

Abstract
Purpose of review
Drugs targeting the renin-angiotensin system (RAS), namely angiotensin converting enzyme (ACE) inhibitors and angiotensin receptor blockers, are the most commonly prescribed drugs for patients with or at risk for cardiovascular events. However, new treatment strategies aimed at mitigating the rise of the heart failure pandemic are warranted because clinical trials show that RAS blockers have limited benefits in halting disease progression. The main goal of this review is to put forward the concept of an intracrine RAS signaling through the novel angiotensin-(1-12)/chymase axis as the main source of deleterious Ang II (Ang II) in cardiac maladaptive remodeling leading to heart failure (HF).
Recent findings
Expanding traditional knowledge, Ang II can be produced in tissues independently from the circulatory renin-angiotensin system. In the heart, angiotensin-(1-12) [Ang-(1-12)], a recently-discovered derivative of angiotensinogen, is a precursor of Ang II, and chymase rather than ACE is the main enzyme contributing to the direct production of Ang II from [Ang-(1-12)]. The Ang-(1-12)/chymase axis is an independent intracrine pathway accounting for the trophic, contractile, and pro-arrhythmic Ang II actions in the human heart. [Ang-(1-12)] expression and chymase activity have been found elevated in the left atrial appendage of heart disease subjects, suggesting a pivotal role of this axis in the progression of HF.
Summary
Recent meta-analysis of large clinical trials on the use of ACE inhibitors and angiotensin receptor blockers in cardiovascular disease has demonstrated an imbalance between patients that significantly benefit from these therapeutic agents and those that remain at risk for heart disease progression. Looking to find an explanation, detailed investigation on the RAS has unveiled a previously-unrecognized complexity of substrates and enzymes in tissues ultimately associated with the production of Ang II that may explain the shortcomings of ACE inhibition and angiotensin receptor blockade. Discovery of the [Ang-(1-12)]/chymase axis in human hearts, capable of producing Ang II independently from the circulatory RAS, has led to the notion that a tissue-delimited RAS signaling in an intracrine fashion may account for the deleterious effects of Ang II in the heart, contributing to the transition from maladaptive cardiac remodeling to heart failure. Targeting intracellular RAS signaling may improve current therapies aimed at reducing the burden of heart failure.
Keywords: intracrine, angiotensin-(1-12), chymase, cardiomyocyte, angiotensin converting enzyme inhibitor, angiotensin receptor blockers
INTRODUCTION
Hypertension is the preeminent risk factor contributing to the development of cardiovascular disease, including heart failure,[1–4] and is thereby considered the leading global mortality hazard by the World Health Organization.[5] In hypertension, the elevated cardiac afterload elicits a series of myocardial responses leading to an initial phase of adaptive hypertrophy aimed at maintaining cardiac output to sustain the body’s elevated metabolic demand.[6] If the external stress persists, myocardial homeostasis becomes compromised preventing maintenance of the initial adaptive response, at which point hypertrophy turns into chamber enlargement and wall thinning with reduced pumping capacity.[6-9] This maladaptive remodeling of the ventricle, characterized by activation of inflammatory processes, replacement of cardiomyocytes with fibrotic tissue, reduced capillary density and overall cellular dysfunction[6] will ultimately progress to heart failure with reduced or preserved left ventricular ejection fraction. As the impact of the hypertension-induced adverse remodeling extends to the atrial chambers it sets the stage for the development of arrhythmias, in particular atrial fibrillation,[10] increasing thereby the predisposition of the cardiac pump to fail.[11] The main events prompting cardiac hypertrophy in the setting of elevated arterial blood pressure are mechanical stress and neurohumoral stimulation, which have been shown to modulate gene expression, protein synthesis, sarcomere assembly and cell metabolism.[12-14] When activated chronically and excessively, mechanotransduction and neurohumoral signaling further contribute to the transition from adaptive hypertrophy to maladaptive cardiac remodeling leading to heart failure. [7, 15]
Current therapeutic interventions aimed at reducing the burden of hypertension are guided by initial evidence suggesting a significant effect on mortality imparted by suppression of neurohumoral signaling of the renin-angiotensin system (RAS) with either angiotensin converting enzyme (ACE) inhibitors or Ang II (Ang II) receptor (AT1R) blockers (ARBs) [16]; randomized clinical trials are published.[17] While the beneficial effects of ACE inhibitors or ARBs in retarding the progression of cardiac dysfunction are documented,[18••] a more critical evaluation of the long-term benefit of high doses of ACE inhibitors and ARBs on cardiovascular mortality in heart failure has found it to be modest.[17] Likewise, recent meta-analyses reveal a suboptimal efficacy of ACE inhibitors or ARBs in reversing or mitigating the progression of cardiovascular disease.[19,20••,21•] Given the vast accumulated evidence demonstrating the contribution of Ang II to adverse cardiovascular remodeling, it is necessary to reconsider whether or not the limited efficacy of current RAS blockers might be partly explained by the inability of these agents to suppress the expression and activity of an independent intracellular RAS.
This review addresses the interplay between the RAS and disease-associated cardiac (mal)adaptive remodeling. We provide a compendium of recent evidence suggesting the need for a shift in the current therapeutic paradigm for hypertension-related cardiovascular disease, and highlighting the pivotal contribution of tissue-compartmentalized RAS and intracrine signaling within cardiac myocytes to the progression from an at-risk hypertensive state to overt heart failure. The recently completed Systolic Blood Pressure Intervention trial (SPRINT) underscores the importance of hypertension as a predictor of heart failure since the rate of heart failure occurrence was almost 40% less in patients assigned to the intensive blood pressure treatment.[22]
THE RENIN-ANGIOTENSIN SYSTEM
Starting with the hallmark discovery of renin by Tigersted and Bergman,[23] extensive knowledge has been accumulated on the nature and characteristics of the RAS over the last 100 years.[24,25•] Several reviews are available for those less familiar with the biochemical mechanisms involved in the biotransformation of angiotensinogen into biologically active peptides.[25•–28•] In the context of this presentation, we place emphasis on regulatory mechanisms participating in the generation of Ang II via enzymatic pathways that are not renin- or ACE-dependent. This non-canonical biotransforming pathway for generation of Ang II and the countervailing actions of angiotensin-(1-7) [Ang-(1-7)] are becoming critically important as the overall foundation of the strategies that led to the development of ACE inhibitors, ARBs, and even direct renin inhibitors, were based on the rationale that renin and ACE were the primary enzymes accounting for Ang II generation. As recounted elsewhere, Dr. Ferrario’s laboratory was the first to put forward the ideas that led to the characterization of an alternate pathway through which angiotensin I (Ang I) or Ang II could lead to the production of Ang-(1-7) via tissue endopeptidases and the monocarboxy peptidase angiotensin converting enzyme 2 (ACE2). [28•–30] The diversity of biologically active angiotensin peptides functions, and the expression of angiotensin producing genes in tissues other than the kidneys endows the RAS with the capacity to modulate morphogenesis, tissue repair and regeneration, as well as immunity. [31]
Endocrine versus Intracrine RAS
Initially described as an exclusively endocrine system regulating blood pressure, the RAS has been recently recognized as a more complex system with extended paracrine and autocrine functions.[18••, 25•, 32] Over the last quarter century, our understanding of the RAS has been further expanded by the characterization of other bioactive derivatives of angiotensinogen, most notably Ang-(1-7)[33] and Ang-(1-9),[34]. More recently, two alternate shorter forms of the angiotensinogen substrate have been identified by Japanese investigators at Miyazaki, Japan. Nagata et al. [35••] first identified the dodecapeptide angiotensin-(1-12) [Ang-(1-12)] in the tissue and blood of a Wistar strain of rats and later reported the detection of angiotensin-(1-25) in human urine.[36•] Ang-(1-12), an extended form of Ang I present in a variety of tissues, has gained interest as an intracellular substrate for Ang II generation.[18••]
The aforementioned alternative biosynthetic pathways seem to be most prominent in tissues rather than in the circulation.[18••] Cytoplasmic levels of Ang-(1-12) in cardiomyocytes are increased in spontaneously hypertensive rats (SHR) compared to Wistar Kyoto (WKY) controls, following an expression pattern similar to Ang I and Ang II but not angiotensinogen.[37] Further studies demonstrated that Ang-(1-12) is indeed an endogenous precursor for intracellular Ang II formation, and that this alternate substrate is regulated independently from the circulating RAS.[18••] Furthermore, Ang-(1-12) metabolism into Ang I or Ang II occurs through a non-renin dependent pathway as documented in anephric rats,[38] or following the administration of a specific rat renin inhibitor in the isolated perfused heart of WKY and SHR.[39] In humans, the expression of Ang-(1-12) has been reported in the left ventricle as well as in the left and right atria.[40–42••] Concomitantly, chymase was found to be expressed in left atrial tissue obtained from patients undergoing open-heart surgery for the correction of resistant atrial fibrillation.[40] Recently, we also showed that left ventricular plasma membranes obtained from normal human subjects metabolized Ang-(1-12) directly into Ang II via chymase. Using normal human left ventricular tissue recovered from vehicular death accidents, reverse phase high performance liquid chromatography in the presence of inhibitors for chymase (chymostatin), ACE (lisinopril), ACE2 (MLN-4760), and neprilysin (SHC39370) demonstrated that almost all of cardiac radiolabeled Ang-(1-12) remained intact, whereas exclusion of chymostatin from the inhibitor cocktail led to significant conversion of radiolabeled Ang-(1-12) into Ang II; negligible Ang-(1-12) hydrolysis occurred by ACE, ACE2, and neprilysin.[41] This predominant role of chymase in the production of Ang II from Ang-(1-12) is consistent with accounts of chymase’s pivotal role in processing Ang I into Ang II in human hearts.[43••–45] Given the lack of chymase activity in the circulation due to the high concentration of circulating endogenous serine protease inhibitors, and the mainly interstitial and intracellular localization of chymase that is upregulated during ACE inhibition, [46••, 47] we have proposed that the Ang-(1-12)/chymase axis constitutes a distinct, non-canonical, tissue-delimited system that accounts for the production of Ang II in cardiomyocytes and its intracrine pathological actions (Figure. 1).[18••] We have extended these findings to demonstrate intracellular chymase in dog cardiomyocytes after ischemia reperfusion injury [48••] and its production by cardiac fibroblasts during heart failure in the rat.[49••] These new findings underscore that mast cells are not the only source of chymase in the heart and that there is a yet to be determined mechanism of chymase uptake and/or synthesis in cardiomyocytes.
Figure 1.
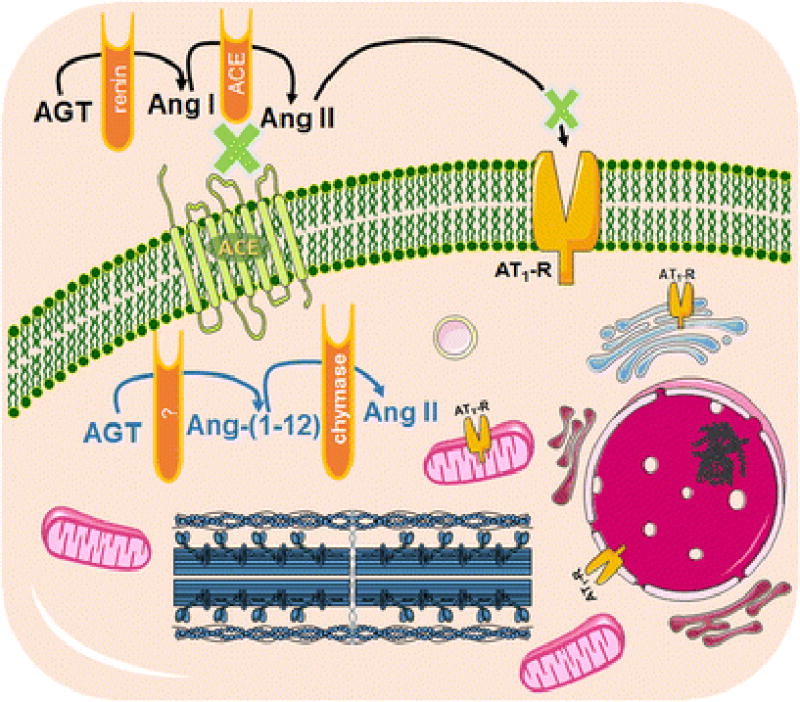
Ang II as an intracrine hormone. Schematic diagram of tissue-delimited RAS depicting the enzymatic action of a yet to be determined enzyme and chymase to form Ang-(1-12) and Ang II, respectively, in a cardiac cell. This intracrine system is independent from circulating RAS, as shown in the presence of ACE inhibition and AT1R blockade, and is capable of cell-contained production of Ang II, which can then interact with intracellular AT1R to exert biological actions. AGT: angiotensinogen; Ang I: angiotensin I; Ang II: Ang II; Ang-(1-12): angiotensin-(1-12); AT1R: Ang II type 1 receptor.
In recent studies, we have extended the importance of chymase as an Ang II forming enzyme through the demonstration that rat chymase catalytic efficiency (ratio of Vmax/Km) for Ang-(1-12) is 15-fold higher than for the Ang I substrate.[50•] Several studies in the rat have documented the functionality of Ang-(1-12) as an Ang II forming substrate [51-58] including the observation of antihypertensive effect through immunoneutralization of cerebrospinal fluid Ang-(1-12) in transgenic hypertensive rats.[59] Studies in intact rats,[35••] the isolated heart,[60] CHO cells transfected with AT1R[54] or intracellular Ang-(1-12) injection into WKY cardiac myocytes[61••] demonstrate that the majority of the cellular responses produced by Ang-(1-12) are mediated through AT1 receptors. Cardiac intracellular Ang-(1-12) actions were manifested as a prolongation of the action potential via a decrease of total potassium current due to activation of protein kinase C.[61••] These findings are of considerable importance as reentrant rhythms are caused by a decline in conduction velocity. Furthermore, our previous study using mRen2 rats indicated that chronic estradiol treatment attenuated ovariectomy-associated increases in cardiac Ang II, chymase gene expression, and mast cell number, in the absence of altered cardiac ACE expression or activity, suggesting that the cardioprotective effects of estradiol may be driven by a local reduction of chymase-dependent Ang II formation.[62] Thus, the characterization of this Ang-(1-12)/chymase axis as an independent intracrine pathway accounting for the trophic, contractile, and pro-arrhythmic Ang II actions in the human heart may explain the less than predicted beneficial actions of ACE inhibitors and ARBs in the evolution of cardiac disease.
An intracrine is defined as a protein or peptide with distinct physiological actions that can traffic between cells and signal intracellularly through interaction with classical receptors or independent thereof.[63••] Intracrines, which can be hormones, growth factors, cytokines, DNA binding proteins or enzymes, act in a feed-forward loop that upregulates their synthesis in target cells (which can include the same cell where they were originally synthesized), or the activity of target signaling cascades, thus exerting their effects even when the initial intracrine signal is no longer present. Several components of the RAS are intracrines, including Ang II, Ang-(1-7), angiotensinogen, ACE, and renin, but it has been suggested that Ang II may be the main effector of intracrine RAS.[63••] If this is the case, development of effective intracellular antagonists of Ang II action should prevent the progression of disease.
Indeed, while Ang II internalization and nuclear binding in cardiac myocytes was demonstrated decades ago, currently available data suggests cardiac intracellular production of both Ang II and Ang-(1-7) as the critical mechanisms accounting for the pathological actions of the RAS.[32, 64, 65] Interestingly, Ang II and Ang-(1-7) have been also shown to exert their local biological activity without mediation of plasma membrane-localized receptors but rather directly inside the cell without intermediate secretion.[25•, 65] It has been reported that intracellular effects of Ang II are not blocked by ARBs. Candesartan does not prevent increased Ang II synthesis in cardiomyocytes exposed to high glucose;[66] while chronic administration of losartan, lisinopril or both had no effect on cardiac Ang II content.[67] Similar conclusions were obtained in the heart of hypertensive rats chronically medicated with olmesartan.[68] On the other hand, a report demonstrating that losartan may be capable of blocking nuclear Ang II receptors following cell-surface receptor-mediated endocytosis and impede Ang II cellular effects,[69] warrants further studies to unveil the intricacies of the tissue RAS.
CURRENT RAS THERAPEUTICS: are we doing all we can?
Captopril[70, 71] was the first ACE inhibitor to become widely available for clinical use in 1981, and the ARB losartan [72] was first introduced into clinical practice in the mid-1990s.[73] Since then, suppressing the activity of the RAS with ACE inhibitors or ARBs has been repeatedly shown to improve the treatment of patients with congestive heart failure, post-myocardial infarction, and chronic renal disease.[21•] Indeed, ACE inhibitors and ARBs have become the cornerstone of treatment for heart failure patients over the past 30 years, as they blunt left ventricular hypertrophy, diminish dilatation of the left ventricle after myocardial infarction, and provide a mortality benefit.[74] A significant number of clinical trials enrolling ≥1,000 patients have been completed on the use of ACE inhibitors and ARBs for cardiovascular disorders (Figure. 2). Although the clinical trial data indicate that ACE inhibitors and ARBs therapies provide a significant benefit compared to placebo (as measured by relative risk reduction for myocardial infarction, stroke, congestive heart failure, cardiovascular hospitalization, and cardiovascular-related mortality), Figure 2 shows that the benefit across all treatment groups is associated with less than a 40% decrease in the primary end-points.[16, 75-78] For example, while treatment with ramipril in the Heart Outcomes Prevention Evaluation (HOPE) trial did reduce the number of high-risk patients experiencing cardiovascular-related mortality, myocardial infarction, or stroke, compared with placebo, 14% of the patients in the treatment group nevertheless experienced a cardiovascular-related event, compared with 17.8% of placebo-treated patients (a 3.8% difference between the two groups).[75] In the SOLVD (Studies of Left Ventricular Dysfunction) trial,[76]; which compared enalapril versus placebo in heart failure, 35.2% (452) of the enalapril patients died, compared with 39.7% (510) of placebo patients. Although the treatment added benefit, a considerable number of treated patients did not survive.[76] In the CHARM-Alternative (Candesartan in Heart failure — Assessment of Reduction in Mortality and Morbidity) trial,[79] in which heart failure patients who did not tolerate ACE inhibitors were randomized to placebo or candesartan, hospitalization or cardiovascular-related death was reported in 33% (n=334) of candesartan patients versus 40% (n=406) of placebo patients during a median follow-up of 33.7 months. Once again, the treatment offered benefit compared with placebo, but a considerable number of patients did not survive or experienced an unfavorable outcome.[77] Only a 5-18% relative reduction in the risk of cardiovascular death in patients with chronic heart failure and reduced ejection fraction has been achieved by treatment with ACE inhibitors and ARBs compared to placebo,[74] and combination therapy provides no additive effect,[17] suggesting that while ACE inhibitor and ARB therapies are effective, there is ample room for improvement. Furthermore, clinical trials have struggled to identify a favorable effect of these drugs on symptoms or quality of life,[80-83] and cardiovascular mortality in heart failure patients remains unacceptably high. Additionally, there is low evidence for superior effectiveness of ACE inhibitors and ARBs in patients with hypertension compared to other therapeutics, suggesting that the benefit of RAS blockade appears to be primarily the result of antihypertensive effects rather than the additional benefit that could be gained from blockade of direct Ang II pathological actions in target organs.[21•,84]
Figure 2.
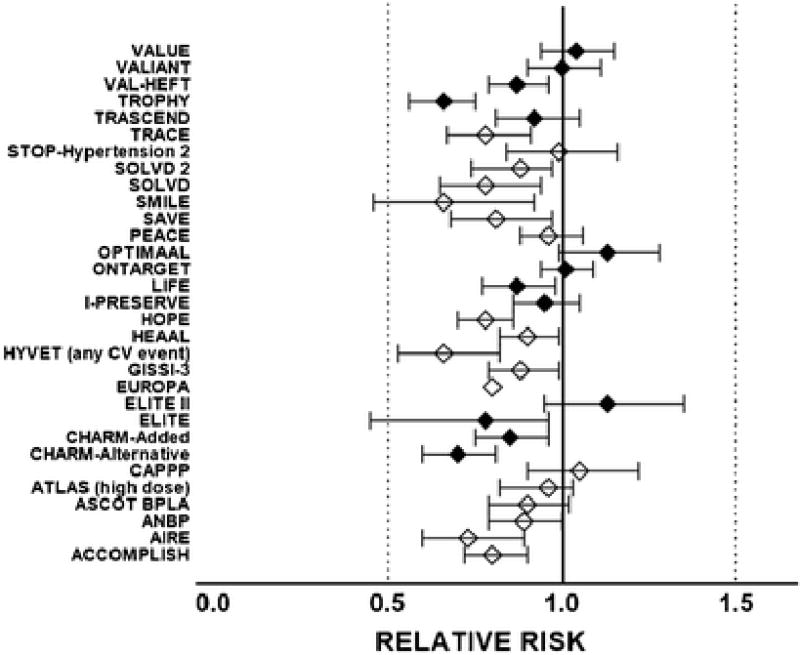
Relative risk and 95 % confidence intervals of the effect of angiotensin converting enzyme inhibitors (open diamonds) or Ang II receptor blockers (closed diamonds) on primary cardiac end points of large randomized clinical trials. Overall, the reduction in the primary end-point across all the trials documented here averaged 0.87 (CI, 0.83 – 0.92). Acronyms are: ACCOMPLISH, Avoiding Cardiovascular Events through Combination Therapy in Patients Living with Systolic Hypertension [96]; AIRE, Acute Infarction Ramipril Efficacy [97]; ANBP-2, Second Australian National Blood Pressure Study Group [98]; ASCOT BPLA, Anglo-Scandinavian Cardiac Outcomes Trial-Blood Pressure Lowering Arm [99]; ATLAS (high dose), Assessment of Treatment with Lisinopril And Survival [100]; CAPPP, Captopril Prevention Project [101]; CHARM-Alternative, Candesartan in Heart failure: Assessment of Reduction in Mortality and morbidity [77]; CHARM-Added, Candesartan in Heart failure: Assessment of Reduction in Mortality and morbidity [79]; ELITE, Evaluation of Losartan in the Elderly Study [102]; ELITE II, the Losartan Heart Failure Survival Study (Evaluation of Losartan in the Elderly Study) [103]; EUROPA, European trial on Reduction Of cardiac events with Perindopril in patients with stable coronary Artery disease [104]; GISSI-3, Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto Miocardico [105]; HYVET, Hypertension in the Very Elderly Trial [106]; HEAAL, Heart failure Endpoint evaluation of Ang II Antagonist Losartan [107]; HOPE, Heart Outcomes Prevention Evaluation Study [108]; I-PRESERVE, Irbesartan in Heart Failure with Preserved Ejection Fraction Study [109]; LIFE, Losartan Intervention For Endpoint reduction Study [78]; ONTARGET, The Ongoing Telmisartan Alone and in Combination with Ramipril Global Endpoint Trial [110]; OPTIMAAL, Optimal Trial in Myocardial Infarction with the Ang II Antagonist Losartan [111]; PEACE, Prevention of Events with Angiotensin Converting Enzyme Inhibition [112]; SAVE, Survival and Ventricular Enlargement trial [113]; SMILE, Survival of Myocardial Infarction Long-Term Evaluation trial [114]; SOLVD, Studies of Left Ventricular Dysfunction [76]; SOLVD 2, Studies of Left Ventricular Dysfunction [115]; STOP-Hypertension 2, Swedish Trial in Old Patients with Hypertension-2 study [116]; TRACE, Trandolapril Cardiac Evaluation [117]; TRASCEND, Telmisartan Randomised Assessmen Study in ACE Intolerant subjects with cardiovascular Disease [118]; TROPHY, Trial Preventing Hypertension [119]; VAL-HEFT, Valsartan Heart Failure Trial [120]; VALIANT, Valsartan in Acute Myocardial Infarction trial [121]; VALUE, Valsartan Antihypertensive Long-term Use Evaluation study [122].
Considering residual cardiovascular risk as the risk of incident cardiovascular events or progression of established cardiovascular damage persisting in patients treated with ACE inhibitors or ARBs, the need for additional, complementary or alternative therapeutics becomes pressing, as patients face residual risks greater than 70% when treated with current standard therapy. The number needed to treat (NNT), defined as the number of patients who need to be treated in order to prevent one additional negative outcome, or alternatively as the inverse of the absolute risk reduction, further highlights the shortcomings of current RAS blockade therapy in cardiovascular disease. A recent meta-analysis of the effectiveness of RAS inhibitors to prevent all-cause and cardiovascular death, myocardial infarction and stroke in hypertensive patients[20••] demonstrated not only that the relative risk reduction by ACE inhibitors and ARBs is modest, but also that the NNT is substantial (116 patients to prevent an additional cardiovascular mortality outcome and 80 to prevent a myocardial infarction for ACE inhibitors; 409 patients to prevent an additional cardiovascular mortality outcome and 336 to prevent a myocardial infarction for ARBs).
Noteworthy, it has been shown that, despite adequate inhibition of ACE, the deleterious effects of Ang II may not always be completely eradicated. Plasma Ang II levels return to normal under long-term ACE inhibition despite a significant drop on treatment initiation[85] while addition of valsartan to the direct renin inhibitor aliskiren augments the antihypertensive effect.[86] Thus, despite significant genetic, molecular, physiological and clinical evidence for a critical participation of the RAS, and in particular Ang II, in the pathogenesis of cardiovascular disease, the long-term effects of RAS blockade using direct renin inhibitors, ACE inhibitors and ARBs falls short of expectations.
Why ACE inhibitors and ARBs are not more effective than other antihypertensive drugs on hard endpoints in clinical trials remains a subject of debate. We have proposed that the discrepancy may be accounted for by the inability of these agents to reach the site(s) at which Ang II is generated (i.e., cardiac intracellular sites).[33, 87] Furthermore, the primary role of chymase rather than ACE[88-93, 46] as an Ang II-forming enzyme in human cardiac and vascular tissues is an additional limiting factor in achieving greater therapeutic benefit. While knowledge as to the importance of chymase as an Ang II-forming enzyme in humans spans over a quarter of a century,[44] the importance of this non-ACE dependent pathway remains grossly underappreciated and blissfully ignored as a topic of relevance in symposia and leading cardiovascular and hypertension journals. A clinical case for studying the chymase/Ang II axis as an additional (potentially additive) target for inhibition of local RAS comes from preclinical studies of chymase inhibition.[94••, 95] In addition, combined chymase and ACE inhibition, compared to ACE inhibition alone, provides an added benefit in terms of left ventricular function, adverse cardiac remodeling and survival after myocardial infarction in hamsters.[46••]
CONCLUSIONS
Recognition that Ang II, the main effector of the RAS, is a potent driver of maladaptive cardiac remodeling increasing the susceptibility to heart failure, has fueled decades of basic, translational and clinical research aimed at blocking Ang II deleterious actions. ACE inhibitors and ARBs have provided relief for a segment of the population with or at risk for heart failure. The impact of these drugs on morbidity and mortality has been widely recognized, yet it is frequently ignored that a vast majority of treated patients remain at risk for disease progression. There is, therefore, a pressing need for research aimed at finding adjuvant or alternative therapies that can fulfill the promise of providing all patients with safe and effective treatment. Targeting the intracellular RAS, in particular the Ang-(1-12)/chymase axis, provides a novel avenue not only for understanding the complex cellular mechanisms mediating the transition from cardiac tissue remodeling to organ failure, but for potentially improving the efficacy of current therapeutic regimens. Promising animal studies demonstrating added benefit of chymase inhibitors when administered in combination with ACE inhibitors[46••] are a solid foundation to build upon for developing clinically-relevant solutions based on interrupting intracrine RAS signaling for the treatment of heart failure.
LIST OF MENTIONED DRUGS.
-
–
Angiotensin Converting Enzyme inhibitors: captopril, lisinopril, ramipril, enalapril, perindopril, trandolapril
-
–
Angiotensin Converting Enzyme 2 inhibitors: MLN-4760
-
–
Angiotensin II Receptor Blockers: losartan, candesartan, olmesartan, irbesartan, telmisartan, valsartan
-
–
Chymase inhibitors: chymostatin
-
–
Neprilysin inhibitors: SHC39370
Acknowledgments
Dr. Ferrario reports grants from National Heart, Lung Blood Institute of the NIH; and personal fees from Sanofi and Daiichi Sankyo.
ABBREVIATIONS
- RAS
renin-angiotensin system
- ACE
angiotensin converting enzyme
- ARB
angiotensin II receptor blocker
- Ang
angiotensin
Footnotes
Compliance with Ethics Guidelines
Conflict of Interest
Drs. Reyes, Varagic, Ahmad, VonCannon, Kon, Wang, Groban, Cheng, and Dell’Italia declare no conflicts of interest relevant to this manuscript.
Human and Animal Rights and Informed Consent
All reported studies/experiments with human or animal subjects performed by the authors have been previously published and complied with all applicable ethical standards (including the Helsinki declaration and its amendments, institutional/national research committee standards, and international/national/institutional guidelines).
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
- 1.Drazner MH. The progression of hypertensive heart disease. Circulation. 2011;123:327–34. doi: 10.1161/CIRCULATIONAHA.108.845792. [DOI] [PubMed] [Google Scholar]
- 2.Khatibzadeh S, Farzadfar F, Oliver J, Ezzati M, Moran A. Worldwide risk factors for heart failure: a systematic review and pooled analysis. Int J Cardiol. 2013;168:1186–94. doi: 10.1016/j.ijcard.2012.11.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O’Donnell CJ, Elosua R. Cardiovascular risk factors. Insights from Framingham Heart Study. Rev Esp Cardiol. 2008;61:299–310. doi: 10.1016/S1885-5857(08)60118-8. [DOI] [PubMed] [Google Scholar]
- 4.Haider AW, Larson MG, Franklin SS, Levy D. Systolic blood pressure, diastolic blood pressure, and pulse pressure as predictors of risk for congestive heart failure in the Framingham Heart Study. Ann Intern Med. 2003;138:10–6. doi: 10.7326/0003-4819-138-1-200301070-00006. [DOI] [PubMed] [Google Scholar]
- 5.Global Health Risks: Mortality and Burden of Disease attributable to Selected Major Risks. www.who.int/healthinfo/global_burden_disease/GlobalHealthRisks_report_full.pdf. Accessed 09-19-2016.
- 6.Shimizu I, Minamino T. Physiological and pathological cardiac hypertrophy. J Mol Cell Cardiol. 2016;97:245–62. doi: 10.1016/j.yjmcc.2016.06.001. [DOI] [PubMed] [Google Scholar]
- 7.Drazner MH. The transition from hypertrophy to failure: how certain are we? Circulation. 2005;112:936–8. doi: 10.1161/CIRCULATIONAHA.105.558734. [DOI] [PubMed] [Google Scholar]
- 8.Kehat I, Molkentin JD. Molecular pathways underlying cardiac remodeling during pathophysiological stimulation. Circulation. 2010;122:2727–35. doi: 10.1161/CIRCULATIONAHA.110.942268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jessup M, Brozena S. Heart failure. N Engl J Med. 2003;348:2007–18. doi: 10.1056/NEJMra021498. [DOI] [PubMed] [Google Scholar]
- 10.Xu Y, Sharma D, Li G, Liu Y. Atrial remodeling: new pathophysiological mechanism of atrial fibrillation. Med Hypotheses. 2013;80:53–6. doi: 10.1016/j.mehy.2012.10.009. [DOI] [PubMed] [Google Scholar]
- 11.Odutayo A, Wong CX, Hsiao AJ, Hopewell S, Altman DG, Emdin CA. Atrial fibrillation and risks of cardiovascular disease, renal disease, and death: systematic review and meta-analysis. Br Med J (Clin Res Ed) 2016;354:i4482. doi: 10.1136/bmj.i4482. [DOI] [PubMed] [Google Scholar]
- 12.Lyon RC, Zanella F, Omens JH, Sheikh F. Mechanotransduction in cardiac hypertrophy and failure. Circ Res. 2015;116:1462–76. doi: 10.1161/CIRCRESAHA.116.304937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Francis GS, McDonald KM, Cohn JN. Neurohumoral activation in preclinical heart failure. Remodeling and the potential for intervention. Circulation. 1993;87:IV90–6. [PubMed] [Google Scholar]
- 14.Usui S, Yao A, Hatano M, Kohmoto O, Takahashi T, Nagai R, et al. Upregulated neurohumoral factors are associated with left ventricular remodeling and poor prognosis in rats with monocrotaline-induced pulmonary arterial hypertension. Circ J. 2006;70:1208–15. doi: 10.1253/circj.70.1208. [DOI] [PubMed] [Google Scholar]
- 15.Hill JA, Olson EN. Cardiac plasticity. N Engl J Med. 2008;358:1370–80. doi: 10.1056/NEJMra072139. [DOI] [PubMed] [Google Scholar]
- 16.The CONSENSUS Trial Study Group. Effects of enalapril on mortality in severe congestive heart failure. Results of the Cooperative North Scandinavian Enalapril Survival Study (CONSENSUS) N Engl J Med. 1987;316:1429–35. doi: 10.1056/NEJM198706043162301. [DOI] [PubMed] [Google Scholar]
- 17.Packer M. Love of angiotensin-converting enzyme inhibitors in the time of cholera. JACC Heart Fail. 2016;4:403–8. doi: 10.1016/j.jchf.2016.02.012. [DOI] [PubMed] [Google Scholar]
- 18••.Ferrario CM, Ahmad S, Varagic J, Cheng CP, Groban L, Wang H, et al. Intracrine Ang II functions originate from noncanonical pathways in the human heart. Am J Physiol Heart Circ Physiol. 2016;311:H404–14. doi: 10.1152/ajpheart.00219.2016. This review summarizes the functional significance of Ang-(1-12) as a primary tissue-borne substrate of the pathological actions of Ang II. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baker WL, Coleman CI, Kluger J, Reinhart KM, Talati R, Quercia R, et al. Systematic review: comparative effectiveness of angiotensin-converting enzyme inhibitors or Ang II-receptor blockers for ischemic heart disease. Ann Intern Med. 2009;151:861–71. doi: 10.7326/0003-4819-151-12-200912150-00162. [DOI] [PubMed] [Google Scholar]
- 20••.Brugts JJ, van Vark L, Akkerhuis M, Bertrand M, Fox K, Mourad JJ, et al. Impact of renin-angiotensin system inhibitors on mortality and major cardiovascular endpoints in hypertension: A number-needed-to-treat analysis. Int J Cardiol. 2015;181:425–9. doi: 10.1016/j.ijcard.2014.11.179. In this study, the NNT metric is used to compare the impact of ACE inhibitors and ARBs on cardiovascular end points. Despite proven benefits, the number of patients needed to be treated to prevent one additional event remains large. [DOI] [PubMed] [Google Scholar]
- 21•.Dusing R. Mega clinical trials which have shaped the RAS intervention clinical practice. Ther Adv Cardiovasc Dis. 2016;10:133–50. doi: 10.1177/1753944716644131. This important review analyzed the outcomes of all clinical trials involving >1000 patients employing ACE inhibitors and ARBs. The article provides a broader perspective on the current knowledge regarding these therapeutic strategies. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wright JTJ, Williamson JD, Whelton PK, Snyder JK, Sink KM, Rocco MV, et al. Sprint Research Group A randomized trial of intensive versus standard blood-pressure control. N Engl J Med. 2015;373:2103–16. doi: 10.1056/NEJMoa1511939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tigerstedt R, Bergman PQ. Niere und Kreislauf. Skand Arch Physiol. 1898;8:223–71. doi: 10.1111/j.1748-1716.1898.tb00272.x. [DOI] [Google Scholar]
- 24.Marks LS, Maxwell MH. Tigerstedt and the discovery of renin. An historical note. Hypertension. 1979;1:384–8. doi: 10.1161/01.hyp.1.4.384. [DOI] [PubMed] [Google Scholar]
- 25•.Abadir PM, Walston JD, Carey RM. Subcellular characteristics of functional intracellular renin-angiotensin systems. Peptides. 2012;38:437–45. doi: 10.1016/j.peptides.2012.09.016. This review focuses on the subcellular localization, distribution and functions of intracellular RAS components, with an emphasis of potential consequences of RAS activation in different organ systems. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chappell MC. Biochemical evaluation of the renin-angiotensin system: the good, bad, and absolute? Am J Physiol Heart Circ Physiol. 2016;310:H137–52. doi: 10.1152/ajpheart.00618.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ferrario CM, Ahmad S, Joyner J, Varagic J. Advances in the renin angiotensin system focus on angiotensin-converting enzyme 2 and angiotensin-(1-7) Adv Pharmacol. 2010;59:197–233. doi: 10.1016/S1054-3589(10)59007-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28•.Ferrario CM, Ahmad S, Nagata S, Simington SW, Varagic J, Kon N, et al. An evolving story of angiotensin-II-forming pathways in rodents and humans. Clin Sci (Lond) 2014;126:461–9. doi: 10.1042/CS20130400. This review summarizes the research done on intermediate shorter forms of angiotensinogen, and highlights the enzymatic production of Ang-(1-12) from cardiac chymase. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ferrario CM, Brosnihan KB, Diz DI, Jaiswal N, Khosla MC, Milsted A, et al. Angiotensin-(1–7): a new hormone of the angiotensin system. Hypertension. 1991;18:III126–33. doi: 10.1161/01.hyp.18.5_suppl.iii126. [DOI] [PubMed] [Google Scholar]
- 30.Ferrario CM, Chappell MC, Tallant EA, Brosnihan KB, Diz DI. Counterregulatory actions of angiotensin-(1-7) Hypertension. 1997;30:535–41. doi: 10.1161/01.HYP.30.3.535. [DOI] [PubMed] [Google Scholar]
- 31.Gomez RA, Belyea B, Medrano S, Pentz ES, Sequeira-Lopez ML. Fate and plasticity of renin precursors in development and disease. Pediatr Nephrol. 2014;29:721–6. doi: 10.1007/s00467-013-2688-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Re RN. Cardiac Ang II: an intracrine hormone? Am J Hypertens. 2003;16:426–7. doi: 10.1016/S0895-7061(02)03265-X. [DOI] [PubMed] [Google Scholar]
- 33.Ferrario CM. New physiological concepts of the renin-angiotensin system from the investigation of precursors and products of angiotensin I metabolism. Hypertension. 2010;55:445–52. doi: 10.1161/HYPERTENSIONAHA.109.145839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ocaranza MP, Michea L, Chiong M, Lagos CF, Lavandero S, Jalil JE. Recent insights and therapeutic perspectives of angiotensin-(1-9) in the cardiovascular system. Clin Sci (Lond) 2014;127:549–57. doi: 10.1042/CS20130449. [DOI] [PubMed] [Google Scholar]
- 35••.Nagata S, Kato J, Sasaki K, Minamino N, Eto T, Kitamura K. Isolation and identification of proangiotensin-12, a possible component of the renin-angiotensin system. Biochem Biophys Res Commun. 2006;350:1026–31. doi: 10.1016/j.bbrc.2006.09.146. Isolation and initial functional characterization of angiotensin-(1-12) as a novel substrate for Ang II production are first described here. [DOI] [PubMed] [Google Scholar]
- 36•.Nagata S, Hatakeyama K, Asami M, Tokashiki M, Hibino H, Nishiuchi Y, et al. Big angiotensin-25: a novel glycosylated angiotensin-related peptide isolated from human urine. Biochem Biophys Res Commun. 2013;441:757–62. doi: 10.1016/j.bbrc.2013.10.124. A 25 amino-acid-long derivative of angiotensinogen, Bang-25, is described as a precursor for Ang II formation by chymase but not renin in human urine. [DOI] [PubMed] [Google Scholar]
- 37.Jessup JA, Trask AJ, Chappell MC, Nagata S, Kato J, Kitamura K, et al. Localization of the novel angiotensin peptide, angiotensin-(1-12), in heart and kidney of hypertensive and normotensive rats. Am J Physiol Heart Circ Physiol. 2008;294:H2614–8. doi: 10.1152/ajpheart.91521.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ferrario CM, Varagic J, Habibi J, Nagata S, Kato J, Chappell MC, et al. Differential regulation of angiotensin-(1-12) in plasma and cardiac tissue in response to bilateral nephrectomy. Am J Physiol Heart Circ Physiol. 2009;296:H1184–92. doi: 10.1152/ajpheart.01114.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Trask AJ, Jessup JA, Chappell MC, Ferrario CM. Angiotensin-(1-12) is an alternate substrate for angiotensin peptide production in the heart. Am J Physiol Heart Circ Physiol. 2008;294:H2242–7. doi: 10.1152/ajpheart.00175.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ahmad S, Simmons T, Varagic J, Moniwa N, Chappell MC, Ferrario CM. Chymase-dependent generation of Ang II from angiotensin-(1-12) in human atrial tissue. PLoS One. 2011;6:e28501. doi: 10.1371/journal.pone.0028501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ahmad S, Wei CC, Tallaj J, Dell’Italia LJ, Moniwa N, Varagic J, et al. Chymase mediates angiotensin-(1-12) metabolism in normal human hearts. J Am Soc Hypertens. 2013;7:128–36. doi: 10.1016/j.jash.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42••.Nagata S, Varagic J, Kon ND, Wang H, Groban L, Simington SW, et al. Differential expression of the angiotensin-(1-12)/chymase axis in human atrial tissue. Ther Adv Cardiovasc Dis. 2015;9:168–80. doi: 10.1177/1753944715589717. Elevated chymase mRNA expression and enzymatic activity, associated increased Ang-(1-12) levels, in left versus right atrial appendages was found to be correlated with left atrial enlargment in humans, suggesting a role for the Ang-(1-12)/chymase axis in adverse heart remodeling. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 43••.Urata H, Healy B, Stewart RW, Bumpus FM, Husain A. Ang II-forming pathways in normal and failing human hearts. Circ Res. 1990;66:883–90. doi: 10.1161/01.RES.66.4.883. This landmark report documents that ACE is not the major Ang II-formaing enzyme in left ventricular tissue from normal and cardiomyopathic patients. [DOI] [PubMed] [Google Scholar]
- 44.Urata H, Kinoshita A, Misono KS, Bumpus FM, Husain A. Identification of a highly specific chymase as the major Ang II-forming enzyme in the human heart. J Biol Chem. 1990;265:22348–57. [PubMed] [Google Scholar]
- 45.Wolny A, Clozel JP, Rein J, Mory P, Vogt P, Turino M, et al. Functional and biochemical analysis of Ang II-forming pathways in the human heart. Circ Res. 1997;80:219–27. doi: 10.1161/01.RES.80.2.219. [DOI] [PubMed] [Google Scholar]
- 46••.Wei CC, Hase N, Inoue Y, Bradley EW, Yahiro E, Li M, et al. Mast cell chymase limits the cardiac efficacy of Ang I-converting enzyme inhibitor therapy in rodents. J Clin Invest. 2010;120:1229–39. doi: 10.1172/JCI39345. This study provides significant evidence for the key role of chymase in cardiac disease. Here, under chronic ACE inhibition conditions, chymase was found to be upregulated in the hamster heart, where it is the predominant Ang II producing enzyme. Chymase inhibition provided added cardiac benefit when administered in combination with an ACE inhibitor. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Urata H, Boehm KD, Philip A, Kinoshita A, Gabrovsek J, Bumpus FM, et al. Cellular localization and regional distribution of an Ang II-forming chymase in the heart. J Clin Invest. 1993;91:1269–81. doi: 10.1172/JCI116325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48••.Zheng J, Wei CC, Hase N, Shi K, Killingsworth CR, Litovsky SH, et al. Chymase mediates injury and mitochondrial damage in cardiomyocytes during acute ischemia/reperfusion in the dog. PLoS One. 2014;9:e94732. doi: 10.1371/journal.pone.0094732. This study provides evidence for interstitial upregulation of chymase activity, as well as intracellular chymase localization in cardiomyocytes of large mammals during ischemia/reperfusion, which could be reduced with an oral cymase inhibitor. Administration of chymase inhibitor protected against mitochondrial damage and cardiomyocyte death. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49••.Fu L, Wei CC, Powell PC, Bradley WE, Ahmad S, Ferrario CM, et al. Increased fibroblast chymase production mediates procollagen autophagic digestion in volume overload. J Mol Cell Cardiol. 2016;92:1–9. doi: 10.1016/j.yjmcc.2016.01.019. The production of chymase by cardiac fibroblasts is demonstrated in this study using the aortocaval fistula model in the rat to induce volume overload. Fibroblast-produced chymase is associated with extracellular-matrix degradation in heart failure. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50•.Ahmad S, Varagic J, VonCannon JL, Groban L, Collawn JF, Dell’Italia LJ, et al. Primacy of cardiac chymase over angiotensin converting enzyme as an angiotensin-(1-12) metabolizing enzyme. Biochem Biophys Res Commun. 2016;478:559–64. doi: 10.1016/j.bbrc.2016.07.100. This study demonstrates that Ang-(1-12) is the preferred substrate for Ang II formation in the adult rat heart, and confirms chymase rather than ACE as the main Ang II-producing enzyme. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Arakawa H, Chitravanshi VC, Sapru HN. The hypothalamic arcuate nucleus: a new site of cardiovascular action of angiotensin-(1-12) and Ang II. Am J Physiol Heart Circ Physiol. 2011;300:H951–60. doi: 10.1152/ajpheart.01144.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Arakawa H, Kawabe K, Sapru HN. Angiotensin-(1-12) in the rostral ventrolateral medullary pressor area of the rat elicits sympathoexcitatory responses. Exp Physiol. 2013;98:94–108. doi: 10.1113/expphysiol.2012.067116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Arnold AC, Isa K, Shaltout HA, Nautiyal M, Ferrario CM, Chappell MC, et al. Angiotensin-(1-12) requires angiotensin converting enzyme and AT1 receptors for cardiovascular actions within the solitary tract nucleus. Am J Physiol Heart Circ Physiol. 2010;299:H763–71. doi: 10.1152/ajpheart.00345.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chan KH, Chen YH, Zhang Y, Wong YH, Dun NJ. Angiotensin-[1-12] interacts with angiotensin type 1 receptors. Neuropharmacology. 2014;81:267–73. doi: 10.1016/j.neuropharm.2013.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chitravanshi VC, Proddutur A, Sapru HN. Cardiovascular actions of angiotensin-(1-12) in the hypothalamic paraventricular nucleus of the rat are mediated via Ang II. Exp Physiol. 2012;97:1001–17. doi: 10.1113/expphysiol.2011.062471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chitravanshi VC, Sapru HN. Cardiovascular responses elicited by a new endogenous angiotensin in the nucleus tractus solitarius of the rat. Am J Physiol Heart Circ Physiol. 2011;300:H230–40. doi: 10.1152/ajpheart.00861.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Moniwa N, Varagic J, Ahmad S, VonCannon JL, Simington SW, Wang H, et al. Hemodynamic and hormonal changes to dual renin-angiotensin system inhibition in experimental hypertension. Hypertension. 2013;61:417–24. doi: 10.1161/HYPERTENSIONAHA.112.201889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nagata S, Kato J, Kuwasako K, Asami M, Kitamura K. Plasma and tissue concentrations of proangiotensin-12 in rats treated with inhibitors of the renin-angiotensin system. Hypertens Res. 2012;35:234–8. doi: 10.1038/hr.2011.165. [DOI] [PubMed] [Google Scholar]
- 59.Isa K, Garcia-Espinosa MA, Arnold AC, Pirro NT, Tommasi EN, Ganten D, et al. Chronic immunoneutralization of brain angiotensin-(1-12) lowers blood pressure in transgenic (mRen2)27 hypertensive rats. Am J Physiol Regul Integr Comp Physiol. 2009;297:R111–5. doi: 10.1152/ajpregu.90588.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Prosser HC, Forster ME, Richards AM, Pemberton CJ. Cardiac chymase converts rat proAngiotensin-12 (PA12) to Ang II: effects of PA12 upon cardiac haemodynamics. Cardiovasc Res. 2009;82:40–50. doi: 10.1093/cvr/cvp003. [DOI] [PubMed] [Google Scholar]
- 61••.De Mello WC, Dell’Itallia LJ, Varagic J, Ferrario CM. Intracellular angiotensin-(1-12) changes the electrical properties of intact cardiac muscle. Mol Cell Biochem. 2016;422:31–40. doi: 10.1007/s11010-016-2801-3. The effects of intracellular Ang-(1-12) on the electrical properties of cardiac tissue are reported for the first time in this study. A decrease in total potassium current mediated by chymase-induced production of Ang II from Ang-(1-12) suggests functional relevance of intracelllular Ang-(1-12) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang H, Jessup JA, Zhao Z, Da Silva J, Lin M, MacNamara LM, et al. Characterization of the cardiac renin angiotensin system in oophorectomized and estrogen-replete mRen2. Lewis rats PLoS One. 2013;8:e76992. doi: 10.1371/journal.pone.0076992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63••.Re RN. A possible mechanism for the progression of chronic renal disease and congestive heart failure. J Am Soc Hypertens. 2015;9:54–63. doi: 10.1016/j.jash.2014.09.016. This review describes how tissue RAS may function in an intracrine fashion, and it proposes that an altered intracrine function of tissue RAS may result in chronic degenerative diseases. [DOI] [PubMed] [Google Scholar]
- 64.Baker KM, Chernin MI, Schreiber T, Sanghi S, Haiderzaidi S, Booz GW, et al. Evidence of a novel intracrine mechanism in Ang II-induced cardiac hypertrophy. Regul Pept. 2004;120:5–13. doi: 10.1016/j.regpep.2004.04.004. [DOI] [PubMed] [Google Scholar]
- 65.Kumar R, Singh VP, Baker KM. The intracellular renin-angiotensin system: a new paradigm. Trends Endocrinol Metab. 2007;18:208–14. doi: 10.1016/j.tem.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 66.Singh VP, Le B, Khode R, Baker KM, Kumar R. Intracellular Ang II production in diabetic rats is correlated with cardiomyocyte apoptosis, oxidative stress, and cardiac fibrosis. Diabetes. 2008;57:3297–306. doi: 10.2337/db08-0805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ferrario CM, Jessup J, Chappell MC, Averill DB, Brosnihan KB, Tallant EA, et al. Effect of angiotensin-converting enzyme inhibition and Ang II receptor blockers on cardiac angiotensin-converting enzyme 2. Circulation. 2005;111:2605–10. doi: 10.1161/CIRCULATIONAHA.104.510461. [DOI] [PubMed] [Google Scholar]
- 68.Varagic J, Ahmad S, VonCannon JL, Moniwa N, Brosnihan KB, Wysocki J, et al. Predominance of AT(1) blockade over mas-mediated angiotensin-(1-7) mechanisms in the regulation of blood pressure and renin-angiotensin system in mRen2.Lewis rats. Am J Hypertens. 2013;26:583–90. doi: 10.1093/ajh/hps090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cook JL, Zhang Z, Re RN. In vitro evidence for an intracellular site of angiotensin action. Circ Res. 2001;89:1138–46. doi: 10.1161/hh2401.101270. [DOI] [PubMed] [Google Scholar]
- 70.Cushman DW, Cheung HS, Sabo EF, Ondetti MA. Design of potent competitive inhibitors of angiotensin-converting enzyme. Carboxyalkanoyl and mercaptoalkanoyl amino acids. Biochemistry. 1977;16:5484–91. doi: 10.1021/bi00644a014. [DOI] [PubMed] [Google Scholar]
- 71.Gavras H, Brunner HR, Turini GA, Kershaw GR, Tifft CP, Cuttelod S, et al. Antihypertensive effect of the oral angiotensin converting-enzyme inhibitor SQ 14225 in man. N Engl J Med. 1978;298:991–5. doi: 10.1056/NEJM197805042981803. [DOI] [PubMed] [Google Scholar]
- 72.Duncia JV, Carini DJ, Chiu AT, Johnson AL, Price WA, Wong PC, et al. The discovery of DuP 753, a potent, orally active nonpeptide Ang II receptor antagonist. Med Res Rev. 1992;12:149–91. doi: 10.1002/med.2610120203. [DOI] [PubMed] [Google Scholar]
- 73.Timmermans PB, Wong PC, Chiu AT, Herblin WF, Benfield P, Carini DJ, et al. Ang II receptors and Ang II receptor antagonists. Pharmacol Rev. 1993;45:205–51. [PubMed] [Google Scholar]
- 74.Re RN, Cook JL. Noncanonical intracrine action. J Am Soc Hypertens. 2011;5:435–48. doi: 10.1016/j.jash.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 75.The Heart Outcomes Prevention Evaluation Study Investigators. Effects of an angiotensin-converting–enzyme inhibitor, Ramipril, on cardiovascular events in high-risk patients. N Engl J Med. 2000;342:145–53. doi: 10.1056/NEJM200001203420301. [DOI] [PubMed] [Google Scholar]
- 76.The SOLVD Investigators. Effect of enalapril on survival in patients with reduced left ventricular ejection fractions and congestive heart failure. N Engl J Med. 1991;325:293–302. doi: 10.1056/NEJM199108013250501. [DOI] [PubMed] [Google Scholar]
- 77.Granger CB, McMurray JJ, Yusuf S, Held P, Michelson EL, Olofsson B, et al. for the CHARM Investigators and Committees Effects of candesartan in patients with chronic heart failure and reduced left-ventricular systolic function intolerant to angiotensin-converting-enzyme inhibitors: the CHARM-Alternative trial. Lancet. 2003;362:772–6. doi: 10.1016/s0140-6736(03)14284-5. [DOI] [PubMed] [Google Scholar]
- 78.Dahlof B, Devereux RB, Kjeldsen SE, Julius S, Beevers G, de Faire U, et al. for the LIFE Study Group Cardiovascular morbidity and mortality in the Losartan Intervention For Endpoint reduction in hypertension study (LIFE): a randomised trial against atenolol. Lancet. 2002;359:995–1003. doi: 10.1016/s0140-6736(02)08089-3. [DOI] [PubMed] [Google Scholar]
- 79.McMurray JJ, Ostergren J, Swedberg K, Granger CB, Held P, Michelson EL, et al. for the CHARM Investigators and Committees Effects of candesartan in patients with chronic heart failure and reduced left-ventricular systolic function taking angiotensin-converting-enzyme inhibitors: the CHARM-Added trial. Lancet. 2003;362:767–71. doi: 10.1016/S0140-6736(03)14283-3. [DOI] [PubMed] [Google Scholar]
- 80.Abdulla J, Abildstrom SZ, Christensen E, Kober L, Torp-Pedersen C. A meta-analysis of the effect of angiotensin-converting enzyme inhibitors on functional capacity in patients with symptomatic left ventricular systolic dysfunction. Eur J Heart Fail. 2004;6:927–35. doi: 10.1016/j.ejheart.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 81.O’Meara E, Solomon S, McMurray J, Pfeffer M, Yusuf S, Michelson E, et al. Effect of candesartan on New York Heart Association functional class. Results of the Candesartan in Heart failure: Assessment of Reduction in Mortality and morbidity (CHARM) programme. Eur Heart J. 2004;25:1920–6. doi: 10.1016/j.ehj.2004.07.025. [DOI] [PubMed] [Google Scholar]
- 82.Cohn JN, Johnson G, Ziesche S, Cobb F, Francis G, Tristani F, et al. A comparison of enalapril with hydralazine-isosorbide dinitrate in the treatment of chronic congestive heart failure. N Engl J Med. 1991;325:303–10. doi: 10.1056/NEJM199108013250502. [DOI] [PubMed] [Google Scholar]
- 83.Majani G, Giardini A, Opasich C, Glazer R, Hester A, Tognoni G, et al. Effect of valsartan on quality of life when added to usual therapy for heart failure: results from the Valsartan Heart Failure Trial. J Card Fail. 2005;11:253–9. doi: 10.1016/j.cardfail.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 84.Turnbull F, Neal B, Ninomiya T, Algert C, Arima H, Barzi F, et al. fo the Blood Pressure Lowering Treatment Trialists’ Collaboration Effects of different regimens to lower blood pressure on major cardiovascular events in older and younger adults: meta-analysis of randomised trials. Br Med J (Clin Res Ed) 2008;336:1121–3. doi: 10.1136/bmj.39548.738368.BE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jin D, Takai S, Yamada M, Sakaguchi M, Kamoshita K, Ishida K, et al. Impact of chymase inhibitor on cardiac function and survival after myocardial infarction. Cardiovasc Res. 2003;60:413–20. doi: 10.1016/S0008-6363(03)00535-2. [DOI] [PubMed] [Google Scholar]
- 86.Oparil S, Yarows SA, Patel S, Zhang J, Satlin A. Dual inhibition of the renin system by aliskiren and valsartan. Lancet. 2007;370:1126–7. doi: 10.1016/S0140-6736(07)61508-6. [DOI] [PubMed] [Google Scholar]
- 87.Ferrario CM. Cardiac remodelling and RAS inhibition. Ther Adv Cardiovasc Dis. 2016;10:162–71. doi: 10.1177/1753944716642677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Balcells E, Meng QC, Johnson WH, Jr, Oparil S, Dell’Italia LJ. Ang II formation from ACE and chymase in human and animal hearts: methods and species considerations. Am J Physiol. 1997;273:H1769–74. doi: 10.1152/ajpheart.1997.273.4.H1769. [DOI] [PubMed] [Google Scholar]
- 89.Dell’Italia LJ, Husain A. Dissecting the role of chymase in Ang II formation and heart and blood vessel diseases. Curr Opin Cardiol. 2002;17:374–9. doi: 10.1097/00001573-200207000-00009. [DOI] [PubMed] [Google Scholar]
- 90.Husain A. The chymase-angiotensin system in humans. J Hypertens. 1993;11:1155–9. [PubMed] [Google Scholar]
- 91.Kinugawa T, Osaki S, Kato M, Ogino K, Shimoyama M, Tomikura Y, et al. Effects of the angiotensin-converting enzyme inhibitor alacepril on exercise capacity and neurohormonal factors in patients with mild-to-moderate heart failure. Clin Exp Pharmacol Physiol. 2002;29:1060–5. doi: 10.1046/j.1440-1681.2002.03779.x. [DOI] [PubMed] [Google Scholar]
- 92.Takai S, Jin D, Miyazaki M. New approaches to blockade of the renin-angiotensin-aldosterone system: chymase as an important target to prevent organ damage. J Pharmacol Sci. 2010;113:301–9. doi: 10.1254/jphs.10R05FM. [DOI] [PubMed] [Google Scholar]
- 93.Urata H, Kinoshita A, Perez DM, Misono KS, Bumpus FM, Graham RM, et al. Cloning of the gene and cDNA for human heart chymase. J Biol Chem. 1991;266:17173–9. [PubMed] [Google Scholar]
- 94••.Takai S, Jin D. Improvement of cardiovascular remodelling by chymase inhibitor. Clin Exp Pharmacol Physiol. 2016;43:387–93. doi: 10.1111/1440-1681.12549. This review describes the preclinical basis for the use of chymase inhibition in cardiovascular diseases, and puts forward the need to clinically test chymase inhibition in combination with RAS-targeting agents to improve current therapeutic strategies for cardiovascular disease. [DOI] [PubMed] [Google Scholar]
- 95.Kanemitsu H, Takai S, Tsuneyoshi H, Yoshikawa E, Nishina T, Miyazaki M, et al. Chronic chymase inhibition preserves cardiac function after left ventricular repair in rats. Eur J Cardiothorac Surg. 2008;33:25–31. doi: 10.1016/j.ejcts.2007.09.040. [DOI] [PubMed] [Google Scholar]
- 96.Jamerson K, Weber MA, Bakris GL, Dahlof B, Pitt B, Shi V, et al. for the ACCOMPLISH Trial Investigators Benazepril plus amlodipine or hydrochlorothiazide for hypertension in high-risk patients. N Engl J Med. 2008;359:2417–28. doi: 10.1056/NEJMoa0806182. [DOI] [PubMed] [Google Scholar]
- 97.The Acute Infarction Ramipril Efficacy (AIRE) Study Investigators. Effect of ramipril on mortality and morbidity of survivors of acute myocardial infarction with clinical evidence of heart failure. Lancet. 1993;342:821–8. [PubMed] [Google Scholar]
- 98.Wing LM, Reid CM, Ryan P, Beilin LJ, Brown MA, Jennings GL, et al. for the Second Australian National Blood Pressure Study Group A comparison of outcomes with angiotensin-converting–enzyme inhibitors and diuretics for hypertension in the elderly. N Engl J Med. 2003;348:583–92. doi: 10.1056/NEJMoa021716. [DOI] [PubMed] [Google Scholar]
- 99.Dahlof B, Sever PS, Poulter NR, Wedel H, Beevers DG, Caulfield M, et al. for the ASCOT Investigators Prevention of cardiovascular events with an antihypertensive regimen of amlodipine adding perindopril as required versus atenolol adding bendroflumethiazide as required, in the Anglo-Scandinavian Cardiac Outcomes Trial-Blood Pressure Lowering Arm (ASCOT-BPLA): a multicentre randomised controlled trial. Lancet. 2005;366:895–906. doi: 10.1016/S0140-6736(05)67185-1. [DOI] [PubMed] [Google Scholar]
- 100.Ryden L, Armstrong PW, Cleland JGF, Horowitz JD, Massie BM, Packer M, et al. on behalf of the ATLAS Study Group Efficacy and safety of high-dose lisinopril in chronic heart failure patients at high cardiovascular risk,including those with diabetes mellitus. Eur Heart J. 2000;21:1967–78. doi: 10.1053/euhj.2000.2311. [DOI] [PubMed] [Google Scholar]
- 101.Hansson L, Lindholm LH, Niskanen L, Lanke J, Hedner T, Niklason A, et al. Effect of angiotensin-converting-enzyme inhibition compared with conventional therapy on cardiovascular morbidity and mortality in hypertension: the Captopril Prevention Project (CAPPP) randomised trial. Lancet. 1999;353:611–6. doi: 10.1016/S0140-6736(98)05012-0. [DOI] [PubMed] [Google Scholar]
- 102.Pitt B, Segal R, Martinez FA, Meurers G, Cowley AJ, Thomas I, et al. on behalf of ELITE Study Investigators Randomised trial of losartan versus captopril in patients over 65 with heart failure (Evaluation of Losartan in the Elderly Study, ELITE) Lancet. 1997;349:747–52. doi: 10.1016/S0140-6736(97)01187-2. [DOI] [PubMed] [Google Scholar]
- 103.Pitt B, Poole-Wilson PA, Segal R, Martinez FA, Dickstein K, Camm AJ, et al. on behalf of the ELITE II investigators Effect of losartan compared with captopril on mortality in patients with symptomatic heart failure: randomised trial–the Losartan Heart Failure Survival Study ELITE II. Lancet. 2000;355:1582–7. doi: 10.1016/S0140-6736(00)02213-3. [DOI] [PubMed] [Google Scholar]
- 104.Fox KM, for the EURopean trial On reduction of cardiac events with Perindopril in stable coronary Artery disease Investigators Efficacy of perindopril in reduction of cardiovascular events among patients with stable coronary artery disease: randomised, double-blind, placebo-controlled, multicentre trial (the EUROPA study) Lancet. 2003;362:782–8. doi: 10.1016/S0140-6736(03)14286-9. [DOI] [PubMed] [Google Scholar]
- 105.GISSI-3: effects of lisinopril and transdermal glyceryl trinitrate singly and together on 6-week mortality and ventricular function after acute myocardial infarction. Lancet. 1994;343:1115–22. [PubMed] [Google Scholar]
- 106.Beckett NS, Peters R, Fletcher AE, Staessen JA, Liu L, Dumitrascu D, et al. Treatment of hypertension in patients 80 years of age or older. N Engl J Med. 2008;358:1887–98. doi: 10.1056/NEJMoa0801369. [DOI] [PubMed] [Google Scholar]
- 107.Konstam MA, Neaton JD, Dickstein K, Drexler H, Komajda M, Martinez FA, et al. for the HEAAL Investigators Effects of high-dose versus low-dose losartan on clinical outcomes in patients with heart failure (HEAAL study): a randomised, double-blind trial. Lancet. 2009;374:1840–8. doi: 10.1016/S0140-6736(09)61913-9. [DOI] [PubMed] [Google Scholar]
- 108.Yusuf S, Sleight P, Pogue J, Bosch J, Davies R, Dagenais G, for the Heart Outcomes Prevention Evaluation Study Investigators Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. N Engl J Med. 2000;342:145–53. doi: 10.1056/NEJM200001203420301. [DOI] [PubMed] [Google Scholar]
- 109.Massie BM, Carson PE, McMurray JJ, Komajda M, McKelvie R, Zile MR, et al. for the for the I-PRESERVE Investigators Irbesartan in patients with heart failure and preserved ejection fraction. N Engl J Med. 2008;359:2456–67. doi: 10.1056/NEJMoa0805450. [DOI] [PubMed] [Google Scholar]
- 110.Yusuf S, Teo KK, Pogue J, Dyal L, Copland I, Schumacher H, et al. for the ONTARGET Investigators Telmisartan, ramipril, or both in patients at high risk for vascular events. N Engl J Med. 2008;358:1547–59. doi: 10.1056/NEJMoa0801317. [DOI] [PubMed] [Google Scholar]
- 111.Dickstein K, Kjekshus J, for the Optimaal Steering Committee, for the OPTIMAAL Study Group Effects of losartan and captopril on mortality and morbidity in high-risk patients after acute myocardial infarction: the OPTIMAAL randomised trial. Optimal Trial in Myocardial Infarction with Ang II Antagonist Losartan. Lancet. 2002;360:752–60. doi: 10.1016/S0140-6736(02)09895-1. [DOI] [PubMed] [Google Scholar]
- 112.Braunwald E, Domanski MJ, Fowler SE, Geller NL, Gersh BJ, Hsia J, et al. Angiotensin-converting-enzyme inhibition in stable coronary artery disease. N Engl J Med. 2004;351:2058–68. doi: 10.1056/NEJMoa042739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Pfeffer MA, Braunwald E, Moye LA, Basta L, Brown EJ, Jr, Cuddy TE, et al. for the SAVE Investigators Effect of captopril on mortality and morbidity in patients with left ventricular dysfunction after myocardial infarction. Results of the survival and ventricular enlargement trial. N Engl J Med. 1992;327:669–77. doi: 10.1056/NEJM199209033271001. [DOI] [PubMed] [Google Scholar]
- 114.Ambrosioni E, Borghi C, Magnani B, for the Survival of Myocardial Infarction Long-Term Evaluation (SMILE) Study Investigators The effect of the angiotensin-converting-enzyme inhibitor zofenopril on mortality and morbidity after anterior myocardial infarction. N Engl J Med. 1995;332:80–5. doi: 10.1056/NEJM199501123320203. [DOI] [PubMed] [Google Scholar]
- 115.The SOLVD Investigators. Effect of enalapril on mortality and the development of heart failure in asymptomatic patients with reduced left ventricular ejection fractions. N Engl J Med. 1992;327:685–91. doi: 10.1056/NEJM199209033271003. [DOI] [PubMed] [Google Scholar]
- 116.Hansson L, Lindholm LH, Ekbom T, Dahlof B, Lanke J, Schersten B, et al. Randomised trial of old and new antihypertensive drugs in elderly patients: cardiovascular mortality and morbidity the Swedish Trial in Old Patients with Hypertension-2 study. Lancet. 1999;354:1751–6. doi: 10.1016/S0140-6736(99)10327-1. [DOI] [PubMed] [Google Scholar]
- 117.Kober L, Torp-Pedersen C, Carlsen JE, Bagger H, Eliasen P, Lyngborg K, et al. for the Trandolapril Cardiac Evaluation (TRACE) Study Group A clinical trial of the angiotensin-converting-enzyme inhibitor trandolapril in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med. 1995;333:1670–6. doi: 10.1056/NEJM199512213332503. [DOI] [PubMed] [Google Scholar]
- 118.Yusuf S, Teo K, Anderson C, Pogue J, Dyal L, Copland I, et al. for the Telmisartan Randomised AssessmeNt Study in, A. C. E. iNtolerant subjects with cardiovascular Disease Investigators. Effects of the angiotensin-receptor blocker telmisartan on cardiovascular events in high-risk patients intolerant to angiotensin-converting enzyme inhibitors: a randomised controlled trial. Lancet. 2008;372:1174–83. doi: 10.1016/S0140-6736(08)61242-8. [DOI] [PubMed] [Google Scholar]
- 119.Julius S, Nesbitt SD, Egan BM, Weber MA, Michelson EL, Kaciroti N, et al. for the Trial of Preventing Hypertension Study Investigators Feasibility of treating prehypertension with an angiotensin-receptor blocker. N Engl J Med. 2006;354:1685–97. doi: 10.1056/NEJMoa060838. [DOI] [PubMed] [Google Scholar]
- 120.Cohn JN, Tognoni G, for the Valsartan Heart Failure Trial Investigators A randomized trial of the angiotensin-receptor blocker valsartan in chronic heart failure. N Engl J Med. 2001;345:1667–75. doi: 10.1056/NEJMoa010713. [DOI] [PubMed] [Google Scholar]
- 121.Pfeffer MA, McMurray JJ, Velazquez EJ, Rouleau JL, Kober L, Maggioni AP, et al. for the Valsartan in Acute Myocardial Infarction Trial Investigators Valsartan, captopril, or both in myocardial infarction complicated by heart failure, left ventricular dysfunction, or both. N Engl J Med. 2003;349:1893–906. doi: 10.1056/NEJMoa032292. [DOI] [PubMed] [Google Scholar]
- 122.Julius S, Kjeldsen SE, Brunner H, Hansson L, Platt F, Ekman S, et al. VALUE trial: Long-term blood pressure trends in 13,449 patients with hypertension and high cardiovascular risk. Am J Hypertens. 2003;16:544–8. doi: 10.1016/S0895-7061(03)00904-X. [DOI] [PubMed] [Google Scholar]