

Abstract
We introduce a simplified computational algorithm for computing isotope distributions (relative abundances and masses) of biomolecules. The algorithm is based on Poisson approximation to binomial and multinomial distributions. It leads to a small number of arithmetic operations to compute isotope distributions of molecules. The approach uses three embedded loops to compute the isotope distributions, as compared with the eight embedded loops in exact calculations. The speed improvement is about 3-fold compared to the fast Fourier transformation-based isotope calculations, often termed as ultrafast isotope calculation. The approach naturally incorporates the determination of the masses of each molecular isotopomer. It is applicable to high mass accuracy and resolution mass spectrometry data. The application to tryptic peptides in a UniProt protein database revealed that the mass accuracy of the computed isotopomers is better than 1 ppm. Even better mass accuracy (below 1 ppm) is achievable when the method is paired with the exact calculations, which we term a hybrid approach. The algorithms have been implemented in a freely available C/C++ code.
Keywords: Poisson distribution, isotope envelope, mass spectrometry, stable isotope labeling
Graphical Abstract
INTRODUCTION
Calculations of isotope distributions of biomolecules are important in diverse areas where biomedical mass spectrometry is used.1,2 Mass spectrometry-based metabolomics and proteomics employ isotope distributions of biomolecules routinely. Recently, stable isotope labeling has been a major element for static and as well as dynamic proteome or metabolome studies. Atom-based labeling approaches, such as 2H,3–6 13C,7 or 15N,8–10 for example, or residue-based labeling, such as SILAC, are popular techniques in quantitative proteomics.11–15 The studies use isotopomer distributions of natural peptides and isotopically labeled peptides16 to determine the relative expression of labeled proteins. Therefore, generating isotope distributions of biomolecules from their elemental composition has been a focus of research for some time.16–20 Two main approaches may be classified. The first approach uses binomial and multinomial distributions to compute the isotope distribution of atoms and then uses their convolution to determine the isotope distribution of a biomolecule.21 The approach can be computationally costly when a molecule is large. Its computational complexity scales with the number of atoms polynomially, and techniques have been developed to speed up the calculations.17,22 The second approach uses fast Fourier transform (FFT)-based convolution to compute the isotope distributions of atoms and then of a molecule.19,20 This approach can be faster, but in general, there are advantages to each approach depending on the molecular weight, number of isotopomers, and desired mass resolution.16 The FFT-based approach has large memory requirements for large molecules and high mass resolution data. Due to the computational speed requirements, some bioinformatics applications needing isotope distributions preprocessed the sequences first and store their distributions.21
In this work, we describe a computationally efficient and conceptually simple approach to calculate isotope distributions of biomolecules using a Poisson approximation of binomial and multinomial distributions. We show that, in most of the cases, a single Poisson formula is enough to compute the isotope distribution. The mass distribution of isotopes can be computed from the same Poisson distribution as an expectation value of a distribution. The paper is organized as follows. First, we introduce the notion of isotope distributions of atoms. Then we show how Poisson distribution approximates the combined isotope distributions of hydrogen (H), carbon (C), and nitrogen (N) atoms. Next, we approximate the multinomial isotope distributions of oxygen (O) and sulfur (S) with multiple Poisson distributions. Single isotope distributions from the O and S atoms are naturally incorporated into the combined Poisson distribution of H, C, and N. The final isotope distribution of a molecule is a convolution of relevant isotope distributions of its atoms. The computations use a number of arithmetic operations which are linear in the sum of the O and S atoms. The mass distributions of isotopes of a biomolecule are obtained as expectation values from Poisson distributions.
METHODS
Isotope Distribution of a Peptide
We will consider isotope distributions of biomolecules made of five types of atoms, H, C, N, O, and S. The first three atoms have two naturally occurring isotopes. Isotope distributions of any of these atoms can be computed using a binomial distribution formula:23
| (1) |
where NA is the number of atoms of type A (any of H, C, and N) and pA is the naturally occurring frequency of the isotope of A. Formula 1 is the probability that there are kA isotopes of NA atoms of A.
In the case of O and S atoms, there are more than two (three for O and five for S) naturally occurring isotopes. The corresponding exact probabilities of isotope distributions for these atoms are calculated using a multinomial distribution. For example, for a sulfur atom with four isotopes the distribution is
| (2) |
where NS is the number of S atoms and k0, k1, k2, and k3 are the number of monoisotopic, first, second, and third isotope atoms of S, with the corresponding probabilities of p0, p1, p2, and p3. The general relationships of multinomial distributions hold:
Computation of the isotope distributions using eq 2 is somewhat computationally expensive, and an approach has been developed to do the computations “recursively”.17 We note that another often occurring in a biomolecular atom, phosphorus, has only one naturally occurring isotope and does not contribute to the isotopic complexity of biomolecules and will not be considered here.
The isotope distribution of a biomolecule comprising H, C, N, O, and S atoms can be thought of as the distribution of a random variable, X, which is the sum of the random variables corresponding to each atomic isotope:
| (3) |
where XH, XC, and XN are the binomial random variables distributed according to eq 1 and XO and XS are distributed according to the multinomial distribution (eq 2). While it is possible to compute the isotope distributions of each atom using eqs 1 and 2, there is no exact (or approximate) closed formula for the isotope distribution of a molecule composed of several different atoms (each with a different isotope distribution). The probability distribution of a sum of random variables is a convolution (in the case of the isotope distributions, a discrete convolution) of probability distributions of each random variable and is formally expressed as
| (4) |
Due to the absence of a closed form formula, it is computationally costly to compute the isotope distributions using eq 4, as the number of atoms in biomolecules is large and the number of possible configurations grows polynomially (different combinations of atomic isotopes may contribute to the same molecular isotope). In addition, in a typical proteomics application, tens of thousands of peptides are analyzed in a single workflow. Each of the peptides may need isotope distributions computed. Equation 4 provides a way to compute the relative abundances of the peaks of the isotope patterns. The sum of all isotope abundances is normalized to be equal to 1. The peaks of the isotope pattern are denoted with integers (0, 1, …, n), counting the mass offset from the light (i = 0) isotope (for the atoms that we consider here, it is termed the monoisotope). The monoisotopic peak is made of the atomic isotopes with highest natural abundance, and it corresponds to the lowest mass-to-charge ratio (m/z) in an isotope pattern. To denote the isotopes of a molecule, we use the following nomenclature: (mi, Mi), where the index i is the mass shift from the monoisotope (i = 0). mi and Mi denote the mass of the ith isotope (also referred here to as isotopomer) and its relative abundance, respectively.
In mass spectrometric applications, in addition to the isotope distribution, it is also important to accurately compute the (averaged) mass of each isotope. High mass accuracy and resolution mass spectrometers allow mass accuracy in few parts per million (ppm) or better. Therefore, it becomes necessary to compute the isotope masses with high mass accuracy to take advantage of the capabilities of modern mass spectrometers and compare the computed isotope masses to the experimentally measured values. The mass of a certain isotopomer, mi, is determined as an expectation value of masses of atomic isotopes whose configuration leads to this mass:
| (5) |
where m0 is the mass of the monoisotope, P(kH,kC,kN,kO,kS) is the probability of the numbers of H, C, N, O, and S atoms that results in the mass mi, mH is the mass difference between the deuterium and hydrogen atoms, and k⃗S*m⃗S denotes the fact that there multiple isotopes of a sulfur atom (the vector m⃗S consists of mass differences for each of its heavy isotopes). In eq 5, the sum is taken over all configurations of number of atoms that lead to the ith isotope of a biomolecule. In Figure 1, we show implementation of eq 5 to compute the relative isotope abundances and the corresponding isotope masses. We termed this approach an exact algorithm.
Figure 1.

Pseudocode of algorithm to compute exact isotope distributions. The inputs are the relevant characteristics of each atom. For example, nO is the number of O atoms, Δ17O and Δ18O are the mass shifts of 17O and 18O isotopes (relative to 16O isotope), respectively, and p17O and p18O are the corresponding (normalized) abundances. M0 is the monoisotopic mass. Binom and Multinom stand for binomial and multinomial distributions, respectively.
Poisson Approximation for Isotope Distributions of Multiatomic Molecules
Poisson distribution has a well-known approximation to the binomial distribution24 when the probability at each trial is small (as is the case with the heavy atom isotopes). For example, the probability to observe kA heavy isotopes in an isotope distribution of the H, C, and N atoms is
| (6) |
where λA is the average number of successes, λA = NA × pA. Note the difference between the capital, PA (probability of a Poisson distribution), and lower case, pA (the probability of success in each trial – Bernoulli distribution). The subscript “A” can be any of H, C, and N. For example, λH for 200 H atoms willbe λH = 200 × 0.000115 = 0.023. To compute probability that among 200 randomly chosen hydrogen atoms there will be (for example) 15 deuteriums, we will set kA = 15 and λA = 0.023 in the eq 6. The probability will be the relative intensity of the 16th isotope in the isotopic distribution of 200 H atoms.
Next, we use the simplifications that are provided in the sum of Poisson random variables. The sum of independent Poisson random variables is also a Poisson variable, with the mean equal to the sum of means of original Poisson variables. This is shown in many different ways, and a simple way to show this is to use the moment generating function of each Poisson random variable and the fact that the moment generating function of a sum of independent random variables is the product of their moment generating functions. The random variables corresponding to H, C, and N atoms are independent. We replace the first three terms in the eq 3 with a single Poisson random variable, XHCN, with the mean, λHCN = λH + λC + λN.
Note that the Poisson distribution, unlike the binomial distribution, does not place a limit on the number successes (mini-isotopes, e.g., 2H, 13C, 15N occurrences). In the latter, there cannot be more success than the number of trials. There is no such limit in Poisson distribution. So potentially an artificial outcome where the number of isotopes is larger than the number of atoms seems to be allowed. However, in practice, when the number of successes is more than the actual number of atoms of a given type, the probability is practically zero (long before reaching the limit). In addition, it is possible to limit the contribution to any isotope from any of the H, C, and N atoms by the corresponding number of atoms.
Multinomial distributions also allow a Poisson approximation.25,26 For example, the distribution of minor (heavy) isotopes of oxygen (17O and 18O isotopes) is approximated with
| (7) |
The above approximation uses the fact the major isotope of oxygen has large abundance (probability) compared to the other two, minor isotope abundances. In eq 7, λ17O = p17ONO is the mean of the Poisson distribution for the 17O isotope, p17O is the frequency of this isotope in the nature, and NO is the number of oxygen atoms in a molecule. Probability properties of the 18O isotope are determined similarly.
Similar approach is extended to the isotope distribution resulting from sulfur atoms. The error of approximations, Δn, between the original multinomial distribution and its approximate Poisson distribution is proportional to the sum of the probabilities of minor isotopes:26,27
where n is the number of atoms, O is a “big O”, e is the Euler’s constant, and the summation is over the elemental isotope probabilities that do not include the main isotope. Thus, for this approximation to work well, the sum of minor isotope relative abundances has to be small. In the case of the sulfur atom, the abundance of the second heavy isotope is relatively large, 0.0421, and the sum becomes relatively large and the accuracy of the approximation reduces. Note that the difference with the exact multinomial distribution is less than 1% (as computed using the probabilities of the heavy isotopes of S, and setting n = 1). For the sake of accuracy, it may be preferable to compute the sulfur isotope distribution separately using the multinomial probabilities. In the freely available C/C++ code (https://ispace.utmb.edu/users/rgsadygo/Proteomics/IsotopeDistributions), we have implemented both options. In this work, we have termed this approach as a hybrid (between exact for S atoms and Poisson approximation for H, C, N, and O atoms) approach.
In practical applications, the masses and relative abundances of the monoisotope (i = 0) and of the first heavy isotope (i = 1) require few numerical operations, and they can be computed exactly. The complexity of the isotope calculations arise for the isotopes with indices i ≥ 2.
Once the Poisson approximations for H, C, N, O, and S atoms are used, we can further simplify the computations by recognizing that the first heavy isotopes from both O (17O) and S (33S) atoms have a common property with the H, C, and N isotopes. They all shift the mass value approximately by 1 Da. Therefore, both of these distributions are integrated into the common distribution with H, C, and N atoms. In addition, the second heavy isotopes of O (18O) and S (34S) can be combined to create a single Poisson distribution. At this point, eq 3 is transformed into the following equation:
where XA is a Poisson random variable which integrates the distribution of 2H, 13C, 15N, 17O, and 33S isotopes, X18O34S is a Poisson random variable of joint O (18O) and S (34S) isotopes, and X36S is a Poisson random variable describing the distributions of 36S isotope. No further simplifications are applicable as each term in eq 7 carries a unique mass shift.
The expected mass of an isotopomer, mi, is calculated using features of the Poisson distribution:
The indices have the same meaning as in eq 7. In Figure 2, we provide an algorithm that implements the relative abundance and mass calculations of isotopes using Poisson approximation.
Figure 2.

Pseudocode of algorithm to compute isotope distributions using Poisson approximations. Poisson denotes a Poisson probability with the specified parameters. The rest of the nomenclature is the same as that in Figure 1.
FFT-Based Computation of Isotope Distributions
In one application of an FFT-based isotope distribution computation, the isotope envelope of a basic unit (such as an atom’s isotopes) is coded by an array, X. If isotope fine structure is not a purpose, the arrays of all elements are one-dimensional. Each of the distributions is fast Fourier transformed to obtain the FFT of the elemental isotopes. Each one of the FFTs of elemental isotope distribution is raised to the corresponding power (the number of atoms of the particular type in the molecule). The product of all FFTs is obtained. The product is inverse Fourier transformed for the isotope distribution of a molecule. The equivalence between the convolution of two vectors and the inverse FFT of a product of forward FFTs of each vector is used. The FFT-based algorithms scale as O(K log(K)), where K is the length of a vector (in this case, the number of isotopes to be computed). The FFT approach to the isotope calculations has been described previously.20 Recent publications have provided codes of applications of this algorithm in R (ecipex algorithm28) and in Matlab (a Matlab version29 of the Taverna workflow in R). These applications are particularly suitable for calculations of isotope fine structure. The mass accuracy and resolution of the modern mass spectrometers continuous to improve. At high mass resolution, it is possible to discern isotope fine structure–isotope distributions of particular isotopologues (molecules that have the same chemical and isotope compositions and differ only on the position of the isotopes). It should be noted the mass resolution requirement to resolve isotope fine structure is higher for resolution of higher isotopologues (isotopologues having more than two or more nonabundant atomic isotopes). In the Supporting Information’s Figure S1, we provide an algorithm and its C/C++ and R implementations.
Recursive Computation of Isotope Distributions
Another approach for generating isotope fine structures computes isotopologue distributions recursively.30,31 This approach uses the fact that the binomial and multinomial probability mass functions can be computed recursively. For example, if two isotopologues (x and y) of an atom differ in exchange of one atomic isotope (assumed natural abundance, p1) to another (assumed natural abundance, p2), then the probability of new isotopologue, Py, resulting from this transition is the same (in terms of Px) for both binomial and multinomial distributions and is equal to
| (8) |
where is the number of first isotope types in the isotopologue x, is the number of the second isotope types in isotopologue y. enviPat uses eq 8 to first estimate the highest probability atomic isotopes, then applies a user-adjustable filter to filter out low probability isotopologues.30 An indexing scheme is used to avoid redundant transitions. The isotopologue probabilities of each atom type computed using eq 8 are combined via the Cartesian product to compute the isotopologue probabilities of molecules.
Note that the recursive calculations based on eq 8 use five arithmetic operations to determine probability of each isotopologues resulting from a single exchange of two different atomic isotopes. The analogous formula for Poisson distribution will have only three arithmetic operations in the case of atoms with only two isotopes (C, H, N):
In the case of the three or more atomic isotopes (O and S), the transition probability formula is similar to the one in eq 8, with p2 and p1 replaced with λy and λx, respectively. Thus, in applications to recursive algorithms, there are computational gains in computing probabilities of isotopologues of atoms with two isotopes. For each atom the total number of isotopologues is computed using a binomial coefficient, , where n is the number of atoms of a given type and i is the number of its isotopes.32 The formula is obtained as a sampling from a set (isotopes of an atom) with replacement and without ordering. The number of isotopologues can become large, and pruning techniques are used filter out low probability isotopologues.30,33
RESULTS AND DISCUSSION
For our calculations, we used atomic isotope distributions recently published in the report of the Institute of Pure and Applied Chemistry.34 In the report, there are two possible isotope distribution choices for each atomic element. One presents the range of values for each isotope, the second is the most accurately determined isotope ratios. We used the most accurately determined isotope ratio values. The natural isotope distributions used in this study are provided in the Supporting Information, Table S1.
To compare the computational speed, we applied Poisson-based and FFT-based isotope distribution to compute isotope distributions of all tryptic peptides in the UniProt database35 downloaded in May of 2015. Three missed cleavages were allowed, and peptides ranging in length between 7 and 41 amino acids were considered. Only 16 isotopes for each tryptic peptide were calculated to exclude the possible effects of memory requirements in the FFT-based method. Theoretical isotope distributions were generated for more than 46 million peptide sequences from 548 454 protein entries. It took the Poisson-based method about 4.8 min to compute isotope distributions of all peptides. The FFT-based method took 14.1 min for the same task (on the same computer). In Figure 3, we show the density of the ratios of relative abundances of the first heavy isotopes as computed by FFT- and Poisson-based methods. As it is seen from this Figure, Poisson-based method accurately predicts the isotope distributions (median and mean of the deviations were the same and equal to 2%; the range of the accuracy was between 0.05 and 7%, and the mean and standard deviation were 0.15 and 0.09%, respectively).
Figure 3.

Ratios of M1 intensities as calculated by Poisson to FFT.
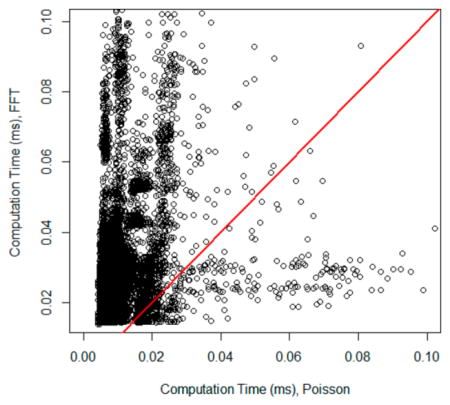
In a separate comparison, we compared the time it took to compute isotope distributions of each peptide sequence by the FFT- and Poisson-based algorithms. Figure 4 shows the detailed comparison of isotope generation times by the FFT-and Poisson-based algorithms. It is a scatter plot of the times for all peptides with each methods. The red line is the line of unity. For less than 0.1% of peptides, the FFT-based algorithm was faster. For the rest of the peptides Poisson-based algorithm performed faster. The time comparison was done sequentially—for each peptide sequence, isotope distribution was first generated by one method and then the other one. We changed the ordering of methods to make sure that there was no dependence on the order in which a method was called. The general features of time distribution for the methods agreed with the overall time comparisons mentioned about (4.8 min versus 14.1 min). Thus, the median of isotope calculation time with the Poisson method was 0.0053 ms, and the FFT was 0.018 ms.
Figure 4.

Scatter plot of computation times of isotope generation using FFT-based (y-axis) and Poisson-based (x-axis) methods. The red line is the line of unity.
We have also compared the running times of our code with those of enviPat. enviPat is available as an R code. R is an interpretable environment, and normally codes take longer to run in this environment. For the comparison, we only recorded the time it takes to run the “isopattern” function in enviPat for atomic composition of each peptide. It took considerably longer to compute the isotope distributions in enviPat (Supporting Information, Figure S2). We believe that the fact the enviPat runs in R environment has contributed to the execution time difference. In addition, enviPat provide fine isotope structure, while our code provided only isotopomer distributions.
Breen et al.36 have used Poisson distribution to predict the isotope distributions of peptides based on the peptide mass. For this purpose, they used the concept of averagine37 amino acid, generated 15 peptides composed of one through 15 averagine amino acids, used a fitting to these data (15 data points) to determine the mean of the Poisson distribution. The mean was a linear function of peptide mass and this information can be used to predict isotope distribution of a peptide based only on peptide mass. Valkenborg et al.38 extended this approach to include sulfur atom, as well. These Poisson model-based approaches predict the isotope distribution of a peptide species based on its mass only. They are important in different applications, for example, for peak picking or deconvolving overlapping isotope profiles36 where the amino acid sequence of a peptide is unknown. Our approach will facilitate the computations of theoretical isotopes in high-throughput data processing of stable isotope labeling proteomics experiments such as experiments with metabolic labeling. As described above our approach has no restrictions in terms of the sulfur atoms.
Patterns of relative isotope abundances are regularly used in interrogation of mass spectrometric data, e.g., for relative quantification or proteome turnover studies.39,40 Some of the application tools use information on both relative isotope abundances and isotope masses.41 The calculations of the masses of isotopes are computationally the most challenging element of isotope generation. The algorithms for relative isotope abundance calculations (such as FFT-based algorithm) implicitly uses the fact that occupational numbers of isotopes are integer values. On the other hand, the masses of isotopes are not integers and therefore may need to be explicitly calculated. In general, there are eight, (3*1 + 2 + 3), embedded loops for computing isotope masses. This makes the computational problem of polynomial complexity. The fully Poisson approach described above avoids most of the embedded loops, and has only three embedded loops. However, the technique did not produce highly accurate masses for all isotopes of peptides. While for the peptides that did not contain sulfur atoms, the largest error in an isotope distribution (of all isotopes with relative abundance large than 0.001 of the tallest isotope peak in the isotope envelope) was less than 1 ppm for peptides that contained sulfur atoms, the mass accuracy was low as 10 ppm (for high mass isotopes). To address this problem, we tested a hybrid approach to simplify the isotope mass calculation and increase the mass accuracy of the calculations by including exact calculations for S atoms. In this approach, we, at first, compute the isotope masses of joint distribution of hydrogen, carbon, and nitrogen atoms using Poisson approximation for these atoms. At the second stage, we compute the isotope masses of joint distribution from the first stage and that of oxygen atoms also computed using Poisson approximation. At the third, final stage, we incorporate the mass shifts due to the isotopes of the sulfur atoms using an exact approach. In this approach, we have three embedded loops (one for each: combined isotope distributions of hydrogen, carbon, and nitrogen atoms; and the isotopes of sulfur atoms). As a result of this “decomposition” of isotope mass calculations into simplified calculations with smaller number of embedded loops, we obtain improved speed of computations. The mass accuracy from this approach was very high—better than 1 ppm for sulfur-containing peptides (the mean of difference was 0.06 ppm and standard deviation 0.012 ppm) and better than 1 part per billion (ppb) for peptides that do not contain a sulfur atom (the mean of difference was 0.4 ppb and standard deviation 0.02 ppb). The hybrid algorithm is shown in Figure 5. The results of its application and the scatter plot of mass accuracy (the worst accuracy for all isotopes that have relative abundance larger than 0.1% of the maximum abundance isotope) against the relative isotope abundance is illustrated in Figure 6.
Figure 5.

Pseudocode of a “hybrid” algorithm to compute isotope distributions using Poisson approximation and exact distributions. The nomenclature is the same as that in Figures 1 and 2. For brevity, we did not show the computations for S atoms (they are similar to the O atom calculations).
Figure 6.

Scatter plot of mass accuracy. Shown are the worst accuracy in each peptide’s isotope cluster. The only filtering used was the requirement that an isotopomer abundance needed to be larger than 0.1% of the maximum abundance isotope for the particular peptide under consideration. Data for 1000 randomly chosen peptides are shown. The x axis shows the absolute values of the relative abundances. The sum of all isotope abundances is normalized to equal 1.
In the case of Poisson approximation, as with many computational approaches, various combinations of approximations may be beneficial dependent on specific needs of an application. Here, we have tested a fully Poisson approximated and hybrid approach to generate isotope distributions of peptides from their amino acid compositions. If only relative abundances are needed, then the fully Poisson approximation is very fast and accurate. If masses of isotopes are also required, then (in particular, for high mass isotopes) a combination of Poisson approximations with exact convolution is a preferable approach.
CONCLUSIONS
We have developed an approximation to accurately generate isotope distributions of biomolecules. Our approach is based on the Poisson approximations to multinomial and binomial distributions. It uses a closed formula and has no requirements for memory storage. We show that the relative isotope abundances are calculated with a high accuracy (Figure 3). The relative isotope calculations are 3-fold faster than the ultrafast isotope calculations using FFT, which are also provided in the freely available code.
The mass accuracy of the masses of isotopomers is in low parts per million. To improve this accuracy, we have proposed and validated a hybrid approach which uses the Poisson approximation for H, C, N, and O atoms and exact calculations for the S atom. In the hybrid approach, we obtain mass accuracy better that 1 ppm.
We conclude that for peptides that do not contain a sulfur atom, the Poisson approximation provides very accurate computation of masses and relative abundances of isotopomers. For peptides containing sulfur atoms, a hybrid approach is suggested, where we use Poisson approximations for all atoms but the sulfur atom. The hybrid approach results in mass accuracy better than 1 ppm for all isotopes with abundance more than 1000th of the maximum abundance isotope (tallest isotope peak in an isotope envelope).
Supplementary Material
Acknowledgments
Research reported in this publication was supported in part by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number R01GM112044.
ABBREVIATIONS
- Binom
binomial distribution
- FFT
fast Fourier transform
- invFFT
inverse Fourier transform
- LC-MS
liquid chromatography–mass spectrometry
- Multinom
multinomial distribution
- MS
mass spectrometry
- ppm
parts per million
- ppb
parts per billion
Footnotes
Notes
The author declares no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jproteome.7b00807.
Table S1 and Figures S1 and S2 (PDF)
References
- 1.Aebersold R, Mann M. Mass-spectrometric exploration of proteome structure and function. Nature. 2016;537:347–355. doi: 10.1038/nature19949. [DOI] [PubMed] [Google Scholar]
- 2.Zhang Y, Fonslow BR, Shan B, Baek MC, Yates JR., III Protein analysis by shotgun/bottom-up proteomics. Chem Rev. 2013;113:2343–2394. doi: 10.1021/cr3003533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kasumov T, Ilchenko S, Li L, Rachdaoui N, Sadygov RG, Willard B, McCullough AJ, Previs S. Measuring protein synthesis using metabolic (2)H labeling, high-resolution mass spectrometry, and an algorithm. Anal Biochem. 2011;412:47–55. doi: 10.1016/j.ab.2011.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim TY, Wang D, Kim AK, Lau E, Lin AJ, Liem DA, Zhang J, Zong NC, Lam MP, Ping P. Metabolic labeling reveals proteome dynamics of mouse mitochondria. Mol Cell Proteomics. 2012;11:1586–1594. doi: 10.1074/mcp.M112.021162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Salisbury JP, Liu Q, Agar JN. QUDeX-MS: hydrogen/deuterium exchange calculation for mass spectra with resolved isotopic fine structure. BMC Bioinf. 2014;15:403. doi: 10.1186/s12859-014-0403-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Naylor BC, Porter MT, Wilson E, Herring A, Lofthouse S, Hannemann A, Piccolo SR, Rockwood AL, Price JC. DeuteRater: a tool for quantifying peptide isotope precision and kinetic proteomics. Bioinformatics. 2017;33:1514–1520. doi: 10.1093/bioinformatics/btx009. [DOI] [PubMed] [Google Scholar]
- 7.Jehmlich N, Fetzer I, Seifert J, Mattow J, Vogt C, Harms H, Thiede B, Richnow HH, von Bergen M, Schmidt F. Decimal place slope, a fast and precise method for quantifying 13C incorporation levels for detecting the metabolic activity of microbial species. Mol Cell Proteomics. 2010;9:1221–1227. doi: 10.1074/mcp.M900407-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guan S, Price JC, Prusiner SB, Ghaemmaghami S, Burlingame AL. A data processing pipeline for mammalian proteome dynamics studies using stable isotope metabolic labeling. Mol Cell Proteomics. 2011;10:M111.010728. doi: 10.1074/mcp.M111.010728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vogt JA, Schroer K, Holzer K, Hunzinger C, Klemm M, Biefang-Arndt K, Schillo S, Cahill MA, Schrattenholz A, Matthies H, Stegmann W. Protein abundance quantification in embryonic stem cells using incomplete metabolic labelling with 15N amino acids, matrix-assisted laser desorption/ionisation time-of-flight mass spectrometry, and analysis of relative isotopologue abundances of peptides. Rapid Commun Mass Spectrom. 2003;17:1273–1282. doi: 10.1002/rcm.1045. [DOI] [PubMed] [Google Scholar]
- 10.Rauniyar N, McClatchy DB, Yates JR., 3rd Stable isotope labeling of mammals (SILAM) for in vivo quantitative proteomic analysis. Methods. 2013;61:260–268. doi: 10.1016/j.ymeth.2013.03.008. [DOI] [PubMed] [Google Scholar]
- 11.Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 2002;1:376–386. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- 12.Li L, Willard B, Rachdaoui N, Kirwan JP, Sadygov RG, Stanley WC, Previs S, McCullough AJ, Kasumov T. Plasma proteome dynamics: analysis of lipoproteins and acute phase response proteins with 2H2O metabolic labeling. Mol Cell Proteomics. 2012;11:M111.014209. doi: 10.1074/mcp.M111.014209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu CC, MacCoss MJ, Howell KE, Matthews DE, Yates JR., III Metabolic labeling of mammalian organisms with stable isotopes for quantitative proteomic analysis. Anal Chem. 2004;76:4951–4959. doi: 10.1021/ac049208j. [DOI] [PubMed] [Google Scholar]
- 14.Zhang T, Price JC, Nouri-Nigjeh E, Li J, Hellerstein MK, Qu J, Ghaemmaghami S. Kinetics of precursor labeling in stable isotope labeling in cell cultures (SILAC) experiments. Anal Chem. 2014;86:11334–11341. doi: 10.1021/ac503067a. [DOI] [PubMed] [Google Scholar]
- 15.Ahmed Z, Zeeshan S, Huber C, Hensel M, Schomburg D, Munch R, Eisenreich W, Dandekar T. Software LS-MIDA for efficient mass isotopomer distribution analysis in metabolic modelling. BMC Bioinf. 2013;14:218. doi: 10.1186/1471-2105-14-218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sperling E, Bunner AE, Sykes MT, Williamson JR. Quantitative analysis of isotope distributions in proteomic mass spectrometry using least-squares Fourier transform convolution. Anal Chem. 2008;80:4906–4917. doi: 10.1021/ac800080v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yergey J, Heller D, Hansen G, Cotter RJ, Fenselau C. Isotopic Distributions in Mass-Spectra of Large Molecules. Anal Chem. 1983;55:353–356. [Google Scholar]
- 18.Yergey JAA. General-Approach to Calculating Isotopic Distributions for Mass-Spectrometry. Int J Mass Spectrom Ion Phys. 1983;52:337–349. doi: 10.1002/jms.4498. [DOI] [PubMed] [Google Scholar]
- 19.Rockwood AL, Van Orden SL, Smith RD. Rapid Calculation of Isotope Dsitributions. Anal Chem. 1995;67:2699–2704. [Google Scholar]
- 20.Rockwood AL, VanOrden SL. Ultrahigh-speed calculation of isotope distributions. Anal Chem. 1996;68:2027–2030. doi: 10.1021/ac951158i. [DOI] [PubMed] [Google Scholar]
- 21.Bocker S, Letzel MC, Liptak Z, Pervukhin A. SIRIUS: decomposing isotope patterns for metabolite identification. Bio-informatics. 2009;25:218–224. doi: 10.1093/bioinformatics/btn603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Claesen J, Dittwald P, Burzykowski T, Valkenborg D. An efficient method to calculate the aggregated isotopic distribution and exact center-masses. J Am Soc Mass Spectrom. 2012;23:753–763. doi: 10.1007/s13361-011-0326-2. [DOI] [PubMed] [Google Scholar]
- 23.Lee WN, Byerley LO, Bergner EA, Edmond J. Mass isotopomer analysis: theoretical and practical considerations. Biol Mass Spectrom. 1991;20:451–458. doi: 10.1002/bms.1200200804. [DOI] [PubMed] [Google Scholar]
- 24.Feller W. An Introduction to Probability Theory and Its Applications. 3. I Wiley & Sons; 1968. [Google Scholar]
- 25.Arenbaev NK. Asymptotic-Behavior of Multinomial Distribution. Theory Probab Its Appl. 1977;21:805–810. [Google Scholar]
- 26.Deheuvels P, Pfeifer D. Poisson Approximations of Multinomial Distributions and Point-Processes. J Multivariate Anal. 1988;25:65–89. [Google Scholar]
- 27.McDonald DR. On the Poisson Approximation to the multinomial distribution. Canadian Journal of Statistics. 1980;8:115–118. [Google Scholar]
- 28.Ipsen A. Efficient calculation of exact fine structure isotope patterns via the multidimensional Fourier transform. Anal Chem. 2014;86:5316–5322. doi: 10.1021/ac500108n. [DOI] [PubMed] [Google Scholar]
- 29.Rockwood AL, Palmblad M. Isotopic distributions. Methods Mol Biol. 2013;1007:65–99. doi: 10.1007/978-1-62703-392-3_3. [DOI] [PubMed] [Google Scholar]
- 30.Loos M, Gerber C, Corona F, Hollender J, Singer H. Accelerated isotope fine structure calculation using pruned transition trees. Anal Chem. 2015;87:5738–5744. doi: 10.1021/acs.analchem.5b00941. [DOI] [PubMed] [Google Scholar]
- 31.Li L, Karabacak NM, Cobb JS, Wang Q, Hong P, Agar JN. Memory-efficient calculation of the isotopic mass states of a molecule. Rapid Commun Mass Spectrom. 2010;24:2689–2696. doi: 10.1002/rcm.4666. [DOI] [PubMed] [Google Scholar]
- 32.Sadygov RG. Using SEQUEST with Theoretically Complete Sequence Databases. J Am Soc Mass Spectrom. 2015;26:1858–1864. doi: 10.1007/s13361-015-1228-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lacki MK, Startek M, Valkenborg D, Gambin A. IsoSpec: Hyperfast Fine Structure Calculator. Anal Chem. 2017;89:3272–3277. doi: 10.1021/acs.analchem.6b01459. [DOI] [PubMed] [Google Scholar]
- 34.Meija J, Coplen TB, Berglund M, Brand WA, De Bievre P, Groning M, Holden NE, Irrgeher J, Loss RD, Walczyk T, Prohaska T. Isotopic compositions of the elements 2013 (IUPAC Technical Report) Pure Appl Chem. 2016;88:293–306. [Google Scholar]
- 35.Bairoch A, Apweiler R, Wu CH, Barker WC, Boeckmann B, Ferro S, Gasteiger E, Huang H, Lopez R, Magrane M, Martin MJ, Natale DA, O’Donovan C, Redaschi N, Yeh LS. The Universal Protein Resource (UniProt) Nucleic Acids Res. 2004;33:D154–D159. doi: 10.1093/nar/gki070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Breen EJ, Hopwood FG, Williams KL, Wilkins MR. Automatic poisson peak harvesting for high throughput protein identification. Electrophoresis. 2000;21:2243–2251. doi: 10.1002/1522-2683(20000601)21:11<2243::AID-ELPS2243>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 37.Senko MW, Beu SC, McLaffertycor FW. Determination of Monoisotopic Massed and Ion Populations for Large Biomolecules from Resolved Isotopic Distributions. J Am Soc Mass Spectrom. 1995;6:229–233. doi: 10.1016/1044-0305(95)00017-8. [DOI] [PubMed] [Google Scholar]
- 38.Valkenborg D, Assam P, Thomas G, Krols L, Kas K, Burzykowski T. Using a Poisson approximation to predict the isotopic distribution of sulphur-containing peptides in a peptide-centric proteomic approach. Rapid Commun Mass Spectrom. 2007;21:3387–3391. doi: 10.1002/rcm.3237. [DOI] [PubMed] [Google Scholar]
- 39.Rahman M, Previs SF, Kasumov T, Sadygov RG. Gaussian Process Modeling of Protein Turnover. J Proteome Res. 2016;15:2115–2122. doi: 10.1021/acs.jproteome.5b00990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lam MP, Wang D, Lau E, Liem DA, Kim AK, Ng DC, Liang X, Bleakley BJ, Liu C, Tabaraki JD, Cadeiras M, Wang Y, Deng MC, Ping P. Protein kinetic signatures of the remodeling heart following isoproterenol stimulation. J Clin Invest. 2014;124:1734–1744. doi: 10.1172/JCI73787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Naylor BC, Porter MT, Wilson E, Herring A, Lofthouse S, Hannemann A, Piccolo SR, Rockwood AL, Price JC. DeuteRater: a Tool for Quantifying Peptide Isotope Precision and Kinetic Proteomics. Bioinformatics. 2017:btx009. doi: 10.1093/bioinformatics/btx009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.