

Abstract
Autism spectrum disorder (ASD) encompasses a group of neurodevelopmental conditions diagnosed solely on the basis of behavioral assessments that reveal social deficits. Progress has been made in understanding its genetic underpinnings, but most ASD-associated genetic variants, which include copy number variants (CNVs) and mutations in ASD-risk genes, account for no more than 1 % of ASD cases. This high level of genetic heterogeneity leads to challenges obtaining and interpreting genetic testing in clinical settings. The traditional definition of syndromic ASD is a disorder with a clinically defined pattern of somatic abnormalities and a neurobehavioral phenotype that may include ASD. Most have a known genetic cause. Examples include fragile X syndrome and tuberous sclerosis complex. We propose dividing syndromic autism into the following two groups: (i) ASD that occurs in the context of a clinically defined syndrome-recognizing these disorders depends on the familiarity of the clinician with the features of the syndrome, and the diagnosis is typically confirmed by targeted genetic testing (eg, mutation screening of FMR1); (ii) ASD that occurs as a feature of a molecularly defined syndrome-for this group of patients, ASD-associated variants are identified by genome-wide testing that is not hypothesis driven (eg, microarray, whole exome sequencing). These ASD groups cannot be easily clinically defined because patients with a given variant have variable somatic abnormalities (dysmorphism and birth defects). In this article, we review common diagnoses from the above categories and suggest a testing strategy for patients, guided by determining whether the individual has essential or complex ASD; patients in the latter group have multiple morphologic anomalies on physical examination. Finally, we recommend that the syndromic versus nonsyndromic designation ultimately be replaced by classification of ASD according to its genetic etiology, which will inform about the associated spectrum and penetrance of neurobehavioral and somatic manifestations.
Keywords: ASD-risk gene, autism spectrum disorder, copy number variant, microarray, syndromic, whole exome sequencing
Abstract
El trastorno del espectro autista (TEA) incluye un grupo diverso de cuadros del neurodesarrollo, diagnosticado por los clínicos únicamente en base a evaluaciones conductuales que revelan déficits sociales. Se ha progresado en la comprensión de sus bases genéticas, pero la mayoría de las variantes genéticas asociadas al TEA dan cuenta de no más del 1 % de los casos, y éstas incluyen variabilidad del número de copias (VNC) y mutaciones en los genes de riesgo para el TEA. Este alto nivel de heterogeneidad genética genera un desafío en la obtención e interpretación de las pruebas genéticas en los ambientes clínicos. La definición tradicional de TEA sindromático se refiere a un trastorno con un patrón clínicamente definido de alteraciones somáticas y un fenotipo neuroconductual que incluye el TEA. La mayoría tiene una causa genéticamente conocida y como ejemplos están el síndrome X frágil y el complejo esclerosis tuberosa. Se propone dividir el autismo sindromático en dos grupos: 1) El TEA que ocurre en el contexto de un síndrome definido clínicamente. El reconocimiento de estos trastornos depende de la familiaridad del clínico con las características del síndrome, y el diagnóstico se confirma típicamente por pruebas genéticas específicas (como la evaluación de FMR1) y 2) El TEA que ocurre como una característica del síndrome definido molecularmente. Para este grupo de pacientes, las variantes asociadas con el TEA se identifican mediante pruebas del genoma completo, que no se basan en una hipótesis (como el estudio de microarray o la secuenciación completa de exoma). Estos grupos de TEA no pueden definirse fácil clínicamente porque los pacientes con una variante determinada tienen alteraciones somáticas variables (dimorfismos y defectos del nacimiento). En este artículo se revisan los diagnósticos comunes a partir de las categorías anteriores y se sugiere una estrategia de evaluación de los pacientes dependiendo de si ellos tienen un TEA esencial o complejo; este último grupo tiene múltiples alteraciones morfológicas al examen físico. Por último, se recomienda que la designación de sindromático versus no-sindromático sea reemplazada finalmente por la clasificación de TEA de acuerdo con su etiología genética, la cual dará cuenta del espectro asociado y de la penetrancia de las manifestaciones neuroconductuales y somáticas.
Abstract
Le trouble du spectre de l'autisme (TSA) est un groupe de maladies neurodéveloppementales dont le diagnostic est établi uniquement sur la base d'évaluations comportementales qui signent des déficits sociaux. La compréhension des fondements génétiques du TSA progresse, mais la plupart des variantes génétiques associées au TSA, comme la variabilité du nombre de copies (VNC) et les mutations des gènes liés au TSA, ne représentent pas plus de 1 % des cas de TSA. Cette hétérogénéité génétique élevée rend difficiles la réalisation et l'interprétation des dépistages génétiques en milieu clinique. La définition traditionnelle du TSA syndromique est un tableau clinique défini, composé d'anomalies somatiques associées à un phénotype neurocomportemental pouvant comprendre le TSA. La plupart ont une cause génétique connue, comme le syndrome de l'X fragile et la sclérose tubéreuse complexe. Nous proposons de diviser l'autisme syndromique en deux groupes : 1) le TSA survenant dans le contexte d'un syndrome cliniquement défini - la reconnaissance de ces troubles dépend de la connaissance du médecin des caractéristiques du syndrome, et le diagnostic est confirmé généralement par des tests génétiques ciblés (par exemple le dépistage d'une mutation du gène FMR1) ; 2) le TSA survenant en tant que caractéristique d'un syndrome moléculairement défini - pour ce groupe de patients, les variantes associées au TSA sont identifiées par un dépistage au niveau du génome entier sans a priori (par exemple puces à ADN, séquençage de l'exome entier). Ces groupes de TSA ne sont pas faciles à définir cliniquement car les patients ayant une variante donnée ont des anomalies somatiques variables (dysmorphisme et anomalies congénitales). Dans cet article, nous examinons les diagnostics courants issus des catégories susmentionnées et suggérons une stratégie de dépistage pour les patients, pour déterminer si leur TSA est essentiel ou complexe, ce dernier groupe ayant des anomalies morphologiques multiples à l'examen clinique. Enfin, nous recommandons que la classification syndromique versus non syndromique soit finalement remplacée par une classification du TSA selon son étiologie génétique, qui renseignera sur le spectre et la pénétrance des manifestations neuro-comportementales et somatiques.
Introduction
Autism spectrum disorder (ASD) encompasses a diverse group of neurodevelopmental conditions diagnosed by clinicians solely on the basis of behavioral assessments that reveal social communication and social interaction deficits. The most recent edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM-5) uses one overarching ASD classification for what were formerly considered distinct clinical entities, including Asperger disorder, autistic disorder, and pervasive developmental disorder not otherwise specified. ASD's clinical heterogeneity includes not just the severity of the core autistic features, but also the presence or absence of neurobehavioral comorbidities, which include intellectual disability, attention-deficit/hyperactivity disorder (ADHD), and anxiety disorders, as well as medical comorbidities. Examples of the latter include the presence of a major congenital anomaly, epilepsy, and/or a broader genetic syndrome of which that individual's ASD is a feature.1,2
The traditional definition of syndromic ASD is a disorder with a clinically defined pattern of somatic abnormalities and a neurobehavioral phenotype that may include ASD. The diagnosis is typically confirmed by targeted genetic testing, eg, for trisomy 21 or fragile X syndrome (FXS).3 Such recognition depends on the clinician's experience with patients with particular groups of clinical findings, particularly for the less common syndromes. Examples where ASD is a variably present feature include (from lowest to highest expertise required for identification) Down syndrome, cases of neurofibromatosis type 1 (NF1) with mild cutaneous involvement, Cohen syndrome, and Potocki-Lupski syndrome.4-7
We propose to differentiate syndromic ASD that occurs in the context of a “phenotype first” clinically defined syndrome, as described above and accounting for 4% to 5% of ASD cases,8,9 from that which is molecularly defined (“genotype first”) (Figure 1). Patients with molecularly defined ASD syndromes are not easily identified clinically because the somatic manifestations of a given genetic change (eg, dysmorphic features, birth defects) vary widely and because, often, the number of reported cases with sufficient accompanying clinical information is low. These syndromes are recognized through testing that is genome-wide and not hypothesis-driven (ie, microarray, whole exome sequencing [WES], or research-based whole genome sequencing). Examples include recurrent deletions and duplications of the 16p11.2 chromosome region10 and pathogenic variants of the ASD-risk gene ADNP (activity-dependent neuroprotective protein).11
Figure 1. Overview of genetic categories of ASD: phenotype versus genotype first. ADNP, activity-dependent neuroprotective protein gene; ANK2, Ankyrin 2, neuronal gene; ARID1B, AT-rich interaction domain 1B gene; ASD, autism spectrum disorder; CNVs, copy number variants; FXS, fragile X syndrome; NF1, Neurofibromatosis, type 1 gene; NRXN1, Neurexin 1 gene; PTEN, Phosphatase and tensin homolog gene; SCN2A, Sodium channel, voltage-gated, type II, a subunit gene; TSC, tuberous sclerosis complex.
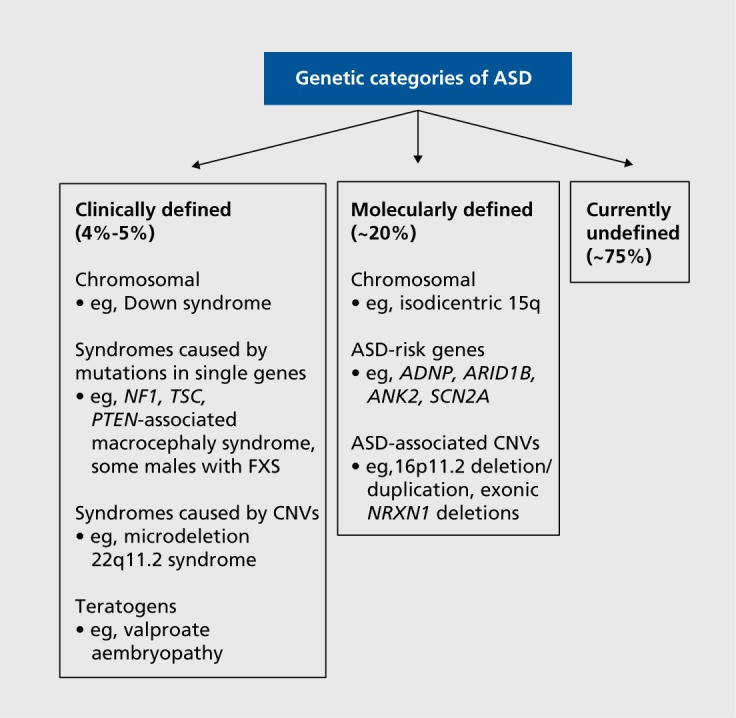
To address the phenotypic complexity of ASD, in 2005, Miles et al suggested physical examination of an affected child for minor physical anomalies (MPAs) and significant anthropometric measurement abnormalities.9 MPAs are morphologic deviations, present in less than 5% of the population (eg, single palmar creases, low-set ears).12 Measurement abnormalities include ocular hypertelorism (inner and outer canthal distances > +2 standard deviations [SD]) and macrocephaly (head circumference > +2SD). Miles and colleagues assigned one point for each MPA or measurement abnormality and two points for each major congenital abnormality (eg, ventricular septal defect) to give a total dysmorphology score for each child. They stratified 260 ASD children into three morphologic categories: essential (score 0-3), equivocal (score 4-5), or complex (score >5). Twenty percent were classified as complex.9
Miles et al hypothesized that, because those with equivocal and complex ASD showed evidence of an insult during early morphogenesis, they would be genetically distinct from those with essential/nondysmorphic ASD. Our group used her classification system to stratify a population-based cohort of 258 ASD children from Newfoundland and Labrador, Canada: 65.1% were classified as essential, 14.3% as equivocal, and 20.5% as complex.8 The combined diagnostic yield from chromosomal microarray (CMA) (for ASD-associated copy number variants [CNVs]) and WES (for pathogenic variants in ASD-risk genes) was 15.8%. Positive genetic tests were enriched in the equivocal and complex groups (diagnostic rates of 28.6% and 37.5% respectively) compared with the essential group (6%).
In this review, we: (i) summarize categories of ASD-associated genetic changes, with an emphasis on those identified by genome-wide testing; (ii) propose an approach to the genetic workup of ASD patients, with a testing strategy guided by morphologic examination; the algorithm is based on the premise that having excluded children with clinically defined syndromes, molecularly defined ASD syndromes are more likely to become apparent among individuals with equivocal or complex morphology; and (iii) use several molecularly defined ASD syndromes to illustrate common themes - such as variable penetrance and expressivity - and overlapping neurobehavioral phenotypes. Those selected are copy number variation at 16p11.2, genomic deletions that include the ASD-risk gene NRXN1 (neurexin 1), and CHD8 (chromosome helicase DNA binding protein 8)-associated ASD.
Box 1. Glossary of genetic terms.
Aneuploicly: The presence of an abnormal number of chromosomes in a cell. The normal number of chromosomes in a human cell is 46.
De novo: A genetic variant that arises for the first time in the proband and is not present in either of the individual's parents.
Haploinsufficiency: This arises when two copies of a gene are ordinarily both transcribed to make protein and one of the two gene copies is inactivated by a mutation. The resulting 50% reduction in the level of the associated protein leads to an abnormal state, for example, to an autosomal dominant disorder.
Loss-of-function (LOF) mutation: A variant that affects a gene in such a way that no functional protein is produced from that copy of the gene.
Nonallelic homologous recombination (NAHR): An abnormal form of recombination (crossing over) that occurs between two stretches of DNA that have high, but not identical, sequence homology. These are typically low copy repeats (LCRS) that have been duplicated through evolution. During cell division (in either meiosis or mitosis), LCRs can misalign and the subsequent crossing over leads to deletion or duplication of segments of DNA. Some of these copy number variants are associated with genetic disorders.
Premutation allele: An allele which, when compared with a normal copy of the gene, contains a higher number of a tandemly repeated nucleotide sequence (eg, CCG repeats in the 5' untranslated region of FMR1 [fragile X mental retardation 1 gene]). The increase in repeat number is not high enough to lead to the severe form of the disorder and may or may not be associated with other milder conditions. A premutation allele can expand to a fully penetrant allele when passed through the germ line.
Supernumerary isodkentric chromosome: An extra chromosome composed of a piece of a chromosome that has been duplicated end-to-end. The duplicated portions exist in a mirror image configuration and the extra chromosome has two centromeres.
Categories of ASD-associated genetic changes
Chromosomal
The first human karyotypes, nine cases of Down syndrome, were published in 1959,13 but chromosome analysis was not routinely used in diagnostic laboratories until the 1970s. Karyotypes identify an ASD-associated chromosomal syndrome in 2% of ASD cases.9,14 Most common are a supernumerary isodicentric chromosome 15 [idic(15)] involving the imprinted Prader-Willi/Angelman syndrome region (such individuals have four copies of the proximal 15q region instead of the usual two copies), Down syndrome, and the sex chromosome aneuploidies.15 Over 85% of maternally derived idic(15) cases develop ASD. For Down syndrome patients, 5% to 15% meet ASD criteria. In one study of Klinefelter syndrome (also known as 47,XXY), 27% (14/51) met ASD criteria.16
Variants identified by genome-wide testing
Most CNVs and mutations in ASD-risk genes identified by genome-wide approaches are characterized by incomplete penetrance for ASD and by variable expressivity, or pleiotropy, with respect to the associated somatic abnormalities.17,18 Very few lead to ASD in every person who carries the variant (ie, are completely penetrant) with the possible exception of truncating mutations in CHD8.19 Also, most are associated with other neurobehavioral disorders, especially intellectual disability. Determining the penetrance of these variants with respect to each associated trait (central nervous system [CNS] and non-CNS abnormalities) would require population screening with deep phenotyping - a daunting task.
Further, few identified variants account for more than 1% of ASD cases.20 There are over 50 ASD-associated CNVs21 and at least 61 ASD-risk genes.22 Many of these converge into shared biological pathways, including neuronal development and axonal guidance, synaptic function, and chromatin remodeling.21 This knowledge holds promise for guided therapeutic interventions.
Copy number variants
The first contiguous gene syndromes—genomic deletions creating haploinsufficiency of multiple neighboring genes—were identified using fluorescence in situ hybridization (FISH) on chromosomes.23 We now recognize some of these as ASD-associated. For example, microdeletion 22q11.l syndrome is typically caused by a recurrent 3-megabase (Mb) deletion of 40 genes, including TBX1 (T-box 1). Twenty percent to 50% of patients with this deletion develop ASD (Table I).49,50 Originally, a clinician might have suspected the diagnosis because of a particular facial appearance (narrow palpebral fissures, tubular nose) and/or the presence of certain birth defects, commonly involving the palate and the conotruncal region of the heart and then have ordered specific FISH testing of chromosome 22. Now, the abnormality would be detected by a genome-wide microarray.
TABLE I. Copy number variants consistently reported in association with autism spectrum disorder. ADHD, attention-deficit/hyperactivity disorder; AS, Angelman syndrome; ASD, autism spectrum disorder; BMI, body mass index; BD, bipolar disorder; BP, breakpoint; btw, between; CHD, congenital heart disease; chr, chromosome; CNV, copy number variant; DCD, developmental coordination disorder; DD, developmental delay; Del, deletion; Dup, duplication; FTT, failure to thrive; GU, genitourinary abnormality; HDAC4, histone deacetylase 4; HNF1B, hepatocyte nuclear factor 1 β; ID, intellectual disability; IQ, intelligence quotient; Kb, kilobase; Mb, megabase; MBD5, methyl- CpG-binding domain 5 gene; MEF2C, myocyte-specific enhancer factor 2C gene; mild-mod ID, mild-to-moderate intellectual disability; MODY, maturity onset diabetes of the young; MRI, magnetic resonance imaging; OCD, obsessive compulsive disorder; ODD, oppositional defiant disorder; PFs, palpebral fissures; PWS, Prader-Willi syndrome; RASA1, RAS p21 protein activator 1; SCZ, schizophrenia; SHANK3, SH3 and multiple ankyrin repeat domains 3 gene; SL disorder, speech and/or language disorder. A Genomic coordinates (hg19) for recurrent CNVs from DECIPHER: Del/Dup1q21.1 chr1:146533376-147883376; Del3q29 chr3:195726835-197344663; Dup7q11.23 chr7:72744455-74142672; Dup15q11-13 chr15:22876632-28557186; Del/Dup15q13.2q13.3 chr15:30910306-32445407; Del/ Dup16p11.2 chr16:29606852-30199855; Dup16p13.11 chr16:14986684-16486684; Dup17p11.2 chr17:16773072-20222149; Del17q12 chr17:34815072-36215917; Del22q11.2 chr22:21917117-23722445; Del22q13.3 chr22:51045516-51187844. Genomic coordinates (hg19) for recurrent CNVs from ClinGen: Del/Dup15q11.2 chr15:22803838-23092697.B This is the frequency of ASD in carriers reported in the referenced manuscript and, because of the influence of ascertainment bias, it should not be equated with the penetrance of the CNV for ASD.C Apparently normal transmitting parents reported.D Includes the imprinted Prader-Willi/Angelman syndrome region.E Nonimprinted region that includes NIPA1 & NIPA2.F Little inheritance data reported due to availability of parental samples, but at least several cases have had de novo duplications.G Nonimprinted region that contains CHRNA7.H Miller et al, 200940: ASD 40% (2/5).I Ben-Schachar et al, 200941: ASD features in 85% (12/14), most not formally tested and one diagnosed with Asperger syndrome.J Greater than 50% by age 7 years.K Causative gene is RAI1 and the reciprocal 17p11.2 deletion causes Smith-Magenis syndrome.L Only one formally assessed for ASD, and this individual met ASD criteria.M HNF1B causes renal cysts and diabetes syndrome (RCAD).N Vortsman et al, 200650: ASD in 30/60 Dutch children.O 50% of these are inherited from parent with a balanced rearrangement.
| CNVA | Size | Rate of ASD in reported carriersB | Other neuropsychiatric phenotypes | Consistently clinically recognizable | Dysmorphic features | Other somatic abnormalities (specific examples) | Inheritance | References |
| Del1q21.1 | 1.35 Mb | <10% | ADHD, mildmod ID (30%), SCZ, seizures (16%), learning disability | No | Variably dysmorphic: microcephaly,short stature, frontal bossing, deep-set eyes, bulbous nose | Ocular (microphthalmia), CHD (tetralogy of Fallot), GU (hydronephrosis), skeletal malformations | Up to 50% inheritedC | 24 |
| Dup1q21.1 | Critical region 1.35 Mb | 36% (21/59) | ID, SCZ, mood disorders (bipolar, depression) | No | Variable mild dysmorphism (1/3): macrocephaly (62%), prominent forehead, hypertelorism | CHD (tetralogy of Fallot), GU (cryptorchidism) | Up to 2/3's inheritedC | 25,26 |
| Del2q23.1 | Various sizes, all including MBD5 | 100% (51/51 had ASD features) | Consistent severe phenotype; all appear to have ID with little/no speech; many ataxic with seizures | Potentially yes (an Angelmanlike syndrome) | Consistently dysmorphic: microcephaly (90%), thick/high-arched eyebrows, wide mouth, small hands & feet | CHD; GU | All presumed de novo due to severity of phenotype | 27,28 |
| Del2q37 | 3-10 Mb (HDAC4 proposed as causative gene) | 30 % | Mild-mod ID (occasionally normal IQ), seizures (20%- 35%) | Potentially yes - initially classified as a form of Albright hereditary osteodystrophy | Characteristic facies: round face, thin high-arched eyebrows, deep-set eyes, upslanting palpebral fissures, hypoplastic nasal alae. Short stature, obesity, short digits | CHD (septal defects), CNS (hydrocephalus) GU (horseshoe kidney); 1% risk of Wilms tumor | 95% de novo deletion; 5% result from balanced parental translocation | 29 |
| Del3q29 | 1.6 Mb | 26% (11/44 patients) | ID; speech delay (60%); Of the non-ASD cases, 28% had another psychiatric phenotype: anxiety disorder, BD, SCZ | No | Paucity of data, subtle dysmorphism in first described patients. | CHD (26%) | Mainly de novo, occasional reports of inherited deletions | 30 |
| Del5q14.3 | Various sizes, all including the ID gene MEF2C | 42% (autistic features in 12/28) | Severe ID syndrome with absent speech. Majority have seizures & hypotonia | No (in the differential of a Rettlike syndrome) | No easily recognizable facial phenotype. Features include broad forehead, small upturned nose, small mouth, large ears. | Brain MRI abnormalities (89%); if the deletion includes RASA1, some have capillary and/or arteriovenous malformations | De novo deletions | 31,32 |
| Dup7q11.23 | 1.5-1.8—Mb dup of the Williams- Beuren syndrome critical region | 20% meet gold standard criteria for ASD | ID (18%), borderline IQ (20%), anxiety disorder (60%), ADHD (35%), oppositional disorders (25%), seizures (19%), hypotonia, ataxia | Potentially yes | Distinctive craniofacial features at all ages: macrobrachycephaly, straight eyebrows, short philtrum, thin upper lip | Dilatation of ascending aorta sometimes requiring surgical correction (46%); 30% have one or more congenital anomalies (CHD, renal, vertebral abnormalities) | 73% de novo; 27% inherited | 33 |
| Dup15q11-13 | Dup of maternal chr 15D | Unknown. ASD is a recognized feature & penetrance appears high | ID, motor coordination difficulties, language disorders including dyspraxia, ADHD | No | Mild or no dysmorphic features (no overt features of PWS or AS) | Joint laxity, occasional congenital abnormalities phenotype ++ variable even within families | De novo & inherited | 34-36 |
| Del15q11.2 | 350-kb del between BP1 and BP2E | 27% (43/161) | ID (37%), speech delay (67%), motor delay (42%), ADHD (35%), SCZ (20%), OCD (26%), ODD (24%), ataxia (28%), seizures (26%) | No | Unspecified dysmorphic features (39%), dysmorphic ears (46%), palatal abnormalities (46%) | Brain MRI abnormalities (43%), congenital heart defect (9%), genital abnormality (7%). | De novo and inheritedC | 37-39 |
| Dup15q11.2 | 350-kb dup between BP1 & BP2E | 43% (20/47) | DD (40%), speech delay (49%), ADHD (38%), ataxia (23%), seizures (12%) | No | Dysmorphic features (42%) - not consistently recognizable | None recorded | UnknownF | 37 |
| Del15q13.2 q13.3 | 1.6-Mb del btw BP4 & 5G | ASD 40%H, ASD features 85%I | DD, ID, ADHD, SL disorder, SCZ, seizures | No | Miller et al,40 2009: Subtle dysmorphic features (5/5); Ben-Schachar et al,41 2009: facies normal to moderately dysmorphic | None recorded | De novo & inheritedC | 40,41 |
| Dup15q13.2 q13.3 | 500 kb-1.98 Mb dup btw/ within BP4 & BP5 | ASD 80% (4/5) | DD (3/3), ID (1/1), SL disorder (3/3) | No | No consistent pattern of dysmorphism (2/2) | None recorded | De novo (2/5); maternally inherited (3/5) | 40 |
| Del16p11.2 Potocki-Lupsky syndrome | 600 kb del btw BP4 & BP5 | ASD 26% (20/78) | ID/borderline IQ (23%), SL disorder (71%), DCD (58%), ADHD (19%), anxiety disorder (6%), seizures (24%) | No | Nonspecific facial dysmorphia (46%, 69/150), some macrocephalic | Wide range of congenital anomalies (most common: vertebral abnormalities [20%] & posterior fossa malformations), obesityJ | Frequently de novo; can be inherited | 42-44 |
| Dup16p11.2 | 600-kb dup btw BP 4 & BP 5 | Unknown | ID, SCZ, bipolar disorder, seizures (overall penetrance 30%- 50%) | No | Not usually dysmorphic, some microcephalic | Low BMI | Usually inheritedC | 45-46 |
| Dup16p13.11 | Dups with varying BPs (790 kb- 2.67 Mb) | ASD 50% (4/8) | DD, ADHD, seizures | No | Not consistently dysmorphic (1/8, severely dysmorphic) | Brain MRI abnormalities, CHD | De novo & inheritedC | 47 |
| Dup17p11.2K Potocki-Lupsky syndrome | 3.7 Mb | 6/7 autistic featuresL | ID, ADHD, severe communication disorder, infant hypotonia | Potentially yes | Shared dysmorphic features: broad forehead, downslanting PFs, long nasal tip | FTT (5/7), CHD | UnknownF | 7 |
| Del17q12 | 1.4-MB del, includes HNF1BM | 6/9 ASD features (4/9 met diagnostic criteria) | SCZ, ID, SL disorder, deficits in motor skills &/ or coordination, anxiety disorders | Potentially yes | Macrodolicocephaly & consistent, mild facial dysmorphism (epicanthal folds, downslanting PFs, arched eyebrows) | Renal abnormalities that can present prenatally (eg, cystic kidneys), risk of diabetes including MODY | De novo & inherited | 48 |
| Del22q11.2 | Common 3-Mb del | 20%- 50%N | Psychiatric diagnosis in 60% of adults (SCZ in 25%) ID, ADHD, SL disorder, anxiety disorders | Yes, in many cases | Characteristic facial features include narrow PFs, hooded eyelids, squared off ear helices | CHD especially conotruncal, palatal abnormalities, renal anomalies | 93% de novo, 7% inherited | 49,50 |
| Del22q13.3 [Phelan- McDermid syndrome] | Terminal deletions, varying sizes, all include SHANK3 | 50% ASD or autistic features | Major features: neonatal hypotonia, moderate- severe ID, severely delayed/absent expressive language, seizures (25%) | No | Common, but subtle facial features: dolichocephaly, wide brow, deep-set eyes, bulbous nose, large poorly formed ears | CHD (>25%), renal anomalies (>25%) | 80%- 85% de novo; 15-20% unbalanced chromosome rearrangementO | 51 |
In 2004, large-scale human genomic variation was recognized.53 A CNV is a segment of DNA, often larger than 1 kilobase (kb) and containing more than one gene, which is deleted or duplicated relative to a reference genome. About 10% of the human genome is affected by copy number variation. Some CNVs are benign (part of normal human variation) and others are pathogenic (associated with a medical phenotype).54
Genome-wide microarrays were introduced into diagnostic laboratories in 2007 and were soon recommended as a first-line test for children with ASD.55 Arrays capture the unbalanced changes detectable by chromosome analysis, which have a minimum resolution of 4 to 5 Mb, as well as smaller genomic deletions and duplications. Current microarrays detect an ASD associated CNV in 7% to 10% of cases.8,22,55
Pathogenic variants in ASD-risk genes
WES documents most of the coding parts of the genome, ie, the exons of 19 000 protein-coding genes (total of 50 Mb), from which a growing list of ASD-risk genes has been identified. In a population-based ASD cohort of 258 children, 9.3% received a molecular diagnosis by microarray and 8.4%, by WES.8
Although still limited to research settings, whole genome sequencing (WGS) offers the advantage of identifying CNVs and DNA sequence changes in a single test. CNVs are more precisely sized by WGS, which helps with clinical interpretation. It offers better screening of the first exon of many genes and better identification of pathogenic variants at the intron/exon boundaries (splice junctions). In our hands, this increases the diagnostic yield by 3% to 5%,56 and we expect WGS to become the first-line test for children with ASDs over the next 5 years.
Approach to an ASD patient in clinic
Figure 2 shows an approach designed for a child who has been diagnosed with ASD by DSM-5 criteria. The child's medical record, including imaging reports, should be reviewed, documenting neurobehavioral comorbidities (eg, intellectual disability, anxiety disorders, sleep disturbance) and medical comorbidities (eg, epilepsy, other neurologic abnormalities, major congenital anomalies).
Figure 2. Clinical flow chart guiding the genetic investigation of individuals with an autism spectrum disorder. ASD, autism spectrum disorder; CNV, copy number variant; ID, intellectual disability; NF1 , neurofibromatosis, type 1 gene; WES, whole exome sequencing; HC, head circumference; SD, standard deviation.
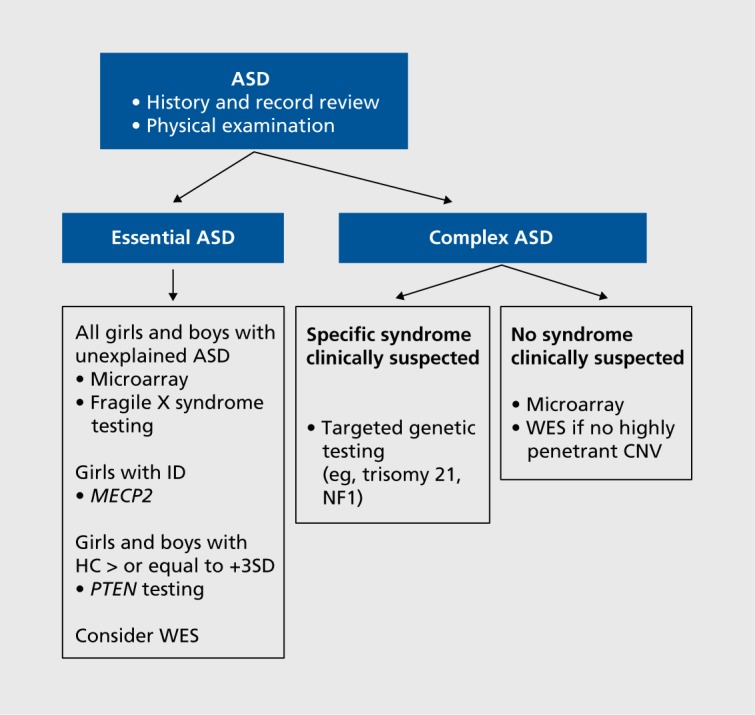
Physical examination should include documentation of height, weight, head circumference, and dysmorphic features. Particular attention should be paid to the face, hands, and feet. The skin should be examined, looking for cutaneous abnormalities associated with NF1 (café-au lait spots, inguinal/axillary freckling, cutaneous/subcutaneous neurofibromas) and with tuberous sclerosis complex (TSC) (hypopigmented macules, facial angiofibromas, ungual fibromas). This may allow the clinician to identify a specific ASD-associated syndrome based on clinical features. For those without expert training in dysmorphology, syndromes that could reasonably be recognized include Down syndrome, NF1,TSC, FXS, and PTEN (phosphatase and tensin homolog gene)-associated ASD in a child with extreme macrocephaly (head circumference greater than 3 SDs above the mean).
If no clinical syndrome is identified, the morphologic exam should be used to classify the child as having either essential (nondysmorphic) or complex ASD (multiple minor physical abnormalities and/or major congenital anomalies). Note that 25% of those with essential ASD are macrocephalic.8
All patients with unexplained essential ASD should have microarray and FXS testing. We also suggest mutation screening of MECP2 (methyl-CpG-binding protein 2 gene) for girls with intellectual disability, particularly if there are other features of Rett syndrome (RTT), such as language regression.57 Some patients with essential ASD, especially those with comorbid intellectual disability, have mutations in ASD-risk genes that will be missed without WES,8 and opportunities for such testing should be considered.
For patients with complex ASD, if a clinically defined syndrome is suspected, targeted genetic testing is indicated. Otherwise, we recommend microarray, followed by WES if no highly penetrant CNV is identified. The caveat to this is that 4% to 8% of children who have an ASD-associated CNV on microarray have a second ASD-associated change identified through WES.8,22
Clinically defined ASD syndromes
Fragile X syndrome (FXS)
FXS is the leading cause of inherited intellectual disability and in some populations, the most common genetic cause of ASD, accounting for 0.5% to 2% of cases.58 Over 98% of mutations are triplet repeat expansions in the 5' untranslated region (5'UTR) of the X-linked gene, FMR1 (fragile X mental retardation 1). The ranges for normal, premutation, and full mutation alleles are 5 to 44 CGG repeats, 55 to 200 repeats, and more than 200 CGG repeats, respectively. FMR1 mutations are dynamic and expand from one generation to the next, particularly when transmitted by a female. Full mutations in males typically lead to methylation of FMR1, inhibiting production of FMR1 messenger RNA (mRNA) and fragile X mental retardation protein (FRMP)—an RNA binding protein that regulates hundreds of proteins, most of which are involved with synaptic plasticity.59
Males with full FMR1 mutations and fully methylated alleles have moderate intellectual disability and a characteristic appearance. Typical facial features are macrocephaly with a prominent forehead, a long face, large protruding ears, and a prominent chin. There may be signs of a connective tissue disorder including a high arched palate, hyperextensible finger joints, pectus excavatum and mitral valve prolapse. Postpubertal macroorchidism also occurs. These findings are age-dependent, and 30% of young children with FXS are not obviously dysmorphic.59 Over 90% of males with full mutations have autistic features, and up to 60% meet diagnostic criteria for ASD.60
In females, the impact of full mutations is buffered by their second X chromosome and the X-inactivation phenomenon, such that 30% to 50% will have the syndrome. Their dysmorphism is milder or less evident than in their male counterparts, the main features being prominent ears and jaw.59 In one review of 31 females with full mutations, 23% met the criteria for ASD on at least one diagnostic test.61
Premutation alleles are paradoxically associated with increased FMR1 transcript levels; this leads to premature ovarian failure in 20% of carrier women and to fragile X-associated tremor ataxia syndrome (FXTAS) in up to 50% of older males.62 Premutation alleles also appear to increase susceptibility to ASD in both sexes. In a study of 50 premutation carriers, 14% of males and 5% of females met Autism Diagnostic Observation Scale-Generic (ADOS-G)63 criteria for ASD.61
In keeping with other reviews,1,58 we recommend FMR1 testing for all males and females with unexplained ASD, even in the absence of typical FXS dysmorphism (Figure 1). Note that trinucleotide repeat expansions, including those in FMR1, are not well detected by WES.
Phosphatase and tensin homolog (PTEN)-associated ASD
This widely expressed tumor suppressor, which maps to 10q23.3, functions as a brake for numerous cellular growth pathways. Its canonical function is downregulation of the phosphoinositol 3-kinase/AKT (or protein kinase B)/mammalian target of rapamycin (mTOR) pathway. Heterozygous PTEN mutations upregulate the AKT pathway leading to decreased apoptosis and increased cell growth.64
PTEN mutations are associated with a family of disorders with overlapping features that include Cowden syndrome (CS), Bannayan-Riley-Ruvalcaba syndrome (BRR), PTEN-related Proteus syndrome, and ASD-macrocephaly syndrome. A patient with any one of these conditions is considered to have PTEN hamartoma tumor syndrome (PTHS). CS patients are at increased risk of certain cancers, mainly breast, thyroid, and uterine. Most are macrocephalic, and by age 30 years, the vast majority develop pathognomonic mucocutaneous lesions (trichilemmomas and papillomatous papules). BRR is characterized by macrocephaly, intestinal polyps, and penile freckling in males. No genotype-phenotype correlations exist, and these phenotypes can be mixed even within the same family with the same mutation.65
In 2001, the first child with autism, macrocephaly, and a PTEN mutation was reported. At 9 years of age, his head circumference was 6 SD above the mean. He inherited the missense mutation from his intellectually and socially normal mother, who was subsequently diagnosed with CS.66 This report was followed by a series of prospective studies, mainly cohorts of individuals with ASD and macrocephaly summarized by Tilot et al,64 and the frequency of PTEN mutations ranged from 1% to 17%.
The prevalence of macrocephaly in ASD is 20% to 25%,8,9 but the degree of macrocephaly in ASD PTEN-mutation-positive patients is higher than in those who are mutation negative. In a cohort of 181 PTEN-mutation-positive individuals, the average head circumference was + 3.5 SD in adults and + 5 SD in children.67 The smallest head circumference reported to date in a macrocephalic child with ASD was + 2.9 SD.68 In our population-based cohort of 258 ASD children, we identified one PTEN mutation (0.4%). The variant was de novo and at age 4.5 years, this female's head circumference was + 4.5 SD.8
We recommend PTEN-mutation screening for all ASD children with a head circumference at or above +3 SD (Figure 2). If a mutation is identified, the child requires tumor surveillance (annual thyroid ultrasound and skin examination) with screening for other tumors beginning at 30 years of age.65 Mutations can be either de novo or inherited, and parents should be checked for the mutation even if they are asymptomatic.
Methyl-CpG-binding protein 2 (MECP2) and its association with ASD
The X-linked MECP2 gene encodes a critical regulator of brain function. It binds to specifically methylated cytosine residues in DNA (5meCyt) leading to chromatin compaction, which regulates the transcription of adjacent genes. MECP2 also influences translation at a global level by enhancing the AKT/mTOR pathway.69
De novo MECP2 mutations are responsible for over 95% of classic Rett syndrome (RTT). Affected girls develop normally for the first 6 to 18 months, followed by a period of regression, with loss of acquired language and replacement of purposeful hand use by stereotypic movements. The regression almost always occurs by age 5 years, and during this phase, some girls meet the criteria for ASD. Regression is followed by a period of stabilization, sometimes with recovery of skills.57
MECP2 mutations were later associated with a broad range of phenotypes including milder or more severe presentations of RTT in girls, and neonatal encephalopathy in males.70
Considerations around MECP2 testing for the ASD population include: (i) some girls first meet diagnostic criteria for ASD and later develop features of classic or variant RTT71; and (ii) MECP2 mutations are found in 2% of girls with ASD with or without intellectual disability and no features of RTT72 Such was first reported when researchers screened 68 autistic females and identified de novo mutations in two (ages 10 and 16 years) who met diagnostic criteria for ASD without features of RTT73 Recently, screening of 120 ASD cases via WES identified three patients (2.5%) with MECP2 mutations previously reported in patients with RTT74
We recommend MECP2 testing in any girl with unexplained ASD, particularly if she has intellectual disability or features to suggest early RTT (Figure 2). If a MECP2 mutation is found in a young girl, she should be monitored until at least age 5 years for the development of RTT features.
Other common clinically defined syndromes
Disorders associated with ASD include those characterized by skin abnormalities (eg, NF1,TSC), intellectual disability coupled with dysmorphic features (eg CHARGE syndrome, Cornelia de Lange syndrome, Sotos syndrome) or particular birth defects (eg, Timothy syndrome, Joubert syndrome and other ciliopathy disorders). For a thorough catalog, see reviews by Carter and Scherer,1 Betancur,16 and Miles.58 The clinically defined syndromes also include several of the genomic disorders listed in Table I that are caused by recurrent CNVs and that have well delineated somatic features.
Molecularly defined ASD syndromes
ASD-associated CNVs
Microarrays identify ASD-associated CNVs in 7% to 10% of ASD cases.8,22,55 Individuals with an ASD have more de novo CNVs than do controls21; this increased level of genomic variation has been documented in other neurodevelopmental disorders,75,76 supporting the concept of overlapping genomic etiologies.
CNVs can be further classified into four categories: (i) large imbalances that would have been detected by routine chromosome analysis, eg, trisomy 21; (ii) large rare deletions or duplications, many of which been catalogued in DECIPHER (DECIPHER consortium, https://decipher.sanger.ac.uk/); (iii) recurrent CNVs involving genomic disorder loci; and (iv) CNVs that involve known ASD-risk genes. Here, we focus on the third and fourth categories.
In general, for neurobehavioral phenotypes, penetrance is greater for de novo than for inherited CNVs, and for genomic deletions than for duplications.21
16p11.2 deletions and duplications
Some regions of the human genome are flanked by lowcopy repeat segments that predispose to nonallelic homologous recombination (NAHR), resulting in recurrent reciprocal deletions and duplications.45
One such region is a 593-kb segment of 16p11.2 that contains 25 annotated genes. First reported in ASD cohorts at 1% frequency,77-79 this CNV's variable expressivity has been recognized, both with respect to the associated neurobehavioral abnormalities, which includes intellectual disability, and to somatic abnormalities.10 Deletions of the 16p11.2 region are the second most common microdeletion identified in clinical laboratories80 and are frequently de novo. Almost all affected individuals are developmentally delayed.
In a review of 85 well-characterized deletion-positive individuals, 93% had at least one psychiatric diagnosis: 24% met ASD diagnostic criteria, and most of the remainder had behavioral traits that overlapped with ASD, including restricted interests and insistence on sameness. Other common diagnoses included speech and language disorders in 71% and developmental coordination disorders.42 Patients with microdeletion 16p11.2 are variably dysmorphic and may have congenital anomalies, the most common of which are posterior fossa malformations of the brain and vertebral anomalies.10,43 Over half are obese by age 7 years—anticipatory steps should be taken to prevent this.43,44
The reciprocal 16p11.2 duplications have a more variable and often milder phenotype and some carriers are nonpenetrant. Penetrance estimates for the associated neuropsychiatric disorders are unknown. Dysmorphology data are sparse, but in our experience, most individuals are not dysmorphic. Although the duplication confers susceptibility to ASD and intellectual disability, its strongest association is with schizophrenia, which has been suggested as ASD's mirror image disorder; duplication-positive patients are at 10- to 14-fold increased risk for psychosis.81,82 Underscoring the pleiotropy that is common to this and other CNVs, among 202 adults with intellectual disability and a comorbid psychiatric diagnosis, the most frequently observed CNV was duplication 16p11.2; of these four individuals, two had ASD and two had psychotic disorders.83
Duplication patients are more likely to have low body mass index (BMI), consistent with this genomic region being dosage-sensitive for weight. They have reduced head circumferences, whereas deletion patients tend to be macrocephalic.44 Another mirror image phenotype was shown among 30 deletion and 25 duplication patients by high-resolution brain magnetic resonance imaging, where clusters of abnormally thick or thin cortex were more extensive in the deletion and duplication patients, respectively.84
Other CNVs that have been consistently associated with ASD are summarized in Table I.
CNVs that involve an ASD-risk gene: Neurexin 1
The list of high confidence ASD-risk genes and of ASD-associated CNVs are largely nonoverlapping; however, some CNVs involve a known ASD-risk gene. In neurobehavioral patient cohorts, most such CNVs appear to be more common than disruptive sequence changes within the associated ASD-risk gene. Examples include deletions that involve NRXN1, NRXN3, ASTN2 (astrotactin 2 gene), DPYD (dihydropyrimidine dehydrogenase gene), and MBD5 (methyl-CpG-binding domain 5 gene), and deletions or duplications that involve AUTS2 (autism susceptibility candidate 2 gene), DMD (dystrophin gene), and NF1 (neurofibromatosis, type 1 gene).22 See Table I for a description of 2q23.1 microdeletion syndrome. The causative overlapping deletions all contain MBD5 and appear to be highly penetrant with respect to ASD.27,28
The human neurexin genes (NRXN1, NRXN2, NRXN3) encode CNS presynaptic scaffolding proteins. They function as cell adhesion molecules and receptors and, through binding neuroligins, mediate synapse formation and function. Each gene has alternative promoters, which direct the expression of longer α-neurexin and shorter β-neurexin transcripts. These transcripts undergo extensive alternative splicing to form over 1000 neurexin isoforms.85,86
NRXN1 maps to 2p16.2 and has 24 exons.87 Heterozygous deletions involving multiple NRXN1 exons, typically at the 5' end of the gene and involving only α-neurexin, were first reported in 2008.88,89 Heterozygous partial gene deletions have been reported in ASD, intellectual disability, and schizophrenia cohorts, as well as in the healthy parents of children with neurobehavioral abnormalities and in healthy controls.90 Among 2620 ASD cases, five had exonic NRXN1 deletions (0.19%).22
The largest published group of exonic-deletion patients with accompanying clinical information includes 24 children ascertained through referral for a clinical microarray.91 Their deletions varied from 40 to 586 kb, and 91% of the children were developmentally delayed. Though not personally examined by the authors, 83% (19/23) were classified as dysmorphic without an overlapping pattern of somatic abnormalities constituting a recognizable syndrome. Forty-three percent (9/21) had a formal diagnosis of ASD and 77% had either ASD or ASD features. On the basis of a review of published cases and the frequency of exonic NRXN1 deletions in controls, the authors suggested that deletion-positive individuals are at a 20-fold increased risk for ASD, and that NRXN1 exonic deletions have an ASD penetrance of 20%.
Heterozygous pathogenic sequence changes in NRXN1 have been reported in ASD cohorts, but appear to be less frequent than exonic deletions.22,92,93
Finally, biallelic disruptions of NRXN1 (exonic deletions and intragenic mutations) are associated with a severe syndromic form of intellectual disability, called Pitt Hopkins-like syndrome 2, which is within the differential diagnosis for a patient with suspected Angelman syndrome. All four reported cases (two sisters and an unrelated male and female) were dysmorphic, and three had severe seizure disorders. One male had a formal diagnosis of ASD,94 and the other three had autistic features.95,96
ASD-risk genes
High-throughput sequencing projects (initially WES and later WGS) have identified genes that are very likely to confer ASD risk, based on the finding of multiple de novo loss-of-function (LOF) mutations in unrelated ASD cases. These high-confidence ASD-risk genes include CHD8,10 ARID1B 98-100 (AT-rich interaction domain 1B), SCN2A (sodium channel, voltage-gated, type II, α subunit gene)52,112-116, DYRK1A (dual-specificity tyrosine-(Y)-phosphorylation-regulated kinase 1A gene), SYNGAP1 (synaptic Ras G TPase activating protein 1 gene),108-110 ADNP and SHANK3 (SH3 and multiple ankyrin repeat domains 3 gene)105-107 (see Table II 11,19,22,56,89,91,119). Of these, CHD8 has the highest number of LOF mutations and accounts for the most de novo mutations among ASD patients.22
Chromosome helicase DNA binding protein 8 (CHD8)-associated ASD
CHD8 maps to chromosome 14q11.2 and is a master transcriptional repressor that acts by remodeling chromatin structure and recruiting histone 1 to target genes. It functions at the center of a complex network of ASD genes. Its targets in human fetal brain include other ASD-risk genes such as ADNP ARID1B, DYRK1A, PTEN, and RAD21 (RAD21 cohesin complex component gene).120,121 In 2015, Cotney et al suggested that mutations in CHD8 “[perturb] an ancient gene regulatory network” critical for normal human brain development.121 This may prove to be an important common underlying mechanism for ASD, at least for a subset of affected individuals.
Heterozygous CHD8 LOF mutations in ASD individuals were first reported in 2012.122,123 In a combined cohort of 6000 patients (40% ASD probands from the Simons Simplex Collection and the remainder, children with ASD or developmental delay), there were 16 LOF CHD8 mutations. Over 90% of the truncating mutations were de novo.19
The group was able to recontact eight families containing 15 individuals with severe truncating CHD8 mutations for structured clinical assessments, including dysmorphology examinations. Of these, 13 met strict criteria for ASD, suggesting that disruptive mutations are possibly specific for ASD. All had low or borderline intelligence quotients (IQs); 80% were macrocephalic, although not to the degree typical of patients with PTEN mutations. The children and adults in this group had strikingly similar facial dysmorphology, including prominent supraorbital ridges, ocular hypertelorism with downslanting palpebral fissures, and pointed chins. The majority had gastrointestinal dysmotility. Both individuals in the cohort who were over age 40 years developed malignancies, including one who died of colorectal cancer at 42.19 Somatic mutations in CHD8 have been associated with gastric cancer.124
This group's work shows the importance of recalling groups of individuals with the same genetic alteration for deep phenotyping. The need to screen over 6000 patients to identify 15 CHD8 patients who could be recontacted underscores one of the biggest challenges in this field, ie, ASD's high level of genetic heterogeneity. The study appears to have identified a genomic marker for an ASD subtype with dysmorphology findings that overlap with FXS, and efforts should continue to characterize independent cohorts with CHD8 truncating mutations, including longitudinal studies to determine whether these patients are at increased risk of gastrointestinal or other malignancies.
Concluding remarks
ASD affects 1 in 68 American children, according to the most recent estimates.125 Providing assessments for such numbers through traditional genetic clinics is not feasible. We present an algorithm that can be followed by other health care providers who care for ASD children and have access to microarray and FXS testing. Children with morphologically complex ASD should be referred to a clinical geneticist, who may undertake targeted genetic testing or WES (Figure 2).
Few studies have examined the diagnostic yield of a thorough medical assessment, coupled with modern genetic testing, for ASD in a typical pediatric developmental clinic; however, a diagnostic rate of 25% for clinically and molecularly defined syndromes has been suggested.20 The availability of microarray and WES to clinicians involved in the care of children with ASD varies widely even within countries that have adopted genomic medicine. For example, in many Canadian provinces, nongeneticist specialists can order microarrays for children with ASD, with access to WES largely limited to geneticists. In the United States, insurance coverage is a major determinant of access to these tests. WGS still mainly occurs through research protocols. The approximate prices in Canadian dollars, which includes the cost of the assay and calling the variants, are as follows: about $500 for a microarray, $750 for WES, and $1250 for WGS. These prices are all based on the experiments being performed in a research setting. As these tests move into a clinical diagnostic realm with increased monitoring and standard operating procedures, the cost often triples compared with the prices listed for research testing.126
Genome-wide testing for the individual child and family is valuable for the following reasons. First, for some variants, sufficient data exist to predict developmental trajectories and provide anticipatory care for known medical comorbidities. For example, almost all patients with PTEN mutations have early motor delay including in independent walking, about one-quarter remain nonverbal or minimally verbal after age 4 years, and another quarter have an intelligence quotient (IQ) that is average or above average. Additionally, these patients need to be enrolled in a tumor surveillance protocol.64 A child with 7q11.23 duplication syndrome requires regular monitoring of the proximal aorta for dilatation,123 and a child with 17q12 deletion should be screened for maturity-onset diabetes of the young (Table I).48 Second, a positive genetic test often leads to improved recurrence risk counseling for parents considering more children. Examples include a full FMR1 mutation in a boy whose mother carries a premutation, or a child with a negative microarray and a de novo truncating mutation in CHD8. In the latter scenario, the risk that a future sibling will develop ASD is very likely less than the empirical recurrence risk of 20%.
Major knowledge gaps remain, including the determinants of penetrance and expressivity associated with the known variants. The proportion of children whose ASD is due to a combination of multiple genetic hits is probably much higher than we currently appreciate, and this will complicate interpretation of their phenotypes and provision of genetic counseling.
Bourgeron hypothesized that individuals with ASD can be divided into three groups with different ASD susceptibilities on the basis of the degree of genetic buffering provided by common low-risk alleles, of which we currently have little knowledge.20 A person with high genetic buffering, and therefore low a priori risk, might only develop ASD in the context of a de novo highly penetrant mutation; these are the changes that we have been most successfully identifying by WES. For a person with a low genetic buffer, accumulation of certain lower risk alleles may be sufficient to lead to ASD; this group probably includes a high proportion of children with essential ASD who have negative genetic testing.
Addressing these issues will require well-defined cohorts who are identified by genotype, clinically characterized, and followed-up longitudinally. Many of the identified ASD variants are in genes whose products converge into shared biologic pathways that ultimately control synaptic plasticity.20 For example, MECP2, MBD5, CHD8, ADNP, ARID1B, and TBR1 (T-box, brain, 1 gene) all encode key regulators of chromatin remodeling (Table II). NF1, PTEN, and SYNGAP1 products all upregulate the mTOR pathway, increasing translation within neurons and at synapses. Once the deregulation of these pathways is better understood, clinical trials will probably ensue, for which well-defined cohorts will be needed.
TABLE II. Examples of ASD-risk genes and their associated additional phenotypes. ADD, attention deficit disorder; ADHD, attention-deficit hyperactivity disorder; ASD, autism spectrum disorder; BAF, BRG1/BRM associated factor (also known as SWI/SNF); CASK, Calcium/calmodulindependent serine protein kinase; CHD, congenital heart disease; CNS, central nervous system; CPVT, catecholaminergic polymorphic ventricular tachycardia; CSS1, Coffin-Siris syndrome 1; DD, developmental delay; GI, gastrointestinal; GU, genitourinary abnormality; het, heterozygous; HVDAS, Helsmoortel-Van der Aa syndrome; ID, intellectual disability; IUGR, intrauterine growth retardation; LD, learning disability; mod-severe ID, moderate-to-severe intellectual disability; MRI, magnetic resonance imaging; NMDAR, N-methyl-Daspartate receptor; OMIM, Online Mendelian Inheritance in Man database; PSD, postsynaptic density; SCZ, schizophrenia; SD, standard deviation; SWI/SNF-A, a chromatin remodeling complex.A SWI/SNF complex in yeast.B Initial ADNP mutations were identified by screening ASD cohorts.C Reports are rare.D ANK2 mutations have been identified in multiple large ASD cohorts with little accompanying phenotypic information.E 16/35 (45.7%) had ASD.119 .
| ASD-risk gene | Chromosomal location/ gene name and function | ASD penetrance | Other neurobehavioral phenotypes | Dysmorphology | Other somatic abnormalities | References |
| Chromatin remodeling | ||||||
| CHD8 | 14q11.2, Chromodomain helicase DNA binding protein 8. Master transcriptional repressor | With truncating mutations, possibly complete | ID | Truncating mutations - common facial dysmorphism: prominent supraorbital ridges, hypertelorism, pointed chin | GI dysmotility, possible increased malignancy risk | 19 |
| ADNP | 20q13.13, Activity-dependent neuroprotective protein. Presumed transcription factor. Cterminus interacts with 3 essential components of BAF complex,A which regulates gene expression by mediating chromatin remodeling | Complete: causes Helsmoortel- Van der Aa syndrome (HVDAS, OMIM ≠615873), which belongs to the group of SWI/SNFrelated ID disordersB | Other features of HVDAS: ID, hypotonia, seizures, ADHD/ ADD, anxiety disorders | Dysmorphology variable. Common features: prominent forehead, high hairline, broad nasal bridge, thin upper lip, long/ smooth philtrum, polydactyly | Feeding problems, CHD, brain MRI abnormalities | 11,97 |
| ARID1B | 6q25.3, AT-rich interactive domain-containing protein 1B. Largest subunit of the mammalian SWI/SNF-A chromatin remodeling complex | Incomplete | AD mutations associated with: SWI/SNF ID syndrome Coffin- Siris syndrome (CSS1, OMIM≠ 135900), apparently nonsyndromic ID, syndromic short stature | Features in some CCS1 patients: hypertrichosis, coarse facies, malformed ears, short stature, small, hypoplastic 5th fingers. Clinical data on phenotypes of apparently nonsyndromic ID/ASD patients is lacking | Documented in CSS1: brain MRI abnormalities (especially agenesis of the corpus callosum), cryptorchidism in males, palatal abnormalities | 98-100 |
| TBR1 | 2q24.2, T-box, brain 1. Coactivated by cask to induce transcription of T-element containing genes, including Reelin, which is essential for cerebrocortical development | Unknown | ID | Unknown - none reported | Growth retardation | 22,101 |
| Synaptic and cytoskeletal proteins | ||||||
| NRXN1 - het missense & truncating variantsC | 2p16.3, Neurexin 1. Cell surface receptor that binds neuroligins to form a complex at CNS synapses | Incomplete | ADHD, LD, ID, SCZ | Mild facial dysmorphism in some (no other details) | Unknown - none reported | 89,92,93,102 |
| NRXN1 - deletions (limited to NRXN1 exons) | 2p16.3, Neurexin 1. Cell surface receptor that binds neuroligins to form a complex at CNS synapses | 20 % | DD, ID, hypotonia, bipolar disorder, ADHD, epilepsy, SCZ | Variably dysmorphic or nondysmorphic | Nonspecific brain MRI abnormalities, ocular abnormalities, other congenital anomalies | 91,103,104 |
| SHANK3 | 22q13.33, SH3 and multiple Ankyrin repeat domain 3. Structural protein of the post-synaptic density (PSD). PSD is responsible for alignment of postsynaptic membrane proteins | Incomplete; penetrance for de novo truncating mutations is high, and most cases also have mod-severe ID | SCZ, ID, epilepsy, speech delay, ADHD/ ADD, hypotonia | Variable facial dysmorphism in some cases similar to Phelan- McDermid syndrome (del22q13.3 syndrome, Table I); some cases nondysmorphic; macrocephaly; large stature | None reported | 105-107 |
| SYNGAP1 | 6p21.3, Synaptic RASGTPase activating protein 1. Part of the N-methyl-d-aspartate receptor (NMDAR) complex located in the PSD of glutamatergic neurons | Incomplete (50%108) | Nonsyndromic ID [MRD5 OMIM≠ 612621]. Appears to be highly penetrant for ID and generalized epilepsy. Other findings: hypotonia, ataxia | Unknown - none reported | Acquired microcephaly. Brain MRI normal or nonspecific features | 108-110 |
| ANK2 | 4q25-q26, Ankyrin 2. Localizes membrane ion channels and transporters | UnknownD | Unknown | None reported | Associated with several cardiac arrhythmia syndromes, including long QT syndrome type 4 & CPVT | 22,111 |
| SCN2A | 2q24.3, Sodium channel, voltage gated, type II alpha subunit. A subunit of a sodium channel | Incomplete | Spectrum of seizure disorders (benign familial neonatal seizures, infantile epileptic encephalopathy, neonatal seizures with later-onset episodic ataxia), SCZ, ID, brain MRI abnormalities | None reported | Optic atrophy, microcephaly | 56,112-116 |
| DYRK1A. Dualspecificity tyrosine phosphorylationregulated kinase 1A. | 21q22.13 (located within the Down syndrome critical region). Protein kinase essential for neurogenesis, neuronal differentiation, synaptic plasticity | IncompleteE | ID, severe speech delay/absent speech, epilepsy, ataxia/ broad-based gait | An Angelman-like syndrome with distinct facial features: sparse scalp hair, deep-set eyes, hooded eyelids, prominent nasal root, pointed nasal tip, short chin (not reminiscent of Down syndrome) | IUGR, congenital microcephaly (—2 SD to —5 SD), brain MRI abnormalities (hypomyelination), eye defects, joint contractures, CHD, GU | 117-119 |
Lastly, informed by better natural history data, it may become appropriate to screen for highly penetrant ASD variants in newborns in order to allow the introduction of behavioral interventions in the first year of life when the brain has its highest level of neuronal plasticity.
Acknowledgments
The authors would like to acknowledge Dr Janet Buchanan, Dr Matthew Deardorff, Ms Carol Negrijn, and Dr Susan Walker. BAF has no conflict of interest. SWS is on the Scientific Advisory Board of Population Diagnostics and Deep Genomics. This work was funded by the University of Toronto McLaughlin Centre, the Canadian Institutes for Advanced Research, the Canadian Institutes of Health Research (CIHR), and The Hospital for Sick Children Foundation. SWS is funded by the GlaxoSmithKline-CIHR Chair in Genome Sciences at the University of Toronto and The Hospital for Sick Children.
Selected abbreviations and acronyms
- FXS
fragile X syndrome
- NF1
neurofibromatosis type 1
- WES
whole exome sequencing
- WGS
whole genome sequencing
Contributor Information
Bridget A. Fernandez, Disciplines of Genetics and Medicine, Faculty of Medicine, Memorial University of Newfoundland, St John's, NL Canada.
Stephen W. Scherer, The Center for Applied Genomics and Program in Genetics and Genomic Biology, The Hospital for Sick Children, Toronto, Ontario, Canada; McLaughlin Center and Department of Molecular Genetics, University of Toronto, Toronto, Ontario, Canada.
REFERENCES
- 1.Carter MT., Scherer SW. Autism spectrum disorder in the genetics clinic: a review. Clin Genet. 2013;83(5):399–407. doi: 10.1111/cge.12101. [DOI] [PubMed] [Google Scholar]
- 2.Anagnostou E., Zwaigenbaum L., Szatmari P., et al. Autism spectrum disorder: advances in evidence-based practice. CMAJ. 2014;186(7):509–519. doi: 10.1503/cmaj.121756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zafeiriou D., Ververi A., Vargiami E. Childhood autism and associated comorbidities. Brain Dev. 2007;29(5):257–272. doi: 10.1016/j.braindev.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 4.Carter JC., Capone GT., Gray RM., Cox CS., Kaufmann WE. Autistic-spectrum disorders in Down syndrome: further delineation and distinction from other behavioral abnormalities. Am J Med Genet B Neuropsychiatr Genet. 2007;144B(1):87–94. doi: 10.1002/ajmg.b.30407. [DOI] [PubMed] [Google Scholar]
- 5.Williams PG., Hersh JH. Brief report: the association of neurofibromatosis type 1 and autism. J Autism Dev Disord. 1998;28(6):567–571. doi: 10.1023/a:1026012414193. [DOI] [PubMed] [Google Scholar]
- 6.Howlin P., Karpf J., Turk J. Behavioural characteristics and autistic features in individuals with Cohen Syndrome. Eur Child Adolesc Psychiatry. 2005;14(2):57–64. doi: 10.1007/s00787-005-0416-4. [DOI] [PubMed] [Google Scholar]
- 7.Potocki L., Bi W., Treadwell-Deering D., et al. Characterization of Potocki-Lupski syndrome (dup(17)(p11.2p11.2)) and delineation of a dosage-sensitive critical interval that can convey an autism phenotype. Am J Hum Genet. 2007;80(4):633–649. doi: 10.1086/512864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tammimies K., Marshall CR., Walker S., et al. Molecular diagnostic yield of chromosomal microarray analysis and whole-exome sequencing in children with autism spectrum disorder. JAMA. 2015;314(9):895–903. doi: 10.1001/jama.2015.10078. [DOI] [PubMed] [Google Scholar]
- 9.Miles JH., Takahashi TN., Bagby S., et al. Essential versus complex autism: definition of fundamental prognostic subtypes. Am J Med Genet A. 2005;135(2):171–180. doi: 10.1002/ajmg.a.30590. [DOI] [PubMed] [Google Scholar]
- 10.Fernandez BA., Roberts W., Chung B., et al. Phenotypic spectrum associated with de novo and inherited deletions and duplications at 16p1 1.2 in individuals ascertained for diagnosis of autism spectrum disorder. J Med Genet. 2010;47(3):195–203. doi: 10.1136/jmg.2009.069369. [DOI] [PubMed] [Google Scholar]
- 11.Helsmoortel C., Vulto-van Silfhout AT., Coe BR., et al. A SWI/SNF-related autism syndrome caused by de novo mutations in ADNP. . Nat Genet. 2014;46(4):380–384. doi: 10.1038/ng.2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ozgen HM., Hop JW., Hox JJ., Beemer FA., van Engeland H. Minor physical anomalies in autism: a meta-analysis. Mol Psychiatry. 2010;15(3):300–307. doi: 10.1038/mp.2008.75. [DOI] [PubMed] [Google Scholar]
- 13.Lejeune J., Turpin R., Gautier M. Chromosomic diagnosis of mongolism [article in French], Arch Fr Pediatr. 1959;16:962–963. [PubMed] [Google Scholar]
- 14.Miles JH. Complex autism spectrum disorders and cutting-edge molecular diagnostic tests. JAMA. 2015;314(9):879–880. doi: 10.1001/jama.2015.9577. [DOI] [PubMed] [Google Scholar]
- 15.Wassink TH., Piven J., Patil SR. Chromosomal abnormalities in a clinic sample of individuals with autistic disorder. Psychiatr Genet. 2001;11(2):57–63. doi: 10.1097/00041444-200106000-00001. [DOI] [PubMed] [Google Scholar]
- 16.Betancur C. Etiological heterogeneity in autism spectrum disorders: more than 100 genetic and genomic disorders and still counting. Brain Res. 2011;1380:42–77. doi: 10.1016/j.brainres.2010.11.078. [DOI] [PubMed] [Google Scholar]
- 17.Cook EH Jr., Scherer SW. Copy-number variations associated with neuropsychiatry conditions. Nature. 2008;455(7215):919–923. doi: 10.1038/nature07458. [DOI] [PubMed] [Google Scholar]
- 18.Vorstman JAS., Parr JR., Moreno-De-Luca D., Anney RJL., Nurnberger J Jr., Hallmayer JF. Autism genetics: opportunities and challenges for clinical translation. Nat Rev Genet. 2017;18(6):362–376. doi: 10.1038/nrg.2017.4. [DOI] [PubMed] [Google Scholar]
- 19.Bernier R., Golzio C., Xiong B., et al. Disruptive CHD8 mutations define a subtype of autism early in development. Cell. 2014;158(2):263–276. doi: 10.1016/j.cell.2014.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bourgeron T. From the genetic architecture to synaptic plasticity in autism spectrum disorder. Nat Rev Neurosci. 2015;16(9):551–563. doi: 10.1038/nrn3992. [DOI] [PubMed] [Google Scholar]
- 21.Pinto D., Delaby E., Merico D., et al. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am J Hum Genet. 2014;94(5):677–694. doi: 10.1016/j.ajhg.2014.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.C Yuen RK., Merico D., Bookman M., et al. Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nat Neurosci. 2017;20(4):602–611. doi: 10.1038/nn.4524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schmickel RD. Contiguous gene syndromes: a component of recognizable syndromes. J Pediatr. 1986;109(2):231–241. doi: 10.1016/s0022-3476(86)80377-8. [DOI] [PubMed] [Google Scholar]
- 24.Haldeman-Englert CR., Jewett T. 1q21.1 Recurrent microdeletion. In: Adam MP, Ardinger HH, Pagon RA, et al, eds. GeneReviews. Seattle, WA: University of Washington, Seattle. 2015 [PubMed] [Google Scholar]
- 25.Dolcetti A., Silversides CK., Marshall CR., et al. 1q21.1 Microduplication expression in adults. Genet Med. 2013;15(4):282–289. doi: 10.1038/gim.2012.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brunetti-Pierri N., Berg JS., Scaglia F., et al. Recurrent reciprocal 1q21.1 deletions and duplications associated with microcephaly or macrocephaly and developmental and behavioral abnormalities. Nat Genet. 2008;40(12):1466–1471. doi: 10.1038/ng.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Talkowski ME., Mullegama SV., Rosenfeld JA., et al. Assessment of 2q23.1 microdeletion syndrome implicates MBD5 as a single causal locus of intellectual disability, epilepsy, and autism spectrum disorder. Am J Hum Genet. 2011;89(4):551–563. doi: 10.1016/j.ajhg.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mullegama SV., Alaimo JT., Chen L., Elsea SH. Phenotypic and molecular convergence of 2q23.1 deletion syndrome with other neurodevelopmental syndromes associated with autism spectrum disorder. Int J Mol Sci. 2015;16(4):7627–7643. doi: 10.3390/ijms16047627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Doherty ES., Lacbawan FL. 2q37 Microdeletion syndrome. In: Adam MP, Ardinger HH, Pagon RA, et al, eds. GeneReviews. Seattle, WA: University of Washington, Seattle. 2013 [PubMed] [Google Scholar]
- 30.Glassford MR., Rosenfeld JA., Freedman AA., Zwick ME., Mulle JG. Unique Rare Chromosome Disorder Support Group. Novel features of 3q29 deletion syndrome: results from the3q29 registry. Am J Med Genet A. 2016;170A(4):999–1006. doi: 10.1002/ajmg.a.37537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zweier M., Rauch A. The MEF2C-related and 5q14.3q1 5 microdeletion syndrome. Mol Syndromol. 2012;2(3-5):164–170. doi: 10.1159/000337496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ilari R., Agosta G., Bacino C. 5q14.3 Deletion neurocutaneous syndrome: contiguous gene syndrome caused by simultaneous deletion of RASA1 and MEF2C. a progressive disease. Am J Med Genet A. 2016;170(3):688–693. doi: 10.1002/ajmg.a.37472. [DOI] [PubMed] [Google Scholar]
- 33.Mervis CB., Morris CA., Klein-Tasman BP., Velleman SL., Osborne LR. 7q11 .23 Duplication syndrome. In: Adam MP, Ardinger HH, Pagon RA, et al, eds. GeneReviews. Seattle, WA: University of Washington, Seattle; 2015 [Google Scholar]
- 34.Thomas JA., Johnson J., Peterson Kraai TL., et al. Genetic and clinical characterization of patients with an interstitial duplication 15q11-q13, emphasizing behavioral phenotype and response to treatment. Am J Med Genet A. 2003;119A(2):111–120. doi: 10.1002/ajmg.a.10176. [DOI] [PubMed] [Google Scholar]
- 35.Depienne C., Moreno-De-Luca D., Heron D., et al. Screening for genomic rearrangements and methylation abnormalities of the 15q11-q13 region in autism spectrum disorders. Biol Psychiatry. 2009;66(4):349–359. doi: 10.1016/j.biopsych.2009.01.025. [DOI] [PubMed] [Google Scholar]
- 36.Hogart A., Wu D., LaSalle JM., Schanen NC. The comorbidity of autism with the genomic disorders of chromosome 15q11.2-q13. Neurobiol Dis. 2010;38(2):181–191. doi: 10.1016/j.nbd.2008.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Burnside RD., Pasion R., Mikhail FM., et al. Microdeletion/microduplication of proximal 15q11.2 between BP1 and BP2: a susceptibility region for neurological dysfunction including developmental and language delay. Hum Genet. 2011;130(4):517–528. doi: 10.1007/s00439-011-0970-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Abdelmoity AT., LePichon JB., Nyp SS., Soden SE., Daniel CA., Yu S. 15q11.2 Proximal imbalances associated with a diverse array of neuropsychiatric disorders and mild dysmorphic features. J Dev Behav Pediatr. 2012;33(7):570–576. doi: 10.1097/DBP.0b013e31826052ae. [DOI] [PubMed] [Google Scholar]
- 39.Cox DM., Butler MG. The 15q11.2 BP1-BP2 microdeletion syndrome: a review. Int J Mol Sci. 2015;16(2):4068–4082. doi: 10.3390/ijms16024068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miller DT., Shen Y., Weiss LA., et al. Microdeletion/duplication at 15q13.2q13.3 among individuals with features of autism and other neuropsychiatric disorders. J Med Genet. 2009;46(4):242–248. doi: 10.1136/jmg.2008.059907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ben-Shachar S., Lanpher B., German JR., et al. Microdeletion 15q13.3: a locus with incomplete penetrance for autism, mental retardation, and psychiatric disorders. J Med Genet. 2009;46(6):382–388. doi: 10.1136/jmg.2008.064378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hanson E., Bernier R., Porche K., et al. The cognitive and behavioral phenotype of the 16p11.2 deletion in a clinically ascertained population. Biol Psychiatry. 2015;77(9):785–793. doi: 10.1016/j.biopsych.2014.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zufferey F., Sherr EH., Beckmann ND., et al. A 600 kb deletion syndrome at 16p1 1 .2 leads to energy imbalance and neuropsychiatric disorders. J Med Genet. 2012;49(10):660–668. doi: 10.1136/jmedgenet-2012-101203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jacquemont S., Reymond A., Zufferey F., et al. Mirror extreme BMI phenotypes associated with gene dosage at the chromosome 16p11.2 locus. Nature. 2011;478(7367):97–102. doi: 10.1038/nature10406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miller DT., Chung W., Nasir R., et al. 16p11.2 Recurrent microdeletion. In: Pagon RA, Adam MP, Ardinger HH, et al, eds. GeneReviews. Seattle, WA: University of Washington, Seattle. 2015 [Google Scholar]
- 46.McCarthy SE., Makarov V., Kirov G., et al. Microduplications of 16p11.2 are associated with schizophrenia. Nat Genet. 2009;41(11):1223–1227. doi: 10.1038/ng.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ramalingam A., Zhou XG., Fiedler SD., et al. 16p13.11 Duplication is a risk factor for a wide spectrum of neuropsychiatric disorders. J Hum Genet. 2011;56(7):541–544. doi: 10.1038/jhg.2011.42. [DOI] [PubMed] [Google Scholar]
- 48.Moreno-De-Luca D., Consortium S., Mulle JG., et al. Deletion 17q12 is a recurrent copy number variant that confers high risk of autism and schizophrenia. Am J Hum Genet. 2010;87(5):618–630. doi: 10.1016/j.ajhg.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McDonald-McGinn DM., Emanuel BS., Zackai EH. 22q11.2 Deletion syndrome. In: Adam MP, Ardinger HH, Pagon RA, et al, eds. GeneReviews. Seattle, Washington: University of Washington, Seattle; 2013 [Google Scholar]
- 50.Vorstman JA., Morcus ME., Duijff SN., et al. The 22q11.2 deletion in children: high rate of autistic disorders and early onset of psychotic symptoms. J Am Acad Child Adolesc Psychiatry. 2006;45(9):1104–1113. doi: 10.1097/01.chi.0000228131.56956.c1. [DOI] [PubMed] [Google Scholar]
- 51.Phelan K., McDermid HE. The 22q13.3 Deletion syndrome (PhelanMcDermid Syndrome). Mol Syndromol. 2012;2(3-5):186–201. doi: 10.1159/000334260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fine SE., Weissman A., Gerdes M., et al. Autism spectrum disorders and symptoms in children with molecularly confirmed 22q11.2 deletion syndrome. J Autism Dev Disord. 2005;35(4):461–470. doi: 10.1007/s10803-005-5036-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Iafrate AJ., Feuk L., Rivera MN., et al. Detection of large-scale variation in the human genome. Nat Genet. 2004;36(9):949–951. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- 54.Lee C., Scherer SW. The clinical context of copy number variation in the human genome. Expert Rev Mol Med. 2010;12:e8. doi: 10.1017/S1462399410001390. [DOI] [PubMed] [Google Scholar]
- 55.Miller DT., Adam MP., Aradhya S., et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86(5):749–764. doi: 10.1016/j.ajhg.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jiang YH., Yuen RK., Jin X., et al. Detection of clinically relevant genetic variants in autism spectrum disorder by whole-genome sequencing. Am J Hum Genet. 2013;93(2):249–263. doi: 10.1016/j.ajhg.2013.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Neul JL., Kaufmann WE., Glaze DG., et al. Rett syndrome: revised diagnostic criteria and nomenclature. Ann Neurol. 2010;68(6):944–950. doi: 10.1002/ana.22124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Miles JH. Autism spectrum disorders-a genetics review. Genet Med. 2011;13(4):278–294. doi: 10.1097/GIM.0b013e3181ff67ba. [DOI] [PubMed] [Google Scholar]
- 59.Lozano R., Rosero CA., Hagerman RJ. Fragile X spectrum disorders. Intractable Rare Dis Res. 2014;3(4):134–146. doi: 10.5582/irdr.2014.01022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Niu M., Han Y., Dy ABC., et al. Autism symptoms in fragile X syndrome. J Child Neurol. 2017;32(10):903–909. doi: 10.1177/0883073817712875. [DOI] [PubMed] [Google Scholar]
- 61.Clifford S., Dissanayake C., Bui QM., Huggins R., Taylor AK., Loesch DZ. Autism spectrum phenotype in males and females with fragile X full mutation and premutation. J Autism Dev Disord. 2007;37(4):738–747. doi: 10.1007/s10803-006-0205-z. [DOI] [PubMed] [Google Scholar]
- 62.Mila M., Alvarez-Mora Ml., Madrigal I., Rodriguez-Revenga L. Fragile X syndrome: an overview and update of the FMR1 gene. Clin Genet. 2017 Jun 15. Epub ahead of print. doi:10.1111/cge.13075. doi: 10.1111/cge.13075. [DOI] [PubMed] [Google Scholar]
- 63.Lord C., Risi S., Lambrecht L., et al. The autism diagnostic observation schedule-generic: a standard measure of social and communication deficits associated with the spectrum of autism. J Autism Dev Disord. 2000;30(3):205–223. [PubMed] [Google Scholar]
- 64.Tilot AK., Frazier TW 2nd., Eng C. Balancing proliferation and connectivity in PTEN-associated autism spectrum disorder. Neurotherapeutics. 2015;12(3):609–619. doi: 10.1007/s13311-015-0356-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ngeow J., Sesock K., Eng C. Clinical implications for germline PTEN spectrum disorders. Endocrinol Metab Clin North Am. 2017;46(2):503–517. doi: 10.1016/j.ecl.2017.01.013. [DOI] [PubMed] [Google Scholar]
- 66.Goffin A., Hoefsloot LH., Bosgoed E., Swillen A., Fryns JP. PTEN mutation in a family with Cowden syndrome and autism. Am J Med Genet. 2001;105(6):521–524. doi: 10.1002/ajmg.1477. [DOI] [PubMed] [Google Scholar]
- 67.Mester JL., Tilot AK., Rybicki LA., Frazier TW 2nd., Eng C. Analysis of prevalence and degree of macrocephaly in patients with germline PTEN mutations and of brain weight in Pten knock-in murine model. Eur J Hum Genet. 2011;19(7):763–768. doi: 10.1038/ejhg.2011.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Varga EA., Pastore M., Prior T., Herman GE., McBride KL. The prevalence of PTEN mutations in a clinical pediatric cohort with autism spectrum disorders, developmental delay, and macrocephaly. Genet Med. 2009;11(2):111–117. doi: 10.1097/GIM.0b013e31818fd762. [DOI] [PubMed] [Google Scholar]
- 69.Ehrhart F., Coort SL., Cirillo E., Smeets E., Evelo CT., Curfs LM. Rett syndrome - biological pathways leading from MECP2 to disorder phenotypes. Orphanet J Rare Dis. 2016;11(1):158. doi: 10.1186/s13023-016-0545-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Christodoulou J., Ho G. MECP2-related disorders. In: Pagon RA, Adam MP, Ardinger HH, et al, eds. GeneReviews. Seattle, WA: University of Washington, Seattle; 2012 [Google Scholar]
- 71.Young DJ., Bebbington A., Anderson A., et al. The diagnosis of autism in a female: could it be Rett syndrome? Eur J Pediatr. 2008;167(6):661–669. doi: 10.1007/s00431-007-0569-x. [DOI] [PubMed] [Google Scholar]
- 72.Zahorakova D., Lelkova P., Gregor V., Magner M., Zeman J., Martasek P. MECP2 mutations in Czech patients with Rett syndrome and Rett-like phenotypes: novel mutations, genotype-phenotype correlations and validation of high-resolution melting analysis for mutation scanning. J Hum Genet. 2016;61(7):617–625. doi: 10.1038/jhg.2016.19. [DOI] [PubMed] [Google Scholar]
- 73.Carney RM., Wolpert CM., Ravan SA., et al. Identification of MeCP2 mutations in a series of females with autistic disorder. Pediatr Neurol. 2003;28(3):205–211. doi: 10.1016/s0887-8994(02)00624-0. [DOI] [PubMed] [Google Scholar]
- 74.Wen Z., Cheng TL., Li GZ., et al. Identification of autism-related MECP2 mutations by whole-exome sequencing and functional validation. Mol Autism. 2017;8:43. doi: 10.1186/s13229-017-0157-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mosca SJ., Langevin LM., Dewey D., et al. Copy-number variations are enriched for neurodevelopmental genes in children with developmental coordination disorder. J Med Genet. 2016;53(12):812–819. doi: 10.1136/jmedgenet-2016-103818. [DOI] [PubMed] [Google Scholar]
- 76.Malhotra D., McCarthy S., Michaelson JJ., et al. High frequencies of de novo CNVs in bipolar disorder and schizophrenia. Neuron. 2011;72(6):951–963. doi: 10.1016/j.neuron.2011.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Marshall CR., Noor A., Vincent JB., et al. Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet. 2008;82(2):477–488. doi: 10.1016/j.ajhg.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kumar RA., KaraMohamed S., Sudi J., et al. Recurrent 16p11.2 microdeletions in autism. Hum Mol Genet. 2008;17(4):628–638. doi: 10.1093/hmg/ddm376. [DOI] [PubMed] [Google Scholar]
- 79.Weiss LA., Shen Y., Korn JM., et al. Association between microdeletion and m icroduplication at 16p11.2 and autism . N Engl J Med. 2008;358(7):667–675. doi: 10.1056/NEJMoa075974. [DOI] [PubMed] [Google Scholar]
- 80.Kaminsky EB., Kaul V., Paschall J., et al. An evidence-based approach to establish the functional and clinical significance of copy number variants in intellectual and developmental disabilities. Genet Med. 2011;13(9):777–784. doi: 10.1097/GIM.0b013e31822c79f9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Giaroli G., Bass N., Strydom A., Rantell K., McQuillin A. Does rare matter? Copy number variants at 16p11.2 and the risk of psychosis: a systematic review of literature and meta-analysis. Schizophr Res. 2014;159(23):340–346. doi: 10.1016/j.schres.2014.09.025. [DOI] [PubMed] [Google Scholar]
- 82.Chang H., Li L., Li M., Xiao X. Rare and common variants at 16p11.2 are associated with schizophrenia. Schizophr Res. 2017;184:105–108. doi: 10.1016/j.schres.2016.11.031. [DOI] [PubMed] [Google Scholar]
- 83.Wolfe K., Strydom A., Morrogh D., et al. Chromosomal microarray testing in adults with intellectual disability presenting with comorbid psychiatric disorders. Eur J Hum Genet. 2016;25(1):66–72. doi: 10.1038/ejhg.2016.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Blackmon K., Thesen T., Green S., et al. Focal cortical anomalies and language impairment in 16p11.2 deletion and duplication syndrome. Cereb Cortex. 2017 Jun 7. Epub ahead of print. doi:10.1093/cercor/bhx143. doi: 10.1093/cercor/bhx143. [DOI] [PubMed] [Google Scholar]
- 85.Sudhof TC. Neuroligins and neurexins link synaptic function to cognitive disease. Nature. 2008;455(7215):903–911. doi: 10.1038/nature07456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Reissner C., Klose M., Fairless R., Missler M. Mutational analysis of the neurexin/neuroligin complex reveals essential and regulatory components. Proc Natl Acad Sci U S A. 2008;105(39):15124–15129. doi: 10.1073/pnas.0801639105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tabuchi K., Sudhof TC. Structure and evolution of neurexin genes: insight into the mechanism of alternative splicing. Genomics. 2002;79(6):849–859. doi: 10.1006/geno.2002.6780. [DOI] [PubMed] [Google Scholar]
- 88.Zahir FR., Baross A., Delaney AD., et al. A patient with vertebral, cognitive and behavioural abnormalities and a de novo deletion of NRXN1α. J Med Genet. 2008;45(4):239–243. doi: 10.1136/jmg.2007.054437. [DOI] [PubMed] [Google Scholar]
- 89.Kim HG., Kishikawa S., Higgins AW., et al. Disruption of neurexin 1 associated with autism spectrum disorder. Am J Hum Genet. 2008;82(1):199–207. doi: 10.1016/j.ajhg.2007.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Woodbury-Smith M., Nicolson R., Zarrei M., et al. Variable phenotype expression in a family segregating microdeletions of the NRXN1 and MBD5 autism spectrum disorder susceptibility genes. NPJ Genom Med. 2017;2:17. doi: 10.1038/s41525-017-0020-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dabell MP., Rosenfeld JA., Bader P., et al. Investigation of NRXN1 deletions: clinical and molecular characterization. Am J Med Genet A. 2013;161A(4):717–731. doi: 10.1002/ajmg.a.35780. [DOI] [PubMed] [Google Scholar]
- 92.Onay H., Kacamak D., Kavasoglu AN., et al. Mutation analysis of the NRXN1 gene in autism spectrum disorders. Balkan J Med Genet. 2016;19(2):17–22. doi: 10.1515/bjmg-2016-0031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gauthier J., Siddiqui TJ., Huashan P., et al. Truncating mutations in NRXN2 and NRXN1 in autism spectrum disorders and schizophrenia. Hum Genet. 2011;130(4):563–573. doi: 10.1007/s00439-011-0975-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Duong L., Klitten LL., Moller RS., et al. Mutations in NRXN1 in a family multiply affected with brain disorders: NRXN1 mutations and brain disorders. Am J Med Genet B Neuropsychiatr Genet. 2012;159B(3):354–358. doi: 10.1002/ajmg.b.32036. [DOI] [PubMed] [Google Scholar]
- 95.Harrison V., Connell L., Hayesmoore J., McParland J., Pike MG., Blair E. Compound heterozygous deletion of NRXN1 causing severe developmental delay with early onset epilepsy in two sisters. Am J Med Genet A. 2011;155A(11):2826–2831. doi: 10.1002/ajmg.a.34255. [DOI] [PubMed] [Google Scholar]
- 96.Zweier C., de Jong EK., Zweier M., et al. CNTNAP2 and NRXN1 are mutated in autosomal-recessive Pitt-Hopkins-like mental retardation and determine the level of a common synaptic protein in Drosophila. Am J Hum Genet. 2009;85(5):655–666. doi: 10.1016/j.ajhg.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Vandeweyer G., Helsmoortel C., Van Dijck A., et al. The transcriptional regulator ADNP links the BAF (SWI/SNF) complexes with autism. Am J Med Genet C Semin Med Genet. 2014;166C(3):315–326. doi: 10.1002/ajmg.c.31413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hoyer J., Ekici AB., Endele S., et al. Haploinsufficiency of ARID1B , a member of the SWI/SNF-A chromatin-remodeling complex, is a frequent cause of intellectual disability. Am J Hum Genet. 2012;90(3):565–572. doi: 10.1016/j.ajhg.2012.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yu Y., Yao R., Wang L., et al. De novo mutations in ARID1B associated with both syndromic and non-syndromic short stature. BMC Genomics. 2015;16:701. doi: 10.1186/s12864-015-1898-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Santen GW., Clayton-Smith J., consortium ABC. The ARID1B phenotype: what we have learned so far. Am J Med Genet C Semin Med Genet. 2014;166C(3):276–289. doi: 10.1002/ajmg.c.31414. [DOI] [PubMed] [Google Scholar]
- 101.Hamdan FF., Srour M., Capo-Chichi JM., et al. De novo mutations in moderate or severe intellectual disability. PLoS Genetics. 2014;10(10):e1004772. doi: 10.1371/journal.pgen.1004772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Feng J., Schroer R., Yan J., et al. High frequency of neurexin 1β signal peptide structural variants in patients with autism. Neurosci Lett. 2006;409(1):10–13. doi: 10.1016/j.neulet.2006.08.017. [DOI] [PubMed] [Google Scholar]
- 103.Schaaf CP., Boone PM., Sampath S., et al. Phenotypic spectrum and genotype-phenotype correlations of NRXN1 exon deletions. Eur J Hum Genet. 2012;20(12):1240–1247. doi: 10.1038/ejhg.2012.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Guilmatre A., Dubourg C., Mosca, et al. Recurrent rearrangements in synaptic and neurodevelopmental genes and shared biologic pathways in schizophrenia, autism, and mental retardation. Arch Gen Psychiatry. 2009;66(9):947–956. doi: 10.1001/archgenpsychiatry.2009.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Boccuto L., Lauri M., Sarasua SM., et al. Prevalence of SHANK3 variants in patients with different subtypes of autism spectrum disorders. Eur J Hum Genet. 2013;21(3):310–316. doi: 10.1038/ejhg.2012.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gauthier J., Champagne N., Lafreniere RG., et al. De novo mutations in the gene encoding the synaptic scaffolding protein SHANK3 in patients ascertained for schizophrenia. Proc Natl Acad Sci U S A. 2010;107(17):7863–7868. doi: 10.1073/pnas.0906232107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Leblond CS., Nava C., Polge A., et al. Meta-analysis of SHANK mutations in autism spectrum disorders: a gradient of severity in cognitive impairments. PLoS Genetics. 2014;10(9):e1004580. doi: 10.1371/journal.pgen.1004580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Mignot C., von Stulpnagel C., Nava C., et al. Genetic and neurodevelopmental spectrum of SYNGAP1-associated intellectual disability and epilepsy. J Med Genet. 2016;53(8):511–522. doi: 10.1136/jmedgenet-2015-103451. [DOI] [PubMed] [Google Scholar]
- 109.Hamdan FF., Gauthier J., Spiegelman D., et al. Mutations in SYNGAP1 in autosomal nonsyndromic mental retardation. N Engl J Med. 2009;360(6):599–605. doi: 10.1056/NEJMoa0805392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Berryer MH., Hamdan FF., Klitten LL., et al. Mutations in SYNGAP1 cause intellectual disability, autism, and a specific form of epilepsy by inducing haploinsufficiency. Hum Mutat. 2013;34(2):385–394. doi: 10.1002/humu.22248. [DOI] [PubMed] [Google Scholar]
- 111.Mohler PJ., Le Scouarnec S., Denjoy I., et al. Defining the cellular phenotype of “ankyrin-B syndrome” variants: human ANK2 variants associated with clinical phenotypes display a spectrum of activities in cardiomyocytes. Circulation. 2007;115(4):432–441. doi: 10.1161/CIRCULATIONAHA.106.656512. [DOI] [PubMed] [Google Scholar]
- 112.Schwarz N., Hahn A., Bast T., et al. Mutations in the sodium channel gene SCN2A cause neonatal epilepsy with late-onset episodic ataxia. J Neurol. 2016;263(2):334–343. doi: 10.1007/s00415-015-7984-0. [DOI] [PubMed] [Google Scholar]
- 113.Sanders SJ., Murtha MT., Gupta AR., et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485(7397):237–241. doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Baasch, Huning I., Gilissen C., et al. Exome sequencing identifies a de novo SCN2A mutation in a patient with intractable seizures, severe intellectual disability, optic atrophy, muscular hypotonia, and brain abnormalities. Epilepsia. 2014;55(4):e25–e29. doi: 10.1111/epi.12554. [DOI] [PubMed] [Google Scholar]
- 115.Fromer M., Pocklington AJ., Kavanagh DH., et al. De novo mutations in schizophrenia implicate synaptic networks. Nature. 2014;506(7487):179–184. doi: 10.1038/nature12929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Nakamura K., Kato M., Osaka H., et al. Clinical spectrum of SCN2A mutations expanding to Ohtahara syndrome. Neurology. 2013;81(11):992–998. doi: 10.1212/WNL.0b013e3182a43e57. [DOI] [PubMed] [Google Scholar]
- 117.O'Roak BJ., Vives L., Fu W., et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science. 2012;338(6114):1619–1622. doi: 10.1126/science.1227764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ji J., Lee H., Argiropoulos B., et al. DYRK1A haploinsufficiency causes a new recognizable syndrome with microcephaly, intellectual disability, speech impairment, and distinct facies. Eur J Hum Genet. 2015;23(11):1473–1481. doi: 10.1038/ejhg.2015.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Luco SM., Pohl D., Sell E., Wagner JD., Dyment DA., Daoud H. Case report of novel DYRK1A mutations in 2 individuals with syndromic intellectual disability and a review of the literature. BMC Med Genet. 2016;17:15. doi: 10.1186/s12881-016-0276-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Breuss MW., Gleeson JG. When size matters: CHD8 in autism. Nat Neurosci. 2016;19(11):1430–1432. doi: 10.1038/nn.4431. [DOI] [PubMed] [Google Scholar]
- 121.Cotney J., Muhle RA., Sanders SJ., et al. The autism-associated chromatin modifier CHD8 regulates other autism risk genes during human neurodevelopment. Nat Commun. 2015;6:6404. doi: 10.1038/ncomms7404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.O'Roak BJ., Vives L., Girirajan S., et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485(7397):246–250. doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Neale BM., Kou Y., Liu L., et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485(7397):242–245. doi: 10.1038/nature11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Sawada G., Ueo H., Matsumura T., et al. CHD8 is an independent prognostic indicator that regulates Wnt/β-catenin signaling and the cell cycle in gastric cancer. Oncol Rep. 2013;30(3):1137–1142. doi: 10.3892/or.2013.2597. [DOI] [PubMed] [Google Scholar]
- 125.Christensen DL., Baio J., Van Naarden Braun K., et al. prevalence and characteristics of autism spectrum disorder among children aged 8 years - autism and developmental disabilities monitoring network, 11 sites. United States, 2012. MMWR Surveill Summ. 2016;65(3):1–23. doi: 10.15585/mmwr.ss6503a1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Tsiplova K., Zur RM., Marshall CR., et al. A microcosting and cost-consequence analysis of clinical genomic testing strategies in autism spectrum disorder. Genet Med. 2017;19(11):1268–1275. doi: 10.1038/gim.2017.47. [DOI] [PubMed] [Google Scholar]