

Abstract
To reveal impacts of sexual mode on genome content, we compared chromosome-scale assemblies of the outcrossing nematode Caenorhabditis nigoni to its self-fertile sibling species, C. briggsae. C. nigoni’s genome resembles outcrossing relatives, but encodes 31% more protein-coding genes than C. briggsae. C. nigoni genes lacking C. briggsae orthologs were disproportionately small and male-biased in expression. These include the male secreted short (mss) gene family, which encodes sperm surface glycoproteins conserved only in outcrossing species. Sperm from mss-null males of outcrossing C. remanei failed to compete with wild-type sperm, despite normal fertility in non-competitive mating. Restoring mss to C. briggsae males was sufficient to enhance sperm competitiveness. Thus, sex has a pervasive influence on genome content that can be used to identify sperm competition factors.
Introduction
Sex between individuals is nearly ubiquitous in eukaryotic life (1). However, in multicellular organisms the costs of sex and scarcity of mates sometimes favor the evolution of uniparental reproduction through asexual parthenogenesis or self-fertilization (2). Such changes in sexual reproduction have consequences for both sexual traits and genome content. Comparative genomics using closely related species with different modes of sexual reproduction can reveal sex-related factors that might otherwise remain cryptic. In the nematode species C. elegans, C. briggsae, and C. tropicalis, animals with two X chromosomes that would normally be female have evolved into self-fertilizing hermaphrodites (Fig. 1a) (3). Nearly all progeny of these selfing XX hermaphrodites are themselves XX. Rare haplo-X (XO) male progeny experience weaker sexual selection than males from outcrossing species, exhibit atrophied traits required for efficient mating (4–7), and are hypersensitive to pheromone-induced mortality (8). Sexually antagonistic sperm-female interactions have also been relaxed in self-fertile Caenorhabditis (9).
Figure 1. The phylogenetic relationship of Caenorhabditis and comparison of the C. nigoni and C. briggsae genome assemblies.

a, Phylogeny of Elegans supergroup Caenorhabditis adapted from (3), with outcrossing species producing XX females indicated in blue, and self-fertile lineages with XX hermaphrodites indicated in red. b, Chromosomal alignments and genomic features over 200-kb chromosomal intervals. Tracks from outside to inside 1, Positions (in Mb) of the six chromosomes of C. nigoni and C. briggsae, 2, gene density heat map (darker shade means higher density), 3, repeat frequency, 4, inversion frequencies, 5, percentage of sequence lacking homology in the other assembly (representing either deletions or species-specific gains), 6, DNA sequence synteny.
Self-fertile Caenorhabditis have smaller genomes and transcriptomes than outcrossing Caenorhabditis (10, 11), as also observed in the selfing plant Arabidopsis thaliana (12). However, comparisons of self-fertilizing to outcrossing Caenorhabditis have involved species as divergent at the nucleotide level as humans are from mice (10, 13), making it unclear how quickly genomic shrinkage occurs. We hypothesized a direct link between the degradation of sexual traits and genome contraction in selfing species. Here we describe new genomic resources and functional experiments that confirm its existence.
Results
Of ~50 known Caenorhabditis species, the most closely related pair with different sexual modes are the outcrossing C. nigoni and the selfing C. briggsae (14–16). They remain partially interfertile, yet have numerous genetic and reproductive incompatibilities (9, 15, 17–19). To compare their genomes, we assembled the C. nigoni genome from 20-kb Pacific Biosciences (PacBio) and Illumina short-read libraries (table S1; (20)). The final C. nigoni chromosome-scale genome assembly totaled 129 Mb with an N50 contig length of 3.3 Mb; it was estimated as 99.6% complete (21). The genome was 19% larger than C. briggsae’s (108 Mb), but similar in size to genomes of the more distantly related outcrossing species C. remanei, C. sinica, C. brenneri, and C. japonica, which range from 131–135 Mb (Fig 1A) (10). Therefore, larger genome sizes were probably the ancestral condition, and genomic shrinkage occurred in the C. briggsae lineage after it diverged from C. nigoni. Over 90% (118 Mb) of the assembly can be aligned to the chromosomes of C. briggsae without large translocations or inversions, despite megabase-sized contigs (fig. S1). Thus, the two genomes are essentially colinear, but differ in many small species-specific segments. C. nigoni’s six chromosomes are 6.6–16.6% larger than their C. briggsae homologs (table S2).
We used whole-genome alignment to identify species-specific genomic segments (20). In C. nigoni, 47.7 Mb (36.9%) did not align with C. briggsae, and C. briggsae had 27.7 Mb (25.6% of 108.4 Mb) that did not align with C. nigoni. This 20.0 Mb difference accounted for 95% of the difference in genome sizes. Non-alignable genomic regions were concentrated on the distal arms of all six holocentric chromosomes, where small inversions and repetitive sequences were abundant and gene densities were low (Fig. 1b). These regions were mostly small (median ~500 bp; Fig. 2a), but larger (1–65 kb) insertions or deletions accounted for 17 Mb (81%) of the genome size difference (Fig. 2b). In both assemblies, non-alignable sequences were most common in intergenic regions and introns (fig. S2). C. nigoni harbored 5.4 Mb more species-specific protein-coding sequences than C. briggsae, consistent with a net loss of genes in C. briggsae (see below). For orthologous genes in both species, exon lengths were highly correlated (Fig. 2c, table S3). In contrast, ortholog intron content was weakly correlated and was significantly larger in C. briggsae. Because both genomes had similar repetitive DNA fractions (C. nigoni 27% versus C. briggsae 25%), disproportionate loss of repetitive sequences (seen in plants) did not contribute to different genome sizes (table S1) (10, 12, 22).
Figure 2. Size distributions of insertion-deletion variants.
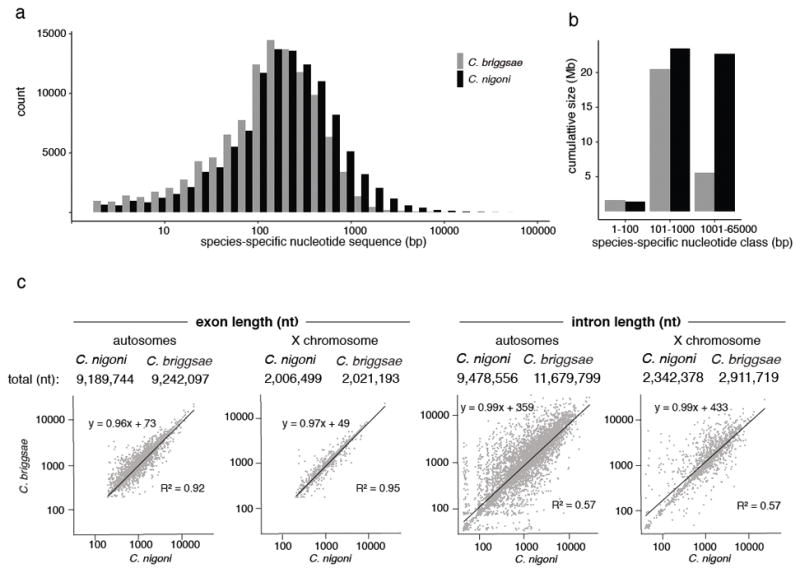
a, Size distribution of species-specific sequences in the C. briggsae-C. nigoni whole-genome alignment. Black, sequences present in C. nigoni alone; grey, sequences present in C. briggsae alone. b, Contribution of different species-specific sequence types to genome size. c, Regression analysis of total exon and intron lengths for 6,404 1:1 C. briggsae-C. nigoni orthologs on autosomes and 1,394 orthologs on the X chromosome. Interspecies differences were insignificant for either exon set (Wilcoxon rank-sum test with Bonferroni correction; p = 0.378 for autosomes; p = 0.668 for X), but introns on both autosomes (p = 1.53·10−10) and the X chromosome (p = 1.2·10−5) were significantly larger in C. briggsae (Wilcoxon rank-sum test with Bonferroni correction).
We predicted 29,167 protein-coding genes for C. nigoni (table S4), with 88.9% (25,929) being expressed in adults (≥0.1 transcripts per million [TPM]). By equivalent methods, we predicted 22,313 genes in C. briggsae (20), 23.5% less than C. nigoni. The published gene annotations for C. briggsae (23) were even fewer (21,814 genes).
This 6,854-gene difference could have several causes, including gene family contraction and loss of sequence classes in C. briggsae, as well as C. nigoni-biased gain of novel sequences. We compared genes of C. briggsae and C. nigoni to genes of the outgroups C. remanei, C. brenneri, and C. elegans (20). In C. nigoni, 24,341 genes (83.5%) were orthologous to 21,124 C. briggsae genes, reflecting larger multigene families in C. nigoni versus C. briggsae (Fig. 3a; table S4) (24). Another 2,949 C. nigoni genes without C. briggsae orthologs (10.1%) represent losses in C. briggsae based on homologs in Caenorhabditis outgroups (fig. S3). Finally, 1,877 C. nigoni genes (6.4%) lacked homologs entirely and were classed as orphans. These genes could be exceptionally divergent, recently arisen in C. nigoni, or arisen shortly before the C. nigoni-C. briggsae split but then lost in C. briggsae. Overall, gene loss in C. briggsae appears to be the primary driver of the gene number difference.
Figure 3. Comparison of the C. nigoni and C. briggsae proteomes.
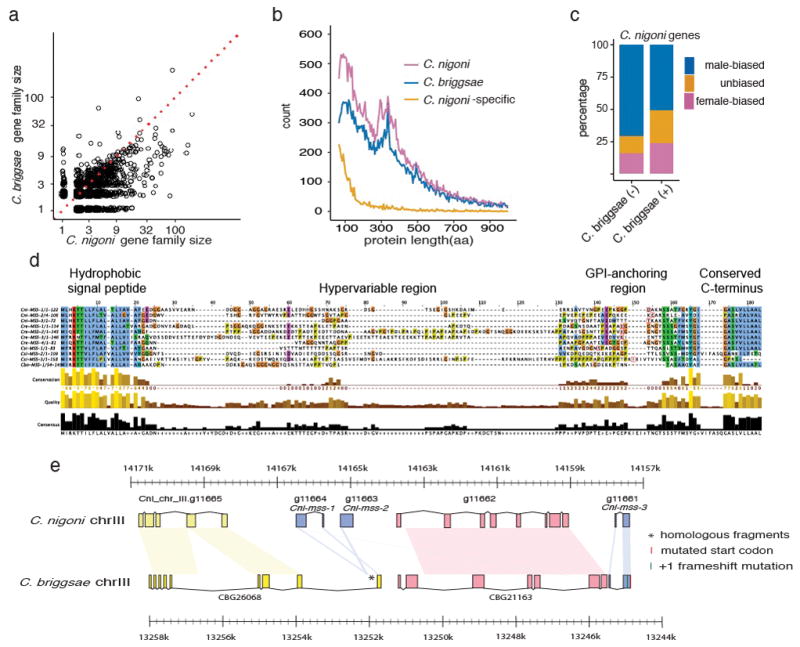
a, Scatter plot of sizes of OrthoFinder gene families, excluding one-to-one orthologs (table S4). Of 2,367 families with unequal numbers of C. nigoni and C. briggsae genes, the majority (1,624) were larger in C. nigoni than in C. briggsae (Wilcoxon signed rank test, p < 2.2·10−16). Dotted line indicates equal family sizes. b, Length distributions of C. nigoni and C. briggsae proteins and of C. nigoni proteins that lack C. briggsae homologs. c, For genes with sex-biased expression, male bias was seen for 50.9% of 6,804 genes with C. briggsae homologs (“C. briggsae (+)”), but significantly overrepresented (70.9%) among 605 genes lacking C. briggsae homologs (“C. briggsae (-)”; Fisher’s exact test, p < 0.0001; table S9). d, Alignment of predicted MSS homologs from outcrossing C. nigoni, C. sinica, C. remanei, and C. brenneri (table S8; (20)), with protein domains indicated above. e, Comparison of mss gene regions in C. nigoni and C. briggsae. Pastel shapes connect homologous sequences. Except for Cni-mss-3, all genes are transcribed from left to right. Genes surrounding the three C. nigoni mss paralogs are conserved in C. briggsae, but only fragments and a pseudogene (Cbr-mss-3-ps) of the mss genes remain. The pseudogene has a lost start codon and a +1 frameshift. CBG26068 has a novel 3′ exon derived from part of the Cni-mss-1 second exon. See fig. S7 and (20) for details.
To characterize genes lost in C. briggsae, we first compared Pfam protein domains encoded by C. nigoni versus C. briggsae. We found 26 Pfam domains that were overrepresented in C. nigoni (fig. S4; table S5); of these, seven were consistently overrepresented in outcrossing C. nigoni, C. remanei, and C. brenneri relative to the selfing species C. briggsae and C. elegans. Three of these domains (F-box, FBA_2/F-box associated, and BTB) are predicted to mediate protein-protein interactions. Male-female Caenorhabditis had 272–1,074 genes in these families, while hermaphroditic Caenorhabditis had only 101–258 genes per family. Two other domains (Peptidase_A17 and DNA_pol_B_2) are associated with repetitive DNA. The final two overrepresented domains were Asp_protease_2 (possibly associated with retroelements) and DUF3557 (a nematode-specific domain, currently of unknown function). One overrepresented domain specific to C. nigoni was zf.RING2_finger; the RING domain gene spe-42 is important for sperm-egg interactions in C. elegans (25).
Because C. nigoni-specific genes might encode fast-evolving proteins that lacked known domains, we compared other gene properties. Genes encoding medium to large proteins (≥200 residues) were similar in frequency in both species, but C. nigoni encodes disproportionately more small proteins (<200 residues) than C. briggsae (Fig. 3b; table S6). As seen in other Caenorhabditis (11), genes with male-biased expression outnumbered female-biased genes (Fig. 3c; table S7). However, even against this background, C. nigoni genes without C. briggsae homologs were disproportionately male-biased in expression. Preferential loss of small and fast-evolving proteins thus occurred in C. briggsae after the adoption of selfing.
We hypothesized that genes with highly male-biased expression that are present in outcrossing species, but lost in selfing species, might function in sexual selection. Among such genes we identified the mss (male secreted short) family. We found one to four mss genes in the outcrossing species C. nigoni, C. sinica, C. remanei, C. brenneri, C. sp. 34, C. japonica, and C. afra, but found none in the selfing C. elegans, C. briggsae, and C. tropicalis. The mss family encodes small proteins (median 111 residues) with N-terminal signal sequences, rapidly evolving central domains with several predicted O-glycosylation sites, and C-terminal glycosylphosphatidylinositol (GPI) anchor membrane attachment signals (Fig. 3d). Enzyme treatments confirmed that MSS proteins were heavily glycosylated (fig. S5).
Although we failed to detect mss genes in selfing species, we did discover a larger family of mss-related protein (msrp) genes, within which mss forms a monophyletic clade (fig. S6; (20)). Notably, msrp genes are found both in outcrossing Caenorhabditis and in the hermaphroditic C. elegans, C. briggsae, and C. tropicalis (fig. S6). Like MSS, MSRP proteins are small, and are predicted to be secreted, O-glycosylated, and (often) GPI-anchored. Both mss and msrp genes show male-biased expression in C. nigoni and other species (table S8). In cases where their chromosomal loci can be identified, mss and msrp genes are autosomal; this linkage fits a general pattern in heterogametic male species of male-biased genes being autosomal rather than X-chromosomal (ref. (26) and references therein).
Because we observed mss genes in two C. elegans outgroups (C. japonica and C. afra, fig. S6, table S8), their absence from hermaphrodites most likely reflects independent gene losses rather than phylogenetic restriction to close relatives of C. nigoni. Examination of the C. briggsae genomic region syntenic to the C. nigoni mss locus revealed fragments of mss-1 and mss-2 coding sequences and a nearly complete mss-3 pseudogene (Fig. 3e, (20)). Mutations that ablate Cbr-mss-3-ps function in the AF16 reference strain also occur in 11 wild isolates that span the known diversity of C. briggsae (fig. S7 (20, 27)). Orthologs of all three C. nigoni mss genes were therefore present in the common ancestor of C. nigoni and C. briggsae, but were lost in C. briggsae before its global diversification.
In the outcrossing species C. remanei, mss transcripts were expressed only in adult males (Fig. 4a), with strongest expression in spermatocytes during mid-pachytene of meiosis I (Fig. 4b). To determine subcellular localization of MSS peptides, we used CRISPR/Cas9 editing to tag the Cre-mss-1 gene of C. remanei with the hemagglutinin (HA) epitope. Crem-MSS-1::HA expression was first detected in large vesicles and on the plasma membrane of spermatocytes, with intensity increasing and localization restricted to secretory vesicles in mature spermatids (Fig. 4c, d, e). The secretory vesicles of nematode sperm, known as membranous organelles (MOs), fuse with the plasma membrane upon ejaculation and sperm activation (28).
Figure 4. C. remanei MSS is a male-specific protein localized to the surface of activated sperm.
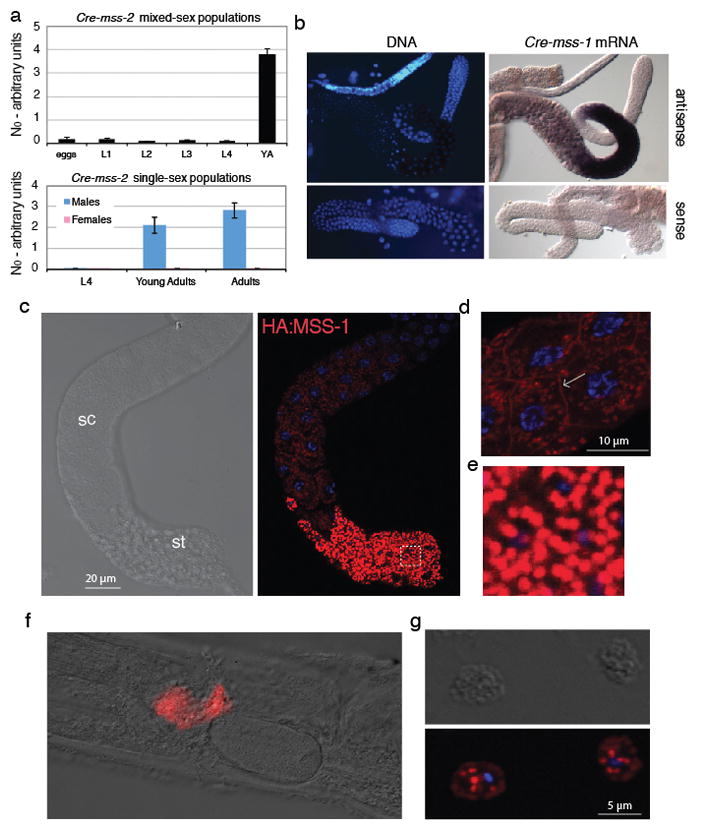
a, qRT-PCR transcript quantification (for Cre-mss-2) of mixed-sex (top) versus larval and adult sex-specific populations (bottom), showing that mss expression is specific to adult males. Mean values are shown with standard error of the mean. Female data are 2–3 orders of magnitude below that for males. b, Cre-mss-1 transcripts are detected in pachytene-stage primary spermatocytes. c, HA-tagged Cre-HA-MSS-1 is first detectable in spermatocytes (sc), and becomes enriched in spermatids (st). d, Some Cre-HA-MSS-1 is localized to the plasma membrane of spermatocytes, as indicated by the arrow. Blue fluorescence: Hoechst-stained DNA. e, Enlarged view of the boxed region in c, showing complete restriction to membranous organelles (MOs). f, Cre-HA-MSS-1 remains attached to sperm after activation and transfer to the female. g, Sperm cells dissected from a female show Cre-HA-MSS-1 in plasma membrane and fused MO remnants.
MSS peptides might be processed by a signal peptidase to release a soluble fragment into the MO lumen, which could then be dumped into seminal fluid upon sperm activation. However, their transient plasma membrane localization in spermatocytes and predicted C-terminal GPI attachment signals (Fig. 3d; table S8) suggested that MSS peptides might instead be attached to membranes. Consistent with this latter hypothesis, Crem-MSS::HA remained associated with activated sperm dissected from inseminated females (Fig. 4f). We observed staining of the plasma membrane and of MO-derived punctae (Fig. 4g), which may be fused vesicles that remain as cup-like invaginations (29). Persistence of MSS on the surface of sperm after activation suggested that MSS acts cell-autonomously, rather than through the seminal fluid.
Because the four C. remanei mss paralogs form a 7-kb tandem array (fig. S8a), we deleted the entire mss cluster via CRISPR/Cas9 editing. To avoid inbreeding depression associated with homozygosity of entire chromosomes (30; fig. S8b), we generated the mss deletion in two different C. remanei strains and crossed them to create hybrid mss-null mutants. The resulting males showed no intrinsic fertility defects as judged by overall brood size (fig. S8c). However, when competing against heterozygous mss(null/+) males, mss mutants sired fewer progeny than non-mutants in both offense (mutant male second) and defense (mutant male first) scenarios (Fig. 5a, b). The mss family is therefore not required for fertility itself, but for male sperm competitiveness in multiple mating situations. Sperm lacking MSS compete poorly even when the female reproductive tract is conditioned by wild-type sperm. Thus, MSS proteins probably do not function as a secreted signal, but instead act cell-autonomously.
Figure 5. mss genes are necessary for sperm competitiveness in an outcrossing species, and sufficient to enhance it in a selfing species.

a, When mated after a wild-type male (“offense”), C. remanei mss(nmDf1/+) males sire more than twice the progeny of nmDf1/nmDf2 mss-null mutants (N = 16 for both). b, When allowed to mate first (“defense”), heterozygous C. remanei mss(nmDf1/+) males have a slight advantage over wild-type males. mss-null mutants, in contrast, do not (N = 15 for both). Heterozygote success is assumed to be double the observed nmDf1 frequency in their progeny. For both defense and offense, p < 0.01 (two-sample Kolmogorov–Smirnov test). c, d, Wild-type young C. briggsae hermaphrodites were mated sequentially (4 h each) with conspecific males carrying either an C. nigoni mss(+) transgene or control mCherry::histone reporter (RW0025). Progeny laid 0–18 h and 18–42 h after the second mating were scored for green (MSS+), red (RW0025), or no (self) fluorescent markers. In both offense (c) and defense (d), MSS+ males sire several-fold more progeny than control males in the first laying window. *, p < 0.001. e, MSS+ C. briggsae males suppress selfing more effectively than control AF16 (wild-type) males. Strain JU936 is a second control strain bearing two transcriptional GFP reporters in the AF16 background. (*, p < 0.001; ns, not significant Kolmogorov–Smirnov test) f, Male frequency in MSS+ and wild-type AF16 C. briggsae populations in which male frequency was artificially elevated to 50% at the start of the experiment.
We then introduced mss-1 and mss-2 genes from C. nigoni into C. briggsae via a low-copy, germline-expressed MSS transgene; this transgene was strongly expressed in C. briggsae males, while also being detectable in hermaphrodites (fig. S9). Remarkably, sperm from transgenic mss(+) C. briggsae males outcompeted those of wild-type males (Fig. 5c, d). After mss(+) sperm were exhausted, however, wild-type mss(null) sperm were still fertilization-competent (Fig. 5c, d). In addition, mss(+) males were more consistently able to suppress use of a hermaphrodite mate’s self-sperm (Fig. 5e).
Because 50% of outcross progeny are male, but selfed progeny are almost exclusively hermaphrodites, we examined the effect of transgenic mss on long-term sex ratios in C. briggsae populations. We started both wild-type and mss(+) C. briggsae populations with a 1:1 male-to-hermaphrodite sex ratio and examined them over time. Wild-type C. briggsae showed a rapid decline of males, as previously seen in C. elegans (7, 31). However, male frequency remained elevated in the mss(+) strain (Fig. 5f), only declining after 12 generations. The expression of MSS proteins was thus sufficient to shift population sex ratios towards parity.
Discussion
Comparison of the C. nigoni and C. briggsae genomes revealed that C. briggsae experienced rapid contraction of chromosomes and loss of protein-coding genes. However, loss of ancestral genomic content in C. briggsae does not fully explain their genomic divergence; the ongoing birth of novel sequences in both species, along with loss of ancestral DNA in C. nigoni, is also important. Net shrinkage of the C. briggsae genome therefore resulted from a substantial increase in the ratio of losses to gains. These losses included many coding sequences, reducing the C. briggsae gene count by nearly one quarter.
Multiple observations implicate the evolution of selfing as the cause of genome shrinkage in C. briggsae. Reduced genome and transcriptome sizes are observed in all three selfing Caenorhabditis (10, 11). Continued interfertility of C. briggsae and C. nigoni (15) indicates that self-fertility and genome shrinkage evolved in quick succession. Genes with male-biased expression, such as the mss family, are disproportionately and consistently lost from selfing species (11). This suggests that genes with male reproductive functions that are either dispensable or maladaptive in the new sexual mode are purged from the genome. Finally, the net genome shrinkage we observed has been predicted to arise from a partially selfing mating system coupled with transmission distortion of autosomal deletion alleles (32, 33). Such distortion is driven by imbalanced chromatin during meiosis I of XO males, and causes preferential inheritance of shorter alleles by hermaphrodite progeny and their increased fixation in the population.
Larger autosomal deletions, influenced most by the deletion segregation distortion mechanism, are primarily responsible for the smaller genome of C. briggsae (Fig. 2). However, such deletions and net shrinkage were also found on the X chromosome (table S2), which should be unaffected. Moreover, orthologous genes have larger introns in C. briggsae than C. nigoni (Fig. 2), and introns comprised a greater fraction of the C. briggsae genome (fig. S2). X-chromosomal C. briggsae introns are also larger than those of the outgroup C. remanei (10; fig. S2c), suggesting that introns of many genes expanded in C. briggsae. Thus, additional processes must also contribute to shrinkage of the C. briggsae genome. Spontaneous short (1–5 nt) mutations in C. elegans are biased towards insertions rather than deletions (34), though biases in formation of larger indels remain uncharacterized. Regardless, the relative rates of insertion and deletion mutations likely evolve too slowly to explain C. briggsae’s reduced genome size, given its recent divergence from C. nigoni (27). Gene loss can sometimes be adaptive (35, 36), and has been proposed as a factor promoting genome shrinkage in selfing Caenorhabditis (10). Our results for the C. nigoni-C. briggsae pair support this hypothesis.
Genes encoding small proteins with male-biased expression are disproportionately lost in C. briggsae, with mss providing an instance affecting reproduction. Unlike comp-1, which encodes a kinase required for male versus hermaphrodite sperm competition in C. elegans (37), and which is conserved regardless of mating system, we found mss orthologs only in outcrossing species. In interspecies matings, sperm from males of outcrossing species rapidly invade the ovaries and body cavities of selfing hermaphrodites, sterilizing or killing them (9). This cryptic toxicity of outcrossing sperm is likely due to ongoing sexual selection in outcrossing species. Given their pronounced role in sperm competition, MSS proteins may contribute to sperm invasiveness.
How MSS improves sperm competitiveness remains unclear, but mature MSS proteins are substantially glycosylated (fig. S5). Such posttranslational modification may impose little constraint on MSS proteins, explaining how they can have weak sequence conservation yet strong functional conservation. Another poorly conserved O-glycosylated protein, the mucin PLG-1, forms a copulatory plug found in all male-female Caenorhabditis species but lost in many wild isolates of C. elegans (4). Glycoproteins form the glycocalyx coat of mammalian sperm, and play important roles in fertility (38). Caenorhabditis provides a useful model for interactions between the glycocalyx and female tissues, and how they affect sperm competition.
Independent loss of mss in the three known hermaphroditic Caenorhabditis species could reflect either relaxed sexual selection coupled with mutation and drift, or adaptive convergence. Other changes in selfing species, such as loss of plg-1 and of plep-1, which mediates reliable male discrimination between the vulva and excretory pore (4, 6), are likely due to relaxed selection. However, restoring mss to C. briggsae enhances male fitness (Fig. 5c,d), and mutations inactivating the Cbr-mss-3-ps pseudogene are not deletions that would be subject to loss via transmission ratio distortion (fig S7). These findings suggest that loss of mss may instead reflect adaptive convergence, permitting proto-hermaphrodites to adapt to a selfing lifestyle and resolve emergent sexual conflicts related to mating (39–41). Selfing Caenorhabditis lack inbreeding depression (42) and reproduce in spatially isolated habitats colonized by small numbers of founders (3). Reduced male mating success creates hermaphrodite-biased sex ratios (Fig. 5f), which may be adaptive under these conditions (41, 43–45). Thus, evolutionary transitions in reproductive mode may produce conditions for selection to rapidly eliminate formerly constrained reproductive genes.
Supplementary Material
Acknowledgments
We thank E. Antoniou, P.W. Sternberg, C.T. Brown, the Michigan State University High-Performance Computing Center (supported by USDA grant 2010-65205-20361 and NIFA-NSF grant IOS-0923812), and the UC Berkeley V.J. Coates Genomics Sequencing Laboratory (supported by NIHS10RR029668) for sequencing and computational support. We thank G. Williams and R.H. Waterston for transcriptome data, the Caenorhabditis Genomes Project for prepublication access to the genomes of C. afra and C. sp. 34, Z. Zhao and M.A. Félix for nematode and plasmid reagents, and S. Mount for helpful discussions. This work was supported by NSF award IOS-1355119 to E.S.H; NIH grant AI111173, Moore Foundation Grant No. 4551, and Cornell University start-up funds to E.M.S.; and NIH grant GM030702 to B.J.M., an investigator of the Howard Hughes Medical Institute. Some strains were provided by the CGC, which is funded by the NIH Office of Research Infrastructure Programs (P40 OD010440).
Footnotes
Data availability and accession codes
C. nigoni genomic and transcriptomic data have been archived as NCBI BioProject accessions PRJNA384657 and PRJNA384658, and the C. nigoni genome assembly as DDBJ/ENA/GenBank accession PDUG00000000. Genome assembly, gene prediction, and gene expression data for C. nigoni, with supplementary datasets for other species, have been archived at the OSF (C. nigoni, https://osf.io/dkbwt and doi:10.17605/osf.io/dkbwt; other species, https://osf.io/b47r8 and doi:10.17605/osf.io/b47r8).
Materials and Methods
References
- 1.Schurko AM, Neiman M, Logsdon JM., Jr Signs of sex: what we know and how we know it. Trends Ecol Evol. 2009;24:208–217. doi: 10.1016/j.tree.2008.11.010. [DOI] [PubMed] [Google Scholar]
- 2.Bell G. The Masterpiece of Nature: The Evolution and Genetics of Sexuality. University of California Press; Berkeley: 1982. [Google Scholar]
- 3.Kiontke K, et al. A phylogeny and molecular barcodes for Caenorhabditis, with numerous new species from rotting fruits. BMC Evol Biol. 2011;11:339. doi: 10.1186/1471-2148-11-339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Palopoli MF, et al. Molecular basis of the copulatory plug polymorphism in Caenorhabditis elegans. Nature. 2008;454:1019–1022. doi: 10.1038/nature07171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garcia LR, LeBoeuf B, Koo P. Diversity in mating behavior of hermaphroditic and male-female Caenorhabditis nematodes. Genetics. 2007;175:1761–1771. doi: 10.1534/genetics.106.068304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Noble LM, et al. Natural variation in plep-1 causes male-male copulatory behavior in C. elegans. Curr Biol. 2015;25:2730–2737. doi: 10.1016/j.cub.2015.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chasnov JR, Chow KL. Why are there males in the hermaphroditic species Caenorhabditis elegans? Genetics. 2002;160:983–994. doi: 10.1093/genetics/160.3.983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shi C, Runnels AM, Murphy CT. Mating and male pheromone kill Caenorhabditis males through distinct mechanisms. Elife. 2017;6:e23493. doi: 10.7554/eLife.23493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ting JJ, et al. Intense sperm-mediated sexual conflict promotes reproductive isolation in Caenorhabditis nematodes. PLoS Biol. 2014;12:e1001915. doi: 10.1371/journal.pbio.1001915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fierst JL, et al. Reproductive mode and the evolution of genome size and structure in Caenorhabditis nematodes. PLoS Genet. 2015;11:e1005323. doi: 10.1371/journal.pgen.1005323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thomas CG, et al. Simplification and desexualization of gene expression in self-fertile nematodes. Curr Biol. 2012;22:2167–2172. doi: 10.1016/j.cub.2012.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hu TT, et al. The Arabidopsis lyrata genome sequence and the basis of rapid genome size change. Nat Genet. 2011;43:476–481. doi: 10.1038/ng.807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kiontke K, et al. Caenorhabditis phylogeny predicts convergence of hermaphroditism and extensive intron loss. Proc Natl Acad Sci U S A. 2004;101:9003–9008. doi: 10.1073/pnas.0403094101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Félix MA, Braendle C, Cutter AD. A streamlined system for species diagnosis in Caenorhabditis (Nematoda: Rhabditidae) with name designations for 15 distinct biological species. PLoS One. 2014;9:e94723. doi: 10.1371/journal.pone.0094723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Woodruff GC, Eke O, Baird SE, Felix MA, Haag ES. Insights into species divergence and the evolution of hermaphroditism from fertile interspecies hybrids of Caenorhabditis nematodes. Genetics. 2010;186:997–1012. doi: 10.1534/genetics.110.120550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cutter AD. Caenorhabditis evolution in the wild. Bioessays. 2015;37:983–995. doi: 10.1002/bies.201500053. [DOI] [PubMed] [Google Scholar]
- 17.Bi Y, et al. A genome-wide hybrid incompatibility landscape between Caenorhabditis briggsae and C. nigoni. PLoS Genet. 2015;11:e1004993. doi: 10.1371/journal.pgen.1004993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li R, et al. Specific down-regulation of spermatogenesis genes targeted by 22G RNAs in hybrid sterile males associated with an X-Chromosome introgression. Genome Res. 2016;26:1219–1232. doi: 10.1101/gr.204479.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bundus JD, Alaei R, Cutter AD. Gametic selection, developmental trajectories, and extrinsic heterogeneity in Haldane’s rule. Evolution. 2015;69:2005–2017. doi: 10.1111/evo.12708. [DOI] [PubMed] [Google Scholar]
- 20.Materials, methods, and datasets are available as supplementary materials.
- 21.Parra G, Bradnam K, Ning Z, Keane T, Korf I. Assessing the gene space in draft genomes. Nucleic Acids Res. 2009;37:289–297. doi: 10.1093/nar/gkn916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wright SI, Nano N, Foxe JP, Dar VU. Effective population size and tests of neutrality at cytoplasmic genes in Arabidopsis. Genet Res (Camb) 2008;90:119–128. doi: 10.1017/S0016672307008920. [DOI] [PubMed] [Google Scholar]
- 23.Coghlan A, et al. nGASP--the nematode genome annotation assessment project. BMC Bioinformatics. 2008;9:549. doi: 10.1186/1471-2105-9-549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Emms DM, Kelly S. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 2015;16:157. doi: 10.1186/s13059-015-0721-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wilson LD, et al. Fertilization in C. elegans requires an intact C-terminal RING finger in sperm protein SPE-42. BMC Dev Biol. 2011;11:10. doi: 10.1186/1471-213X-11-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cassone BJ, Kay RG, Daugherty MP, White BJ. Comparative transcriptomics of malaria mosquito testes: function, evolution, and linkage. G3 (Bethesda) 2017;7:1127–1136. doi: 10.1534/g3.117.040089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thomas CG, et al. Full-genome evolutionary histories of selfing, splitting, and selection in Caenorhabditis. Genome Res. 2015;25:667–678. doi: 10.1101/gr.187237.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ward S, Hogan E, Nelson GA. The initiation of spermiogenesis in the nematode Caenorhabditis elegans. Dev Biol. 1983;98:70–79. doi: 10.1016/0012-1606(83)90336-6. [DOI] [PubMed] [Google Scholar]
- 29.Ward S, Argon Y, Nelson GA. Sperm morphogenesis in wild-type and fertilization-defective mutants of Caenorhabditis elegans. J Cell Biol. 1981;91:26–44. doi: 10.1083/jcb.91.1.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barriére A, et al. Detecting heterozygosity in shotgun genome assemblies: Lessons from obligately outcrossing nematodes. Genome Research. 2009;19:470–480. doi: 10.1101/gr.081851.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stewart A, Phillips P. Selection and maintenance of androdioecy in Caenorhabditis elegans. Genetics. 2002;160:975–982. doi: 10.1093/genetics/160.3.975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang J, Chen PJ, Wang GJ, Keller L. Chromosome size differences may affect meiosis and genome size. Science. 2010;329:293. doi: 10.1126/science.1190130. [DOI] [PubMed] [Google Scholar]
- 33.Le TS, et al. Non-Mendelian assortment of homologous autosomes of different sizes in males is the ancestral state in the Caenorhabditis lineage. Sci Rep. 2017;7:12819. doi: 10.1038/s41598-017-13215-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Denver DR, Morris K, Lynch M, Thomas WK. High mutation rate and predominance of insertions in the Caenorhabditis elegans nuclear genome. Nature. 2004;430:679–682. doi: 10.1038/nature02697. [DOI] [PubMed] [Google Scholar]
- 35.Olson MV. When less is more: gene loss as an engine of evolutionary change. Am J Hum Genet. 1999;64:18–23. doi: 10.1086/302219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cutter AD, Jovelin R. When natural selection gives gene function the cold shoulder. Bioessays. 2015;37:1169–1173. doi: 10.1002/bies.201500083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hansen JM, Chavez DR, Stanfield GM. COMP-1 promotes competitive advantage of nematode sperm. Elife. 2015;4 doi: 10.7554/eLife.05423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tecle E, Gagneux P. Sugar-coated sperm: Unraveling the functions of the mammalian sperm glycocalyx. Mol Reprod Dev. 2015;82:635–650. doi: 10.1002/mrd.22500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Glémin S, Ronfort J. Adaptation and maladaptation in selfing and outcrossing species: new mutations versus standing variation. Evolution. 2013;67:225–240. doi: 10.1111/j.1558-5646.2012.01778.x. [DOI] [PubMed] [Google Scholar]
- 40.Sicard A, Lenhard M. The selfing syndrome: a model for studying the genetic and evolutionary basis of morphological adaptation in plants. Ann Bot. 2011;107:1433–1443. doi: 10.1093/aob/mcr023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chasnov JR. The evolution from females to hermaphrodites results in a sexual conflict over mating in androdioecious nematode worms and clam shrimp. J Evol Biol. 2010;23:539–556. doi: 10.1111/j.1420-9101.2009.01919.x. [DOI] [PubMed] [Google Scholar]
- 42.Dolgin ES, Charlesworth B, Baird SE, Cutter AD. Inbreeding and outbreeding depression in Caenorhabditis nematodes. Evolution. 2007;61:1339–1352. doi: 10.1111/j.1558-5646.2007.00118.x. [DOI] [PubMed] [Google Scholar]
- 43.Hamilton WD. Extraordinary sex ratios. Science. 1967;156:477–488. doi: 10.1126/science.156.3774.477. [DOI] [PubMed] [Google Scholar]
- 44.Lively CM, Lloyd DG. The cost of biparental sex under individual selection. American Naturalist. 1990;135:489–500. [Google Scholar]
- 45.Charnov EL. The Theory of Sex Allocation. In: May RM, editor. Monographs in Population Biology. Vol. 18. Princeton University Press; Princeton, NJ: 1982. p. 355. [PubMed] [Google Scholar]
- 46.Kozlowska JL, Ahmad AR, Jahesh E, Cutter AD. Genetic variation for postzygotic reproductive isolation between Caenorhabditis briggsae and Caenorhabditis sp. 9. Evolution. 2012;66:1180–1195. doi: 10.1111/j.1558-5646.2011.01514.x. [DOI] [PubMed] [Google Scholar]
- 47.Fodor A, Riddle DL, Nelson FK, Golden JW. Comparison of a new wild-type Caenorhabditis briggsae with laboratory strains of C. briggsae and C. elegans. Nematologica. 1983;29:203–217. [Google Scholar]
- 48.Stein L, et al. The genome sequence of Caenorhabditis briggsae: a platform for comparative genomics. PLoS Biology. 2003;1:166–192. doi: 10.1371/journal.pbio.0000045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Baird SE, Fitch DH, Emmons SW. Caenorhabditis vulgaris sp.n. (Nematoda: Rhabditidae): A necromenic associate of pill bugs and snails. Nematologica. 1994;40:1–11. [Google Scholar]
- 50.Sudhaus W, Kiontke K. Phylogeny of Rhabditis subgenus Caenorhabditis (Rhabditidae, Nematoda) J of Zool Syst Evol Res. 1996;34:217–233. [Google Scholar]
- 51.Sudhaus W. Zur Systematik, Verbreitung, Ökologie und Biologie neuer und wenig bekannter Rhabditiden (Nematoda) J Zool Syst Evol Res. 1974;34:217–233. [Google Scholar]
- 52.Baird SE. Natural and experimental associations of Caenorhabditis remanei with Trachelipus rathkii and other terrestrial isopods. Nematology. 1999;1:471–475. [Google Scholar]
- 53.Liu Q, Stumpf C, Thomas C, Wickens M, Haag ES. Context-dependent function of a conserved translational regulatory module. Development. 2012;139:1509–1521. doi: 10.1242/dev.070128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Guo Y, Lang S, Ellis R. Independent recruitment of F-box genes to regulate hermaphrodite development during nematode evolution. Curr Biol. 2009;19:1853–1860. doi: 10.1016/j.cub.2009.09.042. [DOI] [PubMed] [Google Scholar]
- 55.Wood WB, editor. The Nematode Caenorhabditis elegans. Cold Spring Harbor Laboratory; 1988. [Google Scholar]
- 56.Quail MA, et al. A large genome center’s improvements to the Illumina sequencing system. Nature methods. 2008;5:1005–1010. doi: 10.1038/nmeth.1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Berlin K, et al. Assembling large genomes with single-molecule sequencing and locality-sensitive hashing. Nat Biotechnol. 2015;33:623–630. doi: 10.1038/nbt.3238. [DOI] [PubMed] [Google Scholar]
- 58.Sulston J, Hodgkin J. In: The Nematode Caenorhabditis elegans. Wood WB, editor. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1988. pp. 587–606. [Google Scholar]
- 59.Percudani R. A microbial metagenome (Leucobacter sp.) in Caenorhabditis whole genome sequences. Bioinform Biol Insights. 2013;7:55–72. doi: 10.4137/BBI.S11064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fierst JL, Murdock DA, Thanthiriwatte C, Willis JH, Phillips PC. Metagenome-assembled draft genome sequence of a novel microbial Stenotrophomonas maltophilia strain isolated from Caenorhabditis remanei tissue. Genome Announc. 2017;5 doi: 10.1128/genomeA.01646-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kumar S, Jones M, Koutsovoulos G, Clarke M, Blaxter M. Blobology: exploring raw genome data for contaminants, symbionts and parasites using taxon-annotated GC-coverage plots. Front Genet. 2013;4:237. doi: 10.3389/fgene.2013.00237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Camacho C, et al. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.English AC, et al. Mind the gap: upgrading genomes with Pacific Biosciences RS long-read sequencing technology. PLoS One. 2012;7:e47768. doi: 10.1371/journal.pone.0047768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chin CS, et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat Methods. 2013;10:563–569. doi: 10.1038/nmeth.2474. [DOI] [PubMed] [Google Scholar]
- 65.Walker BJ, et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One. 2014;9:e112963. doi: 10.1371/journal.pone.0112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nature methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li H, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Huang S, Kang M, Xu A. HaploMerger2: rebuilding both haploid sub-assemblies from high-heterozygosity diploid genome assembly. Bioinformatics. 2017 doi: 10.1093/bioinformatics/btx220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Morgulis A, Gertz EM, Schaffer AA, Agarwala R. WindowMasker: window-based masker for sequenced genomes. Bioinformatics. 2006;22:134–141. doi: 10.1093/bioinformatics/bti774. [DOI] [PubMed] [Google Scholar]
- 70.Gerstein MB, et al. Integrative analysis of the Caenorhabditis elegans genome by the modENCODE project. Science. 2010;330:1775–1787. doi: 10.1126/science.1196914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Romiguier J, et al. Comparative population genomics in animals uncovers the determinants of genetic diversity. Nature. 2014;515:261–263. doi: 10.1038/nature13685. [DOI] [PubMed] [Google Scholar]
- 72.Grabherr MG, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29:644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Xue W, et al. L_RNA_scaffolder: scaffolding genomes with transcripts. BMC Genomics. 2013;14:604. doi: 10.1186/1471-2164-14-604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhu BH, et al. PEP_scaffolder: using (homologous) proteins to scaffold genomes. Bioinformatics. 2016;32:3193–3195. doi: 10.1093/bioinformatics/btw378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kurtz S, et al. Versatile and open software for comparing large genomes. Genome Biol. 2004;5:R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wootton JC, Federhen S. Analysis of compositionally biased regions in sequence databases. Methods Enzymol. 1996;266:554–571. doi: 10.1016/s0076-6879(96)66035-2. [DOI] [PubMed] [Google Scholar]
- 77.Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573–580. doi: 10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Price AL, Jones NC, Pevzner PA. De novo identification of repeat families in large genomes. Bioinformatics. 2005;21(Suppl 1):i351–358. doi: 10.1093/bioinformatics/bti1018. [DOI] [PubMed] [Google Scholar]
- 79.Wheeler TJ, Eddy SR. nhmmer: DNA homology search with profile HMMs. Bioinformatics. 2013;29:2487–2489. doi: 10.1093/bioinformatics/btt403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rice P, Longden I, Bleasby A. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 2000;16:276–277. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 81.Eddy SR. Accelerated profile HMM searches. PLoS Comput Biol. 2011;7:e1002195. doi: 10.1371/journal.pcbi.1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Finn RD, et al. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 2016;44:D279–285. doi: 10.1093/nar/gkv1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Stanke M, Diekhans M, Baertsch R, Haussler D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics. 2008;24:637–644. doi: 10.1093/bioinformatics/btn013. [DOI] [PubMed] [Google Scholar]
- 84.Hoff KJ, Stanke M. WebAUGUSTUS--a web service for training AUGUSTUS and predicting genes in eukaryotes. Nucleic Acids Res. 2013;41:W123–128. doi: 10.1093/nar/gkt418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kent WJ. BLAT--the BLAST-like alignment tool. Genome research. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nature methods. 2017;14:417–419. doi: 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Schurch NJ, et al. How many biological replicates are needed in an RNA-seq experiment and which differential expression tool should you use? Rna. 2016;22:839–851. doi: 10.1261/rna.053959.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Käll L, Krogh A, Sonnhammer EL. A combined transmembrane topology and signal peptide prediction method. J Mol Biol. 2004;338:1027–1036. doi: 10.1016/j.jmb.2004.03.016. [DOI] [PubMed] [Google Scholar]
- 90.Lupas A. Prediction and analysis of coiled-coil structures. Methods Enzymol. 1996;266:513–525. doi: 10.1016/s0076-6879(96)66032-7. [DOI] [PubMed] [Google Scholar]
- 91.Wootton JC. Non-globular domains in protein sequences: automated segmentation using complexity measures. Comput Chem. 1994;18:269–285. doi: 10.1016/0097-8485(94)85023-2. [DOI] [PubMed] [Google Scholar]
- 92.Eddy SR. A new generation of homology search tools based on probabilistic inference. Genome Inform. 2009;23:205–211. [PubMed] [Google Scholar]
- 93.Finn RD, et al. InterPro in 2017-beyond protein family and domain annotations. Nucleic Acids Res. 2017;45:D190–D199. doi: 10.1093/nar/gkw1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Steentoft C, et al. Precision mapping of the human O-GalNAc glycoproteome through SimpleCell technology. Embo J. 2013;32:1478–1488. doi: 10.1038/emboj.2013.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pierleoni A, Martelli PL, Casadio R. PredGPI: a GPI-anchor predictor. BMC Bioinformatics. 2008;9:392. doi: 10.1186/1471-2105-9-392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Blake JA, et al. Gene Ontology Consortium: going forward. Nucleic Acids Res. 2015;43:D1049–1056. doi: 10.1093/nar/gku1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Götz S, et al. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008;36:3420–3435. doi: 10.1093/nar/gkn176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Alexa A, Rahnenfuhrer J, Lengauer T. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics (Oxford, England) 2006;22:1600–1607. doi: 10.1093/bioinformatics/btl140. [DOI] [PubMed] [Google Scholar]
- 99.Ross J, et al. Caenorhabditis briggsae recombinant inbred line genotypes reveal inter-strain incompatibility and the evolution of recombination. PLoS Genet. 2011;7:e1002174. doi: 10.1371/journal.pgen.1002174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Krzywinski M, et al. Circos: an information aesthetic for comparative genomics. Genome research. 2009;19:1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Capella-Gutierrez S, Silla-Martinez JM, Gabaldon T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009;25:1972–1973. doi: 10.1093/bioinformatics/btp348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Price MN, Dehal PS, Arkin AP. FastTree 2--approximately maximum-likelihood trees for large alignments. PLoS One. 2010;5:e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ. Jalview Version 2--a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25:1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Cutter AD, Yan W, Tsvetkov N, Sunil S, Felix MA. Molecular population genetics and phenotypic sensitivity to ethanol for a globally diverse sample of the nematode Caenorhabditis briggsae. Mol Ecol. 2010;19:798–809. doi: 10.1111/j.1365-294X.2009.04491.x. [DOI] [PubMed] [Google Scholar]
- 107.Ramakers C, Ruijter JM, Deprez RH, Moorman AF. Assumption-free analysis of quantitative real-time polymerase chain reaction (PCR) data. Neurosci Lett. 2003;339:62–66. doi: 10.1016/s0304-3940(02)01423-4. [DOI] [PubMed] [Google Scholar]
- 108.Ruijter JM, et al. Amplification efficiency: linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res. 2009;37:e45. doi: 10.1093/nar/gkp045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hill RC, et al. Genetic flexibility in the convergent evolution of hermaphroditism in Caenorhabditis nematodes. Dev Cell. 2006;10:531–538. doi: 10.1016/j.devcel.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 110.Jones AR, Francis R, Schedl T. GLD-1, a cytoplasmic protein essential for oocyte differentiation, shows stage- and sex-specific expression during Caenorhabditis elegans germline development. Dev Biol. 1996;180:165–183. doi: 10.1006/dbio.1996.0293. [DOI] [PubMed] [Google Scholar]
- 111.Shakes DC, et al. Spermatogenesis-specific features of the meiotic program in Caenorhabditis elegans. PLoS Genet. 2009;5:e1000611. doi: 10.1371/journal.pgen.1000611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Harlow E, Lane D. Using Antibodies: A Laboratory Manual. Cold Spring Harbor Laboratory Press; Cold Spring Harbory, NY: 1999. [Google Scholar]
- 113.Gagnon JA, et al. Efficient mutagenesis by Cas9 protein-mediated oligonucleotide insertion and large-scale assessment of single-guide RNAs. PLoS One. 2014;9:e98186. doi: 10.1371/journal.pone.0098186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Yan C, Bi Y, Yin D, Zhao Z. A method for rapid and simultaneous mapping of genetic loci and introgression sizes in nematode species. PLoS One. 2012;7:e43770. doi: 10.1371/journal.pone.0043770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Altschul SF, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sudhaus W, Kiontke K. Comparison of the cryptic nematode species Caenorhabditis brenneri sp. n. and C. remanei (Nematoda: Rhabditidae) with the stem species pattern of the Caenorhabditis Elegans group. Zootaxa. 2007;1456:45–62. [Google Scholar]
- 117.Slater GS, Birney E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics. 2005;6:31. doi: 10.1186/1471-2105-6-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Howe KL, et al. WormBase 2016: expanding to enable helminth genomic research. Nucleic Acids Res. 2016;44:D774–780. doi: 10.1093/nar/gkv1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Noble WS. How does multiple testing correction work? Nat Biotechnol. 2009;27:1135–1137. doi: 10.1038/nbt1209-1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.