

Abstract
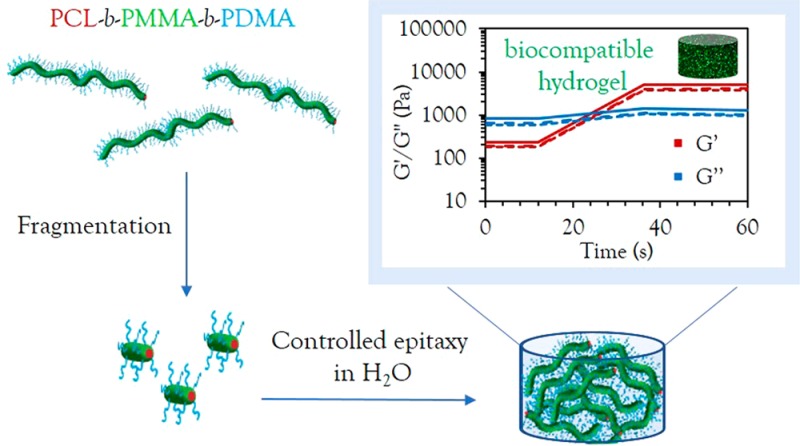
The fabrication of monodisperse nanostructures of highly controlled size and morphology with spatially distinct functional regions is a current area of high interest in materials science. Achieving this control directly in a biologically relevant solvent, without affecting cell viability, opens the door to a wide range of biomedical applications, yet this remains a significant challenge. Herein, we report the preparation of biocompatible and biodegradable poly(ε-caprolactone) 1D (cylindrical) and 2D (platelet) micelles in water and alcoholic solvents via crystallization-driven self-assembly. Using epitaxial growth in an alcoholic solvent, we show exquisite control over the dimensions and dispersity of these nanostructures, allowing access to uniform morphologies and predictable dimensions based on the unimer-to-seed ratio. Furthermore, for the first time, we report epitaxial growth in aqueous solvent, achieving precise control over 1D nanostructures in water, an essential feature for any relevant biological application. Exploiting this further, a strong, biocompatible and fluorescent hydrogel was obtained as a result of living epitaxial growth in aqueous solvent and cell culture medium. MC3T3 and A549 cells were successfully encapsulated, demonstrating high viability (>95% after 4 days) in these novel hydrogel materials.
Introduction
Block copolymer nanostructures have received an increasing amount of interest in the nanomedical field.1,2 The ability to obtain different morphologies with controlled dimensions, from spherical micelles to rods, platelets, vesicles, and more complex structures, opens a wide range of possibilities for potential applications.3 For example, it has been reported that elongated morphologies, such as rod-like particles, not only exhibit better cell uptake rates in comparison to their spherical counterparts4−9 but also show increased blood circulation times on increasing cylindrical micelle length.10
Precise control over the formation of anisotropic materials with biocompatible and biodegradable properties, however, represents a key challenge in enabling their use in nanomedicine. Recent advances in the solution crystallization of polymers have allowed access to a wide range of complex hierarchical structures,11 where the presence of a crystalline core-forming block promotes the formation of morphologies with low interfacial curvature, such as cylinders,12−15 ribbons,16 and platelet micelles.17−21 Despite recent advances in precision design, in particular those using poly(ferrocenyldimethylsilane) (PFS)15,21 and poly(ε-caprolactone) (PCL)22−26 block copolymers, few size-controlled assemblies can be retained in aqueous media, with no reports of direct epitaxial crystallization in water to date. Thus, previously reported controlled crystallization methods are limited by the lack of translation toward simple crystalline growth in aqueous media. Therefore, given the importance of life-essential aqueous environments, the formation of precision nanostructures directly in a biologically relevant solvent remains a key challenge in opening new frontiers for biological applications.
Of the previously reported micelles that can be dispersed in water, few show significant control over dimensions and dispersity. Recently, Manners and co-workers reported that cylindrical micelles could be obtained from PFS-b-poly(allyl glycidyl ether), and grafting modifications to form water-stable micelles via a postpolymerization modification step allowed the micelles to be successfully dialyzed from DMF into water.15 To date, however, such precise control over biologically relevant and degradable polymers has not yet been achieved despite the enhanced micellar stability offered by the crystalline core, the narrow width and length dispersity, and the ability to modulate shape and surface chemistry through living growth, which provides significant potential in advancing a wide range of biomedical applications, from drug delivery to tissue engineering. Furthermore, growth without the need for such postmodification and solvent transfer steps would greatly simplify access to these nanostructures. Previous reports using biocompatible polymers such as polyethylene (PE), poly(ethylene oxide) (PEO), PCL, and poly(l-lactide) (PLLA) have shown that these polymers can form both 1D and 2D assemblies by crystallization-driven self-assembly (CDSA)27−35 and undergo controlled growth from single crystals.36−38 For example, Eisenberg and co-workers reported the formation of cylindrical micelles using CDSA with a PCL core-forming block and a PEO corona.39 The cylindrical micelle formation was ascribed to a crystallinity-driven ripening process of spherical micelles in water, yielding micrometer long cylinders after 2 weeks. However, no control was shown over the cylinders’ growth, and samples aged 3 months or longer were observed to undergo further morphological changes into ribbon-like particles and lamellae. Fan and co-workers also demonstrated that PEO-b-PCL seeds can be elongated into longer fibers via two simultaneous growth regimes, addition of unimers or end-to-end coupling of preformed cylinders. In this example, however, conditions that are not ideal when applied in a biological environment, including H2O/DMF or DMSO assembly solvents, and extended time periods for micellar growth at 4 °C were required.40 Furthermore, PEO-b-PCL copolymers have not enabled full control over particle morphology (a mixture of spheres and cylinders are observed during the formation of short seeds) or precision controlled growth that could be predicted based on the polymer-to-seed ratio.40 An alternative approach of fragmentation and growth of cylinders has also been reported, where the use of small molecule hydrogen-bond donors induce fragmentation (by creating stress in the corona) or dynamic cross-linking of the corona to allow growth.42 However, this method is inherently limited by the demands of the coronal chemistry and provides only moderate length control. To date, only one example of CDSA in aqueous media has been reported using polymers with a poly(2-isopropyl-2-oxazoline) core.41 However, no control over growth has been achieved.
Herein, we present the precise formation of biocompatible PCL block copolymers assembled into cylindrical micelles with unprecedented control over morphology and dimensions in both alcoholic and, for the first time, aqueous media. We report direct epitaxial crystallization in water without the need for postmodification or solvent transfer steps, leading to the formation of strong, biocompatible hydrogel materials capable of >95% cell viability. Furthermore, in contrast to previous work, where changes in block length were necessary to induce transitions from 1D to 2D materials,17,18 we show that block copolymers of the same block lengths but different coronal chemistry can be used to determine different morphologies, including platelet-forming unimers with larger corona blocks.
Results and Discussion
Synthesis and Preparation of PCL Crystalline Seeds
PCL block copolymers were synthesized using a combination of ring-opening polymerization (ROP) and reversible addition–fragmentation chain transfer (RAFT) polymerization (Table S1). Synthesis can be carried out on a large scale with predictable molecular weights and narrow dispersities as determined by NMR spectroscopic and SEC analyses (Figures S1–S5). Polydisperse cylinders of several micrometers in length were prepared by spontaneous nucleation in ethanol, a selective solvent for the corona block,27 at a concentration of 5 mg/mL, when heated at 70 °C for 3 h and subsequently cooled down to room temperature (Figures 1 and S6). Self-assembly was monitored via transmission electron microscopy (TEM), and samples were aged for 5 days to reach well-defined structures. In order to achieve precise control over cylinder length, sonication of the aged micelles was carried out under controlled temperature (0 °C) using a sonication probe. Sonication kinetics revealed controlled fracture of the micelles according to a Gaussian scission model (Figure S7 and Table S2), where preferential fracture occurs toward the center of cylindrical micelles with no recombination of the fragments.43 Uniform crystalline seeds ca. 50 nm in length were obtained as observed by TEM and selected area electron diffraction (SAED) (Figures 1 and S8). Importantly, no side reactions were observed after the heating process in ethanol or after sonication of the crystalline micelles (Figure S9).
Figure 1.
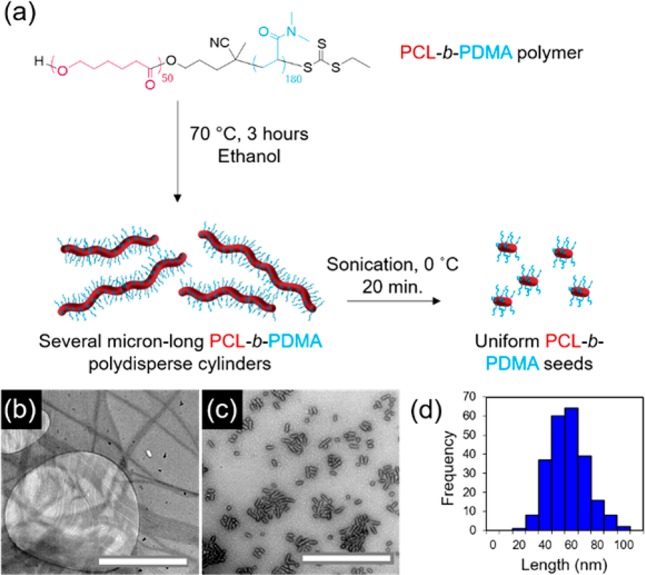
(a) Schematic of self-nucleation of PCL50-b-PDMA180 diblock copolymer followed by sonication of polydisperse cylinders to form uniform seed micelles, TEM micrographs of (b) polydisperse cylinders and (c) seed micelles, and (d) length distribution of seed micelles. Uranyl acetate (1%) was used as a negative stain. Scale bar = 1000 nm.
Epitaxial Growth of PCL Cylinders in an Alcoholic Solvent
A living CDSA process was observed, where the 50 nm crystalline seeds serve as initiation sites for micelle growth on addition of polymer unimers prepared by dissolving the PCL50-b-PDMA180 block copolymer in a miscible solvent, such as tetrahydrofuran (THF). Controlled linear epitaxial growth showed the formation of nearly monodisperse cylindrical micelles up to several micrometers long (Figure S10), where the micelle length was found to be proportional to the amount of unimer added (Figure 2) (Lw/Ln ≤ 1.1 where Ln = number-average length and Lw = weight-average length). In contrast, the addition of PCL50-b-PDMAEMA170 unimers in THF to previously prepared PCL50-b-PDMA180 seeds resulted in the formation of platelet micelles (Figures S11–S14). This can be explained using a unimer solubility approach, as reported previously,27 where more soluble unimers lead to a preference for the crystallization of plates as opposed to an initial aggregation step to form cylinders.
Figure 2.

(a) Schematic of epitaxial growth of PCL50-b-PDMA180 cylindrical micelles in ethanol from 50 nm seeds. TEM micrographs of cylindrical micelles epitaxially grown from seed micelles with a unimer/seed ratio of (b) 1, (c) 2, (d) 3, (e) 5, (f) 7, and (g) 9. Uranyl acetate (1%) was used as a negative stain. Scale bar = 1000 nm. (h) Length dispersity of cylindrical micelles. (i) Plot showing a linear epitaxial growth regime of cylinders with narrow length dispersities (error bars represent the standard deviation, σ, of the length distribution) in comparison to the theoretical length (dashed line).
Controlled Epitaxial Growth in Water
A key challenge in the field of CDSA that currently limits the translation of these methodologies more rapidly into biomedical studies is the inability to apply commonly studied biodegradable polymer-based micelles of controlled length in a biologically relevant solvent. Several methods were attempted to achieve this with our PCL50-b-PDMA180 system, including dialysis of the micelles against water (both directly into pure water and using an ethanol/water gradient, from 9:1 to 1:9), slow addition of water, or fast removal of organic solvent using N2 flow and resuspension in water. However, the structures disassembled in all attempts, leading to rapid polymer precipitation (Figure S15). We attributed this phenomenon to the swelling of the corona block when transferring into water, causing stress to the crystalline structure and subsequent fracture. As such, we concluded that protecting the PCL core with a short block of a glassy, highly hydrophobic polymer would be more efficient in preventing disassembly. Following this strategy, micrometer-long cylindrical micelles were prepared from PCL50-b-PMMA20-b-PDMA200 triblock copolymers (Figures S16–19) using the same methodology described above (ethanol at 70 °C for 3 h and subsequent cooling to room temperature). Similarly to the diblock copolymer system, cylindrical micelles of controlled length were isolated by sonication and subsequent epitaxial growth in ethanol (Figure S20). Notably, the addition of the glassy, hydrophobic midblock enabled the successful transfer of these micellar structures into water by dialysis. We confirmed our methodology by also successfully preparing cylinders of controlled length using an alternative PCL50-b-PS10-b-PDMA200 triblock copolymer (Figures S21–S24).
These polymers show the first example of water-stable precision nanostructures of controllable morphology and dimensions in water using a biocompatible and biodegradable crystalline domain. To simplify our methods further, as well as seek to obtain control directly in aqueous media for the first time, we investigated the self-nucleation, sonication, and epitaxial growth in water. Self-nucleation was carried out in water at room temperature by dissolution of the polymer in a small amount of THF (50 mg/mL), followed by evaporation of the organic solvent to obtain long, polydisperse cylinders (Figure S25). Sonication of the self-nucleated cylinders in water resulted in seeds in a much shorter time scale (ca. 5 min), while still producing a controlled seed length of ca. 40 nm, (Figures S26 and S27). Importantly, the crystalline structure typical of PCL45 was maintained in aqueous media, with the first strongest (110) diffraction at 2θ = 21.41° and the second strongest (200) diffraction at 2θ = 23.76° (Figure S27). Furthermore, the diameter of the cylinders remained constant (ca. 12 nm) for both the long and short micelles, as confirmed by cryogenic-TEM analysis (Figure S28), which is indicative of the controlled nature of this process. Controlled epitaxy was successfully achieved by adding different concentrations of unimers in acetone (a more volatile solvent than THF) to these seeds, followed by fast solvent evaporation to obtain stable structures in aqueous media (Figures 3, S29, and S30), achieving linear epitaxy with a micellar length that is predictable based on the unimer-to-seed ratio. In contrast to other CDSA-formed micelles, these cylinders represent the first example of directly preparing cylindrical micelles of controlled dimensions in water, thus paving the way for their utility in biological applications.
Figure 3.

(a) Schematic representation of epitaxial growth in water using PCL50-b-PMMA20-b-PDMA200 triblock copolymer. TEM micrographs of cylindrical micelles epitaxially grown from 40 nm seed micelles in water with a unimer/seed ratio of (b) 1, (c) 5, (d) 9, and (e) 15, using graphene oxide TEM grids.44 Scale bar = 1000 nm. (f) TEM micrograph (scale bar = 1000 nm) and (g) confocal microscopy image (scale bar = 20 μm) of fluorescently labeled cylindrical micelles epitaxially grown from seed micelles in water with a unimer/seed ratio of 15. Scale bar = 20 μm. (h) Length dispersity of cylindrical micelles. (i) Plot showing a linear epitaxial growth regime of cylinders with narrow length dispersities (error bars represent the standard deviation, σ, of the length distribution) in comparison to the theoretical length (dashed line).
With the success of the above approach to prepare precisely defined cylindrical micelles directly in water, we sought to demonstrate the biorelevance of our cylindrical micelles by investigating cell viability (using cylinders ca. 50 nm in length, as these represent an ideal size for drug delivery purposes).46 MC3T3 (murine preosteoblasts) and A549 (human lung cancer fibroblasts) were treated with increasing concentrations of polymer in water, from 0 to 5 mg/mL. Cell viability was found to be higher than 95% even with the highest concentration of polymer used (Figure S31), suggesting our PCL system is highly biocompatible and can therefore find potential applications as a drug delivery carrier.
Furthermore, labeling of such micellar constructs with functional handles such as fluorescent molecules or radiolabels provides a simple method by which to readily track and image the particles within biological systems. As such, we sought to demonstrate that our water-dispersed cylindrical micelles were easily labeled while retaining the ability to undergo controlled living epitaxial growth in aqueous media. To this end, we took advantage of the ease of postpolymerization modification of the trithiocarbonate RAFT group to attach BODIPY-FL-C5-maleimide by a simple aminolysis and subsequent click reaction (Figures 3, S32, and S33). As expected, the living growth characteristics were retained, demonstrating their utility for further study in this field.
Epitaxy in Water to Form Hydrogel Materials
In order to fully take advantage of the unprecedented control in dimensions achieved in water, we then exploited the triblock copolymer micelle system to create biologically relevant hydrogel materials. Using sequential unimer additions with dye-labeled unimers, a strong hydrogel was obtained upon fast evaporation of acetone, with an overall solid content of 15 wt % (Figure 4a). Interestingly, it was observed that a minimum length of 2 μm was necessary to obtain a hydrogel by living CDSA. Targeting shorter lengths induced a gradual increase in viscosity, until hydrogel formation occurred when enough unimers were added to target the 2 μm length. Oscillatory rheology confirmed the gel-like nature of the material with a storage modulus (G′) of 4 kPa (Figure 4f), around 10 times higher than previously reported worm gels with a similar solid content.47,48 On applied strain (300%), the loss modulus (G″) was measured as greater than G′, indicating that the gel structure had been destroyed; however, the G′ was recovered at rest (0.1% strain) as the gel reformed (Figure 4d,e). The pore structure of the hydrogel was confirmed by cryogenic-scanning electron microscopy (cryo-SEM) analysis (Figure 4c) and, as expected, long, uniform cylinders could be observed by TEM upon freeze-drying a small amount of sample on the grid (Figure 4b).
Figure 4.

(a) Schematic of hydrogel formation via direct epitaxial growth of PCL50-b-PMMA20-b-PDMA200 cylinders in water. (b) TEM micrograph of PCL50-b-PMMA20-b-PDMA200 hydrogel freeze-dried on a TEM grid. Scale bar = 1000 nm. (c) Cryo-SEM image of the cylinder hydrogel (scale bar = 2 μm) with photographs of the hydrogel using BODIPY-tagged (inset, left) and untagged cylinders (inset, right). (d) Step-strain measurements of cylinder hydrogel over three cycles (ω = 10 rad s–1) with (e) enlargement of the recovery of the material properties after each cycle. (f) Strain-dependent oscillatory rheology of the cylinder hydrogel at 293 K and a constant frequency of 10 rad s–1.
Surprisingly, the hydrogel did not swell and could not be redissolved upon addition of water despite the absence of any cross-linking or stabilization by chemical interactions. While the worm-gels as prepared were stable for extended time periods (at least 2 months) in aqueous media, the gel sheared into smaller fragments upon the addition of a larger amount of water, most likely as a consequence of the increasing osmotic pressure leading to fragmentation. This can be supported by TEM analysis of the residual water after fragmentation, where several shorter cylinders were observed (Figure S34).
Finally, the cell viability, using both previously mentioned cell lines, was assessed in 3D by preparing a hydrogel by living CDSA in cell-culture medium (MEM-α with addition of 10% fetal bovine serum and 1% penicillin/streptomycin). Cells were added by injection into the preformed gel and viability was assessed over 4 days. At each time point, high biocompatibility (>95%) was observed in the 3D matrix (Figure S35), opening the door to a new method for biorelevant hydrogel formation. Such systems, already loaded with the desired concentration of a therapeutic agent, would avoid the need for subsequent incorporation of potential drugs after gelation, increasing the control over dose and drug release.
In conclusion, we report the successful formation of crystalline poly(ε-caprolactone)-core 1D and 2D assemblies, achieving unprecedented control in morphology and dimension dispersities by direct epitaxial crystallization in aqueous media for the first time. This critical advance in the preparation of precision nanostructures is crucial to their translation into biological applications, providing a simplified method without the need for postpolymerization or postassembly modifications and solvent transfer steps. Furthermore, the ability to epitaxially grow a strong, biocompatible hydrogel from living CDSA of a biodegradable polymer and encapsulate living cells in its matrix demonstrates the versatility of this technique and opens vast avenues for future biorelevant applications.
Acknowledgments
Zachary P. L. Laker and Neil. R. Wilson are acknowledged for SAED measurements. Nicole Kelly is acknowledged for running WAXD of the samples. Wei Yu is acknowledged for running MALDI-ToF of PCL. Prof. Matthew Gibson is thanked for access to his cell lab facilities. The University of Warwick Advanced BioImaging Research Technology Platform, BBSRC ALERT14 award BB/M01228X/1, is thanked for confocal fluorescence microscopy analysis. The University of Warwick and EPSRC are thanked for the award of a Warwick Chancellors Scholarship (R.K.) and a DTP studentship (M.I. and L.J.M., respectively). ERC is acknowledged for support to M.C.A., A.P.D. (Grant Number 681559), G.C., and R.O.R. (Grant Number 615142).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b10199.
Experimental details and additional results (PDF)
Author Contributions
§ M.C.A. and M.I. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Epps T. H. III; O’Reilly R. K. Chem. Sci. 2016, 7, 1674–1689. 10.1039/C5SC03505H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Reilly R. K.; Hawker C. J.; Wooley K. L. Chem. Soc. Rev. 2006, 35, 1068–1083. 10.1039/b514858h. [DOI] [PubMed] [Google Scholar]
- Nishiyama N. Nat. Nanotechnol. 2007, 2, 203–204. 10.1038/nnano.2007.88. [DOI] [PubMed] [Google Scholar]
- Huang X.; Teng X.; Chen D.; Tang F.; He J. Biomaterials 2010, 31, 438–448. 10.1016/j.biomaterials.2009.09.060. [DOI] [PubMed] [Google Scholar]
- Gratton S. E. A.; Ropp P. A.; Pohlhaus P. D.; Luft J. C.; Madden V. J.; Napier M. E.; DeSimone J. M. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 11613–11618. 10.1073/pnas.0801763105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daum N.; Tscheka C.; Neumeyer A.; Schneider M. Wiley Interdiscip Rev. Nanomed Nanobiotechnol. 2012, 4, 52–65. 10.1002/wnan.165. [DOI] [PubMed] [Google Scholar]
- Caldorera-Moore M.; Guimard N.; Shi L.; Roy K. Expert Opin. Drug Delivery 2010, 7, 479–495. 10.1517/17425240903579971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X.; Wu F.; Tian Y.; Wu M.; Zhou Q.; Jiang S.; Niu Z. Sci. Rep. 2016, 6, 24567. 10.1038/srep24567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raeesi V.; Chou L. Y. T.; Chan W. C. W. Adv. Mater. 2016, 28, 8511–8518. 10.1002/adma.201600773. [DOI] [PubMed] [Google Scholar]
- Geng Y.; Dalhaimer P.; Cai S.; Tsai R.; Tewari M.; Minko T.; Discher D. E. Nat. Nanotechnol. 2007, 2, 249–255. 10.1038/nnano.2007.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crassous J. J.; Schurtenberger P.; Ballauff M.; Mihut A. M. Polymer 2015, 62, A1–A13. 10.1016/j.polymer.2015.02.030. [DOI] [Google Scholar]
- Patra S. K.; Ahmed R.; Whittell G. R.; Lunn D. J.; Dunphy E. L.; Winnik M. A.; Manners I. J. Am. Chem. Soc. 2011, 133, 8842–8845. 10.1021/ja202408w. [DOI] [PubMed] [Google Scholar]
- Schmelz J.; Schedl A. E.; Steinlein C.; Manners I.; Schmalz H. J. Am. Chem. Soc. 2012, 134, 14217–14225. 10.1021/ja306264d. [DOI] [PubMed] [Google Scholar]
- Wang X.; Guérin G.; Wang H.; Wang Y.; Manners I.; Winnik M. A. Science 2007, 317, 644–647. 10.1126/science.1141382. [DOI] [PubMed] [Google Scholar]
- Nazemi A.; Boott C. E.; Lunn D. J.; Gwyther J.; Hayward D. W.; Richardson R. M.; Winnik M. A.; Manners I. J. Am. Chem. Soc. 2016, 138, 4484–4493. 10.1021/jacs.5b13416. [DOI] [PubMed] [Google Scholar]
- Kim Y.-J.; Cho C.-H.; Paek K.; Jo M.; Park M.-k.; Lee N.-E.; Kim Y.-j.; Kim B. J.; Lee E. J. Am. Chem. Soc. 2014, 136, 2767–2774. 10.1021/ja410165f. [DOI] [PubMed] [Google Scholar]
- Mohd Yusoff S. F.; Hsiao M.-S.; Schacher F. H.; Winnik M. A.; Manners I. Macromolecules 2012, 45, 3883–3891. 10.1021/ma2027726. [DOI] [Google Scholar]
- Presa Soto A.; Gilroy J. B.; Winnik M. A.; Manners I. Angew. Chem., Int. Ed. 2010, 49, 8220–8223. 10.1002/anie.201003066. [DOI] [PubMed] [Google Scholar]
- Molev G.; Lu Y.; Kim K. S.; Majdalani I. C.; Guerin G.; Petrov S.; Walker G.; Manners I.; Winnik M. A. Macromolecules 2014, 47, 2604–2615. 10.1021/ma402441y. [DOI] [Google Scholar]
- Cao L.; Manners I.; Winnik M. A. Macromolecules 2002, 35, 8258–8260. 10.1021/ma021068x. [DOI] [Google Scholar]
- Hudson Z. M.; Boott C. E.; Robinson M. E.; Rupar P. A.; Winnik M. A.; Manners I. Nat. Chem. 2014, 6, 893–898. 10.1038/nchem.2038. [DOI] [PubMed] [Google Scholar]
- Du Z. X.; Xu J. T.; Fan Z. Q. Macromolecules 2007, 40, 7633–7637. 10.1021/ma070977p. [DOI] [Google Scholar]
- Du Z. X.; Xu J. T.; Fan Z. Q. Macromol. Rapid Commun. 2008, 29, 467–471. 10.1002/marc.200700795. [DOI] [Google Scholar]
- Su M.; Huang H.; Ma X.; Wang Q.; Su Z. Macromol. Rapid Commun. 2013, 34, 1067–1071. 10.1002/marc.201300218. [DOI] [PubMed] [Google Scholar]
- He W. N.; Xu J. T.; Du B. Y.; Fan Z. Q.; Wang X. Macromol. Chem. Phys. 2010, 211, 1909–1916. 10.1002/macp.201000184. [DOI] [Google Scholar]
- Glavas L.; Olsén P.; Odelius K.; Albertsson A.-C. Biomacromolecules 2013, 14, 4150–4156. 10.1021/bm401312j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inam M.; Cambridge G.; Pitto-Barry A.; Laker Z. P. L.; Wilson N. R.; Mathers R. T.; Dove A. P.; O’Reilly R. K. Chem. Sci. 2017, 8, 4223–4230. 10.1039/C7SC00641A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L.; Petzetakis N.; Pitto-Barry A.; Schiller T. L.; Kirby N.; Keddie D. J.; Boyd B. J.; O’Reilly R. K.; Dove A. P. Macromolecules 2013, 46, 9074–9082. 10.1021/ma401634s. [DOI] [Google Scholar]
- Pitto-Barry A.; Kirby N.; Dove A. P.; O’Reilly R. K. Polym. Chem. 2014, 5, 1427–1436. 10.1039/C3PY01048A. [DOI] [Google Scholar]
- Rizis G.; van de Ven T. G.; Eisenberg A. ACS Nano 2015, 9, 3627–3640. 10.1021/nn505068u. [DOI] [PubMed] [Google Scholar]
- Wang J.; Zhu W.; Peng B.; Chen Y. Polymer 2013, 54, 6760–6767. 10.1016/j.polymer.2013.10.027. [DOI] [Google Scholar]
- Zhu W.; Peng B.; Wang J.; Zhang K.; Liu L.; Chen Y. Macromol. Biosci. 2014, 14, 1764–1770. 10.1002/mabi.201400283. [DOI] [PubMed] [Google Scholar]
- Qi H.; Zhou T.; Mei S.; Chen X.; Li C. Y. ACS Macro Lett. 2016, 5, 651–655. 10.1021/acsmacrolett.6b00251. [DOI] [PubMed] [Google Scholar]
- He X.; He Y.; Hsiao M.-S.; Harniman R. L.; Pearce S.; Winnik M. A.; Manners I. J. Am. Chem. Soc. 2017, 139, 9221–9228. 10.1021/jacs.7b03172. [DOI] [PubMed] [Google Scholar]
- Fan B.; Wang R.-Y.; Wang X.-Y.; Xu J.-T.; Du B.-Y.; Fan Z.-Q. Macromolecules 2017, 50, 2006–2015. 10.1021/acs.macromol.7b00105. [DOI] [Google Scholar]
- Li B.; Wang B.; Ferrier R. C. Jr.; Li C. Y. Macromolecules 2009, 42, 9394–9399. 10.1021/ma902294k. [DOI] [Google Scholar]
- Chen W. Y.; Li C. Y.; Zheng J. X.; Huang P.; Zhu L.; Ge Q.; Quirk R. P.; Lotz B.; Deng L.; Wu C.; Thomas E. L.; Cheng S. Z. D. Macromolecules 2004, 37, 5292–5299. 10.1021/ma0493325. [DOI] [Google Scholar]
- Zheng J. X.; Xiong H.; Chen W. Y.; Lee K.; Van Horn R. M.; Quirk R. P.; Lotz B.; Thomas E. L.; Shi A.-C.; Cheng S. Z. D. Macromolecules 2006, 39, 641–650. 10.1021/ma052166w. [DOI] [Google Scholar]
- Rizis G.; van de Ven T. G. M.; Eisenberg A. Soft Matter 2014, 10, 2825–2835. 10.1039/c3sm52950a. [DOI] [PubMed] [Google Scholar]
- He W.-N.; Zhou B.; Xu J.-T.; Du B.-Y.; Fan Z.-Q. Macromolecules 2012, 45, 9768–9778. 10.1021/ma301267k. [DOI] [Google Scholar]
- Legros C.; De Pauw-Gillet M.-C.; Tam K. C.; Taton D.; Lecommandoux S. Soft Matter 2015, 11, 3354–3359. 10.1039/C5SM00313J. [DOI] [PubMed] [Google Scholar]
- Yang J.-X.; Fan B.; Li J.-H.; Xu J.-T.; Du B.-Y.; Fan Z.-Q. Macromolecules 2016, 49, 367–372. 10.1021/acs.macromol.5b02349. [DOI] [Google Scholar]
- Guérin G.; Wang H.; Manners I.; Winnik M. A. J. Am. Chem. Soc. 2008, 130, 14763–14771. 10.1021/ja805262v. [DOI] [PubMed] [Google Scholar]
- Patterson J. P.; Sanchez A. M.; Petzetakis N.; Smart T. P.; Epps T. H. III; Portman I.; Wilson N. R.; O’Reilly R. K. Soft Matter 2012, 8, 3322–3328. 10.1039/c2sm07040e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nojima S.; Ohguma Y.; Kadena K.-i.; Ishizone T.; Iwasaki Y.; Yamaguchi K. Macromolecules 2010, 43, 3916–3923. 10.1021/ma100236u. [DOI] [Google Scholar]
- Zhang S.; Li J.; Lykotrafitis G.; Bao G.; Suresh S. Adv. Mater. 2009, 21, 419–424. 10.1002/adma.200801393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren N. J.; Rosselgong J.; Madsen J.; Armes S. P. Biomacromolecules 2015, 16, 2514–2521. 10.1021/acs.biomac.5b00767. [DOI] [PubMed] [Google Scholar]
- Simon K. A.; Warren N. J.; Mosadegh B.; Mohammady M. R.; Whitesides G. M.; Armes S. P. Biomacromolecules 2015, 16, 3952–3958. 10.1021/acs.biomac.5b01266. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.