

Abstract
Background
Immunotherapy targeting the amyloid-β (Aβ) peptide is a promising strategy for the treatment of Alzheimer’s disease (AD); however, none of the active or passive vaccines tested have been demonstrated to be effective to date. We have developed the first active vaccine against the C-terminal end of Aβ40, ABvac40, and assessed its safety and tolerability in a phase I clinical trial.
Methods
A randomised, double-blind, placebo-controlled, parallel-group, phase I study of ABvac40 was conducted with patients aged 50–85 years with mild to moderate AD. Participants were entered into three separate groups according to time of study entry and were randomly allocated to receive ABvac40 or placebo (overall ratio 2:1). The first group received two half-doses of ABvac40 or placebo, whereas the second and third groups received two and three full doses, respectively. All treatments were administered subcutaneously at 4-week intervals. Patients, carers and investigators were blind to treatment allocation throughout the study. The primary objective was to assess the safety and tolerability of ABvac40 by registering all adverse events (AEs). All patients who received at least one dose of treatment were included in the safety analysis. The secondary objective was to evaluate the immunogenicity of ABvac40 by titration of specific anti-Aβ40 antibodies in plasma.
Results
Twenty-four patients were randomly allocated: 16 patients to the ABvac40 group and 8 patients to the placebo group. All randomised patients completed the study, therefore the intention-to-treat and safety populations were identical. Overall, 71 AEs affecting 18 patients were recorded: 11 (69%) in the ABvac40 group and 7 (88%) in the placebo group (p = 0.6214). Neither incident vasogenic oedema nor sulcal effusion (amyloid-related imaging abnormalities corresponding to vasogenic oedema and sulcal effusions) nor microhaemorrhages (amyloid-related imaging abnormalities corresponding to microhaemorrhages and hemosiderin deposits) were detected throughout the study period in the ABvac40-treated patients. Eleven of 12 (~92%) individuals receiving three injections of ABvac40 developed specific anti-Aβ40 antibodies.
Conclusions
ABvac40 showed a favourable safety and tolerability profile while eliciting a consistent and specific immune response. An ongoing phase II clinical trial is needed to confirm these results and to explore the clinical efficacy of ABvac40.
Trial registration
ClinicalTrials.gov, NCT03113812. Retrospectively registered on 14 April 2017.
Electronic supplementary material
The online version of this article (10.1186/s13195-018-0340-8) contains supplementary material, which is available to authorized users.
Keywords: Alzheimer’s disease, Amyloid-β, Aβ, Immunotherapy, ABvac40, Phase I
Background
Alzheimer’s disease (AD) is the most common type of dementia, accounting for 50–75% of the estimated 47 million people with dementia worldwide [1]. AD is defined as a neurodegenerative disorder clinically characterised by progressive memory loss and cognitive decline. Currently, there is no effective treatment, and currently approved drugs provide only modest symptomatic benefit. Therefore, development of disease-modifying drugs is of great importance.
The amyloid cascade hypothesis of AD proposes that amyloid-β (Aβ) peptide accumulation in the brain, caused by an imbalance between Aβ production and clearance, is the initiating factor of a cascade of pathogenic events, including the formation of neurofibrillary tangles (NFTs), oxidative stress, neuroinflammation, synaptic dysfunction and neuronal loss, which eventually leads to AD dementia [2, 3]. In recent years, several active immunotherapies targeting Aβ have progressed from preclinical studies in AD mouse models to clinical trials in humans [4]; however, none of the approaches tested have shown clinical efficacy so far [5].
Several isoforms of Aβ are generated from sequential proteolytic cleavage of the amyloid precursor protein (APP), including Aβ40 and Aβ42. Aβ40 is the predominant variant (90%) among the secreted Aβ forms [6–8], and although Aβ42 is more hydrophobic and prone to aggregate, and Aβ42 oligomers are regarded to be the most neurotoxic species, Aβ40 can also produce highly toxic diffusible aggregates [9], which can be prevented in vitro by specific anti-Aβ40 antibodies [10]. Accordingly, researchers in several studies have proposed that a high concentration of Aβ40 in the brain distinguishes patients with AD from those who have senile plaques but are cognitively normal, pointing to the importance of Aβ40 in the onset of dementia, both in AD and in Down syndrome [11–13]. In addition, Aβ40 is the main component of amyloid deposition occurring in cerebral amyloid angiopathy (CAA) [14], which has a prevalence of about 80–90% in patients with AD [15]. In keeping with this, previous studies have demonstrated that specific anti-Aβ40 antibodies label intra- and extra-neuronal NFTs in the entorhinal cortex and the hippocampus of AD brains, and that these do not co-localise with tau NFTs, suggesting the presence of degenerating neuronal populations filled with C-terminal fragments of Aβx-40 [16].
Considering all previous results suggesting that strategies targeting Aβ40 could represent novel disease-modifying therapies, we have developed ABvac40, the first active vaccine targeting the C-terminal end of the Aβ40 peptide. Unlike N-terminal end Aβ-directed antibodies, which could recognise both Aβ and their parental APP while inserted in the cell membrane, anti-C-terminal end Aβ antibodies do not bind to the unprocessed protein, preventing the accumulation of potentially toxic antigen-antibody complexes around neurons and other APP-expressing cells, which further increases the availability of circulating antibodies to interact with Aβ peptides. In addition, C-terminal (and not N-terminal) end Aβ-directed antibodies generated by ABvac40 could provide protection against N-terminally truncated and/or modified Aβ peptides, such as pyroglutamate-3 Aβ, which have been described to be highly toxic and prone to aggregation [17–21].
Therefore, the aim of this study was to assess the safety and tolerability of repeated subcutaneous administrations of an active vaccine against the C-terminal end of Aβ40 in patients with mild to moderate AD. In addition, we evaluated ABvac40 biological activity in terms of the immune response induced in participants by determining the plasma levels of anti-Aβ40 antibodies.
Methods
A randomised, double-blind, placebo-controlled, parallel-group, single-centre, phase I study was done at the Memory Clinic and Research Center of Fundació ACE (Barcelona, Spain) to assess the safety and tolerability of repeated subcutaneous administrations of ABvac40 in patients with mild to moderate AD. The study was initiated upon approval by the independent ethics committee of the Barcelona Hospital Clinic and was conducted in accordance with the ethical and scientific principles described in the Declaration of Helsinki and International Conference on Harmonisation Guideline for Good Clinical Practice (CPMP/ICH/135/95), European guidelines for clinical trials (2001/20/CE) and Spanish legislation (Royal Decree 223/2004 of 6 February, which regulates clinical drug trials). A data and safety monitoring board of medical experts in the fields of neurology and immunology monitored the trial.
Participants
The study population consisted of men and women aged 50–85 years with a clinical diagnosis of probable AD based on National Institute of Neurological and Communicative Diseases and Stroke/Alzheimer’s Disease and Related Disorders Association criteria and a Mini Mental State Examination score of 15–26 (mild to moderate AD). Patients were excluded if they had a history or indications of any other central nervous system disorder that could be the cause of dementia or a history or indications of cerebrovascular disease or diagnosis of possible, probable or definite vascular dementia (National Institute of Neurological Disorders and Stroke-Association Internationale pour la Recherche et l’Enseignement en Neurosciences criteria). Conventional treatments for AD were permitted if they were administered at a stable dose for at least 3 months before screening and were maintained throughout the trial. Further details of the inclusion and exclusion criteria are available in Additional file 1. All participants provided written informed consent before enrolment.
Study design
Participants were randomised into two treatment groups receiving ABvac40 or placebo. To minimise the risk associated with the use of ABvac40 in humans for the first time, a stepped recruiting protocol was followed (Fig. 1). The first four patients were randomised and treated sequentially on consecutive days with half the intended dose (two with ABvac40 and two with placebo). Once these patients successfully completed the safety control after the second injection, a second group of four patients was randomised and treated with the full dose (two with ABvac40 and two with placebo). After these eight patients, following the initial protocol (IP), had completed the safety control after the second dose, an interim analysis was carried out to monitor the immune response, maintaining the double-blinding of the study. Based on the results of this interim analysis, the protocol was amended to introduce an additional third immunisation. Thus, the remaining 16 patients (12 ABvac40 and 4 placebo) following the amended protocol (AP) received three full immunisations. On the whole, the ABvac40/placebo ratio was 2:1. The randomisation lists were prepared by an independent statistician using SAS software (SAS Institute, Cary, NC, USA). Further details about study randomisation are provided in Additional file 2.
Fig. 1.
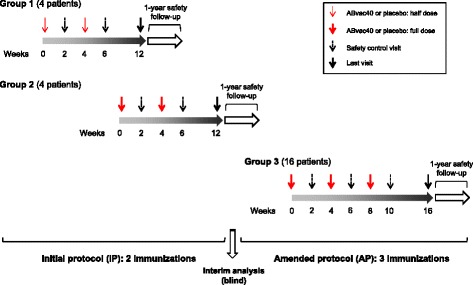
Study design
ABvac40 and placebo treatments were dispensed in identical vials to make them indistinguishable. Only an independent representative of the sponsor worked without blinding to label the treatment kits. Patients, carers, investigators and all staff involved with the trial were blind to treatment allocation throughout the study; however, the principal investigator was permitted to unmask the treatment in case of a medical emergency.
Procedures
In total, over the treatment period spanning 4 or 8 weeks (IP or AP, respectively), the patients received two or three administrations (IP or AP, respectively) at 4-week intervals. The vaccine was administered subcutaneously. Each dose consisted of 1 ml of ABvac40 containing 200 μg of Aβ33–40 peptide coupled to monomeric keyhole limpet haemocyanin suspended in the vaccine vehicle (phosphate buffer with 0.35% aluminium hydroxide in the form of Alhydrogel® [InvivoGen, San Diego, CA, USA] as adjuvant). The placebo consisted of the vaccine vehicle without the immunogenic conjugate.
Study visits were scheduled to follow a logical sequence to monitor patient safety and compliance with trial requirements. Up to 4 weeks before treatment, a screening visit and a baseline visit were carried out to ensure the suitability of the patients for the clinical trial and to define their baseline characteristics. These visits included physical and neurological examinations, blood tests, urinalysis, magnetic resonance imaging (MRI) scans, electrocardiograms (ECGs) and neuropsychological tests. MRI scans were performed in 1.5-Tesla magnets. A standard protocol was used with the following sequence: 3D T1-weighted, Alzheimer’s Disease Neuroimaging Initiative sequence; fluid-attenuated inversion recovery (FLAIR), 2D axial T2-weighted (T2W) FLAIR; T2*-weighted, axial 2D gradient echo; T2W, axial 2D spin echo; diffusion-weighted image and associated apparent diffusion coefficient map. Mesial temporal atrophy was assessed using the Scheltens scale, and detection of amyloid-related imaging abnormalities (ARIAs) was performed according to published criteria [22, 23]. As a safety measure, patients were hospitalised for drug administration at Clínica CIMA in Barcelona and kept under observation for the first 24 h. The patients were discharged from hospital only if stable and there was no reasonable suspicion of a possible allergic reaction. Two or three days later, the status of the participants was checked via a telephone interview. In addition, 2 weeks after each vaccination, the patients underwent a full safety control visit, including a control MRI scan, blood test, urinalysis and a complete physical and neurological examination. Six weeks after the last safety control visit, the final visit took place. After concluding participation in the study, patients were followed for at least 1 additional year for long-term safety control. This open-label follow-up consisted of four additional visits, which included supplementary MRI scans and blood tests.
Outcomes
The primary objective of the study was to evaluate the safety and tolerability of multiple administrations of ABvac40 in patients with mild to moderate AD. The main variable to assess was the frequency (%) of adverse events (AEs). In this regard, special efforts were made to evaluate potential neurological AEs (cerebrovascular events, extrapyramidal symptoms, disorientation, increased gait impairment and occurrence of seizures), psychiatric AEs (hallucinations and other signs and symptoms of affective or psychotic disorders, disorientation, agitation and aggressive behaviour) and cardiovascular AEs (orthostatic hypotension, induced arrhythmias and/or increased risk of myocardial infarction). Safety assessments included the recording of all AEs, regular MRI scans, physical and neurological examinations, laboratory assessments (standard haematology, blood biochemistry and urinalysis panels), ECGs, investigator global evaluation (Clinical Global Impression of Change), assessment of vital signs and body mass index.
The secondary objective of the study was to evaluate ABvac40 biological activity in terms of the immune response induced in the participants by determining the levels of anti-Aβ40 antibodies in plasma, measured as the mean optical density (MOD) signal from three replicated titration enzyme-linked immunosorbent assays (ELISAs) in 96-well plates coated with the Aβ1–40 peptide. Antibodies bound to immobilised Aβ1–40 were detected with anti-human immunoglobulin G (IgG)-specific secondary antibodies coupled to horseradish peroxidase. The MOD of samples with a reported overflow by the ELISA reader was equalled to 4.08 (maximum reading value). The maximal signal increment (MSΔ) was calculated for each subject as the difference between the maximal MOD at any post-baseline visit and the MOD at baseline. To evaluate if the increment of signal was due to specific anti-Aβ40 antibodies, aliquoted parts of the test samples were pre-adsorbed with Aβ33–40 peptide and then processed by titration ELISAs in parallel with the non-pre-adsorbed samples. Patients were classified as positive responders to ABvac40 if, at a 1:10 plasma dilution, the signal increment at any post-treatment visit regarding the baseline was ≥ 3 SD and such increment was reduced in the pre-adsorbed sample by ≥ 50%. The ABvac40 biological activity in the subjects in the ABvac40 group was also expressed in antibody titres, defined as the inverse of the maximal plasma sample dilution which showed an increase in MOD ≥ 3 SD with regard to the baseline sample.
The reactivity of selected plasma samples with amyloid plaques was assessed in brain sections from 9.5-month-old APP/PS1-transgenic mice and patients with AD by immunohistochemistry as described elsewhere (plasma samples diluted 1:500 in 0.5% Triton X-100 PBS were used as the primary antibody) [24]. The ability of the antibodies raised by ABvac40 to target different forms of Aβ was analysed by immunoblotting. Briefly, Aβ1–40 or Aβ1–42 synthetic oligomers were resolved onto Tris-Tricine gels, transferred to nitrocellulose membranes and blotted with diluted plasma samples.
Additional secondary variables were assessed for exploratory purposes. The levels of Aβ peptides in plasma (Aβ40 and Aβ42) were quantified by using an Aβ ELISA kit following the manufacturer’s instructions (Araclon Biotech, Zaragoza, Spain). The levels of cytokines in plasma (interleukin [IL]-6, tumour necrosis factor-α, IL-1β, monocyte chemoattractant protein 1, IL-2 and soluble IL-2 receptor) were determined by a certified clinical analysis laboratory (Laboratorios Echevarne, Barcelona, Spain).
Statistical analysis
Owing to the exploratory nature of the study, a formal statistical estimation of the sample size was not made. In general, categorical data were presented as counts and percentages in each category, and continuous data were reported using number of patients, mean value, SD and SEM.
The number of AEs and the percentage of patients with AEs, overall and grouped as neurological, psychiatric and cardiovascular, were analysed and compared between the active treatment group and the control group with the chi-square test or Fisher’s exact test, as appropriate. The levels of anti-Aβ40 antibodies, Aβ peptides and cytokines in plasma were analysed after each visit using descriptive statistics. Statistical comparison between groups of treatment was done with the Wilcoxon rank-sum test for each time point and for the last follow-up endpoint (defined as the last observation available). The change from baseline in absolute value was analysed in an exploratory manner for each time point and for the last follow-up point. This analysis was performed separately for IP and AP patients. The significance level was set to p ≤ 0.05. All patients who received at least one dose of medication were included in the safety assessment (safety population and intention-to-treat [ITT] population), whereas evaluation of the biological activity was carried out in the ITT and per-protocol (PP) populations (Fig. 2). Statistical analyses were performed using SAS 9.4 software.
Fig. 2.
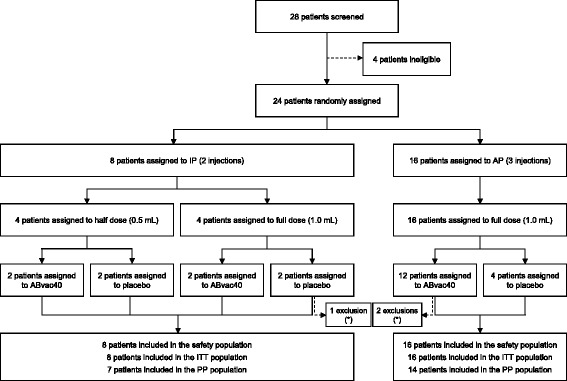
Trial profile. IP Initial protocol, AP Amended protocol, ITT Intention to treat, PP Per protocol, * Major protocol deviations
Results
Participants
Participants were recruited between 20 December 2013 and 30 March 2015. Recruitment was interrupted from 1 July 2014 to 14 January 2015 for interim analysis and submission of an amendment to the IP. A total of 28 patients were initially screened, and 24 were finally enrolled into the study. Of the enrolled patients, 16 were randomly allocated to ABvac40 treatment (2 patients received 2 half-doses, 2 patients received 2 full doses and 12 patients received 3 full doses), and 8 participants were randomly allocated to placebo (Fig. 1). All randomised patients completed the study; therefore, the safety and ITT populations were identical. However, a major protocol deviation was identified in three patients who had been treated with experimental immunotherapies in a previous clinical trial. These three patients (two in the ABvac40 group and one in the placebo group) were excluded from the PP population (Fig. 2).
Baseline patient demographics are summarised in Table 1. Briefly, the ABvac40 and placebo groups were homogeneous concerning most demographic characteristics, including distribution of APOE genotypes, years of education, sex and time from diagnosis; they differed only in age, with the ABvac40 group being 9.6 years older, on average, than the placebo group. All patients received a stable AD medication dose during 3 months prior to screening and throughout the study.
Table 1.
Baseline characteristics
| Safety/ITT population (N = 24) | ||
|---|---|---|
| ABvac40 (n = 16) | Placebo (n = 8) | |
| Age, years | ||
| Mean (SD) | 72.4 (7.2) | 62.8 (6.9) |
| Years of education | ||
| Mean (SD) | 7.1 (3.4) | 8.9 (5.4) |
| Sex | ||
| Male | 8 (50%) | 3 (38%) |
| Female | 8 (50%) | 5 (63%) |
| Time from AD diagnosis, months | ||
| Mean (SD) | 18.3 (17.4) | 13.0 (11.7) |
| APOE genotype | ||
| ε3ε3 | 6 (38%) | 3 (38%) |
| ε3ε4 | 8 (50%) | 3 (38%) |
| ε4ε4 | 2 (13%) | 2 (25%) |
| GDS | ||
| 0–10: Normal | 15 (94%) | 8 (100%) |
| 11–14: Depression | 1 (6%) | 0 (0%) |
| > 14: Depression | 0 (0%) | 0 (0%) |
| Hachinski Ischemic Scale score | ||
| < 4 Suggestive of degenerative disorder | 16 (100%) | 8 (100%) |
| 4–7 Doubtful cases and mixed dementias | 0 (0%) | 0 (0%) |
| > 7 Suggestive of vascular involvement | 0 (0%) | 0 (0%) |
| Leukoaraiosis scale, total | ||
| Mean (SD) | 2.8 (2.7) | 2.6 (4.1) |
| Microhaemorrhage presence | ||
| Yes | 4 (25%) | 2 (25%) |
| CDR | ||
| 0.5 points | 2 (13%) | 4 (50%) |
| 1 point | 14 (88%) | 4 (50%) |
| 2 points | 0 (0%) | 0 (0%) |
| MMSE total score | ||
| Mean (SD) | 19.0 (2.7) | 21.2 (3.4) |
| MMSE total score (by age and schooling) | ||
| Mean (SD) | 20.1 (2.7) | 21.9 (3.3) |
Abbreviations: APOE Apolipoprotein E, AD Alzheimer’s disease, GDS Geriatric Depression Scale, CDR Clinical Dementia Rating, MMSE Mini Mental State Examination, ITT Intention to treat
Data are mean (SD) or number (%)
Safety and tolerability
The primary endpoint to assess the safety and tolerability of the study drug was the frequency of AEs. Overall, 71 AEs were recorded in 18 patients: seven out of the eight patients (88%) in the placebo group suffered at least one AE during the study, compared with 11 out of the 16 patients (69%) in the ABvac40 group (Table 2). There were no significant differences in the incidence of AEs between both groups; neither for the total number of AEs (p = 0.6214 for total AEs occurrence between groups) nor for these grouped as neurological, psychiatric and cardiovascular AEs (p = 0.2038, p = 1.0000 and p = 1.0000, respectively).
Table 2.
Adverse events
| Safety/ITT population | |||||||
|---|---|---|---|---|---|---|---|
| ABvac40 (n = 16) | Placebo (n = 8) | Total (N = 24) | |||||
| AEs (n) | No. of patients (%) | AEs (n) | No. of patients (%) | AEs (n) | No. of patients (%) | p Value | |
| Total AEs | 42 | 11 (69%) | 29 | 7 (88%) | 71 | 18 (75%) | 0.6214 |
| Neurological | 9 | 5 (31%) | 6 | 5 (63%) | 15 | 10 (42%) | 0.2038 |
| Psychiatric | 2 | 2 (13%) | 1 | 1 (13%) | 3 | 3 (13%) | 1.0000 |
| Cardiovascular | 1 | 1 (6%) | 1 | 1 (13%) | 2 | 2 (8%) | 1.0000 |
AE Adverse event, ITT Intention to treat
Analysis was done using Fisher’s exact test. See Additional file 3: Table S1 for a complete list of reported AEs
The most common AEs were headache, which occurred in nine individuals, and urinary tract infection, which occurred in six individuals. No other AE occurred in more than three individuals (13% of the participants). For a complete list of reported AEs and their incidence, see Additional file 3: Table S1. Apart from the urinary tract infections, no other relevant clinical abnormalities or changes from baseline were detected in any participant concerning haematology, blood biochemistry, ECG, vital signs, body mass index and neurological examination explored for complementary assessment of ABvac40 tolerability (data not shown). Most AEs were considered unrelated to the treatment, and only a few were considered possibly or probably related (Additional file 4: Table S2), including one clinically asymptomatic microhaemorrhage detected by MRI after the second immunisation in a patient belonging to the placebo group.
All AEs were classified as mild and did not require modification of the treatment schedule. Of particular relevance, no vasogenic oedema or sulcal effusion (amyloid-related imaging abnormalities corresponding to vasogenic oedema and sulcal effusions [ARIA-E]) was detected throughout the study period or on the four extra MRI scans of the participants taken during the additional 1-year follow-up for long-term safety control. Only one of the participants in the placebo group experienced three simultaneous serious adverse events (SAEs; hypothermia, dehydration and rhabdomyolysis) after escaping from family control and lying overnight in a dry creek. The patient was hospitalised, and the event ended 1 week afterward without sequelae.
Local reactions at the injection point occurred in 13 subjects: 9 patients in the ABvac40 group (56%) and 4 patients in the placebo group (50%). Most reactions disappeared at the safety control visit 2 weeks after the immunisation, and they were limited to redness and slight swelling, except one case followed by itching and erythema that was reported as an AE (Additional file 4: Table S2).
Immune response
The assessment of the biological activity of ABvac40 was achieved by determining the plasma levels of anti-Aβ40 IgG antibodies. Considering the ITT population, the average MSΔ in the ABvac40 group was 1.94 (SD 1.32) optical density (OD) units (Table 3). It should be noted that the third immunisation in the AP dramatically increased the levels of anti-Aβ40 antibodies from an MSΔ of 0.64 (SD 0.81) OD units in the IP patients to 2.37 (SD 1.18) OD units in the subjects following the AP (Fig. 3a). Fourteen of 16 (88%) participants in the ABvac40 group were considered positive responders (3 of 4 patients following the IP and 11 of 12 patients following the AP) (Table 3). On average, > 91% of the signal registered in the native plasma samples from patients treated with ABvac40 disappeared after overnight pre-adsorption of corresponding aliquots of the same samples with the ABvac40 immunogenic peptide (Aβ33–40), indicating that the signal increment was due to the presence of specific anti-Aβ40 antibodies (Table 3 and Fig. 3b). None of the patients receiving placebo had significant specific anti-Aβ40 antibodies. However, it should be noted that a patient in the placebo group (S028) showed a high signal that turned out to be non-specific owing to the low percentage of signal disappearing after overnight pre-adsorption.
Table 3.
Quantification of the immune response
| Treatment | Patient | Protocol | MSΔ | SDp | MSΔ/SDp | Signal adsorbeda (%) | Titres |
|---|---|---|---|---|---|---|---|
| Placebo | S002 | IP | 0.090 | 0.049 | 1.837 | – | – |
| S004 | IP | 0.062 | 0.301 | 0.206 | – | – | |
| S005 | IP | −0.033 | 0.055 | −0.600 | – | – | |
| S010 | IP | 0.086 | 0.036 | 2.380 | – | – | |
| S015 | AP | 0.113 | 0.086 | 1.318 | – | – | |
| S017 | AP | 0.055 | 0.065 | 0.841 | – | – | |
| S020 | AP | −0.076 | 0.118 | −0.644 | – | – | |
| S028 | AP | 0.872 | 0.065 | 13.421 | 12.72b | – | |
| ABvac40 | S001 | IP | 0.249 | 0.071 | 3.507 | 83.00 | 30 |
| S003 | IP | 0.315 | 0.064 | 4.927 | 96.72 | 10 | |
| S008 | IP | 0.129 | 0.397 | 0.326 | – | – | |
| S011 | IP | 1.852 | 0.066 | 28.061 | 90.95 | 810 | |
| S012 | AP | 0.190 | 0.099 | 1.919 | – | – | |
| S013 | AP | 3.635 | 0.097 | 37.474 | 95.54c | 65,610 | |
| S014 | AP | 3.093 | 0.104 | 29.737 | 95.52 | 7290 | |
| S016 | AP | 2.156 | 0.099 | 21.778 | 95.22 | 270 | |
| S018 | AP | 0.626 | 0.037 | 16.298 | 105.22 | 90 | |
| S019 | AP | 3.526 | 0.099 | 35.616 | 99.63c | 21,870 | |
| S021 | AP | 2.265 | 0.156 | 14.519 | 87.90 | 270 | |
| S022 | AP | 2.419 | 0.127 | 19.047 | 63.92 | 810 | |
| S023 | AP | 3.461 | 0.029 | 119.345 | 99.25c | 65,610 | |
| S024 | AP | 1.011 | 0.103 | 9.816 | 72.40 | 90 | |
| S025 | AP | 2.852 | 0.112 | 25.464 | 93.71 | 810 | |
| S026 | AP | 3.230 | 0.104 | 31.058 | 96.81c | 21,870 |
Abbreviations: MSΔ Maximal signal increment (in optical density), SDp Average SD from all visits of each patient, IP Initial protocol, AP Amended protocol, Aβ Amyloid-β
Non-responder patients are shown in bold
aPre-adsorbed with 10−4 M Aβ33–40
bThe low percentage of adsorption of this sample suggests non-specific signal
cPre-adsorbed with 10−3 M Aβ33–40
Fig. 3.
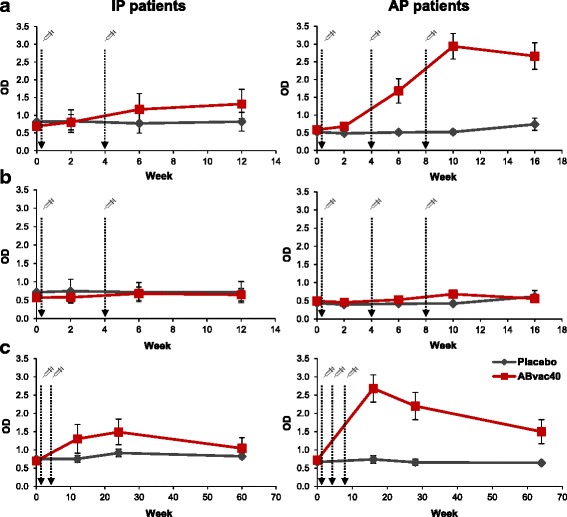
Evolution over time of the immune response of initial protocol (IP) and amended protocol (AP) patients (left and right panels, respectively) from baseline to the final visit (a and b) and during the 1-year open-label follow-up (c). The levels of anti-amyloid-β40 (Aβ40) antibodies in plasma are represented as the optical density (OD) in the titration enzyme-linked immunosorbent assays performed in 96-well plates coated with the Aβ1–40 peptide. Pre-adsorption of plasma samples with Aβ33–40 peptide (b) resulted in a reduction of > 91% of the signal compared with non-pre-adsorbed samples (a), suggesting that the signal corresponded to specific anti-Aβ40 antibodies. The levels of specific anti-Aβ40 antibodies remained elevated in AP patients in the ABvac40 group for up to 56 weeks after the last immunisation (c). Data are mean ± SEM
Interestingly, the levels of specific anti-Aβ40 antibodies in ABvac40-treated patients in the AP subgroup remained significantly higher than pre-immune plasma levels up to 56 weeks after the last immunisation (p = 0.004), as observed during the 1-year follow-up for long-term safety assessment (Fig. 3c). ABvac40-induced antibodies recognised synthetic Aβ1–40, including monomers, dimers, trimers and oligomers; however, they did not label any form of Aβ1–42 (Fig. 4a). The reactivity of plasma samples from ABvac40-treated patients with amyloid brain plaques was confirmed by immunohistochemistry on brain sections from APP/PS1-transgenic mice (Fig. 4b) and patients with AD (Fig. 4c).
Fig. 4.
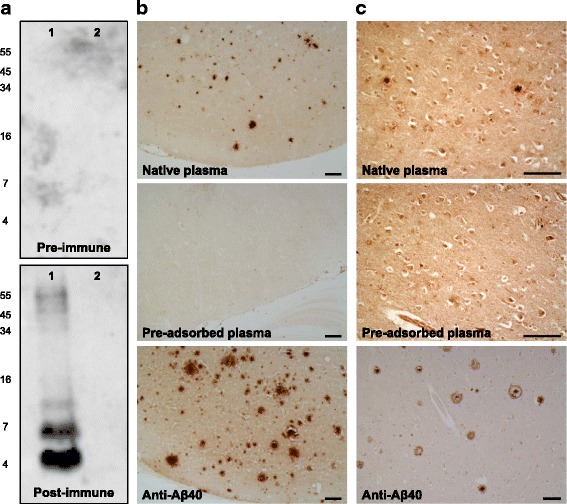
Reactivity of ABvac40-induced anti-amyloid-β (anti-Aβ) antibodies: a Post-immune plasma samples (week 10) from an ABvac40-treated patient (S013) recognised different forms of synthetic Aβ40 peptide (lane 1). In contrast, they did not label any form of synthetic Aβ42 (lane 2). Pre-immune plasma (week 0) did not show reactivity with Aβ40 or Aβ42. b and c Binding of plasma samples from the same patient (S013; week 10) to amyloid plaques in paraffin-embedded brain sections from APP/PS1-transgenic mice (b) and patients with AD (c). Pre-adsorption of plasma with Aβ33–40 peptide prevented plaque staining. Specific anti-Aβ40 polyclonal antibody was used as a positive control. Scale bar = 100 μm
Regarding other exploratory secondary efficacy variables, such as the plasma levels of Aβ peptides (Additional file 5: Table S3) and cytokines (data not shown), no significant differences were found between treatment groups at the end of the clinical trial.
Discussion
Our findings show a good safety and tolerability profile for ABvac40, because upon a relevant and specific immune response in 88% of the participants in the active arm, no SAEs were recorded in the ABvac40 group and no significant differences were found in the frequency of AEs, overall and grouped as neurological, psychiatric and cardiovascular AEs, as compared with the placebo group. All AEs detected throughout the study were classified as mild and did not require changes in treatment schedule; most of them, with the exception of mild and transient local reactions, were considered neither possibly nor probably related to the investigational medical product.
Since it was first reported that active immunisation targeting Aβ halted the progression of AD pathology in transgenic mice [25], numerous studies with promising results in animals have progressed into clinical trials. However, the first clinical trial of active immunotherapy, consisting of repeated administrations of aggregated Aβ42 with QS-21 as an adjuvant (AN1792), was discontinued owing to meningoencephalitis in 6% of treated patients [26]. These AEs were likely caused by an Aβ-specific T-cell-mediated Th1 immune response, which was attributed to the use of QS-21, a strong Th1-type adjuvant, and the use of full-length Aβ1–42 carrying T-cell-activating epitopes. Although the AN1792 clinical trial failed, long-term follow-up of responder patients showed a reduction in brain amyloid burden [27, 28] and attenuated functional decline [29–31], which supports the potential benefits of Aβ immunotherapy, provided that an Aβ-specific T-cell response can be avoided. In this regard, it is important to underline that the T-cell epitopes of the Aβ peptide have been mapped to different regions, including Aβ1–16 [32], Aβ6–28 [33] and Aβ16–25 [34], as well as Aβ16–30, Aβ19–33 and the Aβ28–42 C-terminal fragment of Aβ42 [35]. Nevertheless, it should be noted that those T-cell lines reactive to Aβ28–42 were unreactive to Aβ1–40, suggesting the importance of the two C-terminal amino acid residues [35]. Thus, to minimise the potential risk of T-cell responses, ABvac40 was designed using the C-terminal end of Aβ40 (Aβ33–40) and aluminium hydroxide as an adjuvant to stimulate a Th2-type immune response [36]. In line with this, no cases of meningoencephalitis were found throughout the study.
On one hand, targeting the C-terminal fragment of Aβ40 could have some additional safety advantages over the N-terminal, because the epitope targeted by ABvac40-elicited antibodies is concealed within the transmembrane portion of APP and therefore can be bound to antibodies only after Aβ is cleaved and secreted, avoiding cross-reactions with native APP and the apposition of antigen-antibody complexes on the neuronal cell membrane. On the other hand, after discontinuation of AN1792, passive anti-Aβ immunotherapies were favoured as a better approach to managing undesired immune responses. However, most passive immunotherapy trials have been associated with the highly frequent occurrence of ARIA [37–40], referring to a spectrum of imaging abnormalities detected on MRI scans suggestive of ARIA-E or amyloid-related imaging abnormalities corresponding to microhaemorrhages and hemosiderin deposits (ARIA-H) [41]. ARIA seem to be less frequent after active anti-Aβ immunisation [42–44]. In particular, no incidence of ARIA-E or ARIA-H was associated with ABvac40 during the study period or in the additional 1-year follow-up for long-term safety control. Researchers in a number of studies in transgenic mice have reported an increased incidence of microhaemorrhages following passive anti-Aβ40 immunotherapy [45, 46]. The monoclonal antibody (mAb) ponezumab, which recognises amino acids 33–40 of Aβ40, however, is the only passive immunotherapy that did not increase the incidence of microhaemorrhages or vasogenic oedema when administered to transgenic mice, cynomolgus monkeys or patients with mild to moderate AD [47–50].
The synthesis and kinetics of the different Aβ peptides, namely Aβ40 and Aβ42, and their differential contribution to AD physiopathology have been subject of intensive research but are not yet completely understood. Interestingly, some studies have shown that the proportion of Aβ42 and Aβ40 (the named Aβ42/Aβ40 ratio) may be more crucial for the formation of neurotoxic oligomeric conformations than the total amount of Aβ produced in the brain in the sense that changes in the Aβ42/Aβ40 ratio could favour the stabilisation of highly cytotoxic intermediate oligomers in vitro [51, 52]. These findings suggest that reducing the absolute amount of Aβ in patients with AD, such as with mAbs directed against the N-terminal end of Aβ or the central part of its sequence, could be less effective than trying to restore the appropriate Aβ42/Aβ40 ratio by specifically targeting either Aβ42 or Aβ40 by means of their C-terminal end.
Although Aβ42 is regarded as the most toxic species, other studies have shown that Aβ40 can also form cytotoxic aggregates [9, 53, 54]. Additionally, it has been observed that the levels of insoluble Aβ40 in the brain of patients with AD increase substantially in association with the onset of dementia [11, 12], and we have found large numbers of degenerating neurons filled with C-terminal fragments of Aβx-40 (but not Aβx-42) in the entorhinal cortex of AD brains [16]. These results support the idea that Aβ40 could play a relevant role in the pathophysiology of AD.
Moreover, along what is now described as the AD continuum, pathophysiological mechanisms other than cytotoxicity can be involved in the AD process, such as inflammation and particularly the deposition of Aβ40 in the cerebral blood vessels causing CAA in > 80% of patients with AD. More importantly, Aβ40-targeting therapies could be effective in the treatment of CAA-related inflammation (considered a naturally occurring model of ARIA) because reductions in the rate of Aβ deposition in cerebral vessels and restoration of vascular integrity have been found when anti-Aβ40 mAbs were administered in animal models of CAA [55].
ABvac40 was highly immunogenic because 88% of the patients receiving the vaccine showed specific anti-Aβ40 antibodies that recognised monomeric, oligomeric and insoluble (plaques) forms of Aβ40 peptide. This multi-targeted profile of the polyclonal antibodies generated by active vaccines as ABvac40 may improve their probability of success in patients at different AD pathological stages with regard to single-target mAbs. Thus, a recent phase III clinical trial with an mAb targeting soluble Aβ species (solanezumab; Expedition3 trial) has shown an inability to significantly reduce amyloid cortical burden (although a favourable tendency was apparent) in patients with mild AD [56], whereas mAbs targeting fibrillary Aβ (aducanumab) have produced very promising results [40]. However, it is also possible that the turnover of senile plaques is too slow for treatment during a relatively short period (88 weeks) with an mAb intended to cut the “supply” of soluble Aβ to cortical deposits (known to be accruing for decades before the onset of clinical symptoms), resulting in a reduction of cortical Aβ burden measurable with current neuroimaging techniques. In line with this, the solanezumab Expedition3 trial failure emphasises again the importance of confronting AD from a preventive approach, for which an active vaccine seems to be more suitable than a mAb.
As could be expected, antibody titres showed great variability owing to the individual component of the immune response. Nevertheless, it should be noted that the third immunisation, included in the AP, dramatically increased the levels of anti-Aβ40 antibodies with regard to the two immunisations defined in the IP while maintaining an excellent safety profile. This guarantees moving to a phase II dose-finding study to assess whether immunogenicity can be further increased more robustly across individuals. However, based on available data, no conclusions can currently be drawn about the antibody titres that could be clinically effective [57]. Interestingly, significantly elevated anti-Aβ40 antibody levels persisted in the ABvac40 group for up to 56 weeks after the last immunisation in those patients following the AP, which could offer long-term advantages owing to the continuous production of potentially therapeutic antibodies over time, contributing to the expected benefits of active immunisation as a cost-effective and long-term therapeutic strategy for AD [58].
Besides this, the present study has some limitations intrinsic to this initial stage of development. Because this first-in-human administration of ABvac40 was intended primarily to assess safety and tolerability, we only enrolled a limited number of patients (with unknown amyloid status) from only one centre in one country, and consequently the study was not powered to detect low-incidence AEs or changes in disease biomarkers. Therefore, we considered that in these conditions it was not worthwhile to expose patients to invasive procedures required for the assessment of amyloid biomarkers; nevertheless, these crucial measurements will be approached in an adequately powered phase II trial.
Conclusions
Previous evidence suggests that Aβ40 could have an essential role in AD. Accordingly, in the present work, we have assessed the safety and tolerability of ABvac40, a novel active vaccine against the C-terminal end of Aβ40, in patients with mild to moderate AD. This first-in-class study has shown that ABvac40 elicited a consistent and specific immune response against the C-terminal end of Aβ40 while maintaining a favourable safety and tolerability profile. These results show that active immunisation is a safe therapeutic strategy for AD and also that the C-terminal end of Aβ40 is a promising epitope to be considered in immunotherapy approaches, pointing to ABvac40 as a promising candidate for the treatment of AD. Additional studies including larger cohorts and longer follow-up are warranted to confirm safety assessments and to establish the therapeutic range and clinical efficacy of ABvac40.
Additional files
Inclusion and exclusion criteria (detailed). (DOCX 16 kb)
Randomisation (detailed). (DOCX 13 kb)
Frequency of adverse events (detailed). (DOCX 26 kb)
Relationship of adverse events (AEs) with the treatment. (DOCX 15 kb)
Quantification of Aβ40 and Aβ42 levels in plasma. (DOCX 14 kb)
Acknowledgements
The authors gratefully acknowledge Laura Nuñez and Mireia Torres (Bioscience Clinical and Pharmacovigilance, Grifols S.A., Barcelona, Spain) for their assistance with the administrative work. The authors also thank Dr. Pablo Villoslada Díaz, Dr. Joan Montaner Villalonga and Dr. Joan Costa i Pagès for their participation on the study’s independent data and safety monitoring board. In addition, the authors thank the TFS contract research organisation (Lund, Sweden) for operational support during the study.
Funding
This work was funded by Araclon Biotech Ltd., a subsidiary of Grifols. The sponsor was involved in the study design, data collection, data analysis, data interpretation and writing of the report.
Availability of data and materials
The datasets used and/or analysed during the present study are available from the corresponding author on reasonable request.
Abbreviations
- AD
Alzheimer’s disease
- AE
Adverse event
- AP
Amended protocol
- APOE
Apolipoprotein E
- APP
Amyloid precursor protein
- ARIA
Amyloid-related imaging abnormalities
- ARIA-E
Amyloid-related imaging abnormalities corresponding to vasogenic oedema and sulcal effusions
- ARIA-H
Amyloid-related imaging abnormalities corresponding to microhaemorrhages and hemosiderin deposits
- Aβ
Amyloid-β
- CAA
Cerebral amyloid angiopathy
- CDR
Clinical Dementia Rating
- ECG
Electrocardiogram
- ELISA
Enzyme-linked immunosorbent assay
- FLAIR
Fluid-attenuated inversion recovery
- GDS
Geriatric Depression Scale
- IgG
Immunoglobulin G
- IL
Interleukin
- IP
Initial protocol
- ITT
Intention to treat
- mAb
Monoclonal antibody
- MMSE
Mini Mental State Examination
- MOD
Mean optical density
- MRI
Magnetic resonance imaging
- MSΔ
Maximal signal increment
- NFT
Neurofibrillary tangle
- OD
Optical density
- PP
Per protocol
- SAE
Serious adverse event
- T2W
T2-weighted
Authors’ contributions
PP and MS contributed to the study concept and design and were responsible for major decisions in the study. PP and MS were responsible for the application to the Spanish authorities, contributed to data analysis and interpretation, and contributed to the writing of the manuscript. AML contributed to data collection, analysis and interpretation. MPL contributed to the literature search and drafted the manuscript. DC, VPG, IMC, LS, JC, HB and IM participated in analytical data acquisition. ISJ coordinated the study. JM performed MRI assessments. ORG, CA, AL, MBu, MBo (clinical principal investigator), LT and AR contributed to the study design and to the recruitment and assessment of the patients. All authors read and approved the final manuscript.
Ethics approval and consent to participate
This study was approved by the independent ethics committee of the Barcelona Hospital Clinic and conducted in accordance with the ethical and scientific principles described in the Declaration of Helsinki and the International Conference on Harmonisation Guideline for Good Clinical Practice (CPMP/ICH/135/95), European guidelines for clinical trials (2001/20/CE), and Spanish legislation (Royal Decree 223/2004 of 6 February, which regulates clinical drug trials). All participants provided written informed consent before enrolment.
Consent for publication
Not applicable.
Competing interests
AML, MPL, PP, DC, VPG, IMC, LS, JC, HB, IM, ISJ and MS are employees of Araclon Biotech Ltd. ISJ is a shareholder of Araclon Biotech Ltd. MS holds several patents related to Alzheimer’s disease diagnosis and treatment, and he is the founder, chief executive officer, chief scientific officer and one of the current shareholders of Araclon Biotech Ltd. AR reports receiving personal fees from Landsteiner Genmed, grants from the Innovative Medicines Initiative (IMI) ADAPTED project (European Commission), the IMI MOPEAD project (European Commission), Instituto de Salud Carlos III (ISCIII; Ministry of Health, Spain), Grifols and Fundación Bancaria “La Caixa” outside the submitted work. MBo reports receiving grants from the European Foundation for the Study of Diabetes/Lilly Mental Health and Diabetes Program 2014-2015, and IH2020-JTI-IMI2-2015-05 (European Commission) (Eli Lilly and AstraZeneca) MOPEAD project 2016-2018, as well as personal fees from Grifols, Janssen, Eli Lilly, MSD, Nutricia, Roche and Servier, outside the submitted work. JM, ORG, CA, AL, MBu and LT declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s13195-018-0340-8) contains supplementary material, which is available to authorized users.
Contributor Information
Ana-María Lacosta, Email: alacosta@araclon.com.
María Pascual-Lucas, Email: mpascual@araclon.com.
Pedro Pesini, Phone: +34976796562, Email: ppesini@araclon.com, http://www.araclon.com.
Diego Casabona, Email: dcasabona@araclon.com.
Virginia Pérez-Grijalba, Email: vperez@araclon.com.
Iván Marcos-Campos, Email: imarcos@araclon.com.
Leticia Sarasa, Email: letisarasa@araclon.com.
Jesus Canudas, Email: jcanudas@araclon.com.
Hassnae Badi, Email: hbadi@araclon.com.
Inmaculada Monleón, Email: imonleon@araclon.com.
Itziar San-José, Email: itzsanjose@araclon.com.
Josep Munuera, Email: pepmunuera@hotmail.com.
Octavio Rodríguez-Gómez, Email: orodriguez@fundacioace.com.
Carla Abdelnour, Email: cabdelnour@fundacioace.com.
Asunción Lafuente, Email: alafuente@fundacioace.com.
Mar Buendía, Email: mbuendia@fundacioace.com.
Mercè Boada, Email: mboada@fundacioace.com.
Lluis Tárraga, Email: ltarraga@fundacioace.com.
Agustín Ruiz, Email: aruiz@fundacioace.com.
Manuel Sarasa, Email: msarasa@araclon.com.
References
- 1.Prince M, Wimo A, Guerchet M, Ali GC, Wu YT, Prina M. World Alzheimer report 2015. The global impact of dementia: an analysis of prevalence, incidence, cost and trends. London: Alzheimer’s Disease International; 2015. http://www.alz.co.uk/research/WorldAlzheimerReport2015.pdf. Accessed 25 Apr 2017
- 2.Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992;256:184–5. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 3.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–6. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 4.Wisniewski T, Goni F. Immunotherapeutic approaches for Alzheimer’s disease. Neuron. 2015;85:1162–76. doi: 10.1016/j.neuron.2014.12.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lannfelt L, Relkin NR, Siemers ER. Amyloid-β-directed immunotherapy for Alzheimer’s disease. J Intern Med. 2014;275:284–95. doi: 10.1111/joim.12168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dovey HF, Suomensaari-Chrysler S, Lieberburg I, Sinha S, Keim PS. Cells with a familial Alzheimer’s disease mutation produce authentic β-peptide. Neuroreport. 1993;4:1039–42. doi: 10.1097/00001756-199308000-00011. [DOI] [PubMed] [Google Scholar]
- 7.Vigo-Pelfrey C, Lee D, Keim P, Lieberburg I, Schenk DB. Characterization of β-amyloid peptide from human cerebrospinal fluid. J Neurochem. 1993;61:1965–8. doi: 10.1111/j.1471-4159.1993.tb09841.x. [DOI] [PubMed] [Google Scholar]
- 8.Suzuki N, Cheung TT, Cai XD, Odaka A, Otvos LJ, Eckman C, et al. An increased percentage of long amyloid β protein secreted by familial amyloid β protein precursor (β APP717) mutants. Science. 1994;264:1336–40. doi: 10.1126/science.8191290. [DOI] [PubMed] [Google Scholar]
- 9.Chimon S, Shaibat MA, Jones CR, Calero DC, Aizezi B, Ishii Y. Evidence of fibril-like β-sheet structures in a neurotoxic amyloid intermediate of Alzheimer’s β-amyloid. Nat Struct Mol Biol. 2007;14:1157–64. doi: 10.1038/nsmb1345. [DOI] [PubMed] [Google Scholar]
- 10.Montanes M, Casabona D, Sarasa L, Pesini P, Sarasa M. Prevention of amyloid-β fibril formation using antibodies against the C-terminal region of amyloid-β1-40 and amyloid-β1-42. J Alzheimers Dis. 2013;34:133–7. doi: 10.3233/JAD-120850. [DOI] [PubMed] [Google Scholar]
- 11.Naslund J, Schierhorn A, Hellman U, Lannfelt L, Roses AD, Tjernberg LO, et al. Relative abundance of Alzheimer Aβ amyloid peptide variants in Alzheimer disease and normal aging. Proc Natl Acad Sci U S A. 1994;91:8378–82. doi: 10.1073/pnas.91.18.8378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang J, Dickson DW, Trojanowski JQ, Lee VM. The levels of soluble versus insoluble brain Aβ distinguish Alzheimer’s disease from normal and pathologic aging. Exp Neurol. 1999;158:328–37. doi: 10.1006/exnr.1999.7085. [DOI] [PubMed] [Google Scholar]
- 13.Pesini P, Lacosta AM, Sarasa M. The deposition of Aβ40 in the brain is pathognomonic for Alzheimer-type dementia in Down syndrome [abstract] Alzheimers Dement. 2009;5(4 Suppl):297–8. doi: 10.1016/j.jalz.2009.04.434. [DOI] [Google Scholar]
- 14.Herzig MC, Van Nostrand WE, Jucker M. Mechanism of cerebral β-amyloid angiopathy: murine and cellular models. Brain Pathol. 2006;16:40–54. doi: 10.1111/j.1750-3639.2006.tb00560.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jellinger KA. Alzheimer disease and cerebrovascular pathology: an update. J Neural Transm (Vienna) 2002;109:813–36. doi: 10.1007/s007020200068. [DOI] [PubMed] [Google Scholar]
- 16.Lacosta AM, Insua D, Badi H, Pesini P, Sarasa M. Neurofibrillary tangles of Aβx-40 in Alzheimer’s disease brains. J Alzheimers Dis. 2017;58:661–7. doi: 10.3233/JAD-170163. [DOI] [PubMed] [Google Scholar]
- 17.Schilling S, Lauber T, Schaupp M, Manhart S, Scheel E, Bohm G, et al. On the seeding and oligomerization of pGlu-amyloid peptides (in vitro) Biochemistry. 2006;45:12393–9. doi: 10.1021/bi0612667. [DOI] [PubMed] [Google Scholar]
- 18.D’Arrigo C, Tabaton M, Perico A. N-terminal truncated pyroglutamyl β amyloid peptide Aβpy3-42 shows a faster aggregation kinetics than the full-length Aβ1-42. Biopolymers. 2009;91:861–73. doi: 10.1002/bip.21271. [DOI] [PubMed] [Google Scholar]
- 19.Kummer MP, Hermes M, Delekarte A, Hammerschmidt T, Kumar S, Terwel D, et al. Nitration of tyrosine 10 critically enhances amyloid β aggregation and plaque formation. Neuron. 2011;71:833–44. doi: 10.1016/j.neuron.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 20.Schlenzig D, Ronicke R, Cynis H, Ludwig HH, Scheel E, Reymann K, et al. N-terminal pyroglutamate formation of Aβ38 and Aβ40 enforces oligomer formation and potency to disrupt hippocampal long-term potentiation. J Neurochem. 2012;121:774–84. doi: 10.1111/j.1471-4159.2012.07707.x. [DOI] [PubMed] [Google Scholar]
- 21.Nussbaum JM, Schilling S, Cynis H, Silva A, Swanson E, Wangsanut T, et al. Prion-like behaviour and tau-dependent cytotoxicity of pyroglutamylated amyloid-β. Nature. 2012;485:651–5. doi: 10.1038/nature11060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barakos J, Sperling R, Salloway S, Jack C, Gass A, Fiebach JB, et al. MR imaging features of amyloid-related imaging abnormalities. AJNR Am J Neuroradiol. 2013;34:1958–65. doi: 10.3174/ajnr.A3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barkhof F, Daams M, Scheltens P, Brashear HR, Arrighi HM, Bechten A, et al. An MRI rating scale for amyloid-related imaging abnormalities with edema or effusion. AJNR Am J Neuroradiol. 2013;34:1550–5. doi: 10.3174/ajnr.A3475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Izco M, Martinez P, Corrales A, Fandos N, Garcia S, Insua D, et al. Changes in the brain and plasma Aβ peptide levels with age and its relationship with cognitive impairment in the APPswe/PS1dE9 mouse model of Alzheimer’s disease. Neuroscience. 2014;263:269–79. doi: 10.1016/j.neuroscience.2014.01.003. [DOI] [PubMed] [Google Scholar]
- 25.Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, et al. Immunization with amyloid-β attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400:173–7. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- 26.Orgogozo JM, Gilman S, Dartigues JF, Laurent B, Puel M, Kirby LC, et al. Subacute meningoencephalitis in a subset of patients with AD after Aβ42 immunization. Neurology. 2003;61:46–54. doi: 10.1212/01.WNL.0000073623.84147.A8. [DOI] [PubMed] [Google Scholar]
- 27.Nicoll JA, Wilkinson D, Holmes C, Steart P, Markham H, Weller RO. Neuropathology of human Alzheimer disease after immunization with amyloid-β peptide: a case report. Nat Med. 2003;9:448–52. doi: 10.1038/nm840. [DOI] [PubMed] [Google Scholar]
- 28.Holmes C, Boche D, Wilkinson D, Yadegarfar G, Hopkins V, Bayer A, et al. Long-term effects of Aβ42 immunisation in Alzheimer’s disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet. 2008;372:216–23. doi: 10.1016/S0140-6736(08)61075-2. [DOI] [PubMed] [Google Scholar]
- 29.Hock C, Konietzko U, Streffer JR, Tracy J, Signorell A, Müller-Tillmanns B, et al. Antibodies against β-amyloid slow cognitive decline in Alzheimer’s disease. Neuron. 2003;38:547–54. doi: 10.1016/S0896-6273(03)00294-0. [DOI] [PubMed] [Google Scholar]
- 30.Gilman S, Koller M, Black RS, Jenkins L, Griffith SG, Fox NC, et al. Clinical effects of Aβ immunization (AN1792) in patients with AD in an interrupted trial. Neurology. 2005;64:1553–62. doi: 10.1212/01.WNL.0000159740.16984.3C. [DOI] [PubMed] [Google Scholar]
- 31.Vellas B, Black R, Thal LJ, Fox NC, Daniels M, McLennan G, et al. Long-term follow-up of patients immunized with AN1792: reduced functional decline in antibody responders. Curr Alzheimer Res. 2009;6:144–51. doi: 10.2174/156720509787602852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Das P, Chapoval S, Howard V, David CS, Golde TE. Immune responses against Aβ1-42 in HLA class II transgenic mice: implications for Aβ1-42 immune-mediated therapies. Neurobiol Aging. 2003;24:969–76. doi: 10.1016/S0197-4580(03)00036-8. [DOI] [PubMed] [Google Scholar]
- 33.Cribbs DH, Ghochikyan A, Vasilevko V, Tran M, Petrushina I, Sadzikava N, et al. Adjuvant-dependent modulation of Th1 and Th2 responses to immunization with β-amyloid. Int Immunol. 2003;15:505–14. doi: 10.1093/intimm/dxg049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weiner HL, Frenkel D. Immunology and immunotherapy of Alzheimer’s disease. Nat Rev Immunol. 2006;6:404–16. doi: 10.1038/nri1843. [DOI] [PubMed] [Google Scholar]
- 35.Monsonego A, Zota V, Karni A, Krieger JI, Bar-Or A, Bitan G, et al. Increased T cell reactivity to amyloid β protein in older humans and patients with Alzheimer disease. J Clin Invest. 2003;112:415–22. doi: 10.1172/JCI200318104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wilson-Welder JH, Torres MP, Kipper MJ, Mallapragada SK, Wannemuehler MJ, Narasimhan B. Vaccine adjuvants: current challenges and future approaches. J Pharm Sci. 2009;98:1278–316. doi: 10.1002/jps.21523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ostrowitzki S, Deptula D, Thurfjell L, Barkhof F, Bohrmann B, Brooks DJ, et al. Mechanism of amyloid removal in patients with Alzheimer disease treated with gantenerumab. Arch Neurol. 2012;69:198–207. doi: 10.1001/archneurol.2011.1538. [DOI] [PubMed] [Google Scholar]
- 38.Doody RS, Thomas RG, Farlow M, Iwatsubo T, Vellas B, Joffe S, et al. Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N Engl J Med. 2014;370:311–21. doi: 10.1056/NEJMoa1312889. [DOI] [PubMed] [Google Scholar]
- 39.Salloway S, Sperling R, Fox NC, Blennow K, Klunk W, Raskind M, et al. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N Engl J Med. 2014;370:322–33. doi: 10.1056/NEJMoa1304839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sevigny J, Chiao P, Bussiere T, Weinreb PH, Williams L, Maier M, et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature. 2016;537:50–6. doi: 10.1038/nature19323. [DOI] [PubMed] [Google Scholar]
- 41.Sperling RA, Jack CR, Jr, Black SE, Frosch MP, Greenberg SM, Hyman BT, et al. Amyloid-related imaging abnormalities in amyloid-modifying therapeutic trials: recommendations from the Alzheimer’s Association Research Roundtable Workgroup. Alzheimers Dement. 2011;7:367–85. doi: 10.1016/j.jalz.2011.05.2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Farlow MR, Andreasen N, Riviere ME, Vostiar I, Vitaliti A, Sovago J, et al. Long-term treatment with active Aβ immunotherapy with CAD106 in mild Alzheimer’s disease. Alzheimers Res Ther. 2015;7:23. doi: 10.1186/s13195-015-0108-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pasquier F, Sadowsky C, Holstein A, Leterme GP, Peng Y, Jackson N, et al. Two phase 2 multiple ascending-dose studies of vanutide cridificar (ACC-001) and QS-21 adjuvant in mild-to-moderate Alzheimer’s disease. J Alzheimers Dis. 2016;51:1131–43. doi: 10.3233/JAD-150376. [DOI] [PubMed] [Google Scholar]
- 44.Hull M, Sadowsky C, Arai H, Le Prince LG, Holstein A, Booth K, et al. Long-term extensions of randomized vaccination trials of ACC-001 and QS-21 in mild to moderate Alzheimer’s disease. Curr Alzheimer Res. 2017;14:696–708. doi: 10.2174/1567205014666170117101537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wilcock DM, Rojiani A, Rosenthal A, Subbarao S, Freeman MJ, Gordon MN, et al. Passive immunotherapy against Aβ in aged APP-transgenic mice reverses cognitive deficits and depletes parenchymal amyloid deposits in spite of increased vascular amyloid and microhemorrhage. J Neuroinflammation. 2004;1:24. doi: 10.1186/1742-2094-1-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wilcock DM, Gharkholonarehe N, Van Nostrand WE, Davis J, Vitek MP, Colton CA. Amyloid reduction by amyloid-β vaccination also reduces mouse tau pathology and protects from neuron loss in two mouse models of Alzheimer’s disease. J Neurosci. 2009;29:7957–65. doi: 10.1523/JNEUROSCI.1339-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Freeman GB, Brown TP, Wallace K, Bales KR. Chronic administration of an aglycosylated murine antibody of ponezumab does not worsen microhemorrhages in aged Tg2576 mice. Curr Alzheimer Res. 2012;9:1059–68. doi: 10.2174/156720512803569064. [DOI] [PubMed] [Google Scholar]
- 48.Freeman GB, Lin JC, Pons J, Raha NM. 39-Week toxicity and toxicokinetic study of ponezumab (PF-04360365) in cynomolgus monkeys with 12-week recovery period. J Alzheimers Dis. 2012;28:531–41. doi: 10.3233/JAD-2011-110869. [DOI] [PubMed] [Google Scholar]
- 49.Landen JW, Zhao Q, Cohen S, Borrie M, Woodward M, Billing CB, Jr, et al. Safety and pharmacology of a single intravenous dose of ponezumab in subjects with mild-to-moderate Alzheimer disease: a phase I, randomized, placebo-controlled, double-blind, dose-escalation study. Clin Neuropharmacol. 2013;36:14–23. doi: 10.1097/WNF.0b013e31827db49b. [DOI] [PubMed] [Google Scholar]
- 50.Burstein AH, Zhao Q, Ross J, Styren S, Landen JW, Ma WW, et al. Safety and pharmacology of ponezumab (PF-04360365) after a single 10-minute intravenous infusion in subjects with mild to moderate Alzheimer disease. Clin Neuropharmacol. 2013;36:8–13. doi: 10.1097/WNF.0b013e318279bcfa. [DOI] [PubMed] [Google Scholar]
- 51.Murray MM, Bernstein SL, Nyugen V, Condron MM, Teplow DB, Bowers MT. Amyloid β protein: Aβ40 inhibits Aβ42 oligomerization. J Am Chem Soc. 2009;131:6316–7. doi: 10.1021/ja8092604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kuperstein I, Broersen K, Benilova I, Rozenski J, Jonckheere W, Debulpaep M, et al. Neurotoxicity of Alzheimer’s disease Aβ peptides is induced by small changes in the Aβ42 to Aβ40 ratio. EMBO J. 2010;29:3408–20. doi: 10.1038/emboj.2010.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shin RW, Ogino K, Kondo A, Saido TC, Trojanowski JQ, Kitamoto T, et al. Amyloid β-protein (Aβ) 1–40 but not Aβ1–42 contributes to the experimental formation of Alzheimer disease amyloid fibrils in rat brain. J Neurosci. 1997;17:8187–93. doi: 10.1523/JNEUROSCI.17-21-08187.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Montañés M, Marcos I, Canudas J, Lacosta AM, Pesini P, Sarasa M. Anti-Aβx-40 antibodies exert neuroprotective effect against Aβ oligomer toxicity [abstract] Alzheimers Dement. 2017;13(7 Suppl):1439–40. doi: 10.1016/j.jalz.2017.06.2255. [DOI] [Google Scholar]
- 55.Bales KR, O’Neill SM, Pozdnyakov N, Pan F, Caouette D, Pi Y, et al. Passive immunotherapy targeting amyloid-β reduces cerebral amyloid angiopathy and improves vascular reactivity. Brain. 2016;139:563–77. doi: 10.1093/brain/awv313. [DOI] [PubMed] [Google Scholar]
- 56.Mintun MA, Devous Sr.MD, Lu M, Pontecorvo MJ, Joshi AD, Southekal S, et al. Alzheimers Dementia. 2017;13 (7 supplement): P1452.
- 57.Agadjanyan MG, Petrovsky N, Ghochikyan A. A fresh perspective from immunologists and vaccine researchers: active vaccination strategies to prevent and reverse Alzheimer’s disease. Alzheimers Dement. 2015;11:1246–59. doi: 10.1016/j.jalz.2015.06.1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Winblad B, Graf A, Riviere ME, Andreasen N, Ryan JM. Active immunotherapy options for Alzheimer’s disease. Alzheimers Res Ther. 2014;6:7. doi: 10.1186/alzrt237. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Inclusion and exclusion criteria (detailed). (DOCX 16 kb)
Randomisation (detailed). (DOCX 13 kb)
Frequency of adverse events (detailed). (DOCX 26 kb)
Relationship of adverse events (AEs) with the treatment. (DOCX 15 kb)
Quantification of Aβ40 and Aβ42 levels in plasma. (DOCX 14 kb)
Data Availability Statement
The datasets used and/or analysed during the present study are available from the corresponding author on reasonable request.