

Abstract
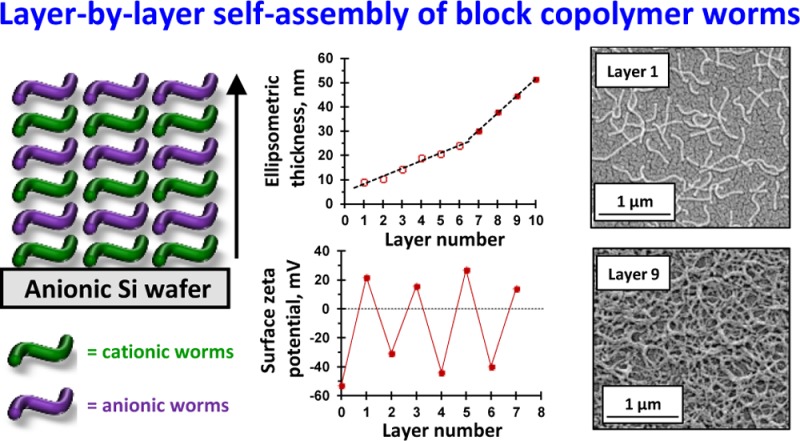
Cationic and anionic block copolymer worms are prepared by polymerization-induced self-assembly via reversible addition–fragmentation chain transfer (RAFT) aqueous dispersion copolymerization of 2-hydroxypropyl methacrylate and glycidyl methacrylate (GlyMA), using a binary mixture of a nonionic poly(ethylene oxide) macromolecular RAFT agent and either a cationic poly([2-(methacryloyloxy)ethyl]trimethylammonium chloride) or an anionic poly(potassium 3-sulfopropyl methacrylate) macromolecular RAFT agent. In each case, covalent stabilization of the worm cores was achieved via reaction of the epoxide groups on the GlyMA repeat units with 3-mercaptopropyltriethoxysilane. Aqueous electrophoresis studies indicated a pH-independent mean zeta potential of +40 mV and −39 mV for the cationic and anionic copolymer worms, respectively. These worms are expected to mimic the rigid rod behavior of water-soluble polyelectrolyte chains in the absence of added salt. The kinetics of adsorption of the cationic worms onto a planar anionic silicon wafer was examined at pH 5 and was found to be extremely fast at 1.0 w/w % copolymer concentration in the absence of added salt. Scanning electron microscopy (SEM) analysis indicated that a relatively constant worm surface coverage of 16% was achieved at 20 °C for adsorption times ranging from just 2 s up to 2 min. Furthermore, the successive layer-by-layer deposition of cationic and anionic copolymer worms onto planar surfaces was investigated using SEM, ellipsometry, and surface zeta potential measurements. These techniques confirmed that the deposition of oppositely charged worms resulted in a monotonic increase in the mean layer thickness, with a concomitant surface charge reversal occurring on addition of each new worm layer. Unexpectedly, two distinct linear regimes were observed when plotting the mean layer thickness against the total number of adsorbed worm layers, with a steeper gradient (corresponding to thicker layers) being observed after the deposition of six worm layers.
Introduction
Following seminal work by Decher,1−3 layer-by-layer (L-b-L) deposition of oppositely charged polyelectrolytes has become increasingly popular for the convenient preparation of functional multilayers at either planar surfaces or colloidal interfaces under exceptionally mild conditions (e.g., aqueous solution, neutral pH, and ambient temperature).4−8 In essence, the L-b-L technique simply involves alternately immersing the desired substrate into successive aqueous solutions of anionic and cationic polyelectrolytes with intermediate washing steps.9 According to Laschewsky and co-workers,5 adsorption of a polyelectrolyte onto an oppositely charged surface is driven by the gain in entropy that results from the release of small molecule counterions (e.g., Na+ or Cl–). Particularly strong adsorption is achieved in the absence of salt, which can otherwise screen the electrostatic interactions. Under such conditions, the adsorbed polyelectrolyte chains adopt a relatively flat conformation on the surface, and the adsorbed amount, Γ, is relatively low (typically Γ ≈ 0.1–0.5 mg m–2). A wide range of thin films comprising polyelectrolyte multilayers (PEMs) have been prepared on planar substrates,10−15 including antimicrobial surfaces.16 However, the design of PEMs is not just restricted to polyelectrolytes. In principle, any charged species can be incorporated into a PEM. For example, composite PEMs have been prepared using polyelectrolytes in combination with oppositely charged inorganic colloids17−20 or biologically active species such as enzymes,21 DNA,22−24 viruses,25,26 or proteins.27−29 Furthermore, the L-b-L protocol has been extended from surfaces to include various planar colloidal substrates30−34 and even human red blood cells.35,36 PEM-modified surfaces have been evaluated not only for biomedical applications9,37,38 but also for corrosion protection39 and for the preparation of electrically conductive films.40 Of particular interest are PEMs comprising block copolymer micelles41−43 and vesicles,32 which have significantly larger dimensions than soluble polyelectrolytes. The scientific literature also contains a few examples of composite PEMs comprising highly anisotropic particles such as cellulose nanocrystals,44−47 microfibrillated cellulose,48 or mixtures of cellulose nanocrystals and single-walled carbon nanotubes.49
It is relatively straightforward to prepare highly functional block copolymers as a result of recent developments in pseudo-living radical polymerization techniques such as atom transfer radical polymerization50,51 and reversible addition–fragmentation chain transfer (RAFT) polymerization.52,53 Furthermore, RAFT-mediated polymerization-induced self-assembly (PISA)54−56 offers a robust strategy for the rational design of highly anisotropic functional block copolymer worms at relatively high copolymer concentrations. Such vermicious particles are typically rather polydisperse in length (although Sumerlin and co-workers have recently claimed to achieve better control over this parameter57) but have relatively well-defined worm widths (≈20 to 40 nm). However, Semsarilar and co-workers reported that the preparation of highly charged worms directly in water can be problematic because a strong electrostatic repulsion between neighboring polyelectrolytic stabilizer chains usually limits the copolymer morphology to kinetically trapped spheres.58,59 Diluting such lateral electrostatic interactions by incorporating a suitable nonionic stabilizer60 can enable convenient access to either cationic or anionic diblock copolymer worms directly in the form of concentrated aqueous dispersions via aqueous PISA.58−62 In principle, this should enable investigation of the L-b-L adsorption of oppositely charged worms onto planar surfaces. It is well-known that polyelectrolyte chains behave as rigid rods in salt-free solutions.63,64 In principle, cross-linking the worm cores should increase their persistent length and rigidity.65 Hence, covalently stabilized block copolymer worms should serve as useful mimics for understanding individual polyelectrolyte chains in terms of their L-b-L behavior in the absence of salt. However, unlike molecularly dissolved polyelectrolytes, it should be possible to visualize each layer of adsorbed worms via electron microscopy. Herein, we investigate the successive L-b-L deposition of cross-linked cationic and anionic block copolymer worms onto planar surfaces using scanning electron microscopy (SEM), ellipsometry, and surface zeta potential measurements.
Results and Discussion
Synthesis of Macromolecular Chain Transfer Agents (CTAs)
The use of poly(ethylene oxide) (PEO) macro-CTAs as a stabilizer block for PISA syntheses has dramatically grown over the past few years.66−73 For example, we recently reported60,70 the preparation of a PEO113 macro-CTA via amidation. However, this synthetic route requires relatively long reaction times. In the present work, a PEO113 macro-CTA is instead prepared via esterification of a hydroxy-capped PEO methyl ether using 4-cyano-4-(2-phenylethanesulfanylthiocarbonyl)sulfanylpentanoic acid (PETTC) (see Scheme S1).67 This synthesis route reduces the total reaction time from approximately 2 weeks to just 3 days. Proton nuclear magnetic resonance (1H NMR) analysis indicated a mean degree of esterification of 95% by comparing the integrated signals associated with the aromatic end groups at 7.2–7.5 ppm with the PEO backbone signals at 3.3–4.6 ppm (see Figure S1a). Tetrahydrofuran (THF) gel permeation chromatography (GPC) analysis indicated an Mn of 5500 g mol–1 and an Mw/Mn of 1.05 against a series of near-monodisperse PEO calibration standards (see Figure S1b).
A poly(2-(methacryloyloxy)ethyl trimethylammonium chloride) (PQDMA) macro-CTA was synthesized by RAFT aqueous solution polymerization, using MPETTC74,75 as the RAFT agent (see Scheme S2). 1H NMR was used to calculate a mean degree of polymerization (DP) of 140, by comparing the integrated aromatic signals at 7.2–7.5 ppm against those assigned to the methacrylic backbone at 0.5–2.5 ppm (see Figure S2). Aqueous GPC studies indicated an Mn of 19 200 g mol–1 and an Mw/Mn of 1.26 (expressed relative to PEO calibration standards) (see Figure S2). Potassium 3-sulfopropyl methacrylate (KSPMA) was selected as the anionic monomer because Semsarilar et al.58 had previously reported the preparation of highly anionic PKSPMA-based block copolymer nanoparticles via RAFT aqueous dispersion polymerization of 2-hydroxypropyl methacrylate (HPMA). Accordingly, a PKSPMA macro-CTA was synthesized by RAFT solution polymerization in a 13:7 v/v methanol/water mixture, using PETTC as the RAFT agent (see Scheme S3). This solvent composition was selected to ensure full solubility of all reagents at both 20 and 70 °C. A mean DP of 150 was targeted using a [PETTC] / [ACVA] molar ratio of 5.0, where ACVA denotes 4,4′-azobis)4-cyanovaleric acid). 1H NMR studies confirmed that a KSPMA conversion of 69% was achieved after heating for 3 h at 70 °C. The purified PKSPMA macro-CTA had a mean DP of 111, as judged by comparing the integrated aromatic signals at 7.2–7.5 ppm to the oxymethylene proton signal at 4.0–4.2 ppm (Figure S3). Aqueous GPC studies at pH 9.8 indicated an Mn of 28 700 g mol–1 and an Mw/Mn of 1.15 (see Figure S3).
Unfortunately, there is no common GPC eluent that dissolves the constituent (co)polymers that make up the polyelectrolytic worms reported in this study. In view of this problem, experiments were performed to investigate the living character and blocking efficiency of the three macro-CTAs. Thus, the PEO113 macro-CTA was chain-extended with 300 units of HPMA via RAFT aqueous dispersion polymerization at 10 w/w % solids to yield diblock copolymer vesicles.70 The final HPMA conversion was determined to be more than 99% by 1H NMR spectroscopy. Importantly, THF GPC analysis indicated an Mn of 53 700 g mol–1, an Mw/Mn of 1.21, and a relatively high blocking efficiency for the PEO113 macro-CTA (see Figure S4a). The cationic PQDMA140 and anionic PKSPMA111 macro-CTAs were subjected to self-blocking experiments via RAFT aqueous solution polymerization of either QDMA or KSPMA, respectively, to yield well-defined PQDMA255 and PKSPMA335 homopolymers at 30 w/w % solids. In both cases, final monomer conversions exceeded 99% as judged by 1H NMR spectroscopy, and aqueous GPC analyses indicated high blocking efficiencies in each case (see Figures S4b and S4c).
Synthesis and Characterization of Core Cross-Linked Polyelectrolytic Worms
As previously described by Penfold et al.,60 core cross-linking of cationic block copolymer worms is essential for retention of the original worm morphology, following adsorption onto micrometer-sized silica spheres. Without such covalent stabilization, the strong torsional forces exerted on the worms by the colloidal silica particles are much greater than the weak hydrophobic forces holding the linear, amphiphilic copolymer chains together, thus resulting in worm dissociation. In contrast, this work is focused on the L-b-L deposition of polyelectrolytic worms onto planar silica surfaces. Nevertheless, core cross-linking was considered desirable to maximize the mean persistence length of the charged worms (cf. the “rigid rod” nature of polyelectrolytes in the absence of added salt)63,64 as well as to prevent potential loss of the worm morphology after adsorption. The polyelectrolytic block copolymer worms were prepared using protocols similar to that previously described:60 RAFT statistical copolymerization of HPMA and glycidyl methacrylate (GlyMA) was conducted using a binary mixture of a nonionic (PEO113) and polyelectrolytic (either PQDMA140 or PKSPMA111) macro-CTAs, as outlined in Scheme 1. In both cases, the core-forming block comprised 80 mol % HPMA and 20 mol % GlyMA.65 A series of exploratory experiments were conducted to identify the precise diblock copolymer compositions required to access well-defined cationic (0.90 PEO113 + 0.10 PQDMA140)-P(HPMA137-stat-GlyMA35) worms and anionic (0.90 PEO113 + 0.10 PKSPMA111)-P(HPMA168-stat-GlyMA39) worms. 3-Aminopropyltriethoxysilane (APTES) has been reported as a suitable cross-linking agent65 for preparing core cross-linked, cationic worms.60 However, the primary amine functionality of APTES is undesirable in this context; secondary amines are formed when this reagent reacts with epoxy groups, which could potentially reduce the negative surface charge on the anionic worms. In contrast, epoxy–thiol chemistry only produces neutral species and hence does not confer cationic character. Cross-linking of the worm cores is achieved by the ring-opening of the epoxy groups in the GlyMA residues using 3-mercaptopropyltriethoxysilane (MPTES). This epoxy–thiol reaction occurs with simultaneous hydrolysis/condensation of the pendent triethoxysilane groups, both with themselves and also with the secondary hydroxyl groups located on the neighboring HPMA residues (see Scheme S4).
Scheme 1. Schematic Representation of the Synthesis of either Cationic or Anionic Core Cross-Linked Block Copolymer Worms via RAFT Aqueous Copolymerization of HPMA and GlyMA Using a Binary Mixture of PEO113 macro-CTA with either a Cationic PQDMA140 Macro-CTA or an Anionic PKSPMA111 Macro-CTA.

Here, n represents the mole fraction of the polyelectrolytic macro-CTA. Core cross-linking is achieved by the post-polymerization addition of MPTES.
Prior to MPTES addition (using a [GlyMA]/[MPTES] molar ratio of 1.0), the worm gels were diluted to 5.0 w/w % (below the critical gelation concentration) using deionized water to aid MPTES dissolution. MPTES was then added, and the worm dispersions were stirred at 20 °C for 24 h. Transmission electron microscopy (TEM) analysis of the core cross-linked polyelectrolytic nanoparticles was performed to confirm that the original worm morphology was preserved (see Figure 1). ImageJ analysis of the core cross-linked cationic and anionic worm TEM images indicated a mean thickness of 27 ± 3 and 31 ± 5 nm, respectively.
Figure 1.

Representative TEM images obtained for (a) cationic (0.90 PEO113 + 0.10 PQDMA140)-P(HPMA137-stat-GlyMA35) and (b) anionic (0.90 PEO113 + 0.10 PKSPMA111)-P(HPMA168-stat-GlyMA39) worms after core cross-linking using MPTES. Images were obtained for 0.1 w/w % aqueous copolymer dispersions dried at pH 5.
The mean worm thickness was calculated from 50 measurements, comprising 5 width measurements equally spaced across the worm length for 10 worms. Aqueous electrophoresis studies were conducted on 0.1 w/w % aqueous dispersions of core cross-linked polyelectrolytic worms from pH 9.5 to pH 3 in the presence of 1 mM KCl (see Figure 2). As expected, the core cross-linked cationic worms exhibited zeta potentials of approximately +41 mV across this pH range.60 Similarly, the core cross-linked anionic worms exhibited a pH-independent mean zeta potential of approximately −39 mV.
Figure 2.
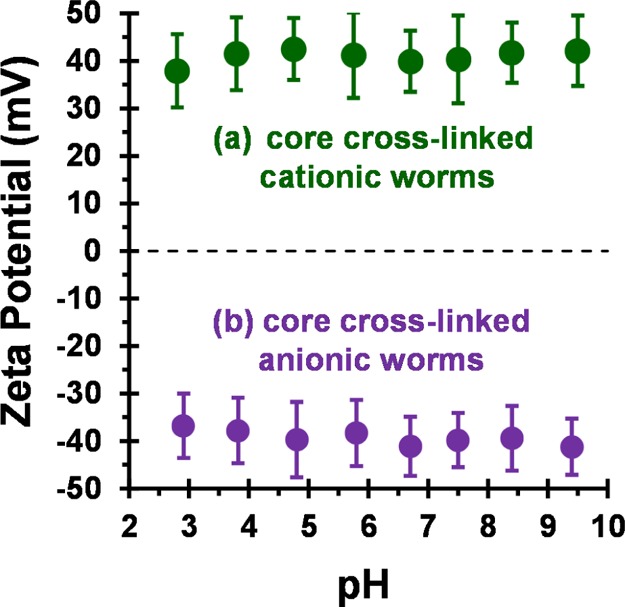
Zeta potential vs pH curves obtained for (a) cationic (0.90 PEO113 + 0.10 PQDMA140)-P(HPMA137-stat-GlyMA35) and (b) anionic (0.90 PEO113 + 0.10 PKSPMA111)-P(HPMA168-stat-GlyMA39) core cross-linked worms. Zeta potentials were determined at 20 °C for 0.1 w/w % aqueous copolymer dispersions in the presence of 1 mM KCl. The aqueous dispersion pH was adjusted using 0.1 M or 1 M HCl. Error bars represent 1 standard deviation.
Adsorption of Core Cross-Linked Cationic Worms onto Planar Silicon Wafers
In initial experiments, a clean silicon wafer was dipped into a dispersion of cationic cross-linked worms at concentrations of 0.1 or 0.5 w/w % for 10 min at pH 5. However, only rather low surface coverages (<5%) were indicated via ImageJ analysis of the corresponding SEM images. Thus, to ensure a relatively high surface coverage, the worm concentration was increased to 1.0 w/w % while the dispersion pH remained at 5. Thus, the kinetics of adsorption of cationic cross-linked worms onto anionic planar silicon wafers was investigated under these conditions. In these experiments, silicon wafers were dipped into the cationic worm dispersion for various time intervals, thoroughly washed with deionized water, and then dried under a stream of nitrogen gas. SEM images of the dried wafers were recorded to visualize the adsorbed cationic worms on the wafer surface. However, an interesting observation was made in these initial experiments: the worm coverage was not uniform across the whole wafer. A significantly higher surface coverage was frequently observed along the wafer edge (see see Figures S5a and S5b), which in principle might be a drying protocol artefact. However, similar observations were also made when drying the wafers in a 25 °C oven overnight (see see Figures S5c and S5d) without nitrogen drying. Thus, this phenomenon may be related to the so-called “coffee ring” effect often observed following evaporation of water from an aqueous dispersion of nanoparticles.76−78 Interestingly, Decher and co-workers reported very similar observations during the alternate adsorption of anionic and cationic polyelectrolytes onto planar silicon wafers.3 The kinetics of adsorption for cationic cross-linked worms onto an anionic silicon wafer was quantified using ImageJ software to analyze SEM images recorded at various time points (see Figure 3a). Only the central section of each wafer was analyzed, thus ignoring any edge effects. Ten separate SEM images were recorded from the central sections of the wafer for each time point at the same magnification; the total surface area analyzed was approximately 900 μm2 per time point.
Figure 3.
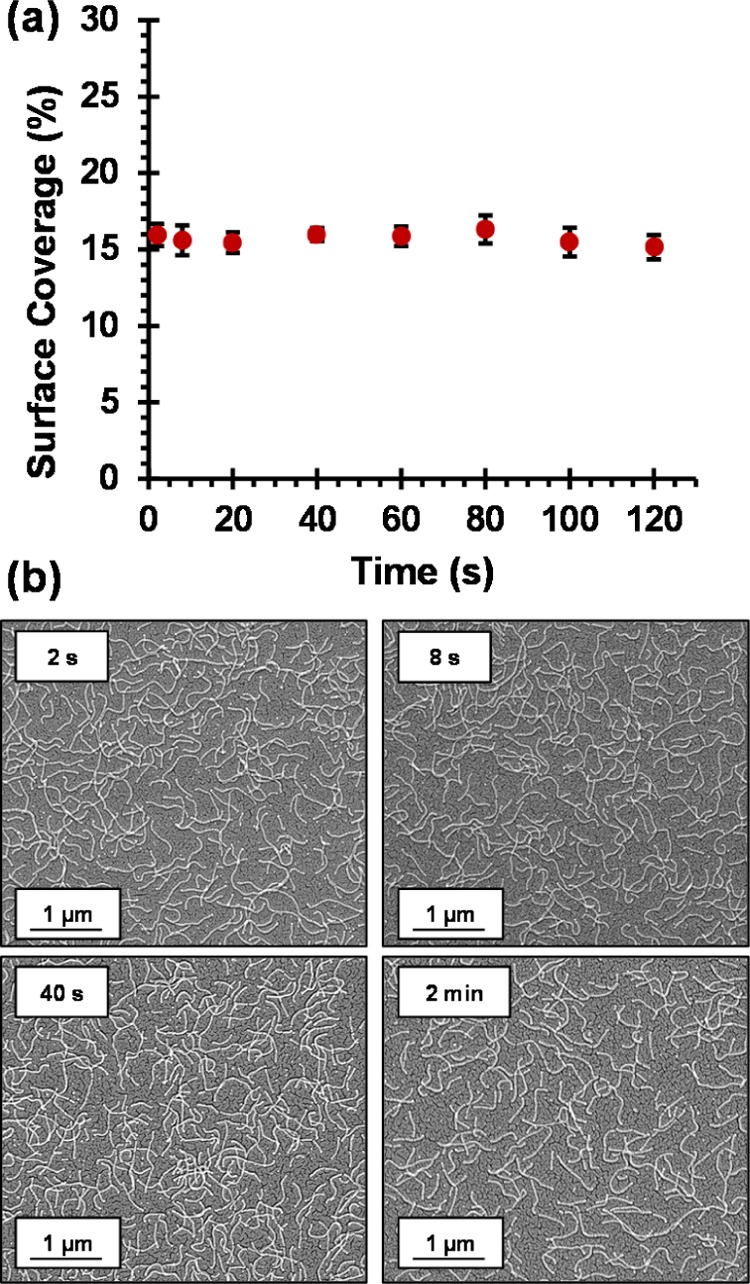
(a) Surface coverage vs adsorption time plot, indicating the remarkably rapid adsorption of cationic cross-linked worms onto the surface of a clean anionic silicon wafer. Surface coverages were determined using ImageJ software threshold analysis to analyze 10 separate areas for each silicon wafer per time point; the total surface area analyzed per time point is approximately 900 μm2. (b) Representative SEM images obtained after dipping an anionic planar silicon wafer into a 1.0 w/w % aqueous dispersion of (0.90 PEO113 + 0.10 PQDMA140)-P(HPMA137-stat-GlyMA35) cationic cross-linked worms at pH 5 for 2, 8, 20, and 40 s. Adsorption conditions: pH 5, no added salt, 1.0 w/w % worms, and 20 °C.
Representative SEM images obtained for wafers dipped into a 1.0 w/w % aqueous dispersion of (0.90 PEO113 + 0.10 PQDMA140)-P(HPMA137-stat-GlyMA35) cationic cross-linked worms at pH 5 for various time periods are shown in Figure 3b. As expected, these worms retained their morphology after adsorption onto anionic silicon wafers. The kinetics of electrostatic adsorption of these worms is remarkably fast, with a surface coverage of approximately 16% obtained within just 2 s under the stated conditions (1.0 w/w % copolymer worms at 20 °C). No further increase in the worm surface coverage occurred on extending the adsorption time up to 2 min or even 24 h (data not shown). An important control experiment was performed to demonstrate that the observed rapid worm adsorption was actually the result of electrostatic interactions, rather than merely gravitational sedimentation. A clean silicon wafer (manipulated using tweezers) was immersed into a 1.0 w/w % aqueous dispersion of cationic worms at pH 5 for either 20 or 60 s, with its anionic surface facing down. SEM analysis of the dried wafers indicated a near-identical surface coverage of approximately 16% for both time periods (see Figure S6).
Ellipsometry is an established technique for determining the mean thickness of thin films.79 It has been widely applied to characterize both polymer brushes80−83 and L-b-L systems.23,48,84 It is a model-dependent technique that assumes a uniform thickness for the adsorbed layer (slab model). This is not strictly the case for these adsorbed multilayers of oppositely charged worms, particularly at lower surface coverages. Nevertheless, ellipsometry is expected to provide complementary information to the SEM analysis and perhaps offers greater reliability for thicker worm layers, where determining the fractional surface coverage by digital image analysis becomes increasingly subjective. Furthermore, the projected ellipsometer beam dimensions on the wafer surface are 8 mm × 3 mm. Thus, the surface area analyzed by ellipsometry (24 mm2) is far greater than that analyzed by SEM (900 μm2). Ellipsometry parameters Ψ and Δ were collected from 370 to 1000 nm. First, analysis of a clean silicon wafer indicated a mean native oxide thickness of 1.97 nm. The mean square error (MSE) of this measurement was relatively low at 1.40, which validates the data fit for the experimental ψ and Δ values against the native oxide model within the CompleteEASE modeling software (MSE values of less than 2 indicate satisfactory fits to the model used).85 Second, the kinetics of cationic worm adsorption onto a clean silicon wafer (1.0 w/w %, pH 5, 20 °C, and no added salt) was monitored via ellipsometry to determine the dry worm layer thickness. The Cauchy model (see eq S1) uses three parameters (An, Bn and Cn) to describe the λ dependence of the refractive index (n) of an optically transparent material. An is a dimensionless parameter describing the refractive index of the material, materials; as λ tends to infinity, n(λ) tends to An. The constants Bn and Cn are parameters that characterize the nonlinear relationship between the refractive index and λ. Figure S7a shows the fitted Ψ and Δ data after adsorption of 1.0 w/w % core cross-linked cationic worms onto the anionic silicon surface for 2 min at pH 5 without added salt. In this case, the refractive index of the surface is not known, but this value must lie between 1.00 and 1.50 as the surface comprises an anionic silicon wafer, block copolymer cationic worms, and air voids within the adsorbed worm layer. Thus, all three Cauchy parameters were fitted to the data, enabling a mean worm layer thickness of 8.9 nm to be calculated. The excellent fit provided by the experimental Ψ and Δ data to the Cauchy model was validated by a low MSE of 1.29 when An = 1.257 (Figure S7b). The latter value is reasonable because the adsorbed worms form a nonuniform patchy layer, rather than a homogeneous thin film. Because these cationic worms exhibit a mean worm width of 27 ± 3 nm and a surface coverage of approximately 16% as judged by the ImageJ threshold analysis, an ellipsometric worm layer thickness of 8.9 nm seems to be physically realistic. Similar worm layer thicknesses were also determined by ellipsometry when anionic silicon wafers were dipped into the cationic worm dispersion for 2, 20, and 40 s, which confirms the remarkably fast kinetics of adsorption of these worms onto the silicon surface. Furthermore, when an inverted bare anionic silicon wafer was immersed into the copolymer worm dispersion, a similar mean worm layer thickness was observed. This suggests that purely electrostatic interaction, rather than gravitational sedimentation, is the primary driving force for worm adsorption. Thus, worm adsorption is essentially complete within a few seconds under the stated conditions. One reviewer of this manuscript has suggested that, if the cationic worms are strongly adsorbed at the air–water interface, then perhaps this could explain our unexpected observation of remarkably rapid cationic worm adsorption onto the anionic silicon wafer. This is an interesting idea that clearly warrants further studies.
L-B-L Deposition of Oppositely Charged Worms onto Planar Surfaces
Formation of polyelectrolytic worm multilayers was achieved by successive adsorption of oppositely charged worms onto a clean anionic silicon wafer using the L-b-L protocol (see Scheme 2). The adsorption conditions were fixed at an arbitrary time of 2 min, pH 5, no added salt, and an aqueous copolymer worm concentration of 1.0 w/w %. SEM, ellipsometry, and surface zeta potential studies were performed for each successive layer. The results obtained from each technique are discussed in turn below. Representative SEM images obtained for layers 1 to 9 are shown in Figure 4. Visual inspection of these images suggests an increase in surface coverage with layer number, which indicates the formation of polyelectrolytic worm multilayers. Threshold analysis using ImageJ software was performed to estimate the increase in surface coverage for each successive worm layer. Adsorption of cationic worms to form the first adsorbed layer only results in a surface coverage of approximately 16% (see Figure 3a).
Scheme 2. Schematic Representation of the L-B-L Protocol Used to Prepare Polyelectrolytic Worm Multilayers.

(a) The first worm layer is prepared by dipping a bare silicon wafer into a dispersion of cationic worms. (b) Second, the cationic worm-coated silicon wafer is dipped into a dispersion of anionic worms to prepare the second worm layer. (c) This protocol is then repeated to fabricate the desired number of polyelectrolytic worm layers by the successive electrostatic adsorption of oppositely charged worms onto the silicon wafer. The green and purple worms represent cationic and anionic worms, respectively. Rinsing steps are performed between the deposition of each worm layer, but these have been omitted from this scheme for clarity.
Figure 4.

Representative SEM images obtained for the gradual build-up of worm multilayers obtained via alternating L-b-L deposition of cationic and anionic cross-linked worms onto a planar anionic silicon wafer. Odd layer numbers correspond to the adsorption of cationic worms and even layer numbers correspond to the adsorption of anionic worms. Adsorption conditions: 1.0 w/w % aqueous worm dispersions, pH 5, 20 °C; time allowed for the adsorption of each worm layer was 2 min.
Formation of further four consecutive worm layers results in an approximate increase in surface coverage of 4% per layer (see Figure 5). Digital image analysis of the corresponding SEM images is straightforward for layers 1 to 5 because it is relatively easy to judge an appropriate threshold cutoff (see Figures S8a and S8b). Increasing the layer number results in a higher surface coverage, as expected. However, it becomes increasingly problematic to judge the appropriate threshold limit to apply when assessing the surface coverage. For example, the two threshold cutoff limits indicated in Figures S8c and S8d for layer 7 appear to be reasonable choices, even though the corresponding worm surface coverages differ significantly. Thus, there is greater experimental uncertainty in the surface coverage as the number of worm layers is increased and this quickly becomes unacceptably large.
Figure 5.
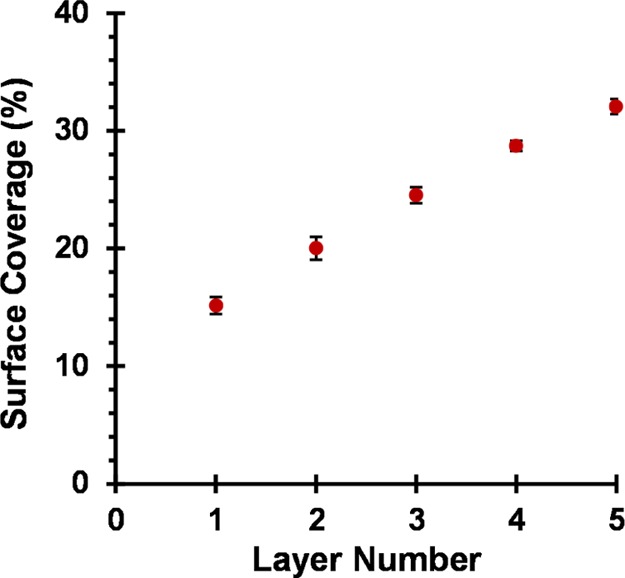
Relationship between the surface coverage of a planar silicon wafer and the layer number for the consecutive deposition of five layers of (0.90 PEO113 + 0.10 PQDMA140)-P(HPMA137-stat-GlyMA35) cationic cross-linked worms (layers 1, 3, and 5) and (0.90 PEO113 + 0.10 PKSPMA111)-P(HPMA168-stat-GlyMA39) anionic cross-linked worms (layers 2 and 4). Surface coverages were determined using the ImageJ software threshold analysis to analyze 10 separate areas per silicon wafer for each layer number; total surface area analyzed per layer number is approximately 900 μm2. Adsorption conditions used for each worm layer: pH 5, 20 °C, 1.0 w/w % worm dispersion, no added salt, and 2 min per adsorption event.
In view of this problem, ellipsometric measurements were also undertaken to assess the extent of worm adsorption. As described earlier, the Cauchy equation provides an appropriate model. The experimental Ψ and Δ data were fitted using the three Cauchy parameters (An, Bn, and Cn). The relationship between the dry layer thickness and the worm layer number is shown in Figure 6.
Figure 6.
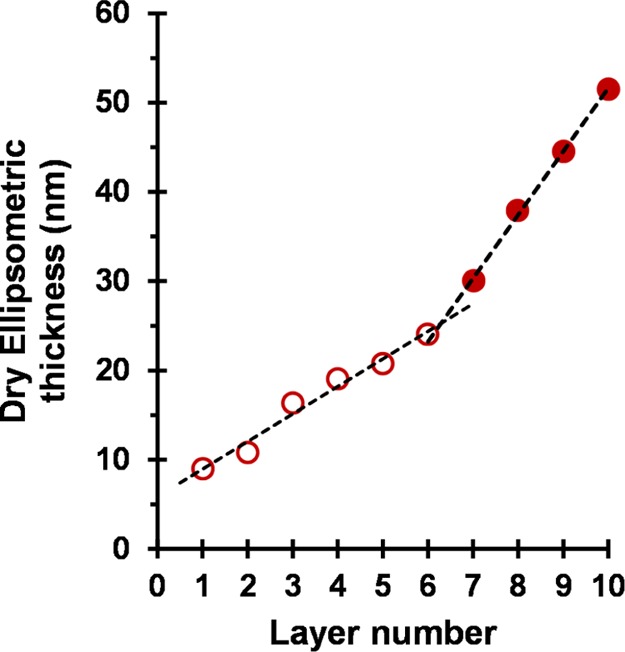
Relationship between the dry ellipsometric thickness of adsorbed worm layers and the layer number for the consecutive deposition of alternating 10 layers of (0.90 PEO113 + 0.10 PQDMA140)-P(HPMA137-stat-GlyMA35) cationic cross-linked worms (layers 1, 3, 5, 7, and 9) and (0.90 PEO113 + 0.10 PKSPMA111)-P(HPMA168-stat-GlyMA39) anionic cross-linked worms (layers 2, 4, 6, 8, and 10). Conditions used for each adsorbed worm layer: pH 5, 20 °C, 1.0 w/w % worms, no added salt, and 2 min per layer. The open circles represent layers 1 to 6 for which the mean increase in the dry film thickness per layer is 3 nm. A change in the gradient is observed for layers 7 to 10 (closed circles), for which the mean increase in the dry film thickness per layer is 7 nm.
According to the literature, the L-b-L deposition of strong polyelectrolyte chains onto a planar surface is typically characterized by a linear increase in the film thickness with the layer number.8,86 However, nonlinear (exponential) growth in the film thickness has been reported in some cases when using weak polyelectrolytes. This has been attributed to either film roughness effects and/or the “in-and-out” diffusion of at least one of the two polyelectrolytes throughout the film.8,87 For example, Yuan and Li prepared relatively thick nanoporous films via L-b-L assembly using poly(ethylene imine) (PEI) and poly(acrylic acid) (PAA).88 Importantly, basic conditions (pH 9.5) were employed for PEI adsorption, whereas acidic solutions (pH 2.9) were utilized for PAA adsorption. This approach reduced the charge density on these two weak polyelectrolytes. Interestingly, increasing the PEI/PAA charge density by adjusting the solution pH suppressed the exponential film growth. Furthermore, Podsiadlo and co-workers reported exponential growth for multilayer films composed of PEI, PAA, and montmorillonite clay particles.87
In the present work, nonlinear film growth is observed for the successive deposition of 10 layers of oppositely charged worms onto a planar silicon surface (see Figure 6). Two growth regimes are observed. For layers 1 to 6 (open circles), the mean increase in the dry film thickness per layer is 3 nm. A change in gradient is observed for layers 7 to 10 (closed circles), for which the mean increase in the dry film thickness per layer is 7 nm. A plausible explanation for this unexpected discontinuity is discussed later.
SEM images recorded for layers 1 to 5 (see Figure 4) confirm the progressive build-up of a relatively rough nanoporous film, with a comparable morphology to that reported by Yuan and Li.88 Presumably, the rigidity and much longer contour lengths of these cross-linked polyelectrolytic worms lead to greater surface roughness, which accounts for the nonlinear growth observed over the whole layer range. On the other hand, it seems rather unlikely that the “in-and-out” diffusion mechanism proposed to account for the enhanced adsorption of water-soluble weak polyelectrolytes is applicable to the present study.
Although higher surface coverages and thicker adsorbed layers are observed with increasing layer number, neither SEM nor ellipsometry can distinguish between the cationic and anionic worms adsorbed at the wafer surface. However, reversal of surface charge with increasing layer number would be expected for the successive adsorption of oppositely charged worms. In 2012, Corbett et al.89 reported a convenient method for determining the surface zeta potential. This new approach enables relatively fragile surfaces to be measured. Furthermore, the planar substrate is inverted relative to the aqueous solution. This means that the tracer particles cannot sediment onto the surface during the measurement, thus minimizing sample contamination. An Uzgiris90 dip cell was used in conjunction with a standard Malvern Zetasizer Nano instrument. To calculate the surface zeta potential, the motion of nonadsorbing tracer particles dispersed in aqueous electrolyte and subjected to an electric field is monitored via phase analysis light scattering.89 Thus, no modification of a conventional Malvern Zetasizer Nano ZS instrument is required.91 One parameter that requires careful consideration in such experiments is the selection of appropriate tracer particles. The sole role of the tracer particles is to scatter light: chemical functionality or surface chemistry does not affect their performance. However, it is essential that the tracer particles do not interact with the sample surface. Typical tracer particles include either sterically stabilized polystyrene latexes92 or food-grade milk substitute emulsions (coffee compliment).89 However, in both cases, such tracer particles possess a non-negligible surface charge at pH 5. This is clearly problematic for the present worm multilayer study because universal tracer particles that are suitable for both anionic and cationic surfaces are desired. Cationic tracer particles would be prone to electrostatic adsorption onto an anionic surface and vice versa. Thus, nonionic spherical nanoparticles exhibiting zero surface charge at pH 5 are required to ensure no interaction with either type of worm layer. Alswieleh and co-workers93 recently reported that sterically stabilized latexes prepared using a zwitterionic macromonomer can be used as tracer particles to determine the surface zeta potentials for poly(cysteine methacrylate) brushes grown from silicon wafers. However, such bespoke latexes require a four-step synthesis.94 On the other hand, the PISA synthesis of poly(glycerol monomethacrylate)–poly(benzyl methacrylate) (PGMA–PBzMA) nanoparticles reported by Cunningham and co-workers offers a relatively straightforward and convenient route to nonionic tracer nanoparticles.95 Thus, a PGMA58 macro-CTA prepared as previously reported75 was chain-extended with BzMA (target DP = 500) at 10 w/w % solids via RAFT aqueous emulsion polymerization (see Scheme S6).951H NMR spectroscopy studies confirmed a monomer conversion of 97% after 5 h at 50 °C (Figure S9a). GPC studies (dimethylformamide eluent) confirmed a high blocking efficiency for the PGMA58 macro-CTA and indicated an Mn of 66 600 g mol–1 and an Mw/Mn of 1.31 (using a series of near-monodisperse poly(methyl methacrylate) calibration standards) for the PGMA58–PBzMA500 copolymer chains (Figure S9b). Furthermore, TEM analysis of the dried dilute aqueous dispersion confirmed a well-defined spherical morphology (Figure S9c). Dynamic light scattering and aqueous electrophoresis studies were performed to examine the effect of varying the solution pH on both the intensity-average particle diameter and zeta potential (Figure S9d). As expected, these spherical nanoparticles exhibited pH-independent behavior: their intensity-average diameter (approximately 120 nm) and zeta potential (around 0 mV) remained essentially constant across a wide pH range.
Corbett et al.89,92 reported that a relatively low derived count rate of 250–500 kcps is required for surface zeta potential measurements. For the PGMA58–PBzMA500 nanoparticles, this corresponds to a concentration of 0.0025 w/w % when the Malvern Zetasizer Nano ZS attenuator is set to 100% light transmittance. All surface zeta potentials were determined at pH 5 using 1 mM KCl as the background electrolyte. Two control experiments were performed to demonstrate that these tracer particles were indeed suitable for surface zeta potential measurements. First, a clean anionic silicon wafer was analyzed at pH 5. The zeta potential versus displacement plot obtained for the tracer nanoparticles and the raw phase data are shown in Figure S10. Figure S10a displays the raw phase plots obtained for slow field reversal (SFR) measurements at four displacements (125, 250, 375, and 500 μm) and the fast field reversal (FFR) measurement made at 1000 μm. High signal-to-noise ratios were obtained in all cases, indicating the expected Doppler shift for the nonionic tracer nanoparticles. From these phase data, the tracer nanoparticle zeta potential was plotted against displacement (Figure S10b). The surface zeta potential for a clean bare anionic silicon wafer is calculated to be −53 ± 4 mV at pH 5 in the presence of 1 mM KCl, using eq S2. This value is comparable to the literature data obtained via streaming potential measurements under the same conditions.96
The same surface zeta potential studies were performed on a worm-coated silicon wafer (layer 1) after immersion of a clean bare anionic silicon wafer into a 1.0 w/w % aqueous dispersion of cationic cross-linked worms for 2 min at pH 5. Figure S11 depicts the raw phase plots obtained for SFR measurements at four displacements (125, 250, 375 and 500 μm) and also the FFR measurement made at 1000 μm for this cationic worm-coated wafer. In this case, the sign of the phase plot has changed, indicating surface charge reversal as the original bare anionic silicon wafer is converted into a cationic worm-coated silicon wafer. A surface zeta potential of +22 ± 1 mV is calculated from this phase data set. These experiments also confirm that the PGMA58–PBzMA500 spheres are appropriate tracer nanoparticles for both cationic and anionic substrates. Surface zeta potential measurements were performed on subsequent worm multilayer films (see Figure 7).
Figure 7.
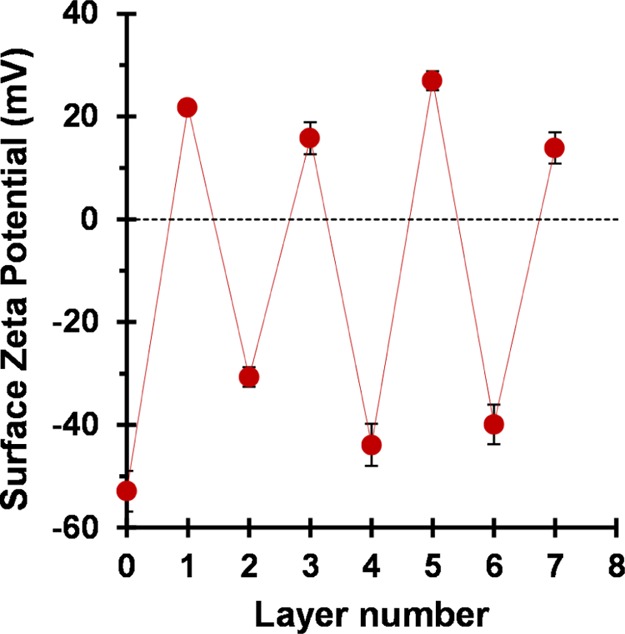
Variation in surface zeta potential with worm layer number for the sequential adsorption of cationic and anionic cross-linked worms onto a planar anionic silicon wafer. Odd layer numbers correspond to the adsorption of cationic worms, while even layer numbers correspond to the adsorption of anionic worms. All measurements were performed at 25 °C with a Malvern ZEN1020 Surface Zeta Potential Dip Cell using a 0.0025 w/w % aqueous dispersion of PGMA58–PBzMA500 nanoparticles as a nonadsorbing tracer to determine surface zeta potentials at pH 5 in the presence of 1 mM KCl.
As previously mentioned, the initial clean anionic silicon wafer (layer 0) exhibits a surface zeta potential of −53 ± 4 mV at pH 5. Surface charge reversal is observed after deposition of the first worm layer (layer 1) to give a surface zeta potential of +22 ± 1 mV. Adsorption of anionic worms (layer 2) onto this cationic worm layer resulted in surface charge reversal, giving a surface zeta potential of −30 ± 2 mV. The sequential adsorption of oppositely charged worms results in successive surface charge reversal, as expected.97 Thus, these surface zeta potential measurements confirm successful deposition of cationic and anionic worms onto an anionic planar silicon wafer and are consistent with the corresponding SEM and ellipsometric data.
In the light of the surface zeta potential data, it is worth reconsidering the ellipsometric data shown in Figure 6. For layer 1, a mean surface coverage of just 16% is sufficient to achieve surface charge reversal. Clearly, although the mean surface zeta potential is now cationic, a substantial proportion of the wafer remains uncoated, and such areas must possess local anionic charge. This means that, during the formation of layer 2, the anionic worms are less likely to adsorb on such bare patches owing to electrostatic repulsion. Such local “patchiness” leads to a relatively slow build-up of surface coverage and worm layer thickness up to layer 6. At this point, the mean thickness of the adsorbed worm layer determined by ellipsometry approximately corresponds to that expected for full monolayer coverage (because 24 nm is comparable to the mean worm cross-sectional diameter of 27 ± 3 nm estimated from TEM studies). Thereafter, the increase in the mean worm layer thickness per layer is significantly greater, presumably because there is no longer any unfavorable electrostatic repulsive interactions.
The anionic and cationic block copolymer worms employed in the present study have been deliberately prepared with covalently cross-linked cores to ensure that they remain intact during electrostatic deposition. In view of this rigidity, they are likely to be useful mimics for understanding the L-b-L behavior of soluble polyelectrolyte chains in the absence of added salt, which is known to adopt a “rigid rod” conformation.63,64 In principle, linear (i.e., non-cross-linked) worms could also be used for such L-b-L experiments. In this case, their greater flexibility should mean that they are more appropriate mimics for understanding the adsorption of polyelectrolytes in the presence of the added salt. This possibility warrants further studies.
Conclusions
RAFT-mediated PISA can be used to prepare cationic and anionic block copolymer worms via RAFT aqueous dispersion polymerization using a judicious binary mixture of a nonionic (PEO) and a polyelectrolytic (PQDMA or PKSPMA) stabilizer macro-CTA. Both types of worms contained reactive epoxy groups located within their core-forming blocks, which enabled their covalent stabilization on addition of MPTES. Kinetic studies indicated that the electrostatic adsorption of cationic worms from an aqueous solution onto a clean bare anionic planar silicon wafer was complete within just a few seconds at 20 °C, although the final surface coverage achieved for this first layer was only 16% as determined by ImageJ analysis. The successive L-b-L deposition of the cationic and anionic polyelectrolytic worms onto a planar anionic silicon wafer was investigated at pH 5. SEM analysis confirmed the gradual build-up of worm multilayers, but assessing the fractional surface coverage via digital image analysis became somewhat subjective after the first few worm layers. Surface zeta potential studies using bespoke nonionic tracer nanoparticles confirmed that surface charge reversal occurred on addition of each successive worm layer. Ellipsometric studies indicated that the worm layer thickness initially increased linearly with the layer number, as expected. However, a second adsorption regime corresponding to a significantly steeper linear gradient was observed after the sixth worm layer. According to the literature, this latter regime could be the result of a surface roughness effect for these relatively large rigid worms. However, this discontinuity occurs at a mean film thickness that corresponds to approximately the monolayer coverage of the silicon wafer. In view of the surface charge reversal observed for each successive worm layer, we attribute the relatively low surface coverages obtained for the first few worm layers to local electrostatic repulsive forces arising from bare anionic patches of the underlying silicon wafer for layers 2, 4, and 6 (or exposed cationic worms for layers 3 and 5). Once monolayer coverage is achieved, this effect is nullified and stronger adsorption per worm layer is observed. Finally, these worms are a useful mimic for understanding the adsorption behavior of soluble “rigid rod” polyelectrolytes because their much larger size facilitates direct visualization via electron microscopy.
Acknowledgments
EPSRC and Procter and Gamble (Brussels Technical Center, Belgium) are thanked for supporting a CASE PhD studentship for N.J.W.P. S.P.A. acknowledges an ERC Advanced Investigator grant (PISA 320372).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.langmuir.7b03571.
Full experimental and analytical details and supplementary schematics and figures (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Decher G.; Hong J.-D. Buildup of ultrathin multilayer films by a self-assembly process. 1 Consecutive adsorption of anionic and cationic bipolar amphiphiles on charged surfaces. Makromol. Chem., Macromol. Symp. 1991, 46, 321–327. 10.1002/masy.19910460145. [DOI] [Google Scholar]
- Decher G.; Hong J. D. Buildup of ultrathin multilayer films by a self-assembly process: II. Consecutive adsorption of anionic and cationic bipolar amphiphiles and polyelectrolytes on charged surfaces. Ber. Bunsen-Ges. 1991, 95, 1430–1434. 10.1002/bbpc.19910951122. [DOI] [Google Scholar]
- Decher G.; Hong J. D.; Schmitt J. Buildup of ultrathin multilayer films by a self-assembly process: III. Consecutively alternating adsorption of anionic and cationic polyelectrolytes on charged surfaces. Thin Solid Films 1992, 210–211, 831–835. 10.1016/0040-6090(92)90417-a. [DOI] [Google Scholar]
- Decher G. Fuzzy nanoassemblies: toward layered polymeric multicomposites. Science 1997, 277, 1232–1237. 10.1126/science.277.5330.1232. [DOI] [Google Scholar]
- Bertrand P.; Jonas A.; Laschewsky A.; Legras R. Ultrathin polymer coatings by complexation of polyelectrolytes at interfaces: suitable materials, structure and properties. Macromol. Rapid Commun. 2000, 21, 319–348. . [DOI] [Google Scholar]
- Dubas S. T.; Farhat T. R.; Schlenoff J. B. Multiple Membranes from “True” Polyelectrolyte Multilayers. J. Am. Chem. Soc. 2001, 123, 5368–5369. 10.1021/ja015774+. [DOI] [PubMed] [Google Scholar]
- Schlenoff J. B.; Dubas S. T.; Farhat T. Sprayed Polyelectrolyte Multilayers. Langmuir 2000, 16, 9968–9969. 10.1021/la001312i. [DOI] [Google Scholar]
- Borges J.; Mano J. F. Molecular Interactions Driving the Layer-by-Layer Assembly of Multilayers. Chem. Rev. 2014, 114, 8883–8942. 10.1021/cr400531v. [DOI] [PubMed] [Google Scholar]
- Hammond P. T. Form and function in multilayer assembly: New applications at the nanoscale. Adv. Mater. 2004, 16, 1271–1293. 10.1002/adma.200400760. [DOI] [Google Scholar]
- Shiratori S. S.; Rubner M. F. pH-Dependent Thickness Behavior of Sequentially Adsorbed Layers of Weak Polyelectrolytes. Macromolecules 2000, 33, 4213–4219. 10.1021/ma991645q. [DOI] [Google Scholar]
- Mendelsohn J. D.; Barrett C. J.; Chan V. V.; Pal A. J.; Mayes A. M.; Rubner M. F. Fabrication of Microporous Thin Films from Polyelectrolyte Multilayers. Langmuir 2000, 16, 5017–5023. 10.1021/la000075g. [DOI] [Google Scholar]
- Dubas S. T.; Schlenoff J. B. Factors Controlling the Growth of Polyelectrolyte Multilayers. Macromolecules 1999, 32, 8153–8160. 10.1021/ma981927a. [DOI] [Google Scholar]
- Dubas S. T.; Schlenoff J. B. Polyelectrolyte Multilayers Containing a Weak Polyacid: Construction and Deconstruction. Macromolecules 2001, 34, 3736–3740. 10.1021/ma001720t. [DOI] [Google Scholar]
- Schwarz B.; Schönhoff M. Surface Potential Driven Swelling of Polyelectrolyte Multilayers. Langmuir 2002, 18, 2964–2966. 10.1021/la015636y. [DOI] [Google Scholar]
- Glinel K.; Moussa A.; Jonas A. M.; Laschewsky A. Influence of Polyelectrolyte Charge Density on the Formation of Multilayers of Strong Polyelectrolytes at Low Ionic Strength. Langmuir 2002, 18, 1408–1412. 10.1021/la0113670. [DOI] [Google Scholar]
- Lichter J. A.; Van Vliet K. J.; Rubner M. F. Design of Antibacterial Surfaces and Interfaces: Polyelectrolyte Multilayers as a Multifunctional Platform. Macromolecules 2009, 42, 8573–8586. 10.1021/ma901356s. [DOI] [Google Scholar]
- Parakhonskiy B. V.; Yashchenok A. M.; Konrad M.; Skirtach A. G. Colloidal micro- and nano-particles as templates for polyelectrolyte multilayer capsules. Adv. Colloid Interface Sci. 2014, 207, 253–264. 10.1016/j.cis.2014.01.022. [DOI] [PubMed] [Google Scholar]
- Kotov N. A.; Dekany I.; Fendler J. H. Layer-by-Layer Self-Assembly of Polyelectrolyte-Semiconductor Nanoparticle Composite Films. J. Phys. Chem. 1995, 99, 13065–13069. 10.1021/j100035a005. [DOI] [Google Scholar]
- Lvov Y.; Ariga K.; Onda M.; Ichinose I.; Kunitake T. Alternate Assembly of Ordered Multilayers of SiO2 and Other Nanoparticles and Polyions. Langmuir 1997, 13, 6195–6203. 10.1021/la970517x. [DOI] [Google Scholar]
- Ariga K.; Lvov Y.; Onda M.; Ichinose I.; Kunitake T. Alternately assembled ultrathin film of silica nanoparticles and linear polycations. Chem. Lett. 1997, 26, 125–126. 10.1246/cl.1997.125. [DOI] [Google Scholar]
- Decher G.; Eckle M.; Schmitt J.; Struth B. Layer-by-layer assembled multicomposite films. Curr. Opin. Colloid Interface Sci. 1998, 3, 32–39. 10.1016/s1359-0294(98)80039-3. [DOI] [Google Scholar]
- Lvov Y.; Decher G.; Sukhorukov G. Assembly of thin films by means of successive deposition of alternate layers of DNA and poly(allylamine). Macromolecules 1993, 26, 5396–5399. 10.1021/ma00072a016. [DOI] [Google Scholar]
- Blacklock J.; Sievers T. K.; Handa H.; You Y.-Z.; Oupický D.; Mao G.; Möhwald H. Cross-Linked Bioreducible Layer-by-Layer Films for Increased Cell Adhesion and Transgene Expression. J. Phys. Chem. B 2010, 114, 5283–5291. 10.1021/jp100486h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren K.; Ji J.; Shen J. Construction and enzymatic degradation of multilayered poly-L-lysine/DNA films. Biomaterials 2006, 27, 1152–1159. 10.1016/j.biomaterials.2005.07.021. [DOI] [PubMed] [Google Scholar]
- Lvov Y.; Haas H.; Decher G.; Moehwald H.; Mikhailov A.; Mtchedlishvily B.; Morgunova E.; Vainshtein B. Successive Deposition of Alternate Layers of Polyelectrolytes and a Charged Virus. Langmuir 1994, 10, 4232–4236. 10.1021/la00023a052. [DOI] [Google Scholar]
- Yoo P. J.; Nam K. T.; Qi J.; Lee S.-K.; Park J.; Belcher A. M.; Hammond P. T. Spontaneous assembly of viruses on multilayered polymer surfaces. Nat. Mater. 2006, 5, 234–240. 10.1038/nmat1596. [DOI] [PubMed] [Google Scholar]
- Ladam G.; Gergely C.; Senger B.; Decher G.; Voegel J.-C.; Schaaf P.; Cuisinier F. J. G. Protein Interactions with Polyelectrolyte Multilayers: Interactions between Human Serum Albumin and Polystyrene Sulfonate/Polyallylamine Multilayers. Biomacromolecules 2000, 1, 674–687. 10.1021/bm005572q. [DOI] [PubMed] [Google Scholar]
- Caruso F.; Möhwald H. Protein Multilayer Formation on Colloids through a Stepwise Self-Assembly Technique. J. Am. Chem. Soc. 1999, 121, 6039–6046. 10.1021/ja990441m. [DOI] [Google Scholar]
- Lvov Y.; Ariga K.; Ichinose I.; Kunitake T. Assembly of Multicomponent Protein Films by Means of Electrostatic Layer-by-Layer Adsorption. J. Am. Chem. Soc. 1995, 117, 6117–6123. 10.1021/ja00127a026. [DOI] [Google Scholar]
- Sukhorukov G. B.; Donath E.; Lichtenfeld H.; Knippel E.; Knippel M.; Budde A.; Möhwald H. Layer-by-layer self assembly of polyelectrolytes on colloidal particles. Colloids Surf., A 1998, 137, 253–266. 10.1016/s0927-7757(98)00213-1. [DOI] [Google Scholar]
- Antipov A. A.; Sukhorukov G. B. Polyelectrolyte multilayer capsules as vehicles with tunable permeability. Adv. Colloid Interface Sci. 2004, 111, 49–61. 10.1016/j.cis.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Coustet M.; Irigoyen J.; Garcia T. A.; Murray R. A.; Romero G.; Cortizo M. S.; Knoll W.; Azzaroni O.; Moya S. E. Layer-by-layer assembly of polymersomes and polyelectrolytes on planar surfaces and microsized colloidal particles. J. Colloid Interface Sci. 2014, 421, 132–140. 10.1016/j.jcis.2014.01.038. [DOI] [PubMed] [Google Scholar]
- Rao J.; Zhang H.; Gaan S.; Salentinig S. Self-Assembly of Polystyrene-b-poly(2-vinylpyridine) Micelles: From Solutions to Silica Particles Surfaces. Macromolecules 2016, 49, 5978–5984. 10.1021/acs.macromol.6b01074. [DOI] [Google Scholar]
- Addison T.; Cayre O. J.; Biggs S.; Armes S. P.; York D. Incorporation of Block Copolymer Micelles into Multilayer Films for Use as Nanodelivery Systems. Langmuir 2008, 24, 13328–13333. 10.1021/la802396g. [DOI] [PubMed] [Google Scholar]
- Estrela-Lopis I.; Leporatti S.; Typlt E.; Clemens D.; Donath E. Small Angle Neutron Scattering Investigations (SANS) of Polyelectrolyte Multilayer Capsules Templated on Human Red Blood Cells. Langmuir 2007, 23, 7209–7215. 10.1021/la700496c. [DOI] [PubMed] [Google Scholar]
- Kreft O.; Georgieva R.; Bäumler H.; Steup M.; Müller-Röber B.; Sukhorukov G. B.; Möhwald H. Red Blood Cell Templated Polyelectrolyte Capsules: A Novel Vehicle for the Stable Encapsulation of DNA and Proteins. Macromol. Rapid Commun. 2006, 27, 435–440. 10.1002/marc.200500777. [DOI] [Google Scholar]
- Boudou T.; Crouzier T.; Ren K.; Blin G.; Picart C. Multiple Functionalities of Polyelectrolyte Multilayer Films: New Biomedical Applications. Adv. Mater. 2010, 22, 441–467. 10.1002/adma.200901327. [DOI] [PubMed] [Google Scholar]
- Skirtach A. G.; Yashchenok A. M.; Möhwald H. Encapsulation, release and applications of LbL polyelectrolyte multilayer capsules. Chem. Commun. 2011, 47, 12736–12746. 10.1039/c1cc13453a. [DOI] [PubMed] [Google Scholar]
- Andreeva D. V.; Skorb E. V.; Shchukin D. G. Layer-by-Layer Polyelectrolyte/Inhibitor Nanostructures for Metal Corrosion Protection. ACS Appl. Mater. Interfaces 2010, 2, 1954–1962. 10.1021/am1002712. [DOI] [Google Scholar]
- Li Y.; Chen S.; Wu M.; Sun J. Polyelectrolyte multilayers impart healability to highly electrically conductive films. Adv. Mater. 2012, 24, 4578–4582. 10.1002/adma.201201306. [DOI] [PubMed] [Google Scholar]
- Xu L.; Zhu Z.; Sukhishvili S. A. Polyelectrolyte Multilayers of Diblock Copolymer Micelles with Temperature-Responsive Cores. Langmuir 2011, 27, 409–415. 10.1021/la1038014. [DOI] [PubMed] [Google Scholar]
- Kim B.-S.; Park S. W.; Hammond P. T. Hydrogen-Bonding Layer-by-Layer-Assembled Biodegradable Polymeric Micelles as Drug Delivery Vehicles from Surfaces. ACS Nano 2008, 2, 386–392. 10.1021/nn700408z. [DOI] [PubMed] [Google Scholar]
- Nguyen P. M.; Zacharia N. S.; Verploegen E.; Hammond P. T. Extended Release Antibacterial Layer-by-Layer Films Incorporating Linear-Dendritic Block Copolymer Micelles. Chem. Mater. 2007, 19, 5524–5530. 10.1021/cm070981f. [DOI] [Google Scholar]
- Moreau C.; Beury N.; Delorme N.; Cathala B. Tuning the Architecture of Cellulose Nanocrystal–Poly(allylamine hydrochloride) Multilayered Thin Films: Influence of Dipping Parameters. Langmuir 2012, 28, 10425–10436. 10.1021/la301293r. [DOI] [PubMed] [Google Scholar]
- Cranston E. D.; Gray D. G. Birefringence in spin-coated films containing cellulose nanocrystals. Colloids Surf., A 2008, 325, 44–51. 10.1016/j.colsurfa.2008.04.042. [DOI] [Google Scholar]
- Cranston E. D.; Gray D. G.; Rutland M. W. Direct Surface Force Measurements of Polyelectrolyte Multilayer Films Containing Nanocrystalline Cellulose. Langmuir 2010, 26, 17190–17197. 10.1021/la1030729. [DOI] [PubMed] [Google Scholar]
- Cranston E. D.; Gray D. G. Morphological and optical characterization of polyelectrolyte multilayers incorporating nanocrystalline cellulose. Biomacromolecules 2006, 7, 2522–2530. 10.1021/bm0602886. [DOI] [PubMed] [Google Scholar]
- Wågberg L.; Decher G.; Norgren M.; Lindström T.; Ankerfors M.; Axnäs K. The build-up of polyelectrolyte multilayers of microfibrillated cellulose and cationic polyelectrolytes. Langmuir 2008, 24, 784–795. 10.1021/la702481v. [DOI] [PubMed] [Google Scholar]
- Olivier C.; Moreau C.; Bertoncini P.; Bizot H.; Chauvet O.; Cathala B. Cellulose Nanocrystal-Assisted Dispersion of Luminescent Single-Walled Carbon Nanotubes for Layer-by-Layer Assembled Hybrid Thin Films. Langmuir 2012, 28, 12463–12471. 10.1021/la302077a. [DOI] [PubMed] [Google Scholar]
- Matyjaszewski K. Atom Transfer Radical Polymerization (ATRP): Current Status and Future Perspectives. Macromolecules 2012, 45, 4015–4039. 10.1021/ma3001719. [DOI] [Google Scholar]
- Matyjaszewski K.; Xia J. Atom Transfer Radical Polymerization. Chem. Rev. 2001, 101, 2921–2990. 10.1021/cr940534g. [DOI] [PubMed] [Google Scholar]
- Moad G. RAFT polymerization to form stimuli-responsive polymers. Polym. Chem. 2017, 8, 177–219. 10.1039/c6py01849a. [DOI] [Google Scholar]
- Moad G.; Rizzardo E.; Thang S. H. Living Radical Polymerization by the RAFT Process? A Third Update. Aust. J. Chem. 2012, 65, 985–1076. 10.1071/ch12295. [DOI] [Google Scholar]
- Warren N. J.; Armes S. P. Polymerization-induced self-assembly of block copolymer nano-objects via RAFT aqueous dispersion polymerization. J. Am. Chem. Soc. 2014, 136, 10174–10185. 10.1021/ja502843f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canning S. L.; Smith G. N.; Armes S. P. A Critical Appraisal of RAFT-Mediated Polymerization-Induced Self-Assembly. Macromolecules 2016, 49, 1985–2001. 10.1021/acs.macromol.5b02602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derry M. J.; Fielding L. A.; Armes S. P. Polymerization-induced self-assembly of block copolymer nanoparticles via RAFT non-aqueous dispersion polymerization. Prog. Polym. Sci. 2016, 52, 1–18. 10.1016/j.progpolymsci.2015.10.002. [DOI] [Google Scholar]
- Figg C. A.; Carmean R. N.; Bentz K. C.; Mukherjee S.; Savin D. A.; Sumerlin B. S. Tuning Hydrophobicity To Program Block Copolymer Assemblies from the Inside Out. Macromolecules 2017, 50, 935–943. 10.1021/acs.macromol.6b02754. [DOI] [Google Scholar]
- Semsarilar M.; Ladmiral V.; Blanazs A.; Armes S. P. Anionic polyelectrolyte-stabilized nanoparticles via RAFT aqueous dispersion polymerization. Langmuir 2012, 28, 914–922. 10.1021/la203991y. [DOI] [PubMed] [Google Scholar]
- Semsarilar M.; Ladmiral V.; Blanazs A.; Armes S. P. Cationic polyelectrolyte-stabilized nanoparticles via RAFT aqueous dispersion polymerization. Langmuir 2013, 29, 7416–7424. 10.1021/la304279y. [DOI] [PubMed] [Google Scholar]
- Penfold N. J. W.; Ning Y.; Verstraete P.; Smets J.; Armes S. P. Cross-Linked Cationic Diblock Copolymer Worms are Superflocculants for Micrometer-sized Silica Particles. Chem. Sci. 2016, 7, 6894–6904. 10.1039/c6sc03732a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams M.; Penfold N. J. W.; Lovett J. R.; Warren N. J.; Douglas C. W. I.; Doroshenko N.; Verstraete P.; Smets J.; Armes S. P. Bespoke cationic nano-objects via RAFT aqueous dispersion polymerisation. Polym. Chem. 2016, 7, 3864–3873. 10.1039/c6py00696e. [DOI] [Google Scholar]
- Williams M.; Penfold N. J. W.; Armes S. P. Cationic and reactive primary amine-stabilised nanoparticles via RAFT aqueous dispersion polymerisation. Polym. Chem. 2016, 7, 384–393. 10.1039/c5py01577d. [DOI] [Google Scholar]
- Holm C.; Rehahn M.; Oppermann W.; Ballauff M. Stiff-chain polyelectrolytes. Adv. Polym. Sci. 2005, 166, 1–27. 10.1007/b11347. [DOI] [Google Scholar]
- Stevens M. J.; Kremer K. The nature of flexible linear polyelectrolytes in salt free solution: a molecular dynamics study. J. Chem. Phys. 1995, 103, 1669–1690. 10.1063/1.470698. [DOI] [Google Scholar]
- Lovett J. R.; Ratcliffe L. P. D.; Warren N. J.; Armes S. P.; Smallridge M. J.; Cracknell R. B.; Saunders B. R. A robust cross-linking strategy for block copolymer worms prepared via polymerization-induced self-assembly. Macromolecules 2016, 49, 2928–2941. 10.1021/acs.macromol.6b00422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackman L. D.; Doncom K. E. B.; Gibson M. I.; O’Reilly R. K. Comparison of photo- and thermally initiated polymerization-induced self-assembly: a lack of end group fidelity drives the formation of higher order morphologies. Polym. Chem. 2017, 8, 2860–2871. 10.1039/c7py00407a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan J.; Sun H.; Yu M.; Sumerlin B. S.; Zhang L. Photo-PISA: Shedding Light on Polymerization-Induced Self-Assembly. ACS Macro Lett. 2015, 4, 1249–1253. 10.1021/acsmacrolett.5b00748. [DOI] [PubMed] [Google Scholar]
- Tan J.; Bai Y.; Zhang X.; Zhang L. Room temperature synthesis of poly(poly(ethylene glycol) methyl ether methacrylate)-based diblock copolymer nano-objects via Photoinitiated Polymerization-Induced Self-Assembly (Photo-PISA). Polym. Chem. 2016, 7, 2372–2380. 10.1039/c6py00022c. [DOI] [Google Scholar]
- Yeow J.; Xu J.; Boyer C. Polymerization-Induced Self-Assembly Using Visible Light Mediated Photoinduced Electron Transfer-Reversible Addition–Fragmentation Chain Transfer Polymerization. ACS Macro Lett. 2015, 4, 984–990. 10.1021/acsmacrolett.5b00523. [DOI] [PubMed] [Google Scholar]
- Warren N. J.; Mykhaylyk O. O.; Mahmood D.; Ryan A. J.; Armes S. P. RAFT aqueous dispersion polymerization yields poly(ethylene glycol)-based diblock copolymer nano-objects with predictable single phase morphologies. J. Am. Chem. Soc. 2014, 136, 1023–1033. 10.1021/ja410593n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren K.; Perez-Mercader J. Thermoresponsive Gels Directly Obtained via Visible Light-Mediated Polymerization-Induced Self-Assembly with Oxygen Tolerance. Polym. Chem. 2017, 8, 3548–3552. 10.1039/c7py00558j. [DOI] [Google Scholar]
- Yuan B.; He X.; Qu Y.; Gao C.; Eiser E.; Zhang W. In situ synthesis of a self-assembled AB/B blend of poly(ethylene glycol)-b-polystyrene/polystyrene by dispersion RAFT polymerization. Polym. Chem. 2017, 8, 2173–2181. 10.1039/c7py00339k. [DOI] [Google Scholar]
- Rieger J.; Stoffelbach F.; Bui C.; Alaimo D.; Jérôme C.; Charleux B. Amphiphilic Poly(ethylene oxide) Macromolecular RAFT Agent as a Stabilizer and Control Agent in ab Initio Batch Emulsion Polymerization. Macromolecules 2008, 41, 4065–4068. 10.1021/ma800544v. [DOI] [Google Scholar]
- Penfold N. J. W.; Lovett J. R.; Verstraete P.; Smets J.; Armes S. P. Stimulus-responsive non-ionic diblock copolymers: protonation of a tertiary amine end-group induces vesicle-to-worm or vesicle-to-sphere transitions. Polym. Chem. 2017, 8, 272–282. 10.1039/c6py01076h. [DOI] [Google Scholar]
- Penfold N. J. W.; Lovett J. R.; Warren N. J.; Verstraete P.; Smets J.; Armes S. P. pH-responsive non-ionic diblock copolymers: protonation of a morpholine end-group induces an order–order transition. Polym. Chem. 2016, 7, 79–88. 10.1039/c5py01510c. [DOI] [Google Scholar]
- Deegan R. D.; Bakajin O.; Dupont T. F.; Huber G.; Nagel S. R.; Witten T. A. Capillary flow as the cause of ring stains from dried liquid drops. Nature 1997, 389, 827–829. 10.1038/39827. [DOI] [Google Scholar]
- Shen X.; Ho C.-M.; Wong T.-S. Minimal Size of Coffee Ring Structure. J. Phys. Chem. B 2010, 114, 5269–5274. 10.1021/jp912190v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigioni T. P.; Lin X.-M.; Nguyen T. T.; Corwin E. I.; Witten T. A.; Jaeger H. M. Kinetically driven self assembly of highly ordered nanoparticle monolayers. Nat. Mater. 2006, 5, 265–270. 10.1038/nmat1611. [DOI] [PubMed] [Google Scholar]
- Keddie J. L. Structural analysis of organic interfacial layers by ellipsometry. Curr. Opin. Colloid Interface Sci. 2001, 6, 102–110. 10.1016/s1359-0294(01)00070-x. [DOI] [Google Scholar]
- Rastogi A.; Paik M. Y.; Tanaka M.; Ober C. K. Direct Patterning of Intrinsically Electron Beam Sensitive Polymer Brushes. ACS Nano 2010, 4, 771–780. 10.1021/nn901344u. [DOI] [PubMed] [Google Scholar]
- Edmondson S.; Vo C.-D.; Armes S. P.; Unali G.-F. Surface Polymerization from Planar Surfaces by Atom Transfer Radical Polymerization Using Polyelectrolytic Macroinitiators. Macromolecules 2007, 40, 5271–5278. 10.1021/ma070876r. [DOI] [Google Scholar]
- Granville A. M.; Boyes S. G.; Akgun B.; Foster M. D.; Brittain W. J. Synthesis and Characterization of Stimuli-Responsive Semifluorinated Polymer Brushes Prepared by Atom Transfer Radical Polymerization. Macromolecules 2004, 37, 2790–2796. 10.1021/ma035915n. [DOI] [Google Scholar]
- Weir M. P.; Heriot S. Y.; Martin S. J.; Parnell A. J.; Holt S. A.; Webster J. R. P.; Jones R. A. L. Voltage-Induced Swelling and Deswelling of Weak Polybase Brushes. Langmuir 2011, 27, 11000–11007. 10.1021/la201343w. [DOI] [PubMed] [Google Scholar]
- Edmondson S.; Vo C.-D.; Armes S. P.; Unali G.-F.; Weir M. P. Layer-by-Layer Deposition of Polyelectrolyte Macroinitiators for Enhanced Initiator Density in Surface-Initiated ATRP. Langmuir 2008, 24, 7208–7215. 10.1021/la7039898. [DOI] [PubMed] [Google Scholar]
- Edmondson S.; Nguyen N. T.; Lewis A. L.; Armes S. P. Co-Nonsolvency Effects for Surface-Initiated Poly(2-(methacryloyloxy)ethyl phosphorylcholine) Brushes in Alcohol/Water Mixtures. Langmuir 2010, 26, 7216–7226. 10.1021/la904346j. [DOI] [PubMed] [Google Scholar]
- Decher G.; Schmitt J. Fine-tuning of the film thickness of ultrathin multilayer films composed of consecutively alternating layers of anionic and cationic polyelectrolytes. Prog. Colloid Polym. Sci. 1992, 89, 160–164. 10.1007/bfb0116302. [DOI] [Google Scholar]
- Podsiadlo P.; Michel M.; Lee J.; Verploegen E.; Kam N. W. S.; Ball V.; Lee J.; Qi Y.; Hart A. J.; Hammond P. T.; Kotov N. A. Exponential Growth of LBL Films with Incorporated Inorganic Sheets. Nano Lett. 2008, 8, 1762–1770. 10.1021/nl8011648. [DOI] [PubMed] [Google Scholar]
- Yuan W.; Li C. M. Exponentially growing layer-by-layer assembly to fabricate pH-responsive hierarchical nanoporous polymeric film and its superior controlled release performance. Chem. Commun. 2010, 46, 9161–9163. 10.1039/c0cc03267k. [DOI] [PubMed] [Google Scholar]
- Corbett J. C. W.; McNeil-Watson F.; Jack R. O.; Howarth M. Measuring surface zeta potential using phase analysis light scattering in a simple dip cell arrangement. Colloids Surf., A 2012, 396, 169–176. 10.1016/j.colsurfa.2011.12.065. [DOI] [Google Scholar]
- Uzgiris E. E. Laser doppler spectroscopy: Applications to cell and particle electrophoresis. Adv. Colloid Interface Sci. 1981, 14, 75–171. 10.1016/0001-8686(81)85015-4. [DOI] [Google Scholar]
- Tucker I. M.; Corbett J. C. W.; Fatkin J.; Jack R. O.; Kaszuba M.; MacCreath B.; McNeil-Watson F. Laser Doppler Electrophoresis applied to colloids and surfaces. Curr. Opin. Colloid Interface Sci. 2015, 20, 215–226. 10.1016/j.cocis.2015.07.001. [DOI] [Google Scholar]
- Thomas T. E.; Aani S. A.; Oatley-Radcliffe D. L.; Williams P. M.; Hilal N. Laser Doppler Electrophoresis and electro-osmotic flow mapping: A novel methodology for the determination of membrane surface zeta potential. J. Membr. Sci. 2017, 523, 524–532. 10.1016/j.memsci.2016.10.029. [DOI] [Google Scholar]
- Alswieleh A. M.; Cheng N.; Canton I.; Ustbas B.; Xue X.; Ladmiral V.; Xia S.; Ducker R. E.; El Zubir O.; Cartron M. L.; Hunter C. N.; Leggett G. J.; Armes S. P. Zwitterionic Poly(amino acid methacrylate) Brushes. J. Am. Chem. Soc. 2014, 136, 9404–9413. 10.1021/ja503400r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson K. L.; Bannister I.; Armes S. P.; Lewis A. L. Preparation of Biocompatible Sterically Stabilized Latexes Using Well-Defined Poly(2-(methacryloyloxy)ethyl phosphorylcholine) Macromonomers. Langmuir 2010, 26, 4693–4702. 10.1021/la903567q. [DOI] [PubMed] [Google Scholar]
- Cunningham V. J.; Alswieleh A. M.; Thompson K. L.; Williams M.; Leggett G. J.; Armes S. P.; Musa O. M. Poly(glycerol monomethacrylate)–poly(benzyl methacrylate) diblock copolymer nanoparticles via RAFT emulsion polymerization: Synthesis, characterization, and interfacial activity. Macromolecules 2014, 47, 5613–5623. 10.1021/ma501140h. [DOI] [Google Scholar]
- Scales P. J.; Grieser F.; Healy T. W.; White L. R.; Chan D. Y. C. Electrokinetics of the silica-solution interface: a flat plate streaming potential study. Langmuir 1992, 8, 965–974. 10.1021/la00039a037. [DOI] [Google Scholar]
- Picart C.; Lavalle P.; Hubert P.; Cuisinier F. J. G.; Decher G.; Schaaf P.; Voegel J.-C. Buildup Mechanism for Poly(L-lysine)/Hyaluronic Acid Films onto a Solid Surface. Langmuir 2001, 17, 7414–7424. 10.1021/la010848g. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.