mutant (Supplemental Fig. S2, right). Rhodopsin, peripherin-2, and CNG channel subunit levels are much reduced in knockouts, which suggests proteasomal or ER-associated degradation. Other components of the rod phototransduction cascade, for example, the peripherally membrane-associated Tα subunit, cGMP phosphodiesterase (PDE6) subunits, and GRK1, also failed to traffic (Fig. 3E–H, right). Rod degeneration was complete at P30 (Fig. 3G, right). Results show that rod OS proteins are unable to traffic as early as P6 in the absence of NPHP5.
Figure 3.

Immunolocalization of rod OS proteins in Nphp5+/− and Nphp5−/− retina. A–D) Expression of rhodopsin (red) at P6 (A), P10 (B), P15 (C), and P30 (D) in Nphp5+/− (left) and Nphp5−/− retina (right). Note that rhodopsin mistraffics in the Nphp5−/− ONL as early as P6. E–J) Localizations of rod Tα (E, F), cGMP phosphodiesterase (PDE6) (G, H) and rhodopsin kinase (GRK1) (I, J) at P10 (E, G, I) and P15 (F, H, J) in Nphp5+/− (left) and Nphp5−/− retina (right). K) Statistical evaluation of Nphp5+/− and Nphp5−/− ONL thickness at P6, P10, P15, and P30. IS, inner segment; ns, not significant; OPL, outer plexiform layer. ***P < 0.001.
Nphp5−/− cones are nonfunctional as judged by a flat photopic ERG at P14 (2 d after eye opening; Fig. 2A, B, right). We used immunohistochemistry to identify the earliest time of cone degeneration. In control mice, nascent cone OSs that are marked by S-opsin antibodies are detected at P6 (Fig. 4A, left). In Nphp5−/− cones at P6, cone pigment trafficking is retained in cone axons and misdirected to cone synaptic pedicles (Fig. 4A, right). As heterozygous control cone OSs continue to develop through P10 and P15 (Fig. 4B, C, left), Nphp5−/− cones degenerate rapidly, with accumulation of cone pigments in the ER and synaptic pedicles (Fig. 4B–D, right). Thus, Nphp5−/− rod degeneration is complete at P30, although cone nuclei are still detectable (Fig. 4F, right). The single ONL retained at P30 most likely represents cone nuclei as ML-opsin is still present perinuclearly. Peripheral membrane proteins, cone PDEα′ (PDE6C), and cone transducin-γ (GNGT2), share the fate of mislocalization in the inner segment, ER-surrounding nuclei, and axons (Fig. 4G–J). Cone arrestin localizations are comparable in Nphp5+/− and Nphp5−/− retinas (Fig. 4K, L).
Figure 4.

Localization of cone OS proteins in Nphp5+/− and Nphp5−/− retina. A–C) Localization of S-opsin in heterozygous controls (left) and knockouts (right) at P6 (A), P10 (B), and P15 (C). D–F) Immunolocalization of ML-opsin in heterozygous controls (left) and knockouts (right) at P10 (D), P15 (E), and P30 (F). G–L) Immunolocalization of cone PDE6 (G, H), cone Tγ (I, J), and cone arrestin (K, L) in heterozygous (left) and homozygous knockouts (right) at P10 (G, I, K) and P15 (H, J, L). IS, inner segment; OPL, outer plexiform layer.
Mature transition zones are malformed in the absence of NPHP5
We investigated transition zone formation by fluorescence labeling of centrosomes with transgenic GFP-tagged centrin 2 (EGFP-CETN2), a centriole/basal body marker (28, 29, 38). In P10 and P15 control mice that express EGFP-CETN2, mother and daughter centrioles are visible as 2 green dots, and each mother centriole (basal body) bears an extension toward the OS that represents the transition zone (Fig. 5A, B, left, insets). At this age, essentially all WT basal bodies carry the elongated, narrow transition zones. By contrast, only very few (∼3%) Nphp5−/− basal bodies are able to extend a transition zone. Normal-length transition zone extensions are absent, but some basal bodies carry short stunted extensions, which suggests aborted transition zone formation (Fig. 5C, D, right, insets). To visualize the proximal portion of the OS immediately adjacent to the transition zone, we used an anti–prominin-1 (Prom1) antibody (Fig. 5E). Prom1 is pentaspan transmembrane glycoprotein known to act as a key regulator of disk morphogenesis in photoreceptors (39, 40). Prom1 normally localizes to the proximal OS disks (Fig. 5E, inset), but in Nphp5−/− photoreceptors, Prom1 is distributed diffusely in the inner segment, which suggests disruption of disk morphogenesis. Taken together, ciliogenesis in Nphp5−/− retina is aborted at a late stage after docking of the basal body to the cell cortex.
Figure 5.
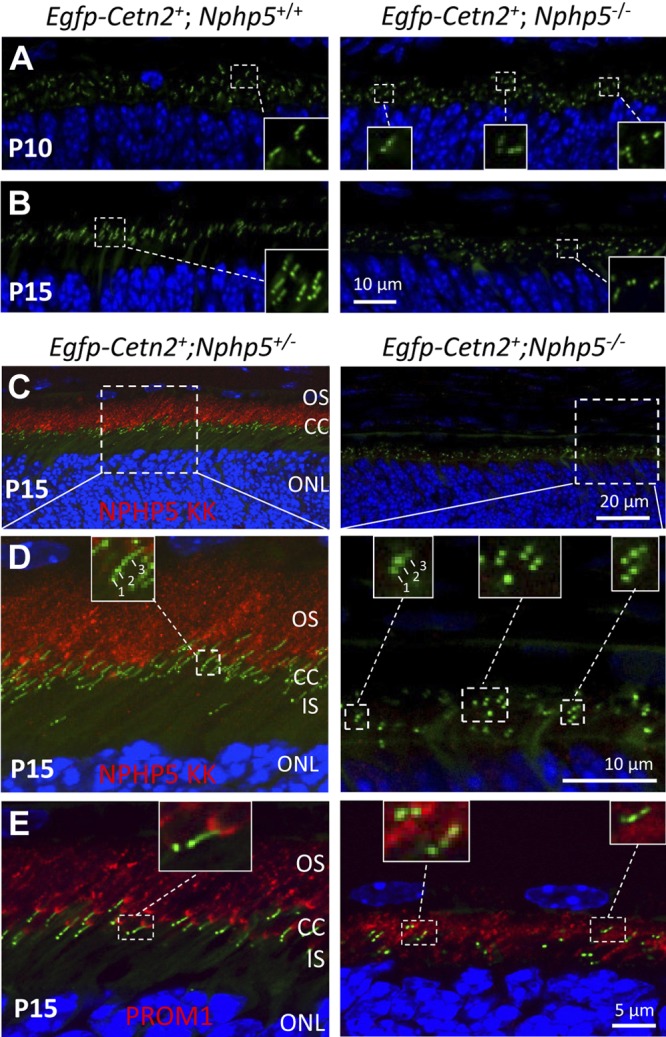
Absence of fully developed transition zones in Nphp5−/− photoreceptors. A, B) WT (left) and Nphp5−/− (right) retina sections expressing EGFP-CETN2 at P10 (A) and P15 (B). CETN2-GFP serves as a centriole and transition zone marker. Insets (A, B) showing enlargements, reveal that WT basal bodies generate transition zones, but transition zones are stunted or absent in Nphp5−/− retina. C) Immunolocalization of NPHP5 in the presence of EGFP-CETN2. P15 EGFP-Cetn2+;Nphp5+/− (left) and EGFP-Cetn2+;Nphp5−/− (right) retina cryosections were probed with anti–NPHP5-KK antibody (red). D) Enlargement of panel C as indicated. In EGFP-Cetn2+;Nphp5+/− photoreceptors, NPHP5 is located in the proximal OS, but is undetectable in the EGFP-Cetn2+;Nphp5−/− retina (right). Arrows 1–3 denote daughter centriole (1), mother centriole (2), and transition zone (3). Note transition zones are absent or stunted in Nphp5−/− photoreceptors (C, right, insets). E) Prominin 1 (PROM1) in heterozygous (left) and homozygous (right) knockouts. PROM1 is primarily present at the proximal Nphp5+/− OS adjacent to the CC (resembling a sickle; inset). In Nphp5−/− photoreceptors, PROM1 mislocalizes in the inner segment (IS).
CEP290/NPHP6 and NPHP5
NPHP5 and CEP290/NPHP6 are known to form a complex and localize to centrosomes of mammalian cell lines (22). In the absence of NPHP6, NPHP5 failed to localize to the centrosome, whereas depletion of NPHP5 had no effect on NPHP6 localization (22). In mice, NPHP6 localized to the transition zone by immunohistochemistry and, more specifically, to the Y-linkers by immuno-electron microscopy (41). In our hands, CEP290 is barely detectable at P6 (Fig. 6A, left), a time point at which transition zones have already formed (28). CEP290 localizes strongly to the transition zone area at P10 and P15 (Fig. 6B, C, left). In Nphp5−/− retina, CEP290/NPHP6 mislocalizes throughout the ONL and synaptic terminals (Fig. 6A–C, right). In transgenic mice that express EGFP-CETN2, CEP290 localizes to CETN2-positive centrosomes and transition zones only occasionally (Fig. 6D, inset), with the bulk of CEP290 located in the proximal outer segment. Results (compare Figs. 5D and 6D) show that CEP290 and NPHP5 both localize to proximal outer segments of control retinas and that NPHP5 is required for extension of an axoneme after docking of the basal body to the cell membrane.
Figure 6.
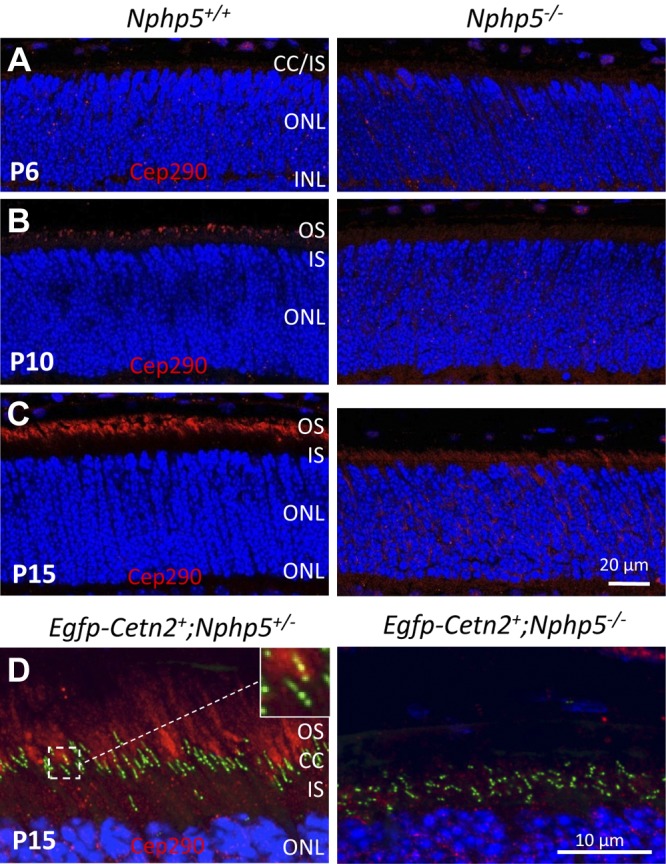
CEP290/NPHP6 in Nphp5−/− retina. A–C) Localization of CEP290/NPHP6 (red) in Nphp5+/− control (left) and Nphp5−/− (right) retinas at P6 (A), P10 (B), and P15 (C). D) EGFP-Cetn2+;Nphp5+/− (left) and EGFP-Cetn2+;Nphp5−/− (right) probed with antibody directed against CEP290. Two sets of centrioles plus transition zone, 1 set colabeled with EGFP-CETN2 and CEP290 (yellow), the other only with CETN2 (left inset). Transition zones are stunted or absent (right inset). IS, inner segment.
Nphp5−/− transition zone ultrastructure
Electron microscopy of WT photoreceptors showed formation of a transition zone and an axoneme as early as P6 (Fig. 7A). Transition zones are also present in heterozygous Nphp5+/− photoreceptors, which indicates haplosufficiency for cilia formation (Fig. 7B). In Nphp5−/− photoreceptors, basal bodies dock to the cell cortex correctly but malformed transition zone remnants are detectable rarely and apparently of reduced cross-sectional area (Fig. 7C). Cross-sections of transition zones revealed a 9:0 MT configuration with reduced diameter (Fig. 7F, inset). At P10, WT and heterozygous photoreceptors developed robust OSs, with evenly stacked disk membranes (Fig. 7D, E), whereas OS formation is inhibited in Nphp5−/− rods and disks are undetectable (Fig. 7F). Photoreceptor degeneration is far advanced at P14 (not shown).
Figure 7.

Transition zone ultrastructure. A–C) At P6, normal basal body docking and connecting cilium structure is observed in the WT (A), heterozygote (B), and knockout (C) mice. D–F) At P10, OSs are formed in the WT (D) and heterozygote (E) animals but not in the knockout mice (F). Insets show transition zone cross-sections with the 9 + 0 microtubule arrangement in all genotypes.
Nphp5−/−;Nrl−/− double-knockout mice
As shown by ERG data (Fig. 2), Nphp5−/− cone photoreceptors are nonfunctional at P15, although Nphp5−/− cone nuclei are still present at P30 (Fig. 4F). As cones are most important for daylight vision, we generated Nphp5−/−;Nrl−/− mice to study photoreceptor fate in an all-cone retina. Nrl−/− ONL thickness is relatively stable from P10 to 3 mo (Fig. 8A–E, left, J), and is comparable in thickness to WT retina that consists mostly of rod nuclei (Fig. 8F–I, left, J). By contrast, Nphp5−/− photoreceptors rapidly degenerate (Fig. 8F–I, right) and, at 2 mo, only a single layer of cone nuclei remains (Fig. 8I, right). Surprisingly, Nphp5−/−;Nrl−/− cone cells survive at least to 3 mo of age, still expressing ML-opsin (Fig. 8D, E, right); however, Nphp5−/−;Nrl−/− cones are nonfunctional at P15 and later (Fig. 8K), which suggests the absence of cone transitional zones (Fig. 8B–E, right), with consequent mislocalization of ML-opsin in surviving cone inner segments. Thus, Nphp5−/−;Nrl−/− mice are excellent candidates for cone-directed gene replacement therapy (13).
Figure 8.

Cone long-term survival in Nphp5−/− ;Nrl−/− retina. A–E) Nphp5−/+;Nrl−/− (left) and Nphp5−/−;Nrl−/− retina sections (right) probed with anti–ML-opsin antibody at P10 (A), P15 (B), 1 mo (C), 2 mo (D), and 3 mo (E) of age. F–I) Nphp5−/+ (left) and Nphp5−/− (right) retina sections probed with anti–ML-opsin antibody at P10 (F), P15 (G), 1 mo (H), and 2 mo (I). Note that cone degeneration in Nrl−/−;Nphp5−/− retina is slowed and mutant cones still express ML-opsin at 3 mo. Panels A–C were from mice expressing EGFP-CETN2. J) Statistical evaluation ONL thickness at P10, P15, 1 mo, 2 mo, and 3 mo. K) Representative ERG traces of Nphp5−/−;Nrl−/− (red), Nphp5+/− (black), and Nrl−/− mice (green) at P15 and 1 mo. Nrl−/− photopic responses are elevated and double-knockout cones are nonfunctional. IS, inner segment; ns, not significant; OPL, outer plexiform layer.
Nphp5−/− kidneys and MEFs form normal cilia
Whereas vision loss in Nphp6rd16/rd16 and Nphp6−/− are accompanied by anosmia/deafness, hydrocephalus, and Joubert syndrome (41–44), other ciliopathies are not apparent in the Nphp5−/− mouse. A previous meeting abstract (45) described renal dysmorphology in the Nphp5-knockout mouse, but that histology result was not substantiated upon repetition and further scrutiny (Fig. 9), and LCA observed in our Nphp5−/− mice (Fig. 2) seems to be nonsyndromic. NPHP-associated changes in kidneys were not observed at 9 mo (not shown), and 12 mo postnatally (Fig. 9A). Nphp5−/− kidneys are well formed and cysts, interstitial fibrosis, tubular atrophy, and basement membrane changes are absent (Fig. 9A, right, insets). Formation of kidney epithelial cilia was normal at 1 and 12 mo of age (Fig. 9B, C, insets). Absence of kidney disease was also reported in other nephrocystin-knockout mice, for example, Nphp1−/− mice (46, 47). We further examined ciliary development in MEFs derived from Nphp5+/− and Nphp5−/− mice that express transgenic EGFP-CETN2; EGFP-CETN2–labeled mother and daughter centrioles as expected (green in Fig. 9D), and colocalized with Ac-α-tubulin in the transition zone (yellow in Fig. 9D). Cilia were of normal length in Nphp5−/− MEFs, which is consistent with normal renal development in Nphp5−/− mice.
Figure 9.
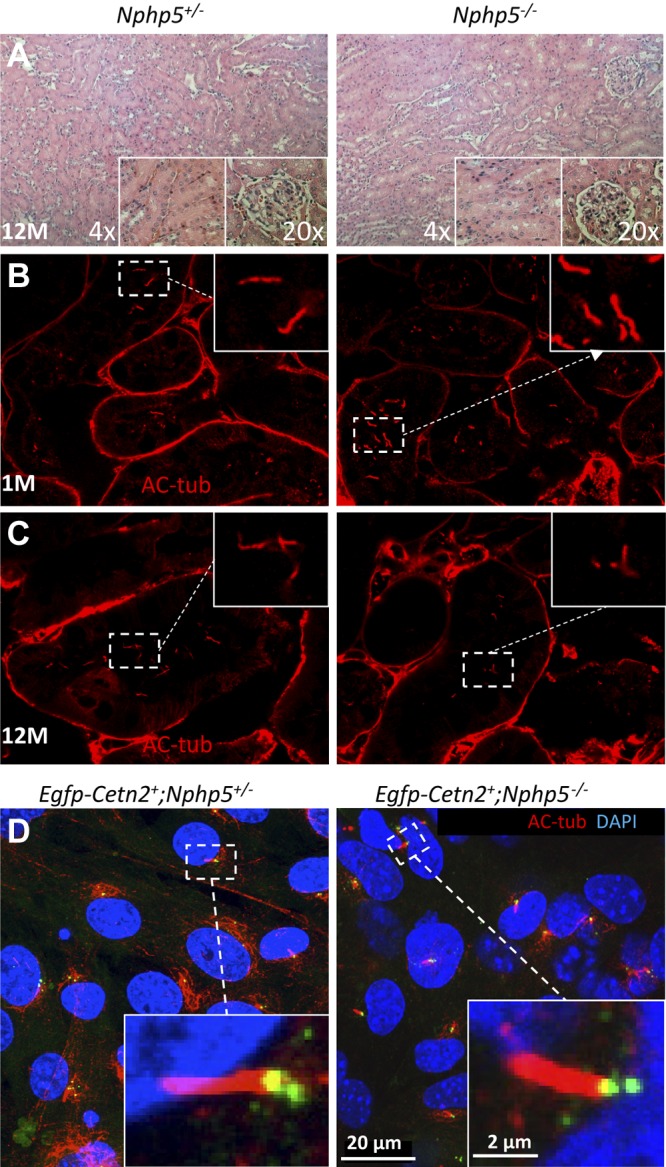
Cilia formation in kidneys and MEFs. A) Light microscopy of Nphp5+/− (left) and Nphp5−/− (right) hematoxylin and eosin–stained kidney sections at 12 mo of age. No morphologic abnormalities were visible in the kidney parenchyma of any knockout mice at 12 mo of age (n = 3) or earlier (not shown). Tubules of heterozygous and homozygous knockouts are well formed. Cyst formation, interstitial fibrosis, and tubular atrophy are not detected. B, C) Confocal microscopy of kidney epithelial cells demonstrate cilia labeled with antiacetylated tubulin antibody (red). D) EGFP-Cetn2+;Nphp5+/− (left) and EGFP-Cetn2+;Nphp5−/− (right) MEFs form cilia of identical length, identified by Ac-α-tubulin (AC-tub) antibody (red). Basal body (yellow) indicates colabeling with CETN2 and Ac-α-tubulin. Daughter centrioles label with GFP-CENT2 only; insets show enlargements of primary cilia.
DISCUSSION
Nphp5−/− mouse carries an N-terminal truncation of NPHP5 that removes all known functional domains of NPHP5. Homozygous knockouts are blind at eye opening and recapitulate human pathology of LCA. At 12 mo of age, there was no evidence of renal cyst formation, interstitial fibrosis, or tubular changes that are characteristic of nephronophthisis (Fig. 9). Deletion of NPHP5 does not prevent anchoring of the basal body to the cell cortex, although development of ciliary transition zones is impaired (Figs. 5–7). As Nphp5−/− mouse rods do not extend an axoneme, OSs are absent and rod nuclei are degenerated at 1 mo of age. In contrast, Nphp5−/− cone nuclei survive to 1 mo (Fig. 5) and their survival can be significantly extended in an Nphp5−/−;Nrl−/− background to at least 3 mo (Fig. 8). NPHP5 absence interferes with late steps of photoreceptor ciliogenesis without affecting cilia formation in MEFs or cells of other organs as the Nphp5−/− mouse develops normally.
C-terminal truncation of NPHP5 in a naturally occurring dog model (crd2) causes a rapidly progressive retinal degeneration in which the 3-wk-old ONL is comparable to age-matched WT dogs (14). Truncation removes the C-terminal IQ, CC, and CEP290 interaction domains (Fig. 1). By 6 wk of age, crd2 rod responses are markedly reduced in amplitude, and cone responses are not recordable (14), which suggests that crd2 rod transition zones and OS remnants are formed. The difference in phenotype between mouse and dog mutant may be explained by stable expression of the truncated N-terminal crd2 fragment that contains the BBSome interaction site, one of the calmodulin binding motifs (IQ), and 2 CC domains (Dr. Gustavo Aguirre, University of Pennsylvania, Philadelphia, PA, USA; personal communication).
In vivo results contrast with those of NPHP5 knockdown in hTertRPE-1 cells in which migration and anchoring of centrioles to the cell cortex is disrupted (8). The discrepancy may be explained, in part, by the observation that mouse photoreceptor NPHP5 is not directly associated with basal bodies and proximal transition zones (Fig. 5C, D, inset) and, therefore, its absence may not prevent docking of the basal body to the cell membrane. Instead, mouse NPHP5 localizes to the distal transition zone and throughout the photoreceptor OS as discrete puncta, most likely caused by interaction with the BBSome or axoneme (17).
Photoreceptor ciliogenesis is presumed to take place in several steps (48). First, photoreceptors exit the cell cycle and become postmitotic, which for rods occurs at ∼P5 (49). Vesicles are recruited at the distal end of the mother centriole, an event that enables docking to the cell membrane. The mother centriole then acquires distal appendages to which several centrosomal proteins (e.g., CEP164, CEP172) have been localized (50, 51). Final steps of ciliogenesis include formation of the transition zone, the axoneme, and assembly of disk membranes. Building of microtubule-stabilized transition zone and axoneme requires intraflagellar transport (IFT) of cargo, for example, axoneme building blocks and tubulin subunits. In organisms from Caenorhabditis elegans to mammals, IFT is essential for axoneme formation and is carried out by heterotrimeric kinesin-2 (KIF3) and homodimeric kinesin-2 (KIF17), together with IFT particles and BBSome. We have shown previously that building the photoreceptor transition zone in mouse requires KIF3 but not KIF17 (28).
Our observation that basal bodies dock to the cell membrane but fail to form a functional transition zone in Nphp5−/− rods reserves a pivotal role for NPHP5 during the last steps of ciliogenesis, that is, axoneme extension and initial disk assembly. As NPHP5 has 2 distinct BBSome binding sites at the N and C termini, it is known to associate with the holo-BBSome complex (17). Loss of NPHP5 leads to mislocalization of BBS2 and BBS5, destroys BBSome integrity, and selectively impairs delivery of cargo in hTert-RPE1 cells (17). Photoreceptor BBSome IFT cargo has not been identified, although rhodopsin and associated OS membrane proteins have been excluded. A possible hypothesis to explain ciliogenesis arrest at this late stage may be that absence of NPHP5 impairs BBSome assembly or renders the BBSome incompetent for IFT-mediated axoneme extension.
NPHP gene family currently consists of 20 members (NPHP1–20) (1, 52–56). Several mouse germline knockouts have been generated (NPHP1, -3, -4, -5, -6, -7, -8, and -14) and naturally occurring loss-of-function models are known (Nphp2/inv, Nphp3/pcy, Nphp6/rd16, Nphp8/ftm, Nphp9/jck, Nphp11/Bpck, Nphp12/aln). None of these loss-of-function models represent a Senior-Løken mouse model. Germline deletions of Nphp3, Nphp8 (ftm,Rpgrip1l), and Nphp12 (Ttc21b) are embryonically lethal. Deletions of Nphp1, Nphp4, and Nphp6 develop retinal degeneration on the basis of overlapping ciliogenesis defects (42, 46, 57). At P14, Nphp1−/− photoreceptors elaborated transition zones and axonemes, but OS disk membranes were severely malformed and rhodopsin and phototransduction components mistrafficked (46). IFT88 mislocalized, which suggests that NPHP1 may regulate a specific subset of IFT particles and trafficking of their associated cargos destined to the OS (46, 58).
A loss-of-function mouse model of Nphp4 (Nphpnmf192) exhibits severe LCA, with mislocalization of rhodopsin and ROM1 to the inner segment (57). Transition zones and axonemes were present, together with rudimentary disk structures, but OS failed to develop and photoreceptors degenerated. Ribbon structures at the synaptic terminals also eventually degenerated. The rd16 mouse, carrying an in-frame deletion of Nphp6 exons 35–39 (13, 42), develops all photoreceptor layers until P12. OS elongation stops at P14 and regresses. Complete extinction of ERG responses in Nphp5−/− mice at P14 contrasts with rd16 mice that exhibit diminished rod and cone ERG amplitudes at P18, with complete loss of ERG responses observed only by P28. A recently generated Nphp6−/− mouse did not survive past 1 mo and developed hydrocephalus and Joubert syndrome (59); mutant photoreceptors fail to generate transition zones and OSs, although basal bodies and associated microtubule assemblies are present. A spontaneous deletion of Nphp11 in mouse (bpck mouse, bilateral polycystic kidneys) (60) with phenotypes that resemble human disease (polycystic kidney and hydrocephalus, Meckel syndrome) causes rapid degeneration with complete absence of OS at P24 and reduction of the transition zone axonemal tip at P14 (61), despite normal basal body and transition zone structure.
In summary, whereas null alleles of NPHP1 (16, 47), NPHP3 (62), NPHP4 (63), NPHP5 (6), and CEP290/NPHP6 (42, 64) are each associated with SLS in humans, gene knockouts of NPHP1, -3, -4, -5, and -6 in mice have not replicated human disease phenotype in entirety. NPHP1, -4, -5, and -6 gene knockouts, however, do associate with recessive retina disease (RP or LCA) and demonstrate important roles of nephrocystins in photoreceptor ciliogenesis.
ACKNOWLEDGMENTS
This work was supported by a U.S. National Institutes of Health (NIH), National Eye Institute (NEI) National Research Service Award Grant 1F31EY021972-01A1 (to C.C.R.), NIH NEI Grants EY08123 and EY019298 (to W.B.) and Grant EY014800-039003 (NEI Core Grant), and unrestricted grants to the Department of Ophthalmology (University of Utah) from Research to Prevent Blindness, the Retina Research Foundation (Houston, TX, USA), and the Foundation for Retina Research. W.B. is a recipient of a Research to Prevent Blindness Senior Investigator and Nelson Trust Award. This work was part of a Ph.D. thesis for the Interdepartmental Graduate Program in Neuroscience (University of Utah Health Science Center) (C.C.R.). The authors declare no conflicts of interest.
Glossary
- CC
coiled coil
- crd2
cone rod dystrophy 2
- ER
endoplasmic reticulum
- ERG
electroretinogram
- β-gal
β-galactosidase
- IFT
intraflagellar transport
- IQCB1
IQ calmodulin-binding motif containing B1
- KIF
kinesin
- LCA
Leber congenital amaurosis
- MEF
mouse embryonic fibroblast
- NPHP
nephrocystin
- ONL
outer nuclear layer
- OS
outer segment
- P
postnatal day
- PFA
paraformaldehyde
- RP
retinitis pigmentosa
- SLS
Senior-Løken syndrome
- WT
wild-type
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
AUTHOR CONTRIBUTIONS
C. C. Ronquillo and W. Baehr designed the experiments; C. C. Ronquillo, C. Hanke-Gogokhia, M. P. Revelo, J. M. Frederick, and L. Jiang performed experiments; and W. Baehr wrote the paper.
REFERENCES
- 1.Ronquillo C. C., Bernstein P. S., Baehr W. (2012) Senior-Løken syndrome: a syndromic form of retinal dystrophy associated with nephronophthisis. Vision Res. 75, 88–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shiba D., Yokoyama T. (2012) The ciliary transitional zone and nephrocystins. Differentiation 83, S91–S96 [DOI] [PubMed] [Google Scholar]
- 3.Satir P., Pedersen L. B., Christensen S. T. (2010) The primary cilium at a glance. J. Cell Sci. 123, 499–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hartong D. T., Berson E. L., Dryja T. P. (2006) Retinitis pigmentosa. Lancet 368, 1795–1809 [DOI] [PubMed] [Google Scholar]
- 5.Den Hollander A. I., Roepman R., Koenekoop R. K., Cremers F. P. (2008) Leber congenital amaurosis: genes, proteins and disease mechanisms. Prog. Retin. Eye Res. 27, 391–419 [DOI] [PubMed] [Google Scholar]
- 6.Otto E. A., Loeys B., Khanna H., Hellemans J., Sudbrak R., Fan S., Muerb U., O’Toole J. F., Helou J., Attanasio M., Utsch B., Sayer J. A., Lillo C., Jimeno D., Coucke P., De Paepe A., Reinhardt R., Klages S., Tsuda M., Kawakami I., Kusakabe T., Omran H., Imm A., Tippens M., Raymond P. A., Hill J., Beales P., He S., Kispert A., Margolis B., Williams D. S., Swaroop A., Hildebrandt F. (2005) Nephrocystin-5, a ciliary IQ domain protein, is mutated in Senior-Loken syndrome and interacts with RPGR and calmodulin. Nat. Genet. 37, 282–288 [DOI] [PubMed] [Google Scholar]
- 7.Wolf M. T., Hildebrandt F. (2011) Nephronophthisis. Pediatr. Nephrol. 26, 181–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barbelanne M., Song J., Ahmadzai M., Tsang W. Y. (2013) Pathogenic NPHP5 mutations impair protein interaction with Cep290, a prerequisite for ciliogenesis. Hum. Mol. Genet. 22, 2482–2494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Halbritter J., Porath J. D., Diaz K. A., Braun D. A., Kohl S., Chaki M., Allen S. J., Soliman N. A., Hildebrandt F., Otto E. A.; GPN Study Group (2013) Identification of 99 novel mutations in a worldwide cohort of 1,056 patients with a nephronophthisis-related ciliopathy. Hum. Genet. 132, 865–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chaki M., Hoefele J., Allen S. J., Ramaswami G., Janssen S., Bergmann C., Heckenlively J. R., Otto E. A., Hildebrandt F. (2011) Genotype-phenotype correlation in 440 patients with NPHP-related ciliopathies. Kidney Int. 80, 1239–1245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Estrada-Cuzcano A., Koenekoop R. K., Coppieters F., Kohl S., Lopez I., Collin R. W., De Baere E. B., Roeleveld D., Marek J., Bernd A., Rohrschneider K., van den Born L. I., Meire F., Maumenee I. H., Jacobson S. G., Hoyng C. B., Zrenner E., Cremers F. P., den Hollander A. I. (2011) IQCB1 mutations in patients with Leber congenital amaurosis. Invest. Ophthalmol. Vis. Sci. 52, 834–839 [DOI] [PubMed] [Google Scholar]
- 12.Stone E. M., Cideciyan A. V., Aleman T. S., Scheetz T. E., Sumaroka A., Ehlinger M. A., Schwartz S. B., Fishman G. A., Traboulsi E. I., Lam B. L., Fulton A. B., Mullins R. F., Sheffield V. C., Jacobson S. G. (2011) Variations in NPHP5 in patients with nonsyndromic Leber congenital amaurosis and Senior-Loken syndrome. Arch. Ophthalmol. 129, 81–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cideciyan A. V., Rachel R. A., Aleman T. S., Swider M., Schwartz S. B., Sumaroka A., Roman A. J., Stone E. M., Jacobson S. G., Swaroop A. (2011) Cone photoreceptors are the main targets for gene therapy of NPHP5 (IQCB1) or NPHP6 (CEP290) blindness: generation of an all-cone Nphp6 hypomorph mouse that mimics the human retinal ciliopathy. Hum. Mol. Genet. 20, 1411–1423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goldstein O., Mezey J. G., Schweitzer P. A., Boyko A. R., Gao C., Bustamante C. D., Jordan J. A., Aguirre G. D., Acland G. M. (2013) IQCB1 and PDE6B mutations cause similar early onset retinal degenerations in two closely related terrier dog breeds. Invest. Ophthalmol. Vis. Sci. 54, 7005–7019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yau K. W., Hardie R. C. (2009) Phototransduction motifs and variations. Cell 139, 246–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hildebrandt F., Otto E., Rensing C., Nothwang H. G., Vollmer M., Adolphs J., Hanusch H., Brandis M. (1997) A novel gene encoding an SH3 domain protein is mutated in nephronophthisis type 1. Nat. Genet. 17, 149–153 [DOI] [PubMed] [Google Scholar]
- 17.Barbelanne M., Hossain D., Chan D. P., Peränen J., Tsang W. Y. (2015) Nephrocystin proteins NPHP5 and Cep290 regulate BBSome integrity, ciliary trafficking and cargo delivery. Hum. Mol. Genet. 24, 2185–2200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Murga-Zamalloa C. A., Desai N. J., Hildebrandt F., Khanna H. (2010) Interaction of ciliary disease protein retinitis pigmentosa GTPase regulator with nephronophthisis-associated proteins in mammalian retinas. Mol. Vis. 16, 1373–1381 [PMC free article] [PubMed] [Google Scholar]
- 19.Anand M., Khanna H. (2012) Ciliary transition zone (TZ) proteins RPGR and CEP290: role in photoreceptor cilia and degenerative diseases. Expert Opin. Ther. Targets 16, 541–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gerner M., Haribaskar R., Pütz M., Czerwitzki J., Walz G., Schäfer T. (2010) The retinitis pigmentosa GTPase regulator interacting protein 1 (RPGRIP1) links RPGR to the nephronophthisis protein network. Kidney Int. 77, 891–896 [DOI] [PubMed] [Google Scholar]
- 21.Schäfer T., Pütz M., Lienkamp S., Ganner A., Bergbreiter A., Ramachandran H., Gieloff V., Gerner M., Mattonet C., Czarnecki P. G., Sayer J. A., Otto E. A., Hildebrandt F., Kramer-Zucker A., Walz G. (2008) Genetic and physical interaction between the NPHP5 and NPHP6 gene products. Hum. Mol. Genet. 17, 3655–3662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sang L., Miller J. J., Corbit K. C., Giles R. H., Brauer M. J., Otto E. A., Baye L. M., Wen X., Scales S. J., Kwong M., Huntzicker E. G., Sfakianos M. K., Sandoval W., Bazan J. F., Kulkarni P., Garcia-Gonzalo F. R., Seol A. D., O’Toole J. F., Held S., Reutter H. M., Lane W. S., Rafiq M. A., Noor A., Ansar M., Devi A. R., Sheffield V. C., Slusarski D. C., Vincent J. B., Doherty D. A., Hildebrandt F., Reiter J. F., Jackson P. K. (2011) Mapping the NPHP-JBTS-MKS protein network reveals ciliopathy disease genes and pathways. Cell 145, 513–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao C., Malicki J. (2011) Nephrocystins and MKS proteins interact with IFT particle and facilitate transport of selected ciliary cargos. EMBO J. 30, 2532–2544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hanke-Gogokhia C., Wu Z., Gerstner C. D., Frederick J. M., Zhang H., Baehr W. (2016) Arf-like protein 3 (ARL3) regulates protein trafficking and ciliogenesis in mouse photoreceptors. J. Biol. Chem. 291, 7142–7155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang H., Hanke-Gogokhia C., Jiang L., Li X., Wang P., Gerstner C. D., Frederick J. M., Yang Z., Baehr W. (2015) Mistrafficking of prenylated proteins causes retinitis pigmentosa 2. FASEB J. 29, 932–942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang L., Tam B. M., Ying G., Wu S., Hauswirth W. W., Frederick J. M., Moritz O. L., Baehr W. (2015) Kinesin family 17 (osmotic avoidance abnormal-3) is dispensable for photoreceptor morphology and function. FASEB J. 29, 4866–4880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mears A. J., Kondo M., Swain P. K., Takada Y., Bush R. A., Saunders T. L., Sieving P. A., Swaroop A. (2001) Nrl is required for rod photoreceptor development. Nat. Genet. 29, 447–452 [DOI] [PubMed] [Google Scholar]
- 28.Jiang L., Wei Y., Ronquillo C. C., Marc R. E., Yoder B. K., Frederick J. M., Baehr W. (2015) Heterotrimeric kinesin-2 (KIF3) mediates transition zone and axoneme formation of mouse photoreceptors. J. Biol. Chem. 290, 12765–12778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ying G., Avasthi P., Irwin M., Gerstner C. D., Frederick J. M., Lucero M. T., Baehr W. (2014) Centrin 2 is required for mouse olfactory ciliary trafficking and development of ependymal cilia planar polarity. J. Neurosci. 34, 6377–6388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang H., Li S., Doan T., Rieke F., Detwiler P. B., Frederick J. M., Baehr W. (2007) Deletion of PrBP/delta impedes transport of GRK1 and PDE6 catalytic subunits to photoreceptor outer segments. Proc. Natl. Acad. Sci. USA 104, 8857–8862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baehr W., Palczewski K. (2007) Guanylate cyclase-activating proteins and retina disease. Subcell. Biochem. 45, 71–91 [DOI] [PubMed] [Google Scholar]
- 32.Avasthi P., Watt C. B., Williams D. S., Le Y. Z., Li S., Chen C. K., Marc R. E., Frederick J. M., Baehr W. (2009) Trafficking of membrane proteins to cone but not rod outer segments is dependent on heterotrimeric kinesin-II. J. Neurosci. 29, 14287–14298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jones B. W., Kondo M., Terasaki H., Watt C. B., Rapp K., Anderson J., Lin Y., Shaw M. V., Yang J. H., Marc R. E. (2011) Retinal remodeling in the Tg P347L rabbit, a large-eye model of retinal degeneration. J. Comp. Neurol. 519, 2713–2733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Takahashi K., Yamanaka S. (2006) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676 [DOI] [PubMed] [Google Scholar]
- 35.Friedel R. H., Seisenberger C., Kaloff C., Wurst W. (2007) EUCOMM--the European conditional mouse mutagenesis program. Brief. Funct. Genomics Proteomics 6, 180–185 [DOI] [PubMed] [Google Scholar]
- 36.Mattapallil M. J., Wawrousek E. F., Chan C. C., Zhao H., Roychoudhury J., Ferguson T. A., Caspi R. R. (2012) The Rd8 mutation of the Crb1 gene is present in vendor lines of C57BL/6N mice and embryonic stem cells, and confounds ocular induced mutant phenotypes. Invest. Ophthalmol. Vis. Sci. 53, 2921–2927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Al-Ubaidi M. R., Pittler S. J., Champagne M. S., Triantafyllos J. T., McGinnis J. F., Baehr W. (1990) Mouse opsin. Gene structure and molecular basis of multiple transcripts. J. Biol. Chem. 265, 20563–20569 [PubMed] [Google Scholar]
- 38.Higginbotham H., Bielas S., Tanaka T., Gleeson J. G. (2004) Transgenic mouse line with green-fluorescent protein-labeled Centrin 2 allows visualization of the centrosome in living cells. Transgenic Res. 13, 155–164 [DOI] [PubMed] [Google Scholar]
- 39.Zacchigna S., Oh H., Wilsch-Bräuninger M., Missol-Kolka E., Jászai J., Jansen S., Tanimoto N., Tonagel F., Seeliger M., Huttner W. B., Corbeil D., Dewerchin M., Vinckier S., Moons L., Carmeliet P. (2009) Loss of the cholesterol-binding protein prominin-1/CD133 causes disk dysmorphogenesis and photoreceptor degeneration. J. Neurosci. 29, 2297–2308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jaszai J., Fargeas C. A., Florek M., Huttner W. B., Corbeil D. (2006) Focus on molecules: prominin-1 (CD133). Exp. Eye Res. 85, 585–586 [DOI] [PubMed] [Google Scholar]
- 41.Rachel R. A., Yamamoto E. A., Dewanjee M. K., May-Simera H. L., Sergeev Y. V., Hackett A. N., Pohida K., Munasinghe J., Gotoh N., Wickstead B., Fariss R. N., Dong L., Li T., Swaroop A. (2015) CEP290 alleles in mice disrupt tissue-specific cilia biogenesis and recapitulate features of syndromic ciliopathies. Hum. Mol. Genet. 24, 3775–3791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chang B., Khanna H., Hawes N., Jimeno D., He S., Lillo C., Parapuram S. K., Cheng H., Scott A., Hurd R. E., Sayer J. A., Otto E. A., Attanasio M., O’Toole J. F., Jin G., Shou C., Hildebrandt F., Williams D. S., Heckenlively J. R., Swaroop A. (2006) In-frame deletion in a novel centrosomal/ciliary protein CEP290/NPHP6 perturbs its interaction with RPGR and results in early-onset retinal degeneration in the rd16 mouse. Hum. Mol. Genet. 15, 1847–1857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McEwen D. P., Koenekoop R. K., Khanna H., Jenkins P. M., Lopez I., Swaroop A., Martens J. R. (2007) Hypomorphic CEP290/NPHP6 mutations result in anosmia caused by the selective loss of G proteins in cilia of olfactory sensory neurons. Proc. Natl. Acad. Sci. USA 104, 15917–15922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rachel R. A., May-Simera H. L., Veleri S., Gotoh N., Choi B. Y., Murga-Zamalloa C., McIntyre J. C., Marek J., Lopez I., Hackett A. N., Zhang J., Brooks M., den Hollander A. I., Beales P. L., Li T., Jacobson S. G., Sood R., Martens J. R., Liu P., Friedman T. B., Khanna H., Koenekoop R. K., Kelley M. W., Swaroop A. (2012) Combining Cep290 and Mkks ciliopathy alleles in mice rescues sensory defects and restores ciliogenesis. J. Clin. Invest. 122, 1233–1245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ronquillo C., Frederick J., Baehr W. (2013) Nephrocystin-5 knockout mice recapitulate retina and kidney pathologies of Senior-Løken syndrome. Invest. Ophthalmol. Vis. Sci. 54, 3149 [Google Scholar]
- 46.Jiang S. T., Chiou Y. Y., Wang E., Lin H. K., Lee S. P., Lu H. Y., Wang C. K., Tang M. J., Li H. (2008) Targeted disruption of Nphp1 causes male infertility due to defects in the later steps of sperm morphogenesis in mice. Hum. Mol. Genet. 17, 3368–3379 [DOI] [PubMed] [Google Scholar]
- 47.Jiang S. T., Chiou Y. Y., Wang E., Chien Y. L., Ho H. H., Tsai F. J., Lin C. Y., Tsai S. P., Li H. (2009) Essential role of nephrocystin in photoreceptor intraflagellar transport in mouse. Hum. Mol. Genet. 18, 1566–1577 [DOI] [PubMed] [Google Scholar]
- 48.Avasthi P., Marshall W. F. (2012) Stages of ciliogenesis and regulation of ciliary length. Differentiation 83, S30–S42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Morrow E. M., Furukawa T., Cepko C. L. (1998) Vertebrate photoreceptor cell development and disease. Trends Cell Biol. 8, 353–358 [DOI] [PubMed] [Google Scholar]
- 50.Graser S., Stierhof Y. D., Lavoie S. B., Gassner O. S., Lamla S., Le Clech M., Nigg E. A. (2007) Cep164, a novel centriole appendage protein required for primary cilium formation. J. Cell Biol. 179, 321–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sillibourne J. E., Hurbain I., Grand-Perret T., Goud B., Tran P., Bornens M. (2013) Primary ciliogenesis requires the distal appendage component Cep123. Biol. Open 2, 535–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chaki M., Airik R., Ghosh A. K., Giles R. H., Chen R., Slaats G. G., Wang H., Hurd T. W., Zhou W., Cluckey A., Gee H. Y., Ramaswami G., Hong C. J., Hamilton B. A., Cervenka I., Ganji R. S., Bryja V., Arts H. H., van Reeuwijk J., Oud M. M., Letteboer S. J., Roepman R., Husson H., Ibraghimov-Beskrovnaya O., Yasunaga T., Walz G., Eley L., Sayer J. A., Schermer B., Liebau M. C., Benzing T., Le Corre S., Drummond I., Janssen S., Allen S. J., Natarajan S., O’Toole J. F., Attanasio M., Saunier S., Antignac C., Koenekoop R. K., Ren H., Lopez I., Nayir A., Stoetzel C., Dollfus H., Massoudi R., Gleeson J. G., Andreoli S. P., Doherty D. G., Lindstrad A., Golzio C., Katsanis N., Pape L., Abboud E. B., Al-Rajhi A. A., Lewis R. A., Omran H., Lee E. Y., Wang S., Sekiguchi J. M., Saunders R., Johnson C. A., Garner E., Vanselow K., Andersen J. S., Shlomai J., Nurnberg G., Nurnberg P., Levy S., Smogorzewska A., Otto E. A., Hildebrandt F. (2012) Exome capture reveals ZNF423 and CEP164 mutations, linking renal ciliopathies to DNA damage response signaling. Cell 150, 533–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Czarnecki P. G., Gabriel G. C., Manning D. K., Sergeev M., Lemke K., Klena N. T., Liu X., Chen Y., Li Y., San Agustin J. T., Garnaas M. K., Francis R. J., Tobita K., Goessling W., Pazour G. J., Lo C. W., Beier D. R., Shah J. V. (2015) ANKS6 is the critical activator of NEK8 kinase in embryonic situs determination and organ patterning. Nat. Commun. 6, 6023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Joo K., Kim C. G., Lee M. S., Moon H. Y., Lee S. H., Kim M. J., Kweon H. S., Park W. Y., Kim C. H., Gleeson J. G., Kim J. (2013) CCDC41 is required for ciliary vesicle docking to the mother centriole. Proc. Natl. Acad. Sci. USA 110, 5987–5992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Halbritter J., Bizet A. A., Schmidts M., Porath J. D., Braun D. A., Gee H. Y., McInerney-Leo A. M., Krug P., Filhol E., Davis E. E., Airik R., Czarnecki P. G., Lehman A. M., Trnka P., Nitschké P., Bole-Feysot C., Schueler M., Knebelmann B., Burtey S., Szabó A. J., Tory K., Leo P. J., Gardiner B., McKenzie F. A., Zankl A., Brown M. A., Hartley J. L., Maher E. R., Li C., Leroux M. R., Scambler P. J., Zhan S. H., Jones S. J., Kayserili H., Tuysuz B., Moorani K. N., Constantinescu A., Krantz I. D., Kaplan B. S., Shah J. V., Hurd T. W., Doherty D., Katsanis N., Duncan E. L., Otto E. A., Beales P. L., Mitchison H. M., Saunier S., Hildebrandt F.; UK10K Consortium (2013) Defects in the IFT-B component IFT172 cause Jeune and Mainzer-Saldino syndromes in humans. Am. J. Hum. Genet. 93, 915–925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bizet A. A., Becker-Heck A., Ryan R., Weber K., Filhol E., Krug P., Halbritter J., Delous M., Lasbennes M. C., Linghu B., Oakeley E. J., Zarhrate M., Nitschké P., Garfa-Traore M., Serluca F., Yang F., Bouwmeester T., Pinson L., Cassuto E., Dubot P., Elshakhs N. A., Sahel J. A., Salomon R., Drummond I. A., Gubler M. C., Antignac C., Chibout S., Szustakowski J. D., Hildebrandt F., Lorentzen E., Sailer A. W., Benmerah A., Saint-Mezard P., Saunier S. (2015) Mutations in TRAF3IP1/IFT54 reveal a new role for IFT proteins in microtubule stabilization. Nat. Commun. 6, 8666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Won J., Marín de Evsikova C., Smith R. S., Hicks W. L., Edwards M. M., Longo-Guess C., Li T., Naggert J. K., Nishina P. M. (2011) NPHP4 is necessary for normal photoreceptor ribbon synapse maintenance and outer segment formation, and for sperm development. Hum. Mol. Genet. 20, 482–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Donaldson J. C., Dise R. S., Ritchie M. D., Hanks S. K. (2002) Nephrocystin-conserved domains involved in targeting to epithelial cell-cell junctions, interaction with filamins, and establishing cell polarity. J. Biol. Chem. 277, 29028–29035 [DOI] [PubMed] [Google Scholar]
- 59.Rachel R. A., Yamamoto E. A., Dewanjee M., Munasinghe J., May-Simera H. L., Dong L., Swaroop A. (2012) CEP290 is required for photoreceptor ciliogenesis and other cilia related functions. Cilia 1, P98 [Google Scholar]
- 60.Cook S. A., Collin G. B., Bronson R. T., Naggert J. K., Liu D. P., Akeson E. C., Davisson M. T. (2009) A mouse model for Meckel syndrome type 3. J. Am. Soc. Nephrol. 20, 753–764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Collin G. B., Won J., Hicks W. L., Cook S. A., Nishina P. M., Naggert J. K. (2012) Meckelin is necessary for photoreceptor intraciliary transport and outer segment morphogenesis. Invest. Ophthalmol. Vis. Sci. 53, 967–974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Olbrich H., Fliegauf M., Hoefele J., Kispert A., Otto E., Volz A., Wolf M. T., Sasmaz G., Trauer U., Reinhardt R., Sudbrak R., Antignac C., Gretz N., Walz G., Schermer B., Benzing T., Hildebrandt F., Omran H. (2003) Mutations in a novel gene, NPHP3, cause adolescent nephronophthisis, tapeto-retinal degeneration and hepatic fibrosis. Nat. Genet. 34, 455–459 [DOI] [PubMed] [Google Scholar]
- 63.Schuermann M. J., Otto E., Becker A., Saar K., Rüschendorf F., Polak B. C., Ala-Mello S., Hoefele J., Wiedensohler A., Haller M., Omran H., Nürnberg P., Hildebrandt F. (2002) Mapping of gene loci for nephronophthisis type 4 and Senior-Løken syndrome, to chromosome 1p36. Am. J. Hum. Genet. 70, 1240–1246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.den Hollander A. I., Koenekoop R. K., Yzer S., Lopez I., Arends M. L., Voesenek K. E., Zonneveld M. N., Strom T. M., Meitinger T., Brunner H. G., Hoyng C. B., van den Born L. I., Rohrschneider K., Cremers F. P. (2006) Mutations in the CEP290 (NPHP6) gene are a frequent cause of Leber congenital amaurosis. Am. J. Hum. Genet. 79, 556–561 [DOI] [PMC free article] [PubMed] [Google Scholar]