

Abstract
Autism spectrum disorder (ASD) is a complex neurodevelopmental disorder characterized by social deficits and repetitive/restrictive interests. ASD is associated with multiple comorbidities, including intellectual disability, anxiety, and epilepsy. Evidence that ASD is highly heritable has spurred major efforts to unravel its genetics, revealing possible contributions from hundreds of genes through rare and common variation and through copy-number changes. In this perspective, we provide an overview of the current state of ASD genetics and of how genetic research has spurred the development of in vivo and in vitro models using animals and patient cells to evaluate the impact of genetic mutations on cellular function leading to disease. Efforts to translate these findings into successful therapies have yet to bear fruit. We discuss how the valuable insight into the disorder provided by these new models can be used to better understand ASD and develop future clinical trials.
Keywords: autism spectrum disorder, genetics, genomics, neurodevelopmental disorder, therapy
Abstract
El trastorno del espectro autista (TEA) es un complejo trastorno del neurodesarrollo caracterizado por déficits sociales e intereses repetitivos/restrictivos. El TEA se asocia con múltiples comorbilidades, incluyendo discapacidad intelectual, ansiedad y epilepsia. La evidencia de la alta heredabilidad del TEA ha estimulado los mayores esfuerzos para descifrar su genética, revelando posibles contribuciones de cientos de genes a través de variaciones raras y comunes, y de la variabilidad en el número de copias. Desde esta perspectiva se entrega una panorámica del estado actual de la genética del TEA y de cómo la investigación genética ha estimulado el desarrollo de modelos in vivo e in vitro que emplean células de animales y de pacientes para evaluar el impacto de las mutaciones genéticas en la función celular que lleva a la enfermedad. Aún no han dado frutos los esfuerzos realizados en la traducción de estos hallazgos en terapias exitosas. Se discute cómo se puede emplear la valiosa información acerca del trastorno, proporcionada por estos nuevos modelos, para una mejor comprensión del TEA y para desarrollar futuros ensayos clínicos.
Abstract
Le trouble du spectre de l'autisme (TSA) représente un trouble complexe neurodéveloppemental caractérisé par des déficits sociaux et des intérêts répétitifs et restreints. Les comorbidités associées au TSA sont multiples, comme la déficience intellectuelle, l'anxiété et l'épilepsie. La transmissibilité élevée démontrée du TSA a encouragé des efforts importants pour décoder sa génétique, des centaines de gènes étant potentiellement impliqués par des variations rares et courantes et par la variabilité du nombre de copies. Dans cette perspective, nous présentons un aperçu de l'état actuel de la génétique du TSA et de la façon dont la recherche génétique a stimulé le développement de modèles in vivo et in vitro utilisant des cellules humaines et animales pour évaluer l'incidence de mutations génétiques sur les fonctions cellulaires responsables de la maladie. Les efforts pour traduire ces résultats en traitements efficaces n'ont pas encore porté leurs fruits. Nous expliquons comment les informations précieuses apportées par ces nouveaux modèles peuvent être utilisées pour mieux comprendre le TSA et développer de futurs essais cliniques.
Introduction
Over the past decade, major advances in genetics and genomics have brought us closer to unraveling the molecular makeup of autism spectrum disorder (ASD). ASD is a constellation of neurodevelopmental abnormalities with social deficits and restrictive/repetitive behaviors as its core features. It affects about 1% of the world population and is three to four times more prevalent in males than in females.1,2 Twin studies and a more recent large population-based epidemiological study of roughly 2.5 million families have provided clear evidence for a genetic component of ASD and estimate the disease heritability at around 50 % to 95 % ,3,4 making it one of the most heritable of neuropsychiatric disorders. Furthermore, in all these studies, the phenotypic concordance between monozygotic twins is incomplete, indicating that nongenetic environmental factors play a role in the etiology of ASD. The diversity of contributing risk factors, degree of impairment, and expressed comorbidities found in individuals with ASD has created significant roadblocks to the identification of therapeutic targets and successful execution of clinical trials. In this review, we highlight how human genetics, induced pluripotent stem-cell (iPSC) -derived neurons from patients, and murine models can be used to develop strategies to stratify patients. Cellular models from patient iPSCs can help identify common biochemical pathways altered in patients, and mouse models replicating different human mutations can define common subsets of affected circuits.
ASD genetics: more patients, more genes
ASD is genetically complex and heterogeneous. To date, the identified genetic contributions to ASD include large chromosomal abnormalities, submicroscopic deletions or duplications (copy number variants [CNVs]), and rare single-nucleotide variants (SNVs), both inherited and de novo (not present in either parent). Common variation is estimated to contribute 40% to 60% of ASD risk with many common variants each providing a very small effect size.5,6 Genomewide association studies (GWAS) remain underpowered to yield reproducible results because of the genetic heterogeneity of ASD patients.7 There is also an additive relationship between ASD risk contributed by rare variants and common polygenic variation.8 Thus, cohorts on the order of tens of thousands of subjects will be needed to identify all common variants, remaining mindful of the possible contribution from rare variants.
CNVs associated with ASD are highly heterogeneous, can be inherited9 or arise de novo,10 and are not specific to the core ASD phenotypes, but are rather associated with a wide range of neuropsychiatric and neurodevelopmental phenotypes.11 The majority of CNVs have very low recurrence in ASD, and a specific CNV can often be unique to a single patient. Although the overall burden of de novo CNVs is higher in affected than in unaffected individuals, many of the same CNVs also occur in the unaffected individuals,12 making it difficult to determine which changes are likely to be disease-causing. The heterogeneity of CNV-associated phenotypes can also manifest within a single family as a result of unidentified modifiers.13
To date, contributions from rare variants in over 700 genes, both de novo and inherited, have been demonstrated in ASD, highlighting its complex genetic architecture. Early studies identified rare ASD-causing variants in established neurodevelopmental disease genes, including mutations in FMR1 (Fragile X syndrome; FXS), TSC (Tuberous Sclerosis Complex; TSC), MECP2 (Rett syndrome), and PTEN among others. Technological advances that enabled cost-effective sequencing of all protein-coding regions of the genome, termed whole exome sequencing (WES), allowed for the identification of coding variants in novel genes associated with “idiopathic,” or nonsyndromic, ASD. WES studies demonstrated that rare de novo, as well as inherited, SNVs contribute to disease risk — with causative de novo mutations identified in about 5% of ASD cases14-17 and inherited complete loss of function (LoF) mutations identified in around 5 % of ASD cases.18 Network analyses using high-confidence ASD risk genes with de novo mutations reveal enrichment for genes encoding chromatin modifiers (eg, CHD8, CHD2, ARID1B), synaptic signaling molecules (e.g. GRIN2B, GABRB3, SHANK), early embryonic development players (eg, TBR1, DYRK1A, PTEN), and fragile X mental retardation protein (FMRP) targets.19-21 Many of these genes control transcriptional or signaling cascades that affect multiple cellular processes.22 Understanding how gene mutations in different networks converge on disrupted pathways will define how cases can be stratified for clinical trials and treatment. For example, if widespread impairments in Wnt and/or AKT/mTOR signaling23,24 are identified in a range of ASD cases, such patients could be sorted into related treatment groups.
The overall impact of rare inherited variants on ASD risk has not been quantified comprehensively, since current studies do not take into account possible contributions from missense variants, whose possible functional impact is difficult to measure. Studies focusing on cohorts of both consanguineous and nonconsanguineous families have shown that rare recessive ASD mutations display similar heterogeneity in molecular pathways as shown by de novo mutations. Examples of ASD genes with recessive LoF mutations identified in families with ASD, intellectual disability, and other neurological and behavioral symptoms include CNTNAP2 24, SLC9A9/NHE9 25, BCKDK 26, and CC2D1A. 28 Some of the established recessive ASD mutations are hypomorphic alleles (retaining partial activity) of genes whose complete inactivation causes severe neurological syndromes.29 Examples of hypomorphic missense variants include those in AMT (encoding aminomethyl transferase), PEX7 (encoding peroxisomal biogenesis factor 7), and VPS13B (encoding vacuolar protein sorting 13 homolog B) that were identified in consanguineous families with ASD. Complete LoF of these genes leads, respectively, to nonketotic hyperglycinemia, rhizomelic chondrodysplasia punctata, and Cohen syndrome.29 UBE3B is another example of a candidate ASD gene in which a missense variant was identified30 and was subsequently associated with a syndrome of intellectual disability, lack of speech, and microcephaly.31 Similar to the spectrum and heterogeneity of ASD-associated CNVs, rare SNVs in “nonsyndromic” ASD genes can be associated with additional neurodevelopmental phenotypes. Thus, mutations in syndromic genes can also contribute to nonsyndromic ASD.
More recent work has focused on whole genome sequencing (WGS) in ASD cohorts to allow interrogation of the remaining (approximately) 99% of the genome that is not covered by WES. These efforts identified de novo CNVs and SNVs in coding regions of the genome that were missed by earlier WES studies, as well as variants in new candidate ASD genes.32 In addition, they suggest a role in ASD risk for de novo variants within noncoding regulatory regions of the genome.33,34 The noncoding variants mapped primarily to the untranslated regions (UTRs) of genes and active cis-regulatory elements.33,34 WGS also suggested a role for paternally inherited structural variants affecting promoter regions and UTRs.35
Large-scale sequencing efforts have contributed significantly to our understanding of the complex genetic architecture of ASD and have begun to define specific molecular pathways and neuronal circuits disrupted in ASD patients with common genetic etiologies. The field is moving toward study of larger cohorts by partnering with families, clinicians, researchers, and community organizations. One such effort is the Simons Foundation Powering Autism Research for Knowledge (SPARK), which aims to build a cohort of 50 000 individuals with ASD. A second collaboration between Autism Speaks, Google, and Toronto's SickKids Hospital has been named MSSNG, and will perform WGS in 10 000 ASD families. Future efforts must focus on high-throughput functional validation to assess the impact of identified variants on protein function and phenotype development, particularly for missense variants. Furthermore, our understanding of how multiple variants might act in concert within an individual and the risk from noncoding variation and gene by environment interactions are still in their infancy.
Preclinical studies in rodents: defining the circuitry controlling behavior
Modeling dysfunction in rodents has proven useful for understanding the impact of mutations associated with neuropsychiatry disorders and for testing possible therapies before use in human studies.36,37 Animal models of human disorders are expected to meet multiple criteria to be considered valid. They need to recapitulate the mechanism triggering the disorder, such as a gene mutation (construct validity), reproduce the symptoms (biochemical, histological, behavioral) of the disorder (face validity), and respond to pharmacological interventions as humans would (predictive validity).37,38 Defining to what extent an animal model of ASD is valid has been difficult.
A primary hurdle in modeling ASD in mice is that there are no molecular diagnostic measures, eg, a simple blood test such as blood sugar for diabetes, or a histological readout such as neurofibrillary tangles for Alzheimer disease. The presence of the disorder must be tested behaviorally in patients. Thus, ethologically appropriate paradigms for rodent or other animal behavior required significant model development to ensure appropriate translation of results. To this end, multiple tests for the core features of ASD — sociability and repetitive behaviors — have been developed for use in conjunction with tests of cognitive deficits and anxiety, which are frequently comorbid.36 Excellent primers with the description of recommended behavioral test batteries for models of ASD and developmental delay have been published.39,40 Briefly, social function is studied by measuring social preference via interaction and/or sniffing time in the following situations: (i) between animals, either stranger or previously encountered, in the three-chamber test; (ii) between a male and a stranger receptive female in mating-related interactions; (iii) between young littermates in social play41; or (iv) with bedding from strangers or cage mates in the social-conditioned place preference (sCPP) test.42
Repetitive behaviors and restricted interests can be measured in rodents by measuring instinctual behaviors, such as grooming and digging.43 Changes in self and reciprocal grooming are a good example of normal behaviors that can become exaggerated to the point of shaving (barbering) or injury that is severe enough to lead to ulcerative lesions. Digging behavior is used as a proxy for compulsive behavior in obsessive -compulsive disorder (OCD) models, and the same paradigms are applied in ASD. One caveat of digging tests is that increased digging is also linked to increased anxiety,44 and it is difficult to determine intent to confirm that the behavior is in fact excessive and compulsive, as opposed to recreational or shelter seeking. Finally, animal models of ASD have demonstrated comorbid anxiety and cognitive deficits that can include impairment in fear memory, cognitive flexibility, memory acquisition, and retention across different contexts as measured with well-established paradigms.45-47
Mouse models with construct validity, ie, engineered to carry the same genetic mutation found in humans, have presented constellations of behavioral impairments that partially overlap with the human condition. As the number of mouse lines with ASD gene mutations is increasing, several comprehensive reviews have been published to summarize and compare findings,38,39,48 often highlighting the variability in outcomes. As with patients, there is not a single clear combination of behavioral deficits caused by mutations in ASD genes. Each mouse line can present with a distinct subset of social and cognitive deficits, and/or anxiety and repetitive behaviors. However, as different mouse lines only partially reflect the features of the human disorder, it is difficult to establish face validity. A recent example of this variability comes from four mouse lines with heterozygous mutations in Chd8, a gene where frequent LoF de novo mutations have been identified in ASD.21 Mild social deficits with fewer and longer social encounters were identified in one study,49 normal interaction with strangers, but no memory of a previously encountered mouse in the three-chamber test, in another line,50 whereas comprehensive sociability analysis found no deficits in a different mouse.51 Additional inconsistencies were found in anxiety and cognitive performance.49-51 The type of genetic mutation, genetic background, and experimental conditions could be responsible for heterogeneity. Even in disorders like FXS and TSC with established mouse models for syndromic ASD,52 translation of pre-clinical work into clinical trials has been challenging.
These results imply limited predictive validity for murine models and have called into question the preclinical use of mice for modeling features of ASD tested in clinical trials. Moreover, they have revealed significant shortcomings in our knowledge of the molecular pathways and circuits that regulate behavior in rodents and humans. We have barely begun to scratch the surface in understanding the circuits involved in the various social paradigms in the mouse, and it remains unclear how tests for repetitive behaviors used to model obsessive-compulsive behavior will translate to ASD. Additional behavioral tests may need to be developed to complement the available ones to reflect other aspects of ASD. Importantly, new assays must demonstrate conservation of underlying molecular processes and circuits that are responsible for the similar function in the human brain. Other components of social interaction, such as cooperation, reciprocity, and empathy may be more relevant to the human disorder, and could be possible to assay in mice and rats.53
Despite these uncertainties, mouse models have been useful in dissecting how specific circuits are regulated and altered by mutations in ASD genes. One example is an elegant analysis of the mechanisms involved in sCPP showing that the preference for bedding linked to social experiences with cage mates depends on the reward circuitry in the nucleus accumbens (NAcc) regulated by both oxytocin and serotonin.54 These findings demonstrate a strong component of reward in remembering the location of a social encounter, and oxytocin has been shown to modulate NAcc connectivity in social motivation and perception tasks in children with ASD.55 Mouse studies have also implicated brain regions that were not traditionally thought to be involved in social behavior, such as the cerebellum. Heterozygous removal of Tsc1 or Tsc2 in cerebellar Purkinje cells affects social behavior.52,56 Comparison of multiple ASD models has shown deficits in cerebellar circuits involved in sensory learning and integration, revealing that sensory processing in the cerebellum could be a shared deficit across multiple types of mutations.57
In summary, work in mice has made strong inroads in understanding how specific genetic mutations alter cellular function, synaptic transmission, and behavior, but coordinated and consistent large-scale comparisons of mutations in multiple genes using consistent experimental conditions is necessary. New behavioral tests may also be important to unravel the intricacies of ASD. As behavioral studies have been often performed in males, particular attention should be placed in comparing males and females in animal models of ASD. Baseline behavioral sex-differences must be considered during interpretation of results, as well as sex-specific changes in mutant mice, leading to the identification of possible models of sex bias.58
Patient-derived neurons point to common cellular and biochemical pathways
Genetics and animal models can provide information about the developmental risk factors contributing to ASD, but do not necessarily reveal which therapeutic targets will improve symptoms after a lifetime of cellular and circuit-driven adaptations to altered neural function. Patient-derived neurons prepared from iPSCs can be used to identify drug targets and patient stratification approaches based on common cellular phenotypes and biochemical pathways. Study of iPSC-derived neurons from syndromic ASD patients has highlighted impairments of neural development including proliferation, differentiation, and synapse formation.59 These dysfunctional cellular processes probably contribute to the changes in neural connectivity described in the context of rodent models and patients. Phenotypes are often associated with disturbances of signal transduction cascades that include mTOR,60 AKT, extracellular-signal-regulated kinase (ERK),61 and/or Wnt signaling.62 In patient-derived neurons from idiopathic autism, evidence has been found for disturbances in synaptic connectivity and function, with changes in expression of ASD-associated genes, including voltage-gated ion channels.63
Through use of patient-derived neurons with specific molecular deficits, investigators can identify targeted therapeutic hypotheses. For example, patient-derived excitatory neurons from Phelan-McDermid syndrome (PMDS) are haploinsufficient for SHANK3 and display defects in synaptic transmission and membrane resistance that was corrected by treating neurons with insulin-like growth factor 1 (IGF1).64 IGF1 treatment was hypothesized to accelerate the maturation of patient-derived excitatory synapses lacking SHANK3 into more mature synapses that are no longer dependent upon SHANK3 because they contain mature expression of postsynaptic density protein 95 (PSD95) and N-methyl-D-aspartate (NMDA) receptors.64 This human developmental sequela was not apparent in murine preparations and may not have been recognized as a potential therapeutic approach if rodent models had been studied exclusively. Furthermore, studies of precisely engineered human neurons bearing heterozygous or homozygous loss of SHANK3 found that the previously observed increases in input resistance in PMDS neurons may be attributable to an impaired I h current that is mediated by hyperpolarization-activated cyclic nucleotide (HCN)-gated channels.65 The finding that PMDS phenotypes are mimicked in control neurons with inhibitors of I h raises the possibility that I h channel potentiators could be a possible draggable target for neurodevelopmental disorders that exhibit impaired I h currents.65 It is worth noting that rodent models of FXS also exhibit dysregulation of h-channel subunits and have impaired homeostatic h-channel plasticity.66 Other disorders that impinge on pathways producing deficits in maturation of glutamatergic synapse formation might yield overlapping subsets of patients that could benefit from similar therapeutic strategies. The development of techniques that promote rapid transdifferentiation67 of patient cells into neurons could, theoretically, allow for prospective stratification of idiopathic ASD patients for future clinical trials on the basis of properties of their transdifferentiated neurons.
Modeling neurodevelopmental disorders through use of human neurons in vitro is creating a valuable substrate for new phenotypic drug screens. The challenges of working with iPSC-derived models have been documented extensively elsewhere68 and include managing variability, determining which neuronal cell type and stage of development to model, and most importantly, determining what types of cellular end points would translate to measurable end points in patients. Use of chemical libraries with annotated mechanisms of action or US Food and Drug Administration (FDA)-approved drugs to identify new targets and repurposing opportunities for approved medicines will be especially powerful. Although access to high-quality small-molecule libraries with well-annotated mechanisms of action can be challenging for academic investigators, these files are available from many industry collaborators. Finding mutually acceptable terms for agreements between academic and industry technology transfer offices is often a hurdle. Furthermore, limited access to pharmacokinetic data and expertise needed to experimentally measure drug levels in animal models remains a critical roadblock to conducting meaningful translational follow-up studies in academic settings and requires new infrastructure.69
Stratification of patients for clinical trials varies by risk factors and therapeutic target
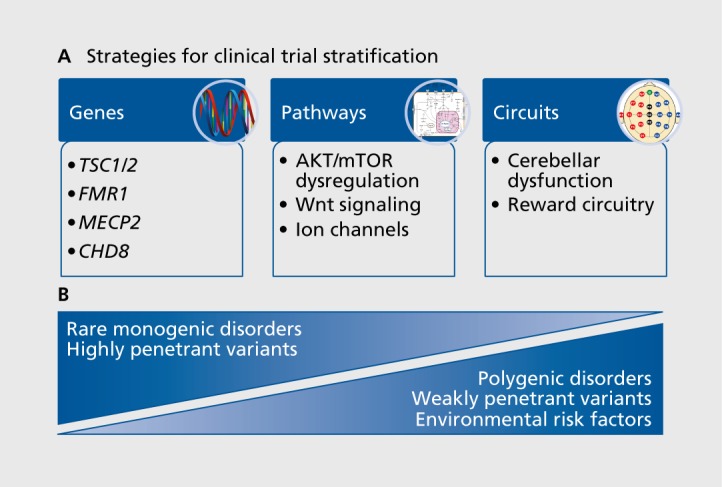
The heterogeneity of genetic and molecular deficits found in ASD suggests that a one-size-fits-all therapy is unlikely. Matching the correct subset of patients to each potential treatment and finding outcome measures that capture the benefit associated with efficacy is the biggest challenge to attracting the interest of commercial drug developers to ASD. Use of genetics as a stratification approach for clinical trials requires that the genetic perturbation confer a high degree of risk to the individual (Figure 1). Monogenic disorders such as FXS, Rett syndrome, or TSC have been chosen as the best candidates for subsets of autism on the basis of the assumption that a common genetic defect will trigger similar compensatory changes. However, even in monogenic disorders with high penetrance, such as FXS, there is a broad range of variation in genotype and phenotype driven by the number of CGG repeats and epigenetic methylation of the FMR1 promoter. Clinical trials for FXS have highlighted that there remains significant variability in the response of individual patients to candidate treatments, even when all patients have the same syndrome.70-73 A comprehensive and thoughtful review of clinical trials conducted in FXS recently highlighted many of the challenges faced by investigators to build the infrastructure to recruit, assess, and enroll patients for the early clinical trials in FXS.74 The remaining challenge for syndromic ASD will be to identify measures that accurately capture benefit for each syndrome and relate them to preclinical data sets.
Figure 1. Strategies for clinical trial stratification. The heterogeneity inherent in autism spectrum disorder (ASD) will require separating patients into more homogeneous subsets to test specific therapeutic hypotheses. (A) Stratification approaches can be developed on the basis of genetic mutations, biochemical pathways, or neural circuits to yield more homogeneous populations. Stratification based on human genetics will require that the genes used to narrow the range of the population confer a very high degree of risk for ASD. This is the case with monogenic disorders or very highly penetrant variants, such as TSC1/2, FMR1, MECP2, or CHD8 (left panel). Stratification based on biochemical pathways will be useful to create subgroups of patients with dysregulation of specific signaling cascades, such as mTOR, Wnt signaling, or impaired ion-channel function that are due to dysregulation from a wide range of de novo or weakly penetrant mutations that impinge on a common pathway (middle panel). (B) Monogenic/syndromic and nonsyndromic ASD may require different strategies for stratification. Nonsyndromic ASD is caused by a mixture of genetic and environmental factors that will be best stratified for clinical trials by monitoring the dysregulation of specific circuits linked to key symptomatic domains. AKT/mTOR, protein kinase B/mechanistic target of rapamycin; CHD8, chromodomain helicase DNA-binding protein 8; FMR1 , fragile X mental retardation protein; MECP2, methyl CpG binding protein 2; TSC1/2, tuberous sclerosis complex.
Preclinical data sets collected with candidate drugs for FXS often use Fmr1-knockout mice to evaluate efficacy against phenotypes observed in mice, which only exhibit a partial overlap with features of the human condition.75 Many preclinical studies focused on reversal of robust mouse deficits observed in measures of hippocampal synaptic plasticity (long-term depression [LTD]) and seizure behavior (audiogenic seizures), which provided little insight into what the most likely benefit would be for a human patient, given that seizures are not a problem for most FXS patients.76 Early clinical trials for FXS used preexisting outcome measures, including the Aberrant Behavior Checklist (ABC) and the Clinical Global Impressions (CGI) scale. Several subsequent studies conducted in small patient populations focused on select subscales of the ABC and expanded the number of clinical scales evaluated in the hope of finding a subset of sensitive measures that would support larger, pivotal trials needed for drug approval.74 Dedicated efforts to develop better clinical tools that are tailored to measure outcomes for these genetically defined patient populations should improve future success for syndromic autism.77
Given that most ASD patients do not have a highly penetrant genetic mutation, nonsyndromic clinical trials will require a biochemical or circuit-driven biomarker to stratify patients into more homogeneous subsets (Figure 1). For example, emerging evidence shows that the activity-dependent refinement of cerebellar circuits is critical to development of appropriate social behavior in rodent models of ASD; this finding is further supported by human studies that show injury to the cerebellum early in development is among the highest risk factor for developing ASD, and developmental malformations are frequently reported in ASD brain (reviewed by Wang and colleagues78). Visual-motor abnormalities in saccadic eye movements reported in ASD patients support the hypothesis that functional integrity of cerebellar circuits is impaired in visually guided tasks in many individuals with ASD.79 As mentioned above, selective removal of Tsc1 and Tsc2 in cerebellar Purkinje cells is sufficient to replicate ASD-like deficits in social approach, social interaction, and water maze reversal learning.52,56 Furthermore, sensory integration deficits in the cerebellum are common to mouse models recapitulating inherited and de novo mutations and CNVs (MECP2, TSC1, SHANK3, CNTNAP2, and 15q11-13 duplication).57
Cerebellar circuitry is highly conserved between rodents and humans, and it can be monitored functionally in animals and humans of all ages through use of classical tests of associative eyeblink conditioning. Associative eyeblink conditioning in normal human babies exhibits a range of efficiency that can be detected as early as at 1 month of age. The efficiency of an individual baby's performance predicts the emergence of social behavior measures at 5 and 9 months of age.80 Furthermore, conditioned eyeblink response has been reported to be impaired in ASD subjects.81,82 Similarly, saccade adaptation abnormalities have been reported in ASD subjects who adapt slower and demonstrate more variability than healthy controls.79,83 It is possible that subsets of ASD patients who exhibit cerebellar circuit dysfunction could represent a similarly impaired subset of idiopathic ASD patients that would benefit from pharmacological treatments that improve activity-dependent plasticity in this circuit during early postnatal development, regardless of their genetic risk factors.
Additional ASD patient subsets defined by minimally invasive and highly translatable circuit biomarkers will be needed in order to design clinical trials for more patients. Consideration for the cost and ease of screening patients for inclusion criteria, as defined by new stratification biomarkers, will be a critical element to paving a path to clinical trials, particularly those conducted over many different sites. For example, electroencephalogram (EEG)-based measures of hemispheric connectivity applied to naturally sleeping children84 may have biomarker potential for subsets of children with similar deficits in connectivity. EEG technology is commonly available at a broad range of medical centers and is not cost-prohibitive for large clinical trials.
Filling the pipeline
Identification of new drug targets to treat ASD is not enough to attract commercial drug development to work in this area. A viable path for conducting hypothesis-based clinical trials must be developed for each patient subset. As additional genes are identified that confer risk for ASD, preclinical models that evaluate their impact on human neurons in vitro and genetically engineered mice in vivo will expand our understanding of common phenotypes. The advent of CRISPR/Cas9 (clustered regularly interspaced short palindromic repeats/CRISPR-associated protein-9 nuclease) genome engineering has further opened up the possibility to study animal species with more complex social behaviors, such as prairie voles or nonhuman primates. Unraveling the mechanistic basis for circuit-level dysfunction across models and identifying the expression of translatable biomarkers that reflect homogenous patient subsets will be required to stratify patient populations for future clinical trials.
Acknowledgments
MC was supported by the University of Texas Southwestern Medical Center and a NARSAD Young Investigator Grant. MCM was supported by the George Washington University and the Clinical and Translational Science Institute at Children's National (NIH UL1 TR001876). RJK was supported by the Boston Children's Hospital Intellectual and Developmental Disabilities Research Center (NIH U54 HD090255) and the LouLou Foundation. The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of the National Center for Advancing Translational Science or the NIH. MCM and MC have no conflicts of interest to declare. RJK has consulted for Ironwood Pharmaceuticals.
Selected abbreviations and acronyms
- ASD
autism spectrum disorder
- CNV
copy number variant
- FXS
fragile X syndrome
- SHANK3
SH3 and multiple ankyrin repeat domains 3
- TSC
tuberous sclerosis complex
Contributor Information
Maria Chahrour, Eugene McDermott Center for Human Growth and Development, Departments of Neuroscience and Psychiatry, University of Texas Southwestern Medical Center, Dallas, Texas, USA.
Robin J. Kleiman, Research and Early Development, Biogen, Cambridge MA, USA.
M. Chiara Manzini, Institute for Neuroscience, Autism and Neurodevelopmental Disorders Institute, and Department of Pharmacology and Physiology, The George Washington University School of Medicine and Health Sciences, Washington, DC, USA.
REFERENCES
- 1.Elsabbagh M., Divan G., Koh YJ., et al. Global prevalence of autism and other pervasive developmental disorders. Autism Res. 2012;5(3):160–179. doi: 10.1002/aur.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Loomes R., Hull L., Mandy WPL. What is the male-to-female ratio in autism spectrum disorder? A systematic review and meta-analysis. J Am Acad Child Adolesc Psychiatry. 2017;56(6):466–474. doi: 10.1016/j.jaac.2017.03.013. [DOI] [PubMed] [Google Scholar]
- 3.Colvert E., Tick B., McEwen F., et al. Heritability of autism spectrum disorder in a UK population-based twin sample. JAMA Psychiatry. 2015;72(5):415–423. doi: 10.1001/jamapsychiatry.2014.3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sandin S., Lichtenstein P., Kuja-Halkola R., et al. The familial risk of autism. JAMA. 2014;311(17):1770–1777. doi: 10.1001/jama.2014.4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Klei L., Sanders SJ., Murtha MT., et al. Common genetic variants, acting additively, are a major source of risk for autism. Mol Autism. 2012;3(1):9. doi: 10.1186/2040-2392-3-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anney R., Klei L., Pinto D., et al. Individual common variants exert weak effects on the risk for autism spectrum disorders. Hum Mol Genet. 2012;21(21):4781–4792. doi: 10.1093/hmg/dds301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anney R., Klei L., Pinto D., et al. A genome-wide scan for common alleles affecting risk for autism. Hum Mol Genet. 2010;19(20):4072–4082. doi: 10.1093/hmg/ddq307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weiner DJ., Wigdor EM., Rjpke S., et al. Polygenic transmission disequilibrium confirms that common and rare variation act additively to create risk for autism spectrum disorders. Nat Genet. 2017;49(7):978–985. doi: 10.1038/ng.3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Levy D., Ronemus M., Yamrom B., et al. Rare de novo and transmitted copy-number variation in autistic spectrum disorders. Neuron. 2011;70(5):886–897. doi: 10.1016/j.neuron.2011.05.015. [DOI] [PubMed] [Google Scholar]
- 10.Pinto D., Pagnamenta AT., Mei L., et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466(7304):368–372. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Han K., Holder JL Jr., Schaaf CP., et al. SHANK3 overexpression causes manic-like behaviour with unique pharmacogenetic properties. Nature. 2013;503(7474):72–77. doi: 10.1038/nature12630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sebat J., Lakshmi B., Troge J., et al. Large-scale copy number polymorphism in the human genome. Science. 2004;305(5683):525–528. doi: 10.1126/science.1098918. [DOI] [PubMed] [Google Scholar]
- 13.Shen Y., Chen X., Wang L., et al. Intra-family phenotypic heterogeneity of 16p11.2 deletion carriers in a three-generation Chinese family. Am J Med Genet B Neuropsychiatr Genet. 2011;156(2):225–232. doi: 10.1002/ajmg.b.31147. [DOI] [PubMed] [Google Scholar]
- 14.Sanders SJ., Murtha MT., Gupta AR., et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485(7397):237–241. doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Neale BM., Kou Y., Liu L., et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485(7397):242–245. doi: 10.1038/nature11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O'Roak BJ., Deriziotis P., Lee C., et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet. 2011;43(6):585–589. doi: 10.1038/ng.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.lossifov I., Ronemus M., Levy M., et al. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012;74(2):285–299. doi: 10.1016/j.neuron.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lim ET., Raychaudhuri S., Sanders SJ., et al. Rare complete knockouts in humans: population distribution and significant role in autism spectrum disorders. Neuron. 2013;77(2):235–242. doi: 10.1016/j.neuron.2012.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Willsey AJ., Sanders SJ., Li M., et al. Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell. 2013;155(5):997–1007. doi: 10.1016/j.cell.2013.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hormozdiari F., Penn O., Borenstein E., Eichler EE. The discovery of integrated gene networks for autism and related disorders. Genome Res. 2015;25(1):142–154. doi: 10.1101/gr.178855.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.O'Roak BJ., Vives L., Girirajan S., et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485(7397):246–250. doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pinto D., Delaby E., Merico D., et al. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am J Hum Genet. 2014;94(5):677–694. doi: 10.1016/j.ajhg.2014.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Packer A. Enrichment of factors regulating canonical Wnt signaling among autism risk genes. Mol Psychiatry. 2016 Dec 13. Epub ahead of print. doi:1Q.1038/mp.2016.228. doi: 10.1038/mp.2016.228. [DOI] [PubMed] [Google Scholar]
- 24.Tsai P., Sahin M. Mechanisms of neurocognitive dysfunction and therapeutic considerations in tuberous sclerosis complex. Curr Opin Neurol. 2011;24(2):106–113. doi: 10.1097/WCO.0b013e32834451c4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Strauss KA., Puffenberger EG., Huentelman MJ., et al. Recessive symptomatic focal epilepsy and mutant contactin-associated protein-like 2. N Engl J Med. 2006;354(13):1370–1377. doi: 10.1056/NEJMoa052773. [DOI] [PubMed] [Google Scholar]
- 26.Morrow EM., Yoo SY., Flavell SW., et al. Identifying autism loci and genes by tracing recent shared ancestry. Science. 2008;321(5886):218–223. doi: 10.1126/science.1157657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Novarino G., El-Fishazy P., Kayserili H., et al. Mutations in BCXD-kinase lead to a potentially treatable form of autism with epilepsy. Science. 2012;338(6105):394–397. doi: 10.1126/science.1224631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Manzini MC., Xiong L., Shaheen R., et al. CC2D1A regulates human intellectual and social function as well as NF-κB signaling homeostasis. Cell Rep. 2014;8(3):647–655. doi: 10.1016/j.celrep.2014.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yu TW., Chahrour MH., Coulter ME., et al. Using whole-exome sequencing to identify inherited causes of autism. Neuron. 2013;77(2):259–273. doi: 10.1016/j.neuron.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chahrour MH., Yu TW., Lim ET., et al. Whole-exome sequencing and homozygosity analysis implicate depolarization-regulated neuronal genes in autism. PLoS Genet. 2012;8(4):e1002635. doi: 10.1371/journal.pgen.1002635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Basel-Vanagaite L., Dallapiccola B., Ramirez-Solis R., et al. Deficiency for the ubiquitin ligase UBE3B in a blepharophimosis-ptosis-intellectual disability syndrome. Am J Hum Genet. 2012;91(6):998–1010. doi: 10.1016/j.ajhg.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.C Yuen RK., Merico D., Bookman M., et al. Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nat Neurosci. 2017;20(4):602–611. doi: 10.1038/nn.4524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Turner TN., Hormozdiari F., Duyzend MH., et al. Genome sequencing of autism-affected families reveals disruption of putative noncoding regulatory DMA. Am J Hum Genet. 2016;98(1):58–74. doi: 10.1016/j.ajhg.2015.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yuen RK., Merico D., Cao H., et al. Genome-wide characteristics of de novo mutations in autism. NPJ Genom Med. 2016;1:160271–1602710. doi: 10.1038/npjgenmed.2016.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brandler WM., Antaki D., Gujral M., et al. Paternally inherited noncoding structural variants contribute to autism [unpublished preprint available online March 29, 2017]. bioRxiv. doi:https://doi.org/10. 1101/102327. [Google Scholar]
- 36.Crawley JN. Translational animal models of autism and neurodevelopmental disorders. Dialogues Clin Neurosci. 2012;14(3):293–305. doi: 10.31887/DCNS.2012.14.3/jcrawley. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nestler EJ., Hyman SE. Animal models of neuropsychiatric disorders. Nat Neurosci. 2010;13(10):1161–1169. doi: 10.1038/nn.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hulbert SW., Jiang, YH Monogenic mouse models of autism spectrum disorders: common mechanisms and missing links. Neuroscience. 2016;321:3–23. doi: 10.1016/j.neuroscience.2015.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kazdoba TM., Leach PT., Crawley JN. Behavioral phenotypes of genetic mouse models of autism. Genes Brain Behav. 2016;15(1):7–26. doi: 10.1111/gbb.12256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Silverman JL., Yang M., Lord C., Crawley JN. Behavioural phenotyping assays for mouse models of autism. Nat Rev Neurosci. 2010;11(7):490–502. doi: 10.1038/nrn2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pellis SM., Pellis VC. Play fighting of rats in comparative perspective: a schema for neurobehavioral analyses. Neurosci Biobehav Rev. 1998;23(1):87–101. doi: 10.1016/s0149-7634(97)00071-7. [DOI] [PubMed] [Google Scholar]
- 42.Trezza V., Campolongo P., Vanderschuren LJ. Evaluating the rewarding nature of social interactions in laboratory animals. Dev Cogn Neurosci. 2011;1(4):444–458. doi: 10.1016/j.dcn.2011.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim H., Lim CS., Kaang BK. Neuronal mechanisms and circuits underlying repetitive behaviors in mouse models of autism spectrum disorder. Behav Brain Funct. 2016;12(1):3. doi: 10.1186/s12993-016-0087-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Njung'e K., Handley SL. Evaluation of marble-burying behavior as a model of anxiety. Pharmacol Biochem Behav. 1991;38(1):63–67. doi: 10.1016/0091-3057(91)90590-x. [DOI] [PubMed] [Google Scholar]
- 45.Dudchenko PA. An overview of the tasks used to test working memory in rodents. Neurosci Biobehav Rev. 2004;28(7):699–709. doi: 10.1016/j.neubiorev.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 46.Haller J., Alicki M. Current animal models of anxiety, anxiety disorders, and anxiolytic drugs. Curr Opin Psychiatry. 2012;25(1):59–64. doi: 10.1097/YCO.0b013e32834de34f. [DOI] [PubMed] [Google Scholar]
- 47.Vorhees CV., Williams MT. Assessing spatial learning and memory in rodents. ILAR J. 2014;55(2):310–332. doi: 10.1093/ilar/ilu013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ogden KK., Ozkan ED., Rumbaugh, G Prioritizing the development of mouse models for childhood brain disorders. Neuropharmacology. 2016;100:2–16. doi: 10.1016/j.neuropharm.2015.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Katayama Y., Nishiyama M., Shoji H., et al. CHD8 haploinsufficiency results in autistic-like phenotypes in mice. Nature. 2016;537(7622):675–679. doi: 10.1038/nature19357. [DOI] [PubMed] [Google Scholar]
- 50.Piatt RJ., Zhou Y., Slaymaker IM., et al. CHD8 mutation leads to autistic-like behaviors and impaired striatal circuits. Cell Rep. 2017;19(2):335–350. doi: 10.1016/j.celrep.2017.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gompers AL., Su-Feher L., Ellegood J., et al. Germline Chd8 haploinsufficiency alters brain development in mouse. Nat Neurosci. 2017;20(8):1062–1073. doi: 10.1038/nn.4592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tsai PT., Hull C., Chu Y., et al. Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature. 2012;488(7413):647–651. doi: 10.1038/nature11310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sapolsky RM. Psychiatric distress in animals versus animal models of psychiatric distress. Nat Neurosci. 2016;19(11):1387–1389. doi: 10.1038/nn.4397. [DOI] [PubMed] [Google Scholar]
- 54.Dolen G., Darvishzadeh A., Huang KW., Malenka RC. Social reward requires coordinated activity of nucleus accumbens oxytocin and serotonin. Nature. 2013;501(7466):179–184. doi: 10.1038/nature12518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gordon I., Jack A., Pretzsch CM., et al. Intranasal oxytocin enhances connectivity in the neural circuitry supporting social motivation and social perception in children with autism. Sci Rep. 2016;6:35054. doi: 10.1038/srep35054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Reith RM., McKenna J., Wu H., et al. Loss of Tsc2 in Purkinje cells is associated with autistic-like behavior in a mouse model of tuberous sclerosis complex. Neurobiol Dis. 2013;51:93–103. doi: 10.1016/j.nbd.2012.10.014. [DOI] [PubMed] [Google Scholar]
- 57.Kloth AD., Badura A., Li A., et al. Cerebellar associative sensory learning defects in five mouse autism models. Elife. 2015;4:e06085. doi: 10.7554/eLife.06085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shansky RM., Woolley CS. Considering sex as a biological variable will be valuable for neuroscience research. J Neurosci. 2016;36(47):11817–11822. doi: 10.1523/JNEUROSCI.1390-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Aigner S., Heckel T., Zhang JD., Andreae LC., Jagasia R. Human pluripotent stem cell models of autism spectrum disorder: emerging frontiers, opportunities, and challenges towards neuronal networks in a dish. Psychopharmacology (Berl). 2014;231(6):1089–1104. doi: 10.1007/s00213-013-3332-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Costa V., Aigner S., Vukcevic M., et al. mTORC1 Inhibition corrects neurodevelopmental and synaptic alterations in a human stem cell model of tuberous sclerosis. Cell Rep. 2016;15(1):86–95. doi: 10.1016/j.celrep.2016.02.090. [DOI] [PubMed] [Google Scholar]
- 61.Mellios N., Feldman DA., Sheridan SD., et al. MeCP2-regulated miRNAs control early human neurogenesis through differential effects on ERK and AKT signaling. Mol Psychiatry. 2017 Apr 25. Epub ahead of print. doi:10.1038/mp.2017.86. doi: 10.1038/mp.2017.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang P., Lin M., Pedrosa E., et al. CRISPR/Cas9-mediated heterozygous knockout of the autism gene CHD8 and characterization of its transcriptional networks in neurodevelopment. Mol Autism. 2015;6:55. doi: 10.1186/s13229-015-0048-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liu X., Campanac E., Cheung HH., et al. Idiopathic autism: cellular and molecular phenotypes in pluripotent stem cell-derived neurons. Mol Neurobiol. 2016;54(6):4507–4523. doi: 10.1007/s12035-016-9961-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shcheglovitov A., Shcheglovitova O., Yazawa M., et al. SHANK3 and IGF1 restore synaptic deficits in neurons from 22q13 deletion syndrome patients. Nature. 2013;503(7475):267–271. doi: 10.1038/nature12618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yi F., Danko T., Botelho SC., et al. Autism-associated SHANK3 haploinsufficiency causes I h channelopathy in human neurons. Science. 2016;352(6286):aaf2669. doi: 10.1126/science.aaf2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Brager DH., Akhavan AR., Johnston D. Impaired dendritic expression and plasticity of h-channels in the fmr1 -ly mouse model of fragile X syndrome. Cell Rep. 2012;1(3):225–233. doi: 10.1016/j.celrep.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rackham OJ., Firas J., Fang H., et al. A predictive computational framework for direct reprogramming between human cell types. Nature Genet. 2016;48(3):331–335. doi: 10.1038/ng.3487. [DOI] [PubMed] [Google Scholar]
- 68.Panchision DM. Concise review: progress and challenges in using human stem cells for biological and therapeutics discovery: neuropsychiatric disorders. Stem Cells. 2016;34(3):523–536. doi: 10.1002/stem.2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kleiman RJ., Ehlers MD. Data gaps limit the translational potential of preclinical research. Sci Transl Med. 2016;8(320):320ps321. doi: 10.1126/scitranslmed.aac9888. [DOI] [PubMed] [Google Scholar]
- 70.Berry-Kravis E., Des Portes V., Hagerman R., et al. Mavoglurant in fragile X syndrome: results of two randomized, double-blind, placebo-controlled trials. Sci Transl Med. 2016;8(321):321ra5. doi: 10.1126/scitranslmed.aab4109. [DOI] [PubMed] [Google Scholar]
- 71.Berry-Kravis E., Hessl D., Coffey S., et al. A pilot open label, single dose trial of fenobam in adults with fragile X syndrome. J Med Genet. 2009;46(4):266–271. doi: 10.1136/jmg.2008.063701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Erickson CA., Mullett JE., McDougle CJ. Open-label memantine in fragile X syndrome. J Autism Dev Disord. 2009;39(12):1629–1635. doi: 10.1007/s10803-009-0807-3. [DOI] [PubMed] [Google Scholar]
- 73.Erickson CA., Wink LK., Ray B., et al. Impact of acamprosate on behavior and brain-derived neurotrophic factor: an open-label study in youth with fragile X syndrome. Psychopharmacology (Berl). 2013;228(1):75–84. doi: 10.1007/s00213-013-3022-z. [DOI] [PubMed] [Google Scholar]
- 74.Erickson CA., Davenport MH., Schaefer TM., et al. Fragile X targeted pharmacotherapy: lessons learned and future directions. J Neurodev Disord. 2016;9:7. doi: 10.1186/s11689-017-9186-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kazdoba TM., Leach PT., Silverman JL., Crawley JN. Modeling fragile X syndrome in the Fmr1 knockout mouse. Intractable Rare Dis Res. 2014;3(4):118–133. doi: 10.5582/irdr.2014.01024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Berry-Kravis E., Raspa M., Loggin-Hester L., et al. Seizures in fragile X syndrome: characteristics and comorbid diagnoses. Am J Intellect Dev Disabil. 2010;115(6):461–472. doi: 10.1352/1944-7558-115.6.461. [DOI] [PubMed] [Google Scholar]
- 77.Budimirovic DB., Berry-Kravis E., Erickson CA., et al. Updated report on tools to measure outcomes of clinical trials in fragile X syndrome. J Neurodev Disord. 2017;9:14. doi: 10.1186/s11689-017-9193-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang SS., Kloth AD., Badura A. The cerebellum, sensitive periods, and autism. Neuron. 2014;83(3):518–532. doi: 10.1016/j.neuron.2014.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mosconi MW., Luna B., Kay-Stacey M., et al. Saccade adaptation abnormalities implicate dysfunction of cerebellar-dependent learning mechanisms in autism spectrum disorders (ASD). PloS One. 2013;8(5):e63709. doi: 10.1371/journal.pone.0063709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Reeb-Sutherland BC., Levitt P., Fox NA. The predictive nature of individual differences in early associative learning and emerging social behavior. PloS One. 2012;7(1):e30511. doi: 10.1371/journal.pone.0030511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Oristaglio J., Hyman West S., Ghaffari M., et al. Children with autism spectrum disorders show abnormal conditioned response timing on delay, but not trace, eyeblink conditioning. Neuroscience. 2013;248:708–718. doi: 10.1016/j.neuroscience.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sears LL., Finn PR., Steinmetz JE. Abnormal classical eye-blink conditioning in autism. J Autism Dev Disord. 1994;24(6):737–751. doi: 10.1007/BF02172283. [DOI] [PubMed] [Google Scholar]
- 83.Schmitt LM., Cook EH., Sweeney JA., Mosconi MW. Saccadic eye movement abnormalities in autism spectrum disorder indicate dysfunctions in cerebellum and brainstem. Mol Autism. 2014;5(1):47. doi: 10.1186/2040-2392-5-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dinstein I., Pierce K., Eyler L., et al. Disrupted neural synchronization in toddlers with autism. Neuron. 2011;70(6):1218–1225. doi: 10.1016/j.neuron.2011.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]