

Abstract
The discovery of noncoding RNAs (ncRNAs) and their importance for gene regulation led us to develop bioinformatics tools to pursue the discovery of novel ncRNAs. Finding ncRNAs de novo is challenging, first due to the difficulty of retrieving large numbers of sequences for given gene activities, and second due to exponential demands on calculation needed for comparative genomics on a large scale. Recently, several tools for the prediction of conserved RNA secondary structure were developed, but many of them are not designed to uncover new ncRNAs, or are too slow for conducting analyses on a large scale. Here we present various approaches using the database RiboGap as a primary tool for finding known ncRNAs and for uncovering simple sequence motifs with regulatory roles. This database also can be used to easily extract intergenic sequences of eubacteria and archaea to find conserved RNA structures upstream of given genes. We also show how to extend analysis further to choose the best candidate ncRNAs for experimental validation.
Keywords: ncRNA, bioinformatics, GraphClust, Infernal, Rfam, RNA secondary structure, riboswitch, methyltransferase, TRAP, CsrA, RsmA
1. Introduction
In the past decade, the availability of a wealth of sequence information was successfully exploited with bioinformatics tools for the discovery of noncoding RNAs (ncRNAs) in bacteria [1]. Because bacterial genomes are dense in information, using comparative genomics to examine intergenic regions (IGRs) has yielded numerous new functional RNA structures. IGRs are sequences that bridge gaps between protein-coding sequences or open reading frames (ORFs). Sequence elements that regulate bacterial gene expression are mostly found in IGRs [2, 3], regardless of whether they are cis or trans-acting [4]. Among the most common ncRNAs known in bacteria are the so-called small RNAs (sRNAs) which are independently transcribed and act in trans by binding mRNA targets, typically to repress expression [5, 6]. There are also many types of cis-acting elements such as ribosomal protein leaders [7], thermoregulators [8] and riboswitches [9, 10].
One way to discover de novo ncRNAs is to look for RNA structures in the IGR upstream of genes with similar activity. While most IGR sequences are poorly conserved compared to coding sequence, conserved sequence and structure can be observed in IGRs where a cis-regulatory RNA element is present. Riboswitches are especially well-suited for such searches because their ability to specifically recognize metabolites to exert gene regulation requires a highly conserved structure.
To embark on a campaign to discover novel ncRNAs by using comparative sequence and structure analysis, it is helpful to pool IGRs associated with genes of related functions. However, finding and extracting all the intergenic sequences for a specific function from general databases like GenBank [11] or Ensembl [12] can be laborious and requires programming skills. There is no simple way to extract intergenic sequences without any prior knowledge of gene positions. Obtaining IGRs associated with all the genes with similar or related annotated activities is even more difficult. Moreover, for large numbers of representatives (more than 500 sequences) and variable length IGRs, prediction of secondary structure is time-consuming and challenging for most available software packages [13].
Since bacterial IGR sequences have important regulatory elements such as promoters, noncoding RNAs and terminators, a tool to easily fetch known information about IGRs would be useful. Here we describe how this can be done with RiboGap by simply choosing the fields you wish to display and the keywords or parameters corresponding to the query. This provides an easy means to look for known RNA structures or sequence motifs associated with genes that are grouped by different annotated functions, taxonomic ensembles or even phenotypic traits, such as concerted regulation under different growth conditions. Moreover, fetching the IGR sequences with RiboGap also facilitate searches for ncRNA elements, notably by comparative genomics.
In this report, we describe such a comparative genomic pipeline which uses RiboGap along with GraphClust [14], a software package for secondary structure alignment prediction that can manage large numbers of sequences. Finally, we describe a method to validate putative ncRNAs with an experimental approach for riboswitch validation, in-line probing [15]. The proposed methodology also relies on other software packages to improve analysis, mainly Infernal, for homology searches, as well as R2R and Emacs (with the Ralee plug-in) for RNA structure and alignment visualization, respectively.
2. Materials
2.1 Bioinformatics
2.1.1 RiboGap
RiboGap is a database (http://ribogap.iaf.inrs.ca) which helps find IGRs from the 5′-UTRs as well as from the 3′-UTR for one or several genes without any previous knowledge of gene positions. RiboGap also provides information on the presence of known noncoding RNAs and Rho-independent transcription terminators [16, 17] in IGRs. Moreover, RiboGap uses operon data from the Operon DataBase: ODB [18] including known and conserved bacterial operons [19], as well as operon predictions based on adjacent genes with the same orientation. RiboGap can carry out keyword searches by using pattern matching. It also offers the possibility to use genome coordinates from hits of the RNA homology search suite Infernal [20] as an input to look for corresponding operons. A simple schematic presentation of RiboGap tables illustrates the potential connections that can be made between different types of data (Figure 1). Despite its great flexibility and ability to permit the equivalent of complex SQL queries, RiboGap has a simple interface (Figure 2).
Figure 1.
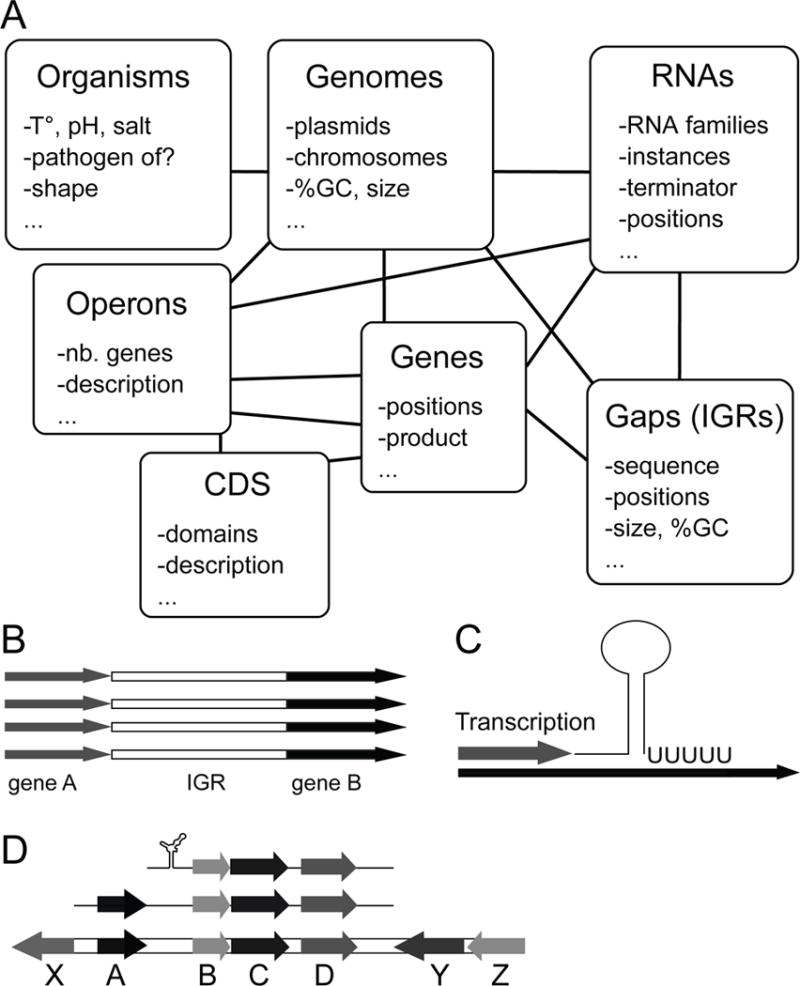
Schematic representation of tables and major applications of RiboGap. (A) Interconnected seven main tables which contain information on chromosomes, genes, proteins, ncRNAs, intergenic sequences and operons, through which defined queries can be defined. (B) Extraction of intergenic sequences from both the 5′-UTR and the 3′-UTR, which is one of the major applications of RiboGap. (C) De novo discovery of Rho independent terminator positions in specific ncRNAs or terminator evaluation for certain intergenic regions. (D) Prediction of genes and operons controlled by specific regulatory elements like riboswitches.
Figure 2.
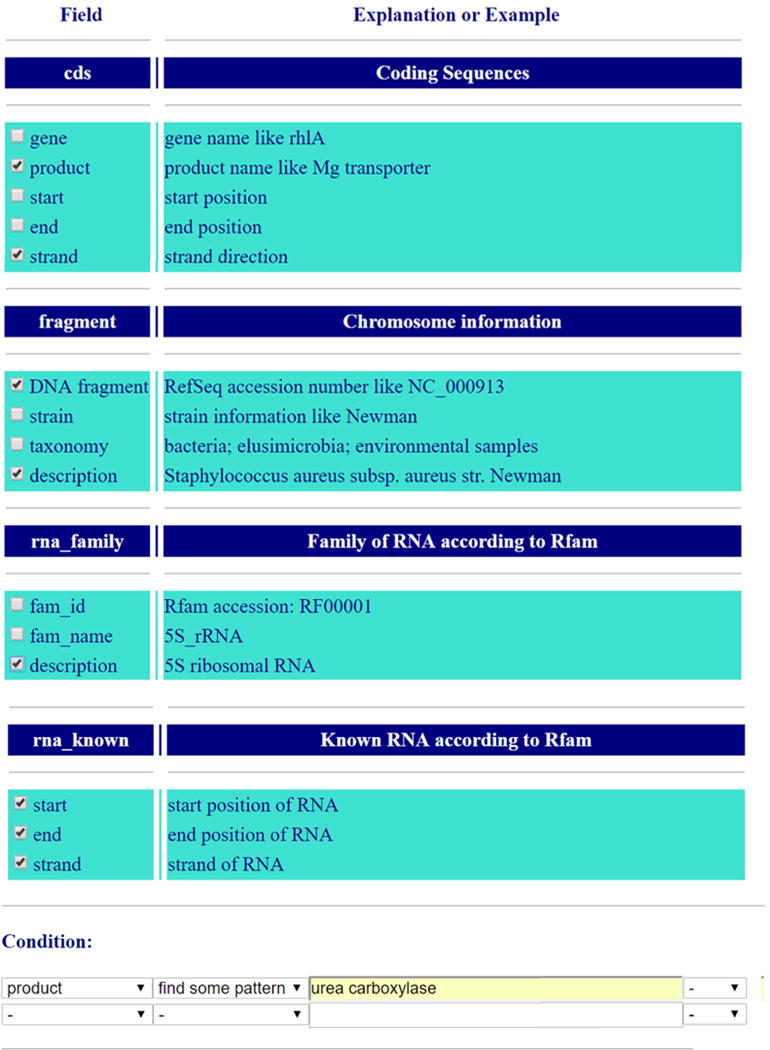
Screenshot of RiboGap interface from the Advanced search page. Some of the empty fields have been cropped to show the full query.
2.1.2 GraphClust
GraphClust requires several algorithms to be installed before using it: LocARNA version 1.7 or more recent [21], Vienna RNApackage version 2.0 or more recent [22], RNAz version 2.1 [23], Infernal version 1.0.2 [24], CMfinder version 0.2 [25], RNAshapes version 2.0.6 [26] and BLASTClust from NCBI [27].
2.1.3 Optional software
The text editor Emacs helps visualize the RNA alignment with the plugin Ralee [28]. R2R is a program useful for displaying RNA secondary structures by summarizing alignment information which can be installed locally [29]. MEME, which can be used locally or on a web server, finds conserved motifs among multiple sequences [30].
2.2 Experimental methods
Buffer mixtures for RNA preparation and analysis are detailed below. 5X transcription buffer: 400 mM HEPES-KOH (pH 7.5 at 23°C), 120 mM MgCl2, 10 mM spermidine, 200 mM DTT. 2X in-line probing buffer:100 mM Tris (pH 8.3 at 23°C), 200 mM KCl). 10X alkaline solution: 1M Na2CO3 at pH 9.2. 2X RNase T1 digest buffer: sodium citrate 75 mM at pH 5.0 and 30% formamide to denature RNA for a more uniform digestion. To stop in-line probing reactions and also to visualize the migration of RNA by denaturing (8 M urea) polyacrylamide gel electrophoresis (PAGE), an equal volume of 2X gel loading buffer (95% formamide, 10 mM EDTA [pH 8], 0.05 % (w/v) bromophenol blue, 0.05% (w/v) xylene cyanol) was added.
3. Methods and results
3.1 Finding known RNA structures
While most proteins are annotated, only a few genomes have a thorough annotation of ncRNAs. Indeed, aside from tRNAs, rRNAs, SRP RNA and tmRNA, RNAs are typically not part of standard annotation pipelines. As such, sRNAs and ncRNAs found in UTR regions need to be found in other databases such as Rfam, which can be especially cumbersome when one wants to search for ncRNAs associated with particular genes. To do this with RiboGap, the user simply needs to choose keywords and fields of interest like the “description” field in the section “rna_family” and position information from the “rna_known” section, combined with “gene product” from the “cds” (coding sequence) section, as well as any other information the user wishes to display, such as the species name obtained with the “description” field from “fragment” (Figure 2). For example, a user who would be interested in the function and regulation of the uca gene could use the keyword “urea carboxylase” in the field “gene product” to look for known RNAs upstream of genes related to the same function, but not necessarily associated with uca. The result of this query shows an association of ykkC-yxkD leader and mini-ykkC RNAs with genes annotated as urea carboxylases (Supplementary Table S1). These RNAs have recently been renamed guanidine-I and guanidine-II riboswitches, respectively [31–33]. Interestingly, a few Rho-independent terminators found in the same UTRs overlap the riboswitch position, providing a clear hypothesis for the expression platform for these representatives (Fig. 3A).
Figure 3.
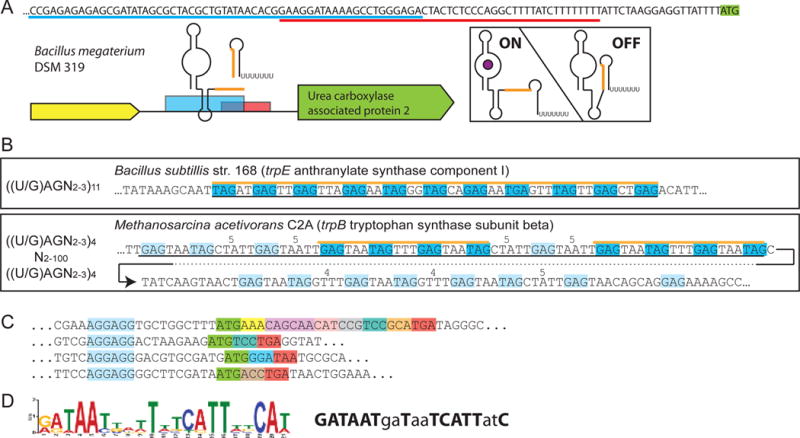
Results from RiboGap queries (A) The expression platform of a ykkC-yxkD (guanidine-I) riboswitch is observed from the overlap of the Rho-independent terminator (sequence underlined in red) and aptamer (partial sequence, underlined in blue). Genetic context, sequence overlap of structures is shown in orange. Hypothetical ON and OFF states are pictured on the right. (B) Examples of TRAP binding motifs found with the patterns used for the search on the left. Top is the canonical pattern, with a hit corresponding to a confirmed TRAP-regulatory site. Bottom, the pattern used for the search was less stringent and revealed a putative archaeon TRAP-like binding site. Black lines under the sequences indicate the putative full TRAP motif. Blue boxes and orange line correspond to binding sites found by the search, pale blue to other potential binding sites. Spacers larger than the typical 2-3 bases are indicated above the sequence. (C) Examples of mini ORFs found by the RiboGap query. Mini-ORF Shine-Dalgarno boxed in blue, start codon in green, stop codon in red and other codons in different colors. (D) Conserved “iron-box” found by MEME and drawn by sequence logo [59] on the left, Fur-box consensus on the right. Positions matching the “iron-box” consensus are in bold.
Inversely, it is possible to query for genes putatively regulated by given riboswitches. For instance, to find the genes that the SAM/SAH riboswitch potentially regulates, we can examine the gene located immediately downstream of the motif, or evaluate multiple genes if there appears to be an operon [34, 35]. Additional details are available in supplementary materials in section S1.1.2 and supplementary table S2.
3.2 Finding sequence motifs
3.2.1 Known sequence motifs
In addition to relatively complex RNA structures, leader regions of mRNAs can harbor simple regulatory sequence motifs bound by proteins. For instance, the Trp RNA-binding Attenuation Protein (TRAP) [36] represses the synthesis pathway of tryptophan when its concentration is high. This repression is achieved when the 11 units of the undecameric TRAP complex bind as many copies of a three nucleotide-motif starting by U or G and followed by A and G. These are separated by two or three nucleotides. The complete motif can be represented as ((U/G)AG(N)2-3)11. Such a motif can easily be searched for in RiboGap with a regular expression (REGEXP, refer to the help page of RiboGap for more information):
[TG]AG[ACGT]{2,3}[TG]AG[ACGT]{2,3}[TG]AG[ACGT]{2,3}[TG]AG[ACGT]{2,3}[TG]AG[ACGT]{2,3}[TG]AG[ACGT]{2,3}[TG]AG[ACGT]{2,3}[TG]AG[ACGT]{2,3}[TG]AG[ACGT]{2,3}[TG]AG[ACGT]{2,3}[TG]AG[ACGT]
Alternatively, to allow for a slight divergence from the known and well-characterized motif, we can look for fewer repeats and allow the insertion of a long sequence, which could hypothetically be looped out to permit the binding of the TRAP complex with multiple copies of the short RNA sequence:
[TG]AG[ACGT]{2,3}[TG]AG[ACGT]{2,3}[TG]AG[ACGT]{2,3}[TG]AG[ACGT]{2,100}[TG]AG[ACGT]{2,3}[TG]AG[ACGT]{2,3}[TG]AG[ACGT]{2,3}[TG]AG[ACGT]
Using this motif to search for TRAP-mediated regulation, we found many of the expected instances of known TRAP binding sites, such as the one upstream of trpE in Bacillus subtilis. We also found an interesting hit upstream of trpB (encoding a subunit of the tryptophan synthase) in the archaea Methanosarcina acetivorans (Fig. 3B). Because the distribution of TRAP is almost exclusively limited to firmicutes, this may look like a spurious hit. However, the presence of a TRAP analog in the genome of this archaea suggests that this example is legitimate and that a horizontal transfer event has occurred. There were, however, numerous hits unlikely to be true TRAP binding sites since the corresponding genomes do not encode the TRAP protein and because the genes downstream do not encode tryptophan-related functions. The complete set of hits is provided in Table S3 and highlights other interesting hits not documented before, such as representatives upstream of genes encoding chorismate-binding-like protein.
As an additional example, the protein RsmA/CsrA is known to bind two stem-loops with the sequence “GGA” in their loops [37, 38]. It was recently shown that both in Escherichia coli and Pseudomonas aeruginosa, the protein regulates itself by binding to its own mRNA, which also harbors the typical binding motif [39, 40]. We assumed this would be the case for more species, and thus we searched upstream of csrA or rsmA for instances of the motif. To do so, we allowed appropriate space for two adjacent stem-loops with a “GGA”, with the second one overlapping the ribosome binding site:
GGA[ACGT]{4,50}GGA[ACGT]{3,10}[ACGT]$
This search query corresponds to “GGA” followed by 4 to 50 nucleotides and a second “GGA” with 4 to 11 nucleotides at the exact end of the IGR (indicated by “$”) and thus directly upstream of the start codon. The proportion of csrA/rsmA IGRs that had this motif was higher than for any other sets of genes we examined: 51% as opposed to 14% on average for other genes (supplementary Table S4). Again, this highlights how simple queries in RiboGap can help to quickly test a hypothesis or to generate data that warrants further analysis. The complete collection of queries and resulting data are available in supplementary materials section S1.2.2 (including an example on how to easily generate a query in MySQL language).
The same principle was used to find potential unannotated small ORFs in IGRs. Given that the optimal ribosome binding site of all proteobacteria is AGGAGG, we looked for this motif followed by a spacer and an AUG start codon, followed by a certain number of “codons” and a stop codon. This query was made more complex by the fact that the coding sequence needs to be a multiple of three, but this can easily be circumvented by copying many times the basic REGEXP pattern:
AGGAGG[ACGT]{6,12}ATG[ACGT]{3}(TAA|TAG|TGA)|
AGGAGG[ACGT]{6,12}ATG[ACGT]{6}(TAA|TAG|TGA)|
AGGAGG[ACGT]{6,12}ATG[ACGT]{9}(TAA|TAG|TGA)|
and so on to 30 nucleotides (10 codons):
|AGGAGG[ACGT]{6,12}ATG[ACGT]{30}(TAA|TAG|TGA)…
Where (TAA|TAG|TGA) corresponds to the three stop codons. This statement thus allowed us to find putative small ORFs in so-called 5′ UTRs encoding between 2 and 29 amino acids. Over 3,000 hits were found in proteobacteria (Fig. 3C and Supplementary Table S5), more details available in supplementary material, section S1.2.3. It has been previously described that such mini-ORFs can affect mRNA stability [41, 42] or efficiency of translation [43] suggesting that, through diverse mechanisms, many of these ~3,000 hits are likely to be involved in regulating expression of the downstream gene. Even if REGEXP requires some knowledge of pattern matching for MySQL, the provided examples can be used as templates for many types of searches.
3.2.2 New sequence motifs
Most regulatory RNAs beyond the simple repeats of a TRAP binding motif usually include secondary structural elements. Nevertheless, it is possible to search for unknown motifs in sequences with readily available tools such as MEME. Extracting IGR sequences from genomes is made particularly easy by the RiboGap database. As an example of such a motif search, we conducted a simple query to fetch the “5′ gap” sequences of genes for which the “product” (in the cds section) had the word “iron”, such as for the gene product “iron-enterobactin transporter subunit”. This was done in the E. coli str. K-12 substr. MG1655 genome (Supplementary Fig. S1) and the sequences provided by RiboGap were submitted to MEME [44]. We found a conserved motif corresponding to the Fur-box (Fig. 3D), which is a 19-base-pair inverted DNA repeat known to allow control of gene expression by binding Fur protein according to iron concentration [45]. While this is not an RNA motif, this search, which took only a few minutes, still yielded an important conserved motif, the Fur-box, from these functionally related sequences (Supplementary Fig. S2). Details are available in Supplementary material, supplementary section S1.2.4.
3.3. Finding new structures
3.3.1. Obtaining intergenic sequences with RiboGap
To discover new ncRNA structures with the pipeline illustrated in Figure 4, the IGR sequences for a chosen gene function or collection need to be extracted. For this purpose, RiboGap is a useful database (available at: ribogap.iaf.inrs.ca). Two fields of the table cds (coding sequence) should be selected: gene and product (Supplementary Fig. S3). Note that more fields can be selected in every step as desired. Second, select the DNA fragment and description fields from Chromosome information to get the plasmid/chromosome accession numbers and the bacteria strain names, respectively. Third, the IGR sequences should be selected from the gap5 table (Supplementary Fig. S3), together with all the fields from gap5. Finally, the search should be narrowed down by using the conditions section. Any keyword can be used for the product or gene fields in the cds sub-section with either “REGEXP” or “find some pattern” (Supplementary Fig. S4). The keywords “methyl” and “RNA” are used to find genes with functions related to RNA methylation, such as genes encoding “tRNA methylases” or “16S rRNA methyltransferases” (Supplementary Fig. S4). The other example uses pattern matching with the REGEXP option. This can be useful for searches that require more complex keywords, which is the case for cation-related genes, such as magnesium, iron or calcium transporters/exporters/efflux pump (Supplementary Fig. S5).
Figure 4.
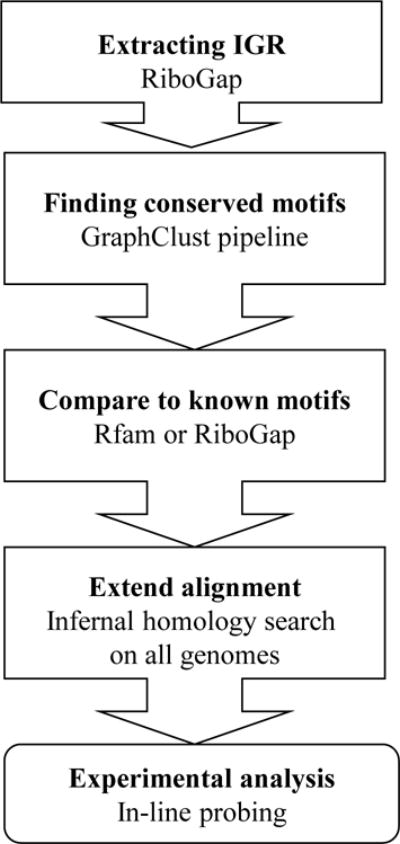
Schematic presentation of the pipeline for novel RNA structures discovery.
The rationale for our focus on “methyl” and “RNA” was that RNA methylases could potentially interact and modify their own mRNA to repress their expression in a classical negative feedback loop. Using RiboGap we extracted IGRs in front of genes annotated as RNA methylases (e.g., tRNA (mnm(5)s(2)U34-methyltransferase). We retrieved 8150 sequences and used this dataset with GraphClust.
In general, to search for ncRNAs in IGRs, one additional condition should be added (Supplementary Figure S4). In the gap5 sub-section, a size bigger than (>=) 25 should be used to filter small IGRs that are unlikely to harbor ncRNAs and otherwise would greatly increase the noise and false positives in the search for conserved structured RNAs. Indeed, in many species, the vast majority of IGRs are smaller than 25 nucleotides, while most ncRNAs are observed in IGRs of at least 100 nucleotides [46]. A cut-off of 100 nucleotides would thus be appropriate, but by being very conservative with a 25-nucleotide lower limit, we still eliminate a large number of IGRs, while retaining the possibility of finding a particularly small structured RNA in noncoding sequences between genes of the same operon. Otherwise, 25 base-pairs are not even enough to accommodate a typical promoter.
3.3.2. Finding conserved elements and predicting RNA secondary structures
In this critical step, the GraphClust package is used to compare all the sequences of the FASTA file generated by the query to group in clusters the sequences that have homology to their primary sequence and secondary structure. This package generates many alignments, as well as figures and graphs that are used to help discriminate ncRNAs from false positives. To predict new structured RNAs, the FASTA file should be provided as input for GraphClust. GraphClust uses the BLASTClust [47] package to remove sequences that are too closely related. The default filter screens at 90%, which permits it to automatically discard sequences that are more than 90% related. This ensures that conservation is due to function, rather than close phylogenetic relationship. It also favors alignments with more covariation.
To run GraphClust, some preinstalled software is required, and more information is provided with the GraphClust package. Several parameters can be modified, but the default command line for GraphClust is:
MASTER_GraphClust.pl --root run_test_1 --fasta my_seqs.fasta --config config.default_global –verbose
Other default parameters include a window size of 150 nucleotides, minimum sequence length of 100 nucleotides and two iterations. These parameters provided excellent ncRNA candidates containing secondary structures very well supported by covariation. To verify whether the candidate ncRNA may already be known, it is useful to do the same query, but with more information from tables such as RNA_family and RNA_known for the query (Supplementary Fig. S6) as described in section 3.1. These fields provide information on ncRNAs from the Rfam database [48], which is particularly useful for identifying known RNAs and avoiding the “discovery” of an already well-known ncRNAs. By selecting both the boxes of Known RNA and RNA family sub-sections (Supplementary Fig. S6) all the intergenic sequences containing known RNAs will be shown. If no results are returned, it means that there is no known RNA assigned to the IGR. For our “RNA” and “methyl” query, all the good candidates obtained with the described settings corresponded to known ncRNAs: tRNA, RNase P, cobalamin riboswitch and SAM riboswitch (SAM example in Supplementary Fig. S8). Indeed, the more widespread a ncRNA is, and the more covariation it has, the more it is likely that it has already been found. We thus executed the GraphClust command using 97% to filter out only very closely related sequences with BLASTClust and using 15 iterations to get more candidates (details on how to use different parameters in Supplementary Data section S2).
3.3.3. How to analyze results
3.3.3.1. Analyzing the structure alignments
By running GraphClust with default parameters, 20 clusters of sequences with predicted RNA secondary structures can be obtained. Two folders are interesting for analysis, CLUSTER and RESULT. These folders contain numerous files, and so we will mention only the most useful files for candidate analysis. In the CLUSTER folder, there are three subfolders, but the most important is the MODEL subfolder where the sequence alignments and predicted secondary structures can be found. MODEL has the clusters with high scores. The RESULT folder contains the final refined results from CLUSTER but sometimes omits useful information by discarding sequences from other intermediate alignments. For this reason, it can be useful to examine the model.tree.aln.ps, model.tree.aln.alirna.ps, best_subtree.aln.ps and model.tree.aln.rel_plot .pdf files as well (Supplementary Fig. S8).
To analyze the predicted structure, several criteria should be taken into account: sequence alignment quality, sequence conservation, covariation and structure likelihood. Since the sequences were aligned based on structure conservation, it is important to look at sequence alignments both in predicted models by CMfinder [49] and LocARNA [21]. It is worthwhile to visualize alignments with more than 10 sequences with Emacs in Ralee-mode [28] with the cmfinder.stk file, this allows easier inspection of base-pairs for which mis-pairs occur in a few sequences of the alignment (Supplementary Fig. S9). Indeed, LocARNA settings cause mispairs to dim color coding of base-pairs, which can lead to the base-pair not being highlighted at all in large and diverse alignments. Note that alignments with at least three sequences can be considered as candidates, depending on the criteria met.
The first criterion is sequence conservation. An alignment with too much conservation is not ideal because in such cases, apparent structure conservation could result from nearly identical sequences from phylogenetically close strains (Supplementary Fig. S10). However, some level of sequence conservation is expected (Supplementary Fig. S11), otherwise, spurious conserved structures can result from alignments of unrelated stem-loops for instance. Depending on the type of ncRNA, double-stranded regions are often more conserved because mutations that disrupt structure are not tolerated. However, in the case of ncRNAs with a very wide phylogenetic distribution, positions in stems can vary considerably and in such cases, critical single stranded regions can have a higher level of conservation, which can be a good indicator for quality of sequence alignment.
The second criterion is covariation of sequences. Since RNA structures are conserved evolutionarily, then mutations at positions where base-pairing need to be preserved should be compensated by a mutation of the opposite nucleotide that restores the base-pairing to maintain RNA structure [50]. More covariation in the alignment indicates a higher probability of a structured functional RNA. Covariation is the strongest indicator that helps support and confirm ncRNA candidates. While more likely to occur by chance, compatible variations are often observed in alignments of structured RNAs and also support the model. Compatible variations correspond to instances when only one position of the base-pair varies, but still permits base-pairing. For example, in the case of an A-U base-pair compared to a G-U base-pair, only the purine changes, but the U stays the same while still permitting a base-pair at that position. Several examples are provided to illustrate how to distinguish good (Supplementary Fig. S12), ambiguous (Supplementary Fig. S13), partially good (Supplementary Fig. S14 and S15), and poor sequence alignments or covariation (Supplementary Fig. S16 and 17).
The third criterion is the reliability of structures and alignments. To evaluate this, we use the files model.tree.aln.ps, bestsubtree.ps and model.tree.aln.rel_plot.pdf. The latter indicates the reliability of the predicted structure at given positions and is generated by LocARNA-P [51]. This file shows the degree of reliability of sequence alignment and structure. The more it tends towards “1”, the higher the probability of forming the corresponding base-pair at each given position. Note that this prediction is made from the model.tree.aln.ps file, which is created by LocARNA. (Supplementary Fig S18-S21). The file model.tree.aln.ps is an initial model prediction of ncRNA. LocARNA uses this file as a guide for making secondary structure predictions for more sequences. The file of bestsubtree.ps groups the best sequences for the predicted model. Reliability of the predicted structure for model.tree.aln.ps is evaluated by LocARNA-P and presented in the tree.aln.rel_plot.pdf file. This file indicates the boundary of ncRNAs as well as the reliability of the structure and sequence alignment. The boundaries of the ncRNA are very useful to locate more precisely the ncRNA position in the IGR.
As a result of this procedure, we selected the “RNA-methyl-28” (cluster number 28) from the RESULTS folder as an example of a potential candidate for further analysis (Fig. 5). The alignment has many positions with compatible variation and all the sequences clearly belong to the alignment, as illustrated by the highly-conserved stem around position 30 (Fig. 5 and Supplementary Fig. S22). Note that RNA-methyl-28 has no hits in Rfam, indicating that this putative ncRNA is likely a novel RNA family.
Figure 5.
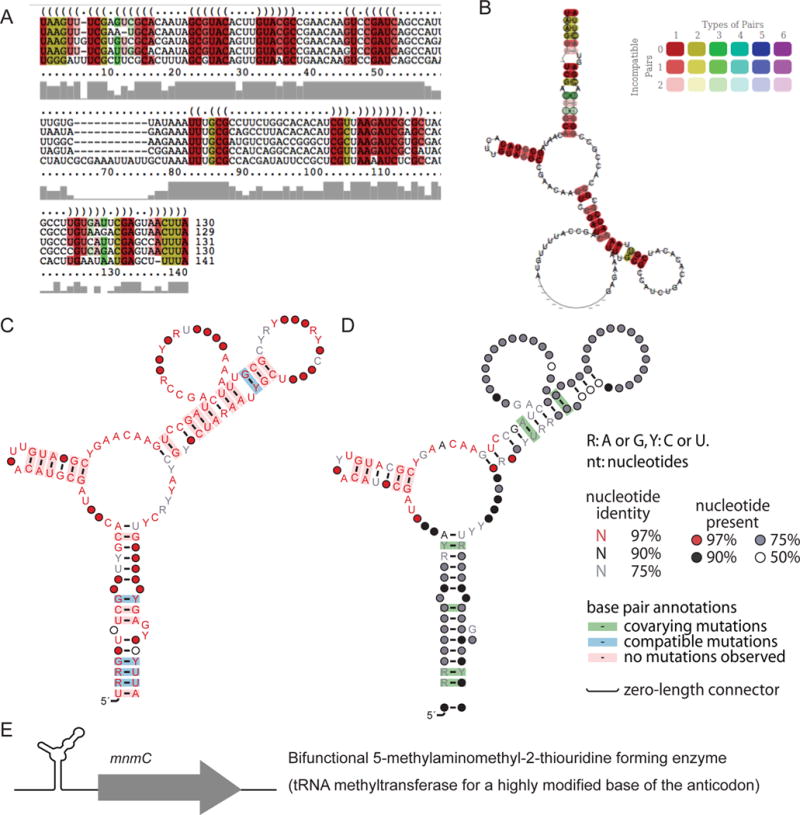
RNA methylation-related candidate “RNA-methyl-28”. (A) Sequence alignment of predicted structure for the candidate. (B) Prediction of secondary structure by GraphClust for the candidate. GraphClust uses the RNAalifold [60] option of the ViennaRNA Package [22] for motif drawing. (C) Consensus secondary structures for the RNA-methyl-28 candidate drawn by R2R [29], a software package that draws consensus motifs and annotations in one figure: secondary structure, sequence conservation, covariation and compatible variation. (D) R2R structure of the Infernal-extended alignment. (E) Genetic context of RNA-methyl-28.
3.3.3.2. Analyzing potential riboswitches
To selectively bind metabolites, riboswitches must fold in defined binding pockets which are determined by precise secondary and tertiary structures that are very well conserved. As a consequence, riboswitches often have a “conservation and structural signature”. Many functional RNAs can have several elements in their structure: loops, bulges or multistem junctions. In general, functional RNAs such as riboswitches or ribozymes have several of these structural elements with conserved nucleotides (examples of a candidate unlikely to be a riboswitch, Supplementary Fig S23; of the Mg2+-II riboswitch [Mg-sensor], S24-S27; and of the Mg2+-I riboswitch [ykoK], S28-S31). When they are phylogenetically widespread, the base-pairing regions often harbor a lot of covariation and thus have low sequence conservation. In contrast, nucleotides in single-stranded regions involved in ligand binding can be highly conserved, and are typically in multistem junctions. These features can help to distinguish between different types of ncRNAs and to choose candidate clusters more likely to be riboswitches for further analysis.
3.3.3.3. Additional considerations
The proximity of the candidate structure to a potential expression platform can also be a good indication that it acts as a regulatory RNA. This can easily be evaluated with the positions of the putative ncRNA within the IGR (which are the numbers in the name of the sequence in the alignment). Subtracting the end position from the size of the IGR gives the number of nucleotides separating the RNA structure from the start codon. Hence, if this number is less than 10, chances are that the structure overlaps the Shine-Dalgarno sequence [52, 53] and thus blocks translation, at least in some conditions. Additional criteria include whether the structure is already known (see section 3.1.) or if it is close to a Rho-independent transcription terminator, which would also suggest a potential mode of regulation, through transcription termination in this case. In the case of the candidate RNA-methyl-28, we evaluated the distance of the RNA structure from the start codon. In all the sequences from the alignment, the short distance separating the ncRNA from the start codon (1-2 nucleotides) implies that the Shine-Dalgarno is sequestered in the RNA structure. This further supports a regulatory function for this putative RNA (Supplementary Fig. S32).
3.3.3.4. Performing a global homology search of candidate motifs by Infernal
Once a potential candidate is found, GraphClust does a search for each model to extend from the initial “cluster alignment” through all sequences provided as input. However, the aligned structures can be used to evaluate the distribution of the motif in the whole bacterial genomes as well as in additional databases if desired. To do so, the stockholm file, (model.cmfinder.stk) in the CLUSTER/MODEL folder of the candidate should be chosen to do a cmbuild for Infernal. The procedure has been described in more detail previously [54]. Briefly, the steps to be executed are as follow: cmbuild and cmcalibrate to prepare the infernal covariance model, cmsearch to do a homology search in the chosen target database, and cmalign to consolidate the hits in a new alignment. If the ncRNA candidate is real, the new alignment is likely to provide more support for the conserved structure, with more covariation notably. It may also highlight which portions of the initially predicted structure are more important, since the other portions might not be present in all sequences of the post infernal alignment.
We applied this analysis to the RNA-methyl-28 motif. Using Infernal, we did a global homology search to evaluate the distribution of the motif in all bacterial genomes (NCBI bacteria genomes version 2015). We found 264 hits in different bacteria (Supplementary information and Table S6). We used R2R to draw the RNA-methyl-28 motif based on information from all 264 hits found with Infernal, and found that the RNA structure consensus derived from the 264 hits is similar to the initial GraphClust structure-alignment (Fig. 5). One small stem appears as highly conserved, but not the other stems, which are nevertheless supported by additional covariation in this extended alignment, as opposed to merely a compatible variation in the initial alignment. Note that these two types of base-pair variations are not distinguished by the analyses of GraphClust, but can be observed directly in the alignments or with the help of a R2R summarized consensus (Fig. 5). The results from any cmsearch can be uploaded with the tabfile format in RiboGap to get information on each hit.
4. In-line probing for experimental validation
Once a potential ncRNA candidate is chosen, it has to be experimentally validated to prove that it is a genuine ncRNA and to decipher its function. The sequence to be analyzed can be chosen by comparing the SCORE of cmsearch, which is indicated in the cluster.all.fa file in the RESULT folder. The top score indicates that the sequence fits well with the predicted model. This does not mean that sequences with lower scores are not good. For instance, if the motif is already known, the lower scoring sequences could actually represent distal variants worth investigating. Indeed, the deoxyguanosine riboswitch was revealed by looking at unusual hits for guanine riboswitches [55]. Based on this we also decided to look at outliers of Mg2+-II riboswitch alignments (Supplementary data section S4.1).
4.1. PCR to construct the template for RNA production
The first step to start in vitro assays is the preparation of a PCR template. For this purpose, the complete intergenic sequence can be selected from RiboGap and positions of the candidate ncRNA should be determined within this IGR. About 20 to 50 nucleotides can be added to each extremity of the candidate sequence, i.e. the portion of the sequence corresponding to the alignment, which is smaller than the full IGR. Alternatively, if the transcription start site is known, it can be used to determine the 5′ end. Several PCR templates can be constructed for the in vitro assays. If genomic DNA is readily available, preparing the transcription template simply requires addition of a T7 RNA polymerase promoter to a DNA primer used to amplify the fragment of interest. For RNA-methyl-28, the template for transcription could be amplified from E. coli JM109 genomic DNA with the following forward 5′-TTCTAATACGACTCACTATAGGTAAGTTTCGAATGCACAATA-3′ and reverse 5′-TAAGTTACTCGTCTTACAGG-3′ primers. However, when doing global genomics screens, it is common to end up with sequences from species for which assembly PCR is a more practical option (example of a Mg2+-II riboswitch in Supplementary material section S5.2). Overlapping oligonucleotides can be designed with primerize (https://primerize.stanford.edu/) [56]. Two or three G nucleotides should be added in 5′ of the sequence for efficient RNA transcription. Note that to do assembly PCR, the concentration of first and last oligonucleotide should be 1 μM and the other oligonucleotides concentration should be 0.1 μM.
4.2. RNA transcription
After producing double-stranded DNA templates, RNA can be transcribed from DNA. There are several commercial kits for this purpose. We use 10 μL DNA template (~10 pmoles) with 10 μL of 5X transcription buffer, 15 μL 10 mM rNTPs, 1 μL 0.1 U/μL pyrophosphatase (Roche), 1μL 40 U/μL RNase inhibitor (Roche) and 2 μL T7 RNA polymerase (10 U/μL final concentration) in a final volume of 50 μL. After incubating at 37°C for 2 h and degrading the template with 1 μL 2 U/μL RQ1 DNase, the RNA product is purified by denaturing 6% PAGE for 2 h. RNA is then eluted and dissolved in 21 μL RNase-free water. 1 μL of this sample is used to determine the concentration of RNA by using a Nanodrop spectrophotometer.
4.3. Dephosphorylation and Labeling
Dephosphorylation of RNA is performed by following the manufacturer’s instructions for Antarctic phosphatase (NEB). To label RNA, 2 μL radioactive ATP (γ-32-P), 3 to 10 pmoles of dephosphorylated RNA, 1 μL of 10 U/μL polynucleotide T4 kinase and PNK buffer (NEB) in 20 μL is incubated at 37°C for 1 h. The labeled product is purified on denaturing 6% PAGE.
4.4. Determination of candidate RNA structure and potential modulation by in-line probing
To determine the activity of an RNA suspected to be a riboswitch, RNA is incubated in conditions favoring a structure-dependent degradation pattern with in-line probing. In these conditions, different concentrations of ligand can be assayed to test ligand binding and get data necessary for KD calculation. In-line reactions are carried out for 40 h at room temperature. Standard in-line reactions are 50mM Tris pH 8.3, 100 mM KCl and 20 mM MgCl2, but in the case of metal ion ligands, the in-line reaction can be carried with varying concentrations of Mg2+. To be able to determine RNA structure, two types of ladder are prepared. Up to three times as much of labeled RNA can be digested by T1 enzyme and alkaline digestion. T1 reactions are carried out with the radiolabeled RNA and 1.5 μL of T1 RNAse 1 U/μL in T1 solution incubated at 56°C for 5 minutes. Alkaline digestion is conducted with the RNA being incubated in 20 μL of 1X alkaline solution at 90° C for 1 minute and 20 seconds. The samples are then run on 10% denaturing PAGE for approximately 3 hours at 70 W, exposed with phosphorimaging screens and scanned by a Typhoon FL9500. The technique has been described in more detail by Regulski and Breaker [57].
In-line probing was carried out on a construct of the IGR upstream of mnmC in E. coli str. JM109 to confirm the structure of the RNA. We prepared a control in-line probing reaction containing a spontaneous digested RNA without metabolite, a no-reaction sample of undigested RNA, RNA subjected to partial digestion by RNase T1, and a partial alkaline digestion (Na2CO3). The labeled RNA was incubated for 35 hours and then the pattern of RNA degradation was examined by denaturing 10% PAGE (Fig. 6). The small highly conserved stem is not supported by the in-line probing data in this construct. This could be due to inappropriate structure prediction, especially considering that the stem is not supported by covariation. Alternatively, since riboswitches and many types of regulatory RNA elements have at least two mutually exclusive structures, in-line probing (Fig. 6B) may not represent the conserved RNA motif candidate RNA-methyl-28, but rather an alternative structure. An additional example for the Mg2+-II riboswitch is provided (Supplementary material section S4.1).
Figure 6.
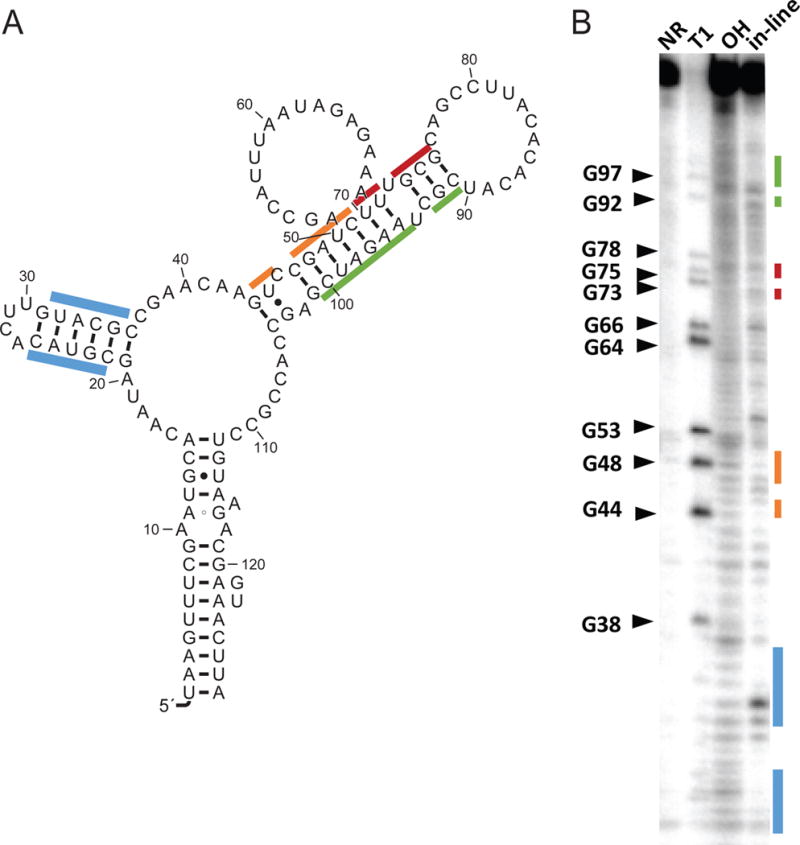
In-line probing of the RNA-methyl-28 candidate. (A) Secondary structure of the sequence used for the in-line probing experiment. (B) In-line probing of the RNA illustrated in B. RNA was incubated for 40 h. The colored lines along the gel correspond to regions indicated with the same colors on the secondary structure representation.
5. Discussion
Targeted comparative genomics, where sequences upstream of homologous proteins are aligned to look for conserved RNA structures, have been fruitful in the past [58]. However, these studies were undertaken with data extracted from databases such as “NCBI Nucleotide” using scripts or homemade programs and could only be performed by someone with programming skills. Here we demonstrate the accessibility and usefulness of RiboGap in extracting and exploring intergenic sequences for ncRNA. Beyond its ease of use, RiboGap extends the types of sequence ensembles the user can make by allowing function-based queries, rather than protein domain-based queries, providing additional data useful for downstream analysis. RiboGap can be used on a regular basis by most genomics researchers interested in obtaining results from simple or complex queries. Since RiboGap centralizes data from many different databases, it permits users to optimize their research by querying the combined data from various original databases. This cuts down on the laborious compilation and parsing of multiple sets of data required prior to analysis, associated with drawing information from regular databases.
The extraction of intergenic sequences is an important part of the pipeline which can lead to de novo predictions of ncRNAs with GraphClust. Even if the latter is relatively efficient to compare sequences on a large scale, comparing all IGRs of all sequenced bacteria and archaea would require considerable computing resources, as opposed to targeting limited datasets provided by RiboGap. This targeted strategy focuses on sets of genes which have a higher likelihood of harboring regulating ncRNAs. Alternatively, the users can choose from multiple sets of functions to explore less obvious associations which may link more subtle regulatory mechanisms related to the ncRNA candidate structures. In both cases, using RiboGap can greatly reduce computing time as well as the number of candidate ncRNAs to evaluate, which is even more time consuming. Perhaps even more critical, choosing which candidate ncRNA to study from the large number of putative ncRNAs requires the analysis of countless alignments and structures, many of which might be interesting, but most of which would not be.
While RiboGap is a powerful tool for extracting intergenic sequences associated with chosen gene functions, it is limited by gene annotations. Poorly annotated genes can either prevent the user from getting a set of sequences corresponding to the chosen function, or lead to the prediction of ncRNAs associated with another unrelated function. Here, we show an example of the latter. Intergenic sequences associated with genes annotated as “urea carboxylases” were searched for the presence of known ncRNAs. As expected, this led us to find ykkC and mini-ykkC guanidine riboswitches [31–33]. In this case, the annotation of uca is likely wrong due to a lack of knowledge regarding guanidine biology, resulting in missannotation of uca as encoding urea decarboxylase enzyme, rather than a guanidine decarboxylase.
Our initial screen of magnesium-related genes only identified members of one of the two known Mg2+ riboswitches. We thus adjusted parameters of BLASTClust from 90% to 98% to find the Mg2+-I (ykoK) riboswitch as we had previously done with our RNA-methyl-28 search. Analysis of the GraphClust results should be performed with precaution as none of the currently available software can comprehensively predict the existing RNA secondary structures. Yet, because GraphClust uses both CMfinder and LocARNA, it benefits from different covariation model predictions and alignments of secondary structure instead of merely sequence. Finally, one should not forget that even though some very powerful tools are available for ncRNA prediction, the inspection of the candidate motifs is necessary to appropriately evaluate them and decide which ones to prioritize for experimental validation.
Supplementary Material
Acknowledgments
J.P. thanks support from Natural Sciences and Engineering Council of Canada (NSERC) [418240 to J.P.]. J.P. is a junior 1 FRQS research scholar. R.R.B. is supported by the NIH (GM022778) and by the Howard Hughes Medical Institute. The authors wish to thank J. Lajoie, V. Korniakova, R. Walsh and E. Boutet for helpful discussions. Computations and data extraction were made on the supercomputer Mammouth parallèle 2, managed by Calcul Québec and Compute Canada (funded by CFI, NanoQuébec, RMGA and FRQ-NT).
Footnotes
Database URL: http://ribogap.iaf.inrs.ca/
References
- 1.Backofen R, Amman F, Costa F, Findeiss S, Richter AS, Stadler PF. Bioinformatics of prokaryotic RNAs. RNA biology. 2014;11(5) doi: 10.4161/rna.28647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Breaker RR. Prospects for riboswitch discovery and analysis. Molecular cell. 2011;43(6):867–79. doi: 10.1016/j.molcel.2011.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gossringer M, Hartmann RK. 3′-UTRs as a source of regulatory RNAs in bacteria. The EMBO journal. 2012;31(20):3958–60. doi: 10.1038/emboj.2012.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Serganov A, Nudler E. A decade of riboswitches. Cell. 2013;152(1-2):17–24. doi: 10.1016/j.cell.2012.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Masse E, Majdalani N, Gottesman S. Regulatory roles for small RNAs in bacteria. Current opinion in microbiology. 2003;6(2):120–4. doi: 10.1016/s1369-5274(03)00027-4. [DOI] [PubMed] [Google Scholar]
- 6.Mizuno T. Regulation of gene expression by a small RNA transcript (micRNA), Tanpakushitsu kakusan koso. Protein, nucleic acid, enzyme. 1984;29(11):908–13. [PubMed] [Google Scholar]
- 7.Meyer MM. The role of mRNA structure in bacterial translational regulation, Wiley interdisciplinary reviews. RNA. 2016 doi: 10.1002/wrna.1370. [DOI] [PubMed] [Google Scholar]
- 8.Morita MT, Tanaka Y, Kodama TS, Kyogoku Y, Yanagi H, Yura T. Translational induction of heat shock transcription factor sigma32: evidence for a built-in RNA thermosensor. Genes & development. 1999;13(6):655–65. doi: 10.1101/gad.13.6.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nahvi A, Sudarsan N, Ebert MS, Zou X, Brown KL, Breaker RR. Genetic control by a metabolite binding mRNA. Chemistry & biology. 2002;9(9):1043. doi: 10.1016/s1074-5521(02)00224-7. [DOI] [PubMed] [Google Scholar]
- 10.Winkler WC, Nahvi A, Sudarsan N, Barrick JE, Breaker RR. An mRNA structure that controls gene expression by binding S-adenosylmethionine. Nature structural biology. 2003;10(9):701–7. doi: 10.1038/nsb967. [DOI] [PubMed] [Google Scholar]
- 11.Benson DA, Clark K, Karsch-Mizrachi I, Lipman DJ, Ostell J, Sayers EW. GenBank. Nucleic acids research. 2015;43:D30–5. doi: 10.1093/nar/gku1216. (Database issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cunningham F, Amode MR, Barrell D, Beal K, Billis K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fitzgerald S, Gil L, Giron CG, Gordon L, Hourlier T, Hunt SE, Janacek SH, Johnson N, Juettemann T, Kahari AK, Keenan S, Martin FJ, Maurel T, McLaren W, Murphy DN, Nag R, Overduin B, Parker A, Patricio M, Perry E, Pignatelli M, Riat HS, Sheppard D, Taylor K, Thormann A, Vullo A, Wilder SP, Zadissa A, Aken BL, Birney E, Harrow J, Kinsella R, Muffato M, Ruffier M, Searle SM, Spudich G, Trevanion SJ, Yates A, Zerbino DR, Flicek P. Ensembl 2015. Nucleic acids research. 2015;43:D662–9. doi: 10.1093/nar/gku1010. (Database issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seetin MG, Mathews DH. RNA structure prediction: an overview of methods. Methods Mol Biol. 2012;905:99–122. doi: 10.1007/978-1-61779-949-5_8. [DOI] [PubMed] [Google Scholar]
- 14.Heyne S, Costa F, Rose D, Backofen R. GraphClust: alignment-free structural clustering of local RNA secondary structures. Bioinformatics. 2012;28(12):i224–32. doi: 10.1093/bioinformatics/bts224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Regulski EE, Moy RH, Weinberg Z, Barrick JE, Yao Z, Ruzzo WL, Breaker RR. A widespread riboswitch candidate that controls bacterial genes involved in molybdenum cofactor and tungsten cofactor metabolism. Molecular microbiology. 2008;68(4):918–32. doi: 10.1111/j.1365-2958.2008.06208.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Farnham PJ, Platt T. Rho-independent termination: dyad symmetry in DNA causes RNA polymerase to pause during transcription in vitro. Nucleic acids research. 1981;9(3):563–77. doi: 10.1093/nar/9.3.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wilson KS, von Hippel PH. Transcription termination at intrinsic terminators: the role of the RNA hairpin. Proceedings of the National Academy of Sciences of the United States of America. 1995;92(19):8793–7. doi: 10.1073/pnas.92.19.8793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Okuda S, Yoshizawa AC. ODB: a database for operon organizations, 2011 update. Nucleic acids research. 2011;39:D552–5. doi: 10.1093/nar/gkq1090. (Database issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tatusova T, Ciufo S, Fedorov B, O’Neill K, Tolstoy I. RefSeq microbial genomes database: new representation and annotation strategy. Nucleic acids research. 2015;43(7):3872. doi: 10.1093/nar/gkv278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nawrocki EP, Eddy SR. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics. 2013;29(22):2933–5. doi: 10.1093/bioinformatics/btt509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Will S, Reiche K, Hofacker IL, Stadler PF, Backofen R. Inferring noncoding RNA families and classes by means of genome-scale structure-based clustering. PLoS computational biology. 2007;3(4):e65. doi: 10.1371/journal.pcbi.0030065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lorenz R, Bernhart SH, Honer Zu Siederdissen C, Tafer H, Flamm C, Stadler PF, Hofacker IL. ViennaRNA Package 2.0. Algorithms Mol Biol. 2011;6:26. doi: 10.1186/1748-7188-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gruber AR, Findeiss S, Washietl S, Hofacker IL, Stadler PF. RNAz 2.0: improved noncoding RNA detection. Pac Symp Biocomput. 2010:69–79. [PubMed] [Google Scholar]
- 24.Nawrocki EP, Kolbe DL, Eddy SR. Infernal 1.0: inference of RNA alignments. Bioinformatics. 2009;25(10):1335–7. doi: 10.1093/bioinformatics/btp157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yao Z, Weinberg Z, Ruzzo WL. CMfinder–a covariance model based RNA motif finding algorithm. Bioinformatics. 2006;22(4):445–52. doi: 10.1093/bioinformatics/btk008. [DOI] [PubMed] [Google Scholar]
- 26.Janssen S, Giegerich R. The RNA shapes studio. Bioinformatics. 2015;31(3):423–5. doi: 10.1093/bioinformatics/btu649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. BLAST+: architecture and applications. BMC bioinformatics. 2009;10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Griffiths-Jones S. RALEE–RNA ALignment editor in Emacs. Bioinformatics. 2005;21(2):257–9. doi: 10.1093/bioinformatics/bth489. [DOI] [PubMed] [Google Scholar]
- 29.Weinberg Z, Breaker RR. R2R–software to speed the depiction of aesthetic consensus RNA secondary structures. BMC bioinformatics. 2011;12:3. doi: 10.1186/1471-2105-12-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS. MEME SUITE: tools for motif discovery and searching. Nucleic acids research. 2009;37:W202–8. doi: 10.1093/nar/gkp335. (Web Server issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sherlock ME, Breaker RR. Biochemical Validation of a Third Guanidine Riboswitch Class in Bacteria. Biochemistry. 2017 doi: 10.1021/acs.biochem.6b01271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Breaker RR, Atilho RM, Malkowski SN, Nelson JW, Sherlock ME. The Biology of Free Guanidine As Revealed by Riboswitches. Biochemistry. 2017 doi: 10.1021/acs.biochem.6b01269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sherlock ME, Malkowski SN, Breaker RR. Biochemical Validation of a Second Guanidine Riboswitch Class in Bacteria. Biochemistry. 2017 doi: 10.1021/acs.biochem.6b01270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Montange RK, Batey RT. Structure of the S-adenosylmethionine riboswitch regulatory mRNA element. Nature. 2006;441(7097):1172–5. doi: 10.1038/nature04819. [DOI] [PubMed] [Google Scholar]
- 35.Wang JX, Breaker RR. Riboswitches that sense S-adenosylmethionine and S-adenosylhomocysteine. Biochem Cell Biol. 2008;86(2):157–68. doi: 10.1139/O08-008. [DOI] [PubMed] [Google Scholar]
- 36.Gollnick P, Babitzke P, Antson A, Yanofsky C. Complexity in regulation of tryptophan biosynthesis in Bacillus subtilis. Annual review of genetics. 2005;39:47–68. doi: 10.1146/annurev.genet.39.073003.093745. [DOI] [PubMed] [Google Scholar]
- 37.Valverde C, Lindell M, Wagner EG, Haas D. A repeated GGA motif is critical for the activity and stability of the riboregulator RsmY of Pseudomonas fluorescens. J Biol Chem. 2004;279(24):25066–74. doi: 10.1074/jbc.M401870200. [DOI] [PubMed] [Google Scholar]
- 38.Lapouge K, Schubert M, Allain FH, Haas D. Gac/Rsm signal transduction pathway of gamma-proteobacteria: from RNA recognition to regulation of social behaviour. Mol Microbiol. 2008;67(2):241–53. doi: 10.1111/j.1365-2958.2007.06042.x. [DOI] [PubMed] [Google Scholar]
- 39.Jean-Pierre F, Perreault J, Deziel E. Complex autoregulation of the post-transcriptional regulator RsmA in Pseudomonas aeruginosa. Microbiology. 2015;161(9):1889–96. doi: 10.1099/mic.0.000140. [DOI] [PubMed] [Google Scholar]
- 40.Romeo T, Vakulskas CA, Babitzke P. Post-transcriptional regulation on a global scale: form and function of Csr/Rsm systems. Environmental microbiology. 2013;15(2):313–24. doi: 10.1111/j.1462-2920.2012.02794.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lodato PB, Hsieh PK, Belasco JG, Kaper JB. The ribosome binding site of a mini-ORF protects a T3SS mRNA from degradation by RNase E. Molecular microbiology. 2012;86(5):1167–82. doi: 10.1111/mmi.12050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mathy N, Benard L, Pellegrini O, Daou R, Wen T, Condon C. 5′-to-3′ exoribonuclease activity in bacteria: role of RNase J1 in rRNA maturation and 5′ stability of mRNA. Cell. 2007;129(4):681–92. doi: 10.1016/j.cell.2007.02.051. [DOI] [PubMed] [Google Scholar]
- 43.Qiao H, Lu N, Du E, Yao L, Xiao H, Lu S, Qi Y. Rare codons in uORFs of baculovirus p13 gene modulates downstream gene expression. Virus research. 2011;155(1):249–53. doi: 10.1016/j.virusres.2010.10.016. [DOI] [PubMed] [Google Scholar]
- 44.Bailey TL, Elkan C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol. 1994;2:28–36. [PubMed] [Google Scholar]
- 45.de Lorenzo V, Giovannini F, Herrero M, Neilands JB. Metal ion regulation of gene expression. Fur repressor-operator interaction at the promoter region of the aerobactin system of pColV-K30. Journal of molecular biology. 1988;203(4):875–84. doi: 10.1016/0022-2836(88)90113-1. [DOI] [PubMed] [Google Scholar]
- 46.Meyer MM, Ames TD, Smith DP, Weinberg Z, Schwalbach MS, Giovannoni SJ, Breaker RR. Identification of candidate structured RNAs in the marine organism ‘Candidatus Pelagibacter ubique’. BMC genomics. 2009;10:268. doi: 10.1186/1471-2164-10-268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. Journal of molecular biology. 1990;215(3):403–10. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 48.Nawrocki EP, Burge SW, Bateman A, Daub J, Eberhardt RY, Eddy SR, Floden EW, Gardner PP, Jones TA, Tate J, Finn RD. Rfam 12.0: updates to the RNA families database. Nucleic acids research. 2015;43:D130–7. doi: 10.1093/nar/gku1063. (Database issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yao ZZ, Weinberg Z, Ruzzo WL. CMfinder - a covariance model based RNA motif finding algorithm. Bioinformatics. 2006;22(4):445–452. doi: 10.1093/bioinformatics/btk008. [DOI] [PubMed] [Google Scholar]
- 50.Fox GE, Woese CR. 5S RNA secondary structure. Nature. 1975;256(5517):505–7. doi: 10.1038/256505a0. [DOI] [PubMed] [Google Scholar]
- 51.Will S, Joshi T, Hofacker IL, Stadler PF, Backofen R. LocARNA-P: accurate boundary prediction and improved detection of structural RNAs. RNA. 2012;18(5):900–14. doi: 10.1261/rna.029041.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Curry KA, Tomich CS. Effect of ribosome binding site on gene expression in Escherichia coli. DNA. 1988;7(3):173–9. doi: 10.1089/dna.1988.7.173. [DOI] [PubMed] [Google Scholar]
- 53.Chen H, Bjerknes M, Kumar R, Jay E. Determination of the optimal aligned spacing between the Shine-Dalgarno sequence and the translation initiation codon of Escherichia coli mRNAs. Nucleic acids research. 1994;22(23):4953–7. doi: 10.1093/nar/22.23.4953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.El Korbi A, Ouellet J, Naghdi MR, Perreault J. Finding instances of riboswitches and ribozymes by homology search of structured RNA with Infernal. Methods Mol Biol. 2014;1103:113–26. doi: 10.1007/978-1-62703-730-3_9. [DOI] [PubMed] [Google Scholar]
- 55.Kim JN, Roth A, Breaker RR. Guanine riboswitch variants from Mesoplasma florum selectively recognize 2′-deoxyguanosine. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(41):16092–7. doi: 10.1073/pnas.0705884104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tian S, Yesselman JD, Cordero P, Das R. Primerize: automated primer assembly for transcribing non-coding RNA domains. Nucleic acids research. 2015;43(W1):W522–6. doi: 10.1093/nar/gkv538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Regulski EE, Breaker RR. In-line probing analysis of riboswitches. Methods Mol Biol. 2008;419:53–67. doi: 10.1007/978-1-59745-033-1_4. [DOI] [PubMed] [Google Scholar]
- 58.Weinberg Z, Barrick JE, Yao Z, Roth A, Kim JN, Gore J, Wang JX, Lee ER, Block KF, Sudarsan N, Neph S, Tompa M, Ruzzo WL, Breaker RR. Identification of 22 candidate structured RNAs in bacteria using the CMfinder comparative genomics pipeline. Nucleic acids research. 2007;35(14):4809–19. doi: 10.1093/nar/gkm487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome research. 2004;14(6):1188–90. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bernhart SH, Hofacker IL, Will S, Gruber AR, Stadler PF. RNAalifold: improved consensus structure prediction for RNA alignments. BMC bioinformatics. 2008;9:474. doi: 10.1186/1471-2105-9-474. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.