

Abstract
We analyzed two West African samples (Guinea-Bissau: n = 289 cases, 322 controls; The Gambia: n = 240 cases, 248 controls) to evaluate single nucleotide polymorphisms (SNPs) in Epiregulin (EREG) and V-ATPase (T cell immune regulator 1, TCIRG1) using single and multi-locus analyses to determine whether previously described associations with pulmonary tuberculosis (PTB) in Vietnamese and Italians would replicate in African populations. We did not detect any significant single locus or haplotype associations in either sample. We also performed exploratory pairwise interaction analyses using Visualization of Statistical Epistasis Networks (ViSEN), a novel method to detect only interactions among multiple variables, to elucidate possible interaction effects between SNPs and demographic factors. Although we found no strong evidence of marginal effects, there were several significant pairwise interactions that were identified in either the Guinea-Bissau or The Gambia samples, two of which replicated across populations. Our results indicate that the effects of EREG and TCIRG1 variants on PTB susceptibility, to the extent that they exist, are dependent on gene-gene interactions in West African populations as detected with ViSEN. In addition, epistatic effects are likely to be influenced by inter- and intra-population differences in genetic or environmental context and/or the mycobacterial lineages causing disease.
Keywords: Tuberculosis, EREG, TCIRG1, V-ATPase, infectious disease, genetic epidemiology
INTRODUCTION
With an estimated 8.6 million incident cases in the world in 2012, tuberculosis (TB) caused by M. tuberculosis (Mtb), represents a major global health problem. Endemic regions in Asia and Africa carry the highest burden of disease, with 29% and 27% of global cases, respectively. According to the World Health Organization, in 2012 the prevalence of TB in Africa was 303/100 000, the incidence 255/100 000 and the mortality rate 26/100 000. These numbers reflect the high percentage of HIV co-infection in Africa (37% of active TB cases).1 Nevertheless the biological bases of TB are complex; the vast majority of those infected with Mtb maintain the bacterium in a latent state and fail to develop overt clinical disease, even if they remain at higher risk of progressing to disease later in their lives.2 The factors determining risk of infection and progression to active disease are multifactorial, involving innate and adaptive immunity, host–pathogen interactions, and environmental components.3-6
Host genetic variation has been shown to influence individual responses to Mtb, and candidate gene, as well as genome-wide association studies, have identified several genes involved in modulating susceptibility to TB.7 However, results for candidate genes or loci have been inconsistent, possibly due to heterogeneity in phenotype definitions, study populations, or undefined variables interacting with the genetic background. Recently, two genes, Epiregulin (EREG) and V-ATPase (T cell immune regulator 1, TCIRG1) were found to be associated with susceptibility to TB in Vietnamese8 and Italian9 subjects, respectively. To our knowledge, these associations have not yet been replicated. In the Vietnamese study, which included 332 cases and 380 controls in the discovery dataset, a significant association was found between SNP rs7675690 in EREG and pulmonary TB (PTB) (OR = 4.95) as well as meningeal disease, the latter being more significant when infection by the Mtb Beijing lineage was taken into account (OR = 7.81).8 The involvement of TCIRG1 genetic variation in host susceptibility to Mtb has been shown in an Italian case-control study where patients heterozygous at either rs2075609 or rs4147780 were protected against active disease in genotypic tests. Also, haplotype analyses revealed that heterozygosity at either or both TCIRG1 sites protected from active disease when compared to doubly homozygous individuals, regardless of the allele.9
EREG, located on chromosome 4q13.3 (OMIM 602061), is composed of 5 exons and spans 23.6 kb. The gene is a member of the epidermal growth factor (EGF) family and acts both as an EGF receptor ligand and in a regulatory capacity by inhibiting the growth of several epithelial cell lines and stimulating the growth of fibroblasts and various other cell types.10 Epiregulin expression can be stimulated in human monocytes, macrophages, and peripheral blood mononuclear cells by Mtb, TLR4 and TLR2/1/6 ligands.8, 11 In a murine system, EREG expression was stimulated in Mtb infected macrophages and, in vivo, mRNA was expressed by interstitial macrophages within TB lung lesions, suggesting a role of EREG in TB.12
TCIRG1 (T-cell immune regulator 1; OMIM 604592) is located on chromosome 11q13.2 and encodes a vacuolar ATPase (V-ATPase) that is a member of a family of ATP-dependent proton pumps localized in the membranes of several eukaryotic cells, such as lysosomes and endosomes. This family regulates the pH of intracellular compartments, cytoplasm and extracellular space.13, 14 Two overlapping TCIRG1 transcripts have been identified, OC116 (or transcript variant 1, TV1, containing 20 exons) and TIRC7 (or transcript variant 2, TV2, containing 15 exons), which share 14 introns and exons.15 OC116 and TIRC7 code for proteins referred to as isoform “a” and “b”, respectively. Isoform a, the a3 subunit of the V-ATPase enzyme, is ubiquitously expressed in human tissue but its highest expression is in osteoclastic cells.16, 17 Mutations in TCIRG1 have been associated with infantile malignant autosomal recessive osteopetrosis, a heterogeneous bone disease characterized by osteoclast failure and impaired bone resorption,18 providing evidence for potential pleiotropic effects. Isoform b, TIRC7, has a role in early events of T cell activation.19
During infection, Mtb can survive and replicate in macrophages by preventing the fusion of mycobacterial phagosomes with lysosomes by inhibiting the accumulation of V-ATPase, an essential step in the acidification of phagosomes by macrophages.20-22 The lack of V-ATPase accumulation is directly caused by a specific mycobacterial protein, PtpA (protein tyrosine phosphatase) that inhibits V-ATPase trafficking by binding one of its subunits.23 Moreover, V-ATPase is involved in macrophage processing of Mtb antigens, and in infected macrophages the lack of V-ATPase impedes cellular function at antigen presenting cells.23 V-ATPase also negatively regulates pro-inflammatory cytokines in macrophages, in particular TNFα, the pro-inflammatory cytokine essential for Mtb infection control.24
The aim of our paper was to evaluate EREG and TCIRG1 associations with PTB in two samples from West Africa, 321 cases and 346 controls from Guinea-Bissau and 257 cases and 272 controls from The Gambia. We genotyped eight SNPs in EREG and six SNPs in TCIRG1 to assess associations (Table S1–S3 and Figure 1).
Figure 1. EREG and TCIRG1 gene structures and SNPs positions.
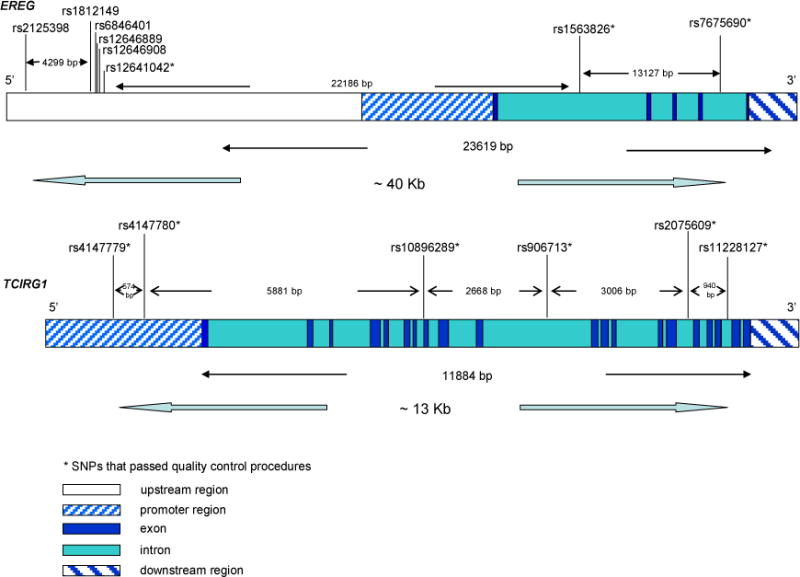
The figure shows the structures of the two genes and the SNPs genotyped. Both genes are oriented 5′ to 3′.
RESULTS
After quality control (QC) procedures, 289 cases and 322 controls from Guinea-Bissau and 248 cases and 240 controls from The Gambia were included in the analysis, as were nine SNPs, three in EREG and six in TCIRG1 (Methods and Table S2). Differences between cases and controls in median age, gender distribution, and ethnic group distribution were assessed in each sample (Table 1a–b). A significant difference in age between cases and controls was observed in both samples (Guinea-Bissau p = 0.0017, The Gambia p < 0.0001), although in opposite directions, and in Guinea-Bissau there was a significant difference in gender distribution (p = 0.0068) not seen in The Gambia (p = 0.6617). There were no differences in ethnic group distributions between cases and controls in either sample (Guinea-Bissau p = 0.112, The Gambia p = 0.263).
Table 1.
| a. Demographic information for Guinea-Bissau sample* | |||
|---|---|---|---|
| Guinea-Bissau sample | |||
| Case (n = 289) | Control (n = 322) | P-Value | |
| Age (Median, SE)1 | (33, 1.36) | (37, 0.89) | 0.0017 |
| Sex (% Female, SE)2 | (39.1%, 0.03) | (50%, 0.03) | 0.0068 |
| Ethnic Group: | 14.4% | 18.6% | 0.1124 |
| Balanta | |||
| Fula | 15.1% | 12.4% | |
| Manjago | 19.3% | 9.9% | |
| Pepel | 21.1% | 29.5% | |
| Mancanha | 8.1% | 11.8% | |
| Mandinka | 7.0% | 7.2% | |
| Other3 | 15.1% | 10.6% | |
| b. Demographic Information for The Gambia sample* | |||
|---|---|---|---|
| The Gambia sample | |||
| Case (n = 240) | Control (n = 248) | P-value | |
| Age (Median, SE)1 | (29, 1.11) | (25, 0.60) | <0.0001 |
| Sex (% Female, SE)2 | (31.7%, 0.03) | (29.8%, 0.03) | 0.6617 |
| Ethnic Group: | 5.0% | 5.2% | 0.2634 |
| Serahuli | |||
| Jola | 21.3% | 32.7% | |
| Wolof | 10.8% | 13.7% | |
| Fula | 12.9% | 8.1% | |
| Manjago | 2.1% | 4.0% | |
| Mandinka | 39.6% | 32.3% | |
| Other3 | 8.3% | 4.0% | |
Age is presented in years; Standard Error (SE) of the median was calculated using bootstrapping with 1000 repetitions. P-value presented for this variable is from the Wilcoxon Rank Sum Test.
SE=Standard Error of the proportion of females in each group; p-value presented for this variable is from the test of difference in proportions
Other: this group contains individuals whose ethnic groups made up less than 5% of the total sample of participants. These groups were collapsed into this category.
The p-value presented for the variable “Ethnic Group” is from the Kolmogorov-Smirnov test for equality of distributions. Due to a large number of ties in the data, p-values presented are permutation p-values (n = 1000).
Values in bold are statistically significant (p ≤ 0.05)
Single locus analyses revealed an association in Guinea-Bissau between rs10896289, located in TCIRG1, and TB (allelic p-value = 0.045) (Table 2). The EREG SNP rs1563826 was almost significant in this sample as well (allelic p-value = 0.053) (Table 2). After correction for multiple testing, neither of these associations remained significant; nor was either association detected in The Gambia (Table 2).
Table 2.
Single locus associations between EREG and TCIRG1 SNPs and TB*
| Guinea-Bissau sample | The Gambia sample | |||||
|---|---|---|---|---|---|---|
| Chr | Gene | SNP | Allelic1 | Genotypic2 | Allelic1 | Genotypic2 |
| 4 | EREG | rs12641042 | 0.933 | 0.516 | 0.182 | 0.424 |
| 4 | EREG | rs1563826 | 0.053 | 0.163 | 0.918 | 0.944 |
| 4 | EREG | rs7675690 | 0.739 | 0.864 | 0.432 | 0.650 |
| 11 | TCIRG1 | rs10896289 | 0.045 | 0.130 | 0.382 | 0.666 |
| 11 | TCIRG1 | rs11228127 | 0.406 | 0.694 | 0.602 | 0.316 |
| 11 | TCIRG1 | rs2075609 | 0.461 | 0.749 | 0.278 | 0.434 |
| 11 | TCIRG1 | rs4147779 | 0.844 | 0.976 | 0.510 | 0.793 |
| 11 | TCIRG1 | rs4147780 | 0.575 | 0.279 | 0.382 | 0.497 |
| 11 | TCIRG1 | rs906713 | 0.960 | 0.177 | 0.450 | 0.772 |
P-value calculated from allelic chi-square test of association; Fisher’s Exact P-value is presented where appropriate.
P-value calculated from genotypic chi-square test of association.
Values in bold are statistically significant (p ≤ 0.05)
To further investigate the possibility of single locus effects on TB susceptibility, we examined both samples using PRAT, a single locus analysis of association that is methodologically different from the chi-square test and has been shown to be as or more powerful than standard tests under most genetic models.25 PRAT analysis of cases and controls in Guinea-Bissau and The Gambia samples did not provide evidence for any significant single SNP associations with TB, with the exception of TCIRG1 SNP rs10896289 in the Guinea-Bissau sample (PRAT case p-value = 0.044) (Table S4).
To explore the possibility that significant or nearly significant variants from the Guinea-Bissau sample shared the same direction of effect in The Gambia, we performed logistic regression analysis for all variants with a p-value ≤ 0.20 in chi-square or PRAT analyses. Following this criteria, four SNPs, two in EREG and two in TCIRG1, were selected for further study. Logistic regression models were adjusted for age, sex, and ethnicity. Logistic regression analysis revealed that rs1563826, in EREG, was almost significant in Guinea-Bissau sample (OR = 0.799, p-value = 0.056). However, in The Gambia sample, this SNP was not significant and trended in the opposite direction (OR = 1.038, p-value = 0.769) (Table 3). Rs10896289 in TCIRG1 showed a significant effect in Guinea-Bissau sample (OR = 1.300, p-value = 0.042), which was not replicated in The Gambia sample (p-value = 0.466). Neither of the other variants showed evidence for association in either sample after adjustment for demographic variables.
Table 3.
Logistic regression results for selected EREG and TCIRG1 variants and TB susceptibility*
| Guinea-Bissau sample | The Gambia sample | |||||||
|---|---|---|---|---|---|---|---|---|
| Chr | Gene | SNP | Odds ratio | 95% confidence interval | P-value | Odds ratio | 95% confidence interval | P-value |
| 4 | EREG | rs1563826 | 0.799 | 0.635 – 1.005 | 0.056 | 1.038 | 0.809 – 1.332 | 0.769 |
| 4 | EREG | rs12641042 | 1.000 | 0.796 – 1.262 | 0.985 | 1.23 | 0.95 – 1.591 | 0.109 |
| 11 | TCIRG1 | rs10896289 | 1.300 | 1.009 – 1.673 | 0.042 | 0.894 | 0.661 – 1.208 | 0.466 |
| 11 | TCIRG1 | rs906713 | 1.1591 | 0.837 – 1.5641 | 0.3591 | 1.102 | 0.813 – 1.495 | 0.531 |
Logistic Regression Model was adjusted for age, sex, and ethnicity
Odds ratio, Confidence Interval, and P-value were produced using Exact Logistic Regression
Values in bold are statistically significant (p ≤ 0.05)
In order to account for the difference in study design between Guinea Bissau and The Gambia regarding inclusion of individuals with HIV, we performed additional stratified analyses in the Bissau sample (which did not exclude HIV positive TB cases). Chi-square analyses comparing HIV positive and HIV negative TB cases to Bissau sample controls did not reveal any significant effect of HIV status on association between EREG/TCIRG1 variants and PTB (Table S5).
Due to the possible clinical importance of identifying significant gene by Mtb lineage interaction effects, we conducted Mtb lineage specific exploratory analyses. However, Mtb lineage data was only available for TB cases from The Gambia. Mtb lineage based subset analysis revealed a significant association between rs11228127 in TCIRG1 (p=0.03) and PTB (Table S11) when assessed using only TB cases infected with M. africanum. It should be noted that these results are based on a small number of TB cases (n=83) and that the association loses significance when corrected for multiple testing. Due to the limitations of assessing this putatively associated interaction effect in our study due to small sample size, we are unable to determine if this is a true association or merely a statistical artifact; further evaluation in a larger sample is needed to confirm the validity of this effect. In addition, we note that there is virtually no M. africanum in Italy and therefore this should not have played any role in the prior paper by Capparelli et al.(REF).
Interpreted together, our single locus results indicate there are no strong additive main effects for any of the interrogated EREG or TCIRG1 variants.
Additional genotype and haplotype analyses for rs2075609 and rs4147780 in EREG were performed in both The Gambia and Guinea-Bissau to test the models of Capparelli et al.9 No association with PTB in either West African sample was detected. Even when not significant, the direction of effect for all tested haplotypes was discordant between the three (Guinea-Bissau, The Gambia, and Italian) samples (Table S3a–b). In addition, we explicitly tested for support of the heterozygote protective advantage shown in Capparelli et al. (Table 4, Table S8). We did not detect any significant associations between haplotypes at rs2075609/rs4147780 in either West African sample and TB. In the single instance in which the two West African populations displayed the same effect direction (heterozygotes for rs2075609 only vs. double homozygotes) the trend was in the opposite direction in Italians. These negative results are not unexpected as in Capparelli et al. the impetus to perform haplotype analyses arose from a deviation from HWE in cases only and an association between rs2075609/rs4147780 and TB at the genotypic level, neither of which was observed in our study (Table S6a–b).
Table 4.
Test of heterozygote protection at rs2075609 and/or rs4147780
| Guinea-Bissau | The Gambia | Italy (Capparelli et al, 2009) |
||||
|---|---|---|---|---|---|---|
| Tested Haplotype Model | OR (95% CI)2 | P3 | OR (95% CI)2 | P3 | OR (95% CI)2 | P3 |
| Heterozygous at only rs2075609 | 1.16 (0.76–1.77) | 0.492 | 1.17 (0.73–1.90) | 0.499 | 0.38 (0.22–0.66) | 0.001 |
| Heterozygous at only rs4147780 | 0.86 (0.49–1.49) | 0.580 | 1.11 (0.64–1.91) | 0.712 | 0.35 (0.31–0.54) | 2×10−6 |
| Heterozygous at both loci | 0.80 (0.54–1.18) | 0.264 | 1.16 (0.73–1.83) | 0.539 | 0.26 (0.16–0.41) | 7×10−9 |
| All heterozygous1 | 0.93 (0.67–1.28) | 0.641 | 1.15 (0.79–1.67) | 0.457 | 0.32 (0.23–0.45) | 3.3×10−11 |
Double homozygotes (individuals with haplotypes: 13/13, 23/23, 14/14, 24/24) were used as the reference group for all models tested above.
Individuals who are heterozygous at either rs2075609 or rs4147780 or both
OR: Odds ratio, 95% CI: 95% Confidence Interval
P: p-value for specified logistic regression model
ViSEN analysis was employed to investigate the possibility of pairwise interactions without the presence of significant single-locus effects.26, 27 Entropy-based pairwise interaction models were constructed containing all SNPs analyzed, as well as all demographic covariates from Table 1, to determine if interactions played a role in TB susceptibility in either sample. Eight pairwise interactions with information gain (IG) greater than 1% between EREG/TCIRG1 SNPs and biological variables were significant in the Guinea-Bissau sample (Table 5a, Figure 2a). The most significant pairwise model contained only biological variables, age and sex (IG = 5.02%, p < 0.001). The most significant pairwise model containing a SNP and a demographic variable was rs4147779 in TCIRG1 and ethnic group (IG = 2.27%, p = 0.014), and the most significant model containing only SNPs included rs12641042 in EREG and rs906713 in TCIRG1 (IG = 1.06%, p = 0.008). In The Gambia (Table 5b, Figure 1b), the most significant model contained ethnic group and rs7675690 in EREG (IG = 3.41%, p = 0.010) and the most significant model containing only SNPs included rs7675690 in EREG and rs10896289 in TCIRG1 (IG = 1.85%, p < 0.001). There were no significant models containing only demographic variables in the Gambian sample. Of the 19 significant two-way interaction effects identified in either Guinea-Bissau or The Gambia, two interactions replicated across the samples (Table 6, Figure 3); these models both contained ethnic group and a single SNP. One was between ethnicity and rs1563826 in EREG (Guinea-Bissau: IG = 1.53%, p = 0.044, The Gambia: IG = 2.60%, p = 0.032), and the other was between ethnicity and rs4147779 in TCIRG1 (Guinea-Bissau IG = 2.27%, p = 0.014, The Gambia IG = 2.06%, p = 0.049). Although there were no single locus effects identified in The Gambia, this sample had more significant pairwise interactions than Guinea-Bissau (eleven significant effects vs. eight significant effects out of 66 total comparisons in each sample), and displayed an enrichment of significant pairwise interaction effects compared to that expected by chance alone (expected number ~3.3 at p = 0.05).
Table 5.
| a. Top Visualization of Statistical Epistasis Networks (ViSEN) results for EREG/TCIRG1 SNPs and demographic variables for TB susceptibility in Guinea-Bissau | |||
|---|---|---|---|
| Predictor 1 | Predictor 2 | IG1 | IG P-value2 |
| AGEQRT3 | SEX | 5.02% | <0.001 |
| AGEQRT | ETHGRP4 | 3.50% | 0.010 |
| ETHGRP | rs4147779 | 2.27% | 0.014 |
| ETHGRP | rs906713 | 1.93% | 0.015 |
| ETHGRP | rs1563826 | 1.53% | 0.044 |
| AGEQRT | rs7675690 | 1.46% | 0.006 |
| AGEQRT | rs12641042 | 1.17% | 0.012 |
| rs12641042 | rs906713 | 1.06% | 0.008 |
| b. Top Visualization of Statistical Epistasis Networks (ViSEN) results for EREG/TCIRG1 SNPs and demographic variables for TB susceptibility in The Gambia | |||
|---|---|---|---|
| Predictor 1 | Predictor 2 | IG1 | IG P-value2 |
| ETHGRP | rs7675690 | 3.41% | 0.010 |
| ETHGRP | rs1563826 | 2.60% | 0.032 |
| ETHGRP | rs11228127 | 2.19% | 0.038 |
| ETHGRP | rs4147780 | 2.17% | 0.043 |
| ETHGRP | rs4147779 | 2.06% | 0.049 |
| rs7675690 | rs10896289 | 2.03% | <0.001 |
| AGEQRT | rs10896289 | 1.85% | 0.011 |
| rs1563826 | rs10896289 | 1.82% | 0.005 |
| rs7675690 | rs1122812 | 1.54% | 0.003 |
| rs4147779 | rs906713 | 1.18% | 0.006 |
| rs1563826 | rs906713 | 1.07% | 0.022 |
Note: Pairwise interactions presented above are comprised of all statistically significant pairwise interactions with IG > 1.00%.
Information Gain (IG) = Percentage of information gained about TB Status from considering Predictor 1 and Predictor 2 together after controlling for main effects from both variables
Information Gain p-value; p-value is based on 1000 permutations
AGEQRT = Age (quartile)
ETHGRP = Ethnicity
Figure 2. Significant pairwise interactions from ViSEN analysis in Guinea-Bissau (a) and in The Gambia (b).
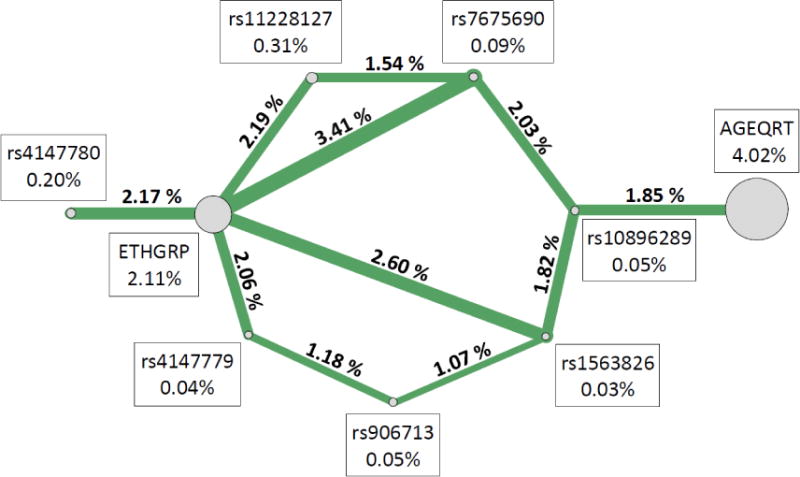
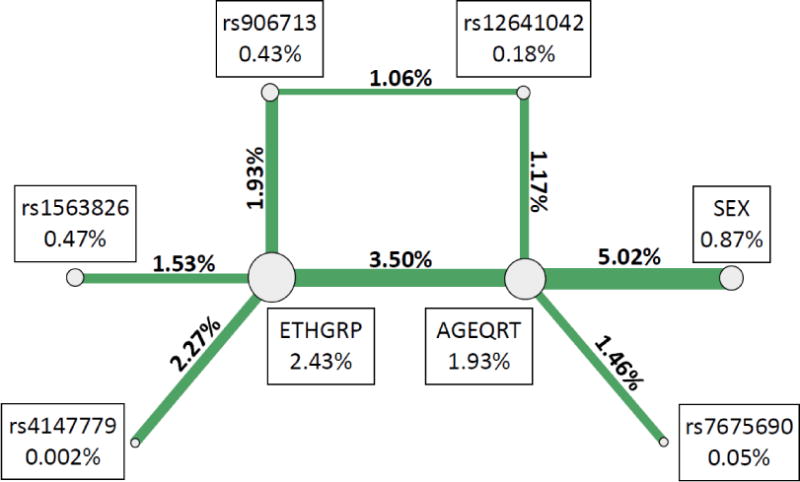
Shown above are all pairwise interactions that were statistically significant with IG > 1.00%. Circles represent single variables, and connecting lines represent pairwise interactions between specified nodes. Line thickness corresponds to interaction strength (IG), with thicker lines representing stronger interactions. Percentages presented above represent ViSEN (IG) for either main effects (shown in boxes adjacent to each circle) or pairwise interaction effects (shown above the connecting lines).
Table 6.
Pairwise interactions identified by ViSEN that replicated between The Gambia and Guinea-Bissau for association with TB Status
| Guinea-Bissau | The Gambia | ||||
|---|---|---|---|---|---|
| Predictor 1 | Predictor 2 | IG1 | IG P-value2 | IG1 | IG P-value2 |
| ETHGRP3 | rs1563826 | 1.53% | 0.044 | 2.60% | 0.032 |
| ETHGRP | rs4147779 | 2.27% | 0.014 | 2.06% | 0.049 |
Information Gain (IG) = Percentage of information gained about TB Status from considering Predictor 1 and Predictor 2 together after controlling for main effects from both variables
Information Gain p-value; p-value is based on 1000 permutations
ETHGRP = Ethnicity
Figure 3. Replicated significant pairwise interaction effects for TB susceptibility in The Gambia and Guinea-Bissau.
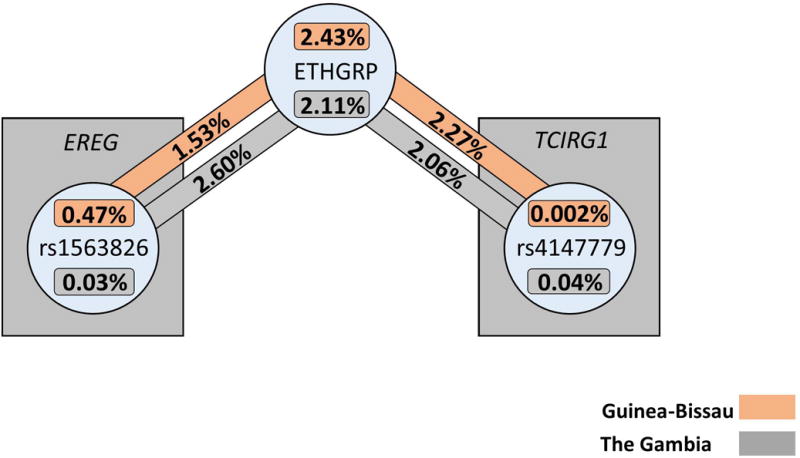
Percentages shown above are Information Gain measurements produced by ViSEN analysis; analyses in separate samples are indicated by corresponding colors.
DISCUSSION
Although we were adequately powered to detect the large effect sizes for the EREG and TCIRG1 SNPs genotyped by both the previous authors and us,8, 9 we failed to replicate associations with TB in our study for both marginal and haplotype effects. We could only detect a marginal association between SNP rs1089689 in TCIRG1 and TB in Guinea-Bissau, but this did not pass correction for multiple testing and in The Gambia it was not close to significant. In addition, this SNP was not one shown to associate in the previous work as it was not genotyped by Capparelli et al. Our negative findings could be explained by several factors, including the interaction of putative susceptibility SNPs with demographic or environmental variables as well as interactions with other, ungenotyped, SNPs elsewhere in the genome, i.e. epistasis.28 Interestingly, the only SNP providing evidence of marginal association, rs10896289, identified in Guinea-Bissau was not detected in any of the significant two way interactions there. It was, however, present in three significant interaction models with demographic variables in The Gambia. Differences across models and populations could indicate different distributions of interacting variables.
Because Mtb lineage can affect disease risk, as shown in Thuong et al., and differences in host allele/haplotype frequencies can affect statistical association, our negative findings may also be explained by the complex interaction between human population genetic structure and Mtb lineage. Specifically, the data from Capparelli et al. when compared to the West African data, showed significantly different allele and genotype/haplotype frequencies. Previous genetic and epidemiological studies present evidence of an interaction between ethnicity and Mtb phylogenetic lineage that influences both susceptibility to active disease as well as its clinical presentation.29-31 With respect to Mtb lineage (spoligotype data), which was only available in The Gambia, we found no evidence that lineage heterogeneity strongly affected association with TB, but this will require further evaluation, especially with respect to M. africanum.
In previous candidate gene studies using the same West African samples, we have shown that ethnicity has a significant impact on genetic risk.32, 33 Our ViSEN results revealed that the two interactions that replicated across populations both contained “ethnic group”, confirming that ethnicity is a key factor in modulating TB risk. The effect we detected is due to as yet undefined genetic and/or environmental factor(s) for which ethnicity serves as a proxy.
In addition, the overall similarity of LD patterns and minor allele frequencies (MAF) of the evaluated variants in EREG and TCIRG1 between the two populations (Table S1) and the extreme differences displayed in association with TB in marginal and interaction effects is consistent with the hypothesis that, if these SNPs associate at all with TB, they do so by interacting with other unmeasured factors to affect risk, but the nature of the key context(s) is as yet undefined.
We note that due to differences in LD between the West African populations we studied with those previously studied for EREG and TCIRG1, it is important to assess whether or not we are covering the same parts of each gene. LD plots show that the genic regions we studied overlap with almost all of the regions previously studied in the two genes, suggesting that if there are any associations they are not in these genes per se in our West African populations (Figure S5–S6). However, we do recognize that if long range LD differs among study populations, we would not have detected it. Hence, we cannot definitively exclude the loci or regions flanking these genes.
In conclusion, our data do not provide evidence for marginal effects in either gene but do support the theory that inter- and intragenic interactions or context, more broadly speaking, plays a substantial role in susceptibility to TB. Our results, interpreted alongside those of Thuong et al. and Capparelli et al., are suggestive that the effect of EREG and TCIRG1 on susceptibility to TB is population specific, and that in the West African populations we analyzed, these are not major independent susceptibility loci but these genes may affect TB risk as part of complex multi-variable models.
MATERIALS AND METHODS
Study Populations
Detailed clinical and demographic information for subjects from Guinea-Bissau and The Gambia has been previously published.32, 33
The Gambia
Between June 2002 and October 2004, PTB cases and their household contacts were enrolled in a prospective study in the Greater Banjul region of The Gambia, at the major government TB clinic and the Medical Research Council (MRC) outpatient clinic. Cases defined as patients with two sputum smear-positive for acid-fast bacilli by Ziehl-Neelsen stains and with M. tuberculosis isolated upon culture. HIV positive patients were excluded from the study. Household contacts were included if they lived the majority of the time within the same compound as the case patient.33
The Gambian sample consisted of 257 cases and 272 controls. Information on age, sex and ethnic group for cases and controls in our sample are in table 1b.
This study was approved by The Gambia Government/MRC joint Ethics Committee. All adults and children’s guardians signed a written informed consent to the study.
Guinea-Bissau
This case-control study was conducted at the Bandim Health Project (BHP), a demographic surveillance site in Bissau, the capital of Guinea-Bissau. During the inclusion period from November 2003 to November 2005 patients were included in the study if they were residents or long-term guests of Bissau, aged greater than 15 years and newly diagnosed with PTB using three sputum examinations for acid fast bacteria or clinical criteria by the World Health Organization’s definition of active PTB.34 No culture confirmation of TB was available in Bissau during the study period, as facilities were destroyed during the civil war of 1998–1999.32
Healthy controls were recruited, from May 2005 to November 2005, from a random sample of 200 houses in the study area, excluding those houses with a recorded case of TB within the past 2 years. Exclusion criteria for controls included the presence of cough for more than 2 weeks and history of TB.32
While the Gambian case-control study was designed to recruit both index cases and their contacts and included a genetic component from its conception, the Bissau genetic study was nested into a large prospective community study at a much later stage. The original Bissau design involved recruitment of TB cases, but not household contacts and their follow-up. As the Bissau sample does not include household contacts, we used randomly selected healthy population based controls. Healthy controls that were not household contacts of TB cases were chosen to minimize two confounding factors: 1) the development of active disease among controls, which would have negatively affected the power of our study and; 2) relatedness to cases, which would have been too complex to ascertain, given the field-work resources available at the time.
To further reduce the probability of recruiting related individuals among TB cases and controls we sampled husband and wife pairs from houses with no TB cases. In general these were unrelated individuals, but in rare instances a family relation within couples was claimed, most frequently being 2nd or 3rd degree cousins, although this degree of relationship is very difficult to ascertain in Africa. As this rare consanguinity did not affect Hardy-Weinberg equilibrium at any of the loci tested we are confident that this had no effect on our results.
The Guinea-Bissau sample consists of 321 cases and 346 controls. Information on age, sex and ethnic group for cases and controls are in table 1a.
Ethical approval was granted by the ‘Unidade de Coordenacao de Estudos e Pesquisas em material de Saude’ (Ministry of Health) in Guinea-Bissau. All adults and children’s guardians signed a written informed consent to the study.
DNA extraction, SNPs selection and genotyping
DNA samples were extracted using a standard salting-out procedure, DNA purities were estimated spectrophotometrically and final concentrations were determined by PicoGreen (Invitrogen – Molecular Probes, Eugene, OR, USA).
We selected a total of fourteen SNPs in EREG (n = 8) and TCIRG1 (n = 6) based on previously published association studies and/or location. Of the selected SNPs, six variants in EREG (rs2125398, rs1812149, rs6846401, rs12646889, rs12646908, rs12641042) were previously associated with susceptibility to TB in Vietnam in a genome-wide association study, and two SNPs (rs1563826, rs7675690) were associated with susceptibility to TB in a case-control study in the same population, with rs7675690 (a tag SNP) remaining significant after Bonferroni correction (Table S1).8
Among the six SNPs selected in TCIRG1 (rs4147779, rs4147780, rs10896289, rs906713, rs2075609, rs11228127, all tagSNPs), three were previously assessed in an Italian population (rs4147779, rs4147780, and rs2075609); two SNPs, rs4147780 and rs2075609, were found to be significantly associated with PTB (Table S1).9
Selected SNPs were genotyped using the Sequenom MASSarray system (Sequenom, San Diego, CA) in the Vanderbilt University Center for Human Genetics Research Core Laboratory according to the manufacturer’s protocol.
Statistical methods
Quality control
The markers were tested for genotyping efficiency, allele frequency, and deviation from Hardy-Weinberg Equilibrium (HWE) in each sample, independently; HWE was assessed in cases and controls separately. If cases or controls deviated from HWE, the inbreeding coefficient, f, was calculated to determine if this deviation was in the same direction in both groups, which might indicate genotyping error. If concordant deviation was detected then the affected marker was removed from analysis, if deviation from equilibrium was discovered to be in opposite directions in cases and controls, then the marker was flagged and included in analysis. Quality control parameters included a genotyping efficiency ≥ 0.95, minor allele frequency (MAF) ≥ 0.5, HWE p-value ≥ 0.001, and Linkage disequilibrium (LD) ≥ 0.8 (r2). To minimize multiple testing of correlated SNPs, we assessed LD among all SNPs in each sample based on our data. If a set of markers had r2 ≥ 0.8 in the entire sample, then the LD was assessed in each ethnicity separately to determine if the level of high LD was similar among ethnic groups. If the overall, and ethnicity specific, LD were concordant for highly correlated SNPs one marker from the high LD ”bin” was selected to “tag” the other markers in the block to reduce redundancy in statistical analyses. QC parameters were assessed for participants as well as EREG/TCIRG1 SNPs using the PLINK software package,35 with the exception of LD measurements. LD between markers was determined using Haploview (Figure S1A – B, Figure S2A – B, Figure S3A – G, Figure S4A–G).36 After QC, nine SNPs, three in EREG and six in TCIRG1, were assessed in Guinea-Bissau (cases, n = 289; controls n = 322) and the Gambia (cases, n = 248; controls n = 240) (Table S2, Table S3). At the completion of QC procedures, all nine remaining SNPs met the LD cutoff in both samples.
Percentage of female participants, ethnic group distribution, and age at study enrollment were compared between cases and controls in each sample, separately. For continuous variables, such as age at enrollment, the Shapiro-Wilkes test was used to determine if the distribution was normally distributed. If normal then a Student’s T-test was used to compare cases and controls, if the variable distribution was non-normal, then the non-parametric Wilcoxon Rank Sum test was applied instead. To analyze the difference in percentage of female participants between cases and controls, a standard difference in proportions test was used. The ethnic group distribution in cases and controls was compared using the Kolmogorov-Smirnov test. All participant demographic analyses were performed using STATA 11,37 with the exception of the Kolmogorov-Smirnov (KS) test. The KS test was performed using the KS. Test command in R;38, 39 due to a large number of ties in the dataset bootstrapping (n = 1000) was employed to produce a more accurate p-value.
Single Locus Association Analyses
Chi-square analysis was performed for all markers using PLINK, and results were confirmed using STATA 11. In instances where n < 5 for any genotype group, the Fisher’s Exact test was used instead of chi-square test for analysis (Table 2). Correction for multiple testing was performed using false discovery rate (FDR) q = 0.1.
Prevalence based association testing (PRAT)25 was performed using the PLATO (PLatform for the Analysis, Translation, and Organization of large-scale data) software package (Table S4).40 PRAT is a single locus association analysis that is based on the principles of HWE. PRAT assesses cases and controls separately for deviations from HWE taking into account the population prevalence of the phenotype being tested; under instances of genotype-phenotype association deviation from HWE at the associated marker may be expected. Simulation tests have shown that PRAT analysis is as powerful, and in several instances more powerful, than standard single locus tests of association under various genetic models; tests also showed that PRAT was robust to misspecification of the population prevalence. Correction for multiple testing was performed using permutations (n = 1000), and population prevalence was set at 0.10.
Markers that were marginally significant (p ≤ 0.2) in chi-square or PRAT analysis were evaluated for direction of effect using logistic regression analysis in STATA 11 (Table 3). In instances where sample size was small (n < 5 in any genotype group), exact logistic regression analysis was performed, using the elrm (Exact-like inference for regression models) package in the R software suite.38 This method is more appropriate for smaller datasets as it does not rely on the asymptotic approximations based on the unconditional likelihood that are unreliable and inappropriate when sample size is small.
Recognizing the difference in study design between the Guinea-Bissau and Gambian samples regarding inclusion of individuals with HIV, we performed additional analyses in the Guinea-Bissau sample to determine if HIV status significantly modified the observed associations between TCIRG1/EREG SNPs and TB. HIV status information was only available on TB cases in this sample. To assess the significance of HIV status as an effect modifier or confounder, chi-square analyses were performed in subsets of the Guinea-Bissau sample defined by TB case – HIV status; in two separate analyses HIV positive TB cases and HIV negative TB cases were compared to all Guinea-Bissau TB controls (Table S5). TB cases with HIV-1, HIV-2, or a combination of HIV-1 and HIV-2 were classified as HIV positive.
Additional exploratory analyses were performed to identify possible Mtb lineage by gene interaction effects by performing Mtb lineage-stratified Chi-square tests. Where Mtb lineage data was available, TB cases were stratified by Mtb lineage and then compared to TB controls, separately using the Chi-square test. In cases of small sample size, the Fisher’s exact test was substituted. Mtb lineage was determined using spoligotype data; spoligotype data was only available for TB cases in the Gambian sample.
TCIRG1 Haplotype Analysis (rs2075609 and rs4147780)
Genotype and haplotype models, containing rs2075609 and rs4147780 in TCIRG1, were constructed according to the scheme of Capparelli et al. Genotype (Table S6a) and haplotype (Table S7a) frequencies were measured in Guinea-Bissau, The Gambia, and the Italian samples, in cases and controls, separately. Logistic regression analyses were used to assess association with PTB for all common genotypes and haplotypes (frequency ≥ 5% for each in the full sample) (Table S6b, Table S7b).
Additional haplotype analyses were employed to explicitly test the hypothesis of heterozygote protection. Haplotypes were divided into four categories defined by heterozygosity at rs2075609 and rs4147780 as follows: 1.) heterozygous at rs2075609 only, 2.) heterozygous at rs4147780 only, 3.) double heterozygotes, and 4.) summation of all heterozygotes (Table S8). Each category was compared against a common reference group that contained all rs2075609/rs4147780 double homozygotes, regardless of allele status (major/minor) using logistic regression (Table 4). These analyses were performed in the Guinea-Bissau, The Gambia, and the Italian samples separately. All haplotype analyses were conducted using STATA 11, and for each analysis only common haplotypes (frequency ≥ 5% in the full sample) were analyzed.
Exploratory Pairwise Interaction Analyses
ViSEN provides a quantification of the percentage of disease status (case, control) explained by the additional information due to interactions among variables as compared to the additive main effects of the variables combined linearly. The program constructs an interaction network of genetic and environmental factors organized around the strongest interaction effects as opposed to the strongest main effects; main effects are measured by percentage of mutual information (I) between the uncertainty of disease status (case, control) and individual variables. Strength of interaction effects are measured by percentage of IG. ViSEN is optimized to calculate “pure” interaction effects. Specifically, to determine the strength of only the pairwise interaction effect being evaluated, the calculation of interaction IG explicitly excludes the main effect determined for each variable, ensuring that strong pairwise effects are not simply artifacts of strong main effects.
All possible pairwise interactions between analyzed EREG/TCIRG1 SNPs and ethnic group, age quartile, and sex were evaluated for percentage IG in TB susceptibility status in both Guinea-Bissau and The Gambia samples, separately (Table 5a–b, Tables S9–S10). Results were assessed to determine if there were any significant pairwise interactions that replicated across samples (Table 6). Pairwise interactions were considered statistically significant if they had an IG > 1.00% and a p-value < 0.05. P-values were determined in ViSEN by creating a permutation distribution and determining the number of permutation-based IG statistics for each pair of variables that was stronger than or equal to the observed IG statistic (permutation n = 1000).
It should be noted that the ViSEN software is unable to accommodate missingness in the data; therefore, any individuals who were missing demographic (age, sex, ethnic group) or genotypic data for any EREG/TCIRG1 SNPs were excluded from interaction analyses. A total of 279 cases and 318 controls, and 218 cases and 224 controls, were included in interaction analyses for Guinea-Bissau and The Gambia samples, respectively.
Supplementary Material
Acknowledgments
This work was supported by the MRC (UK) award G0000690 to GS and by grants from the Danish Medical Research Council, the Danish society of respiratory medicine, the Danish Council of Development Research to CW and LO. MJW and SMW were partially supported by NIH grant P20 GM103534.
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supplementary information is available at Genes and Immunity’s website.
Reference List
- 1.Global Tuberculosis Report. WHO; 2013. [Google Scholar]
- 2.Frieden TR, Sterling TR, Munsiff SS, Watt CJ, Dye C. Tuberculosis. Lancet. 2003;362:887–99. doi: 10.1016/S0140-6736(03)14333-4. [DOI] [PubMed] [Google Scholar]
- 3.Sirugo G, Hennig BJ, Adeyemo AA, Matimba A, Newport MJ, Ibrahim ME, et al. Genetic studies of African populations: an overview on disease susceptibility and response to vaccines and therapeutics. Hum Genet. 2008;123:557–98. doi: 10.1007/s00439-008-0511-y. [DOI] [PubMed] [Google Scholar]
- 4.Stein CM, Baker AR. Tuberculosis as a complex trait: impact of genetic epidemiological study design. Mamm Genome. 2010;22:91–99. doi: 10.1007/s00335-010-9301-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Caws M, Thwaites G, Dunstan S, Hawn TR, Lan NT, Thuong NT, et al. The influence of host and bacterial genotype on the development of disseminated disease with Mycobacterium tuberculosis. PLoS Pathog. 2008;4:e1000034. doi: 10.1371/journal.ppat.1000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Salie M, van der ML, Moller M, Daya M, van der Spuy GD, van Helden PD, et al. Associations Between Human Leukocyte Antigen Class I Variants and the Mycobacterium tuberculosis Subtypes Causing Disease. J Infect Dis. 2013;209:216–223. doi: 10.1093/infdis/jit443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thye T, Meyer CG. Chapter 4. Human genetic variability and susceptibility to pulmonary TB. In: Lange C, Migliori GB, editors. Tuberculosis [58] 2012. pp. 38–58. [Google Scholar]
- 8.Thuong NT, Hawn TR, Chau TT, Bang ND, Yen NT, Thwaites GE, et al. Epiregulin (EREG) variation is associated with susceptibility to tuberculosis. Genes Immun. 2012;13:275–81. doi: 10.1038/gene.2011.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Capparelli R, Palumbo D, Iannaccone M, Iannelli D. Human V-ATPase gene can protect or predispose the host to pulmonary tuberculosis. Genes Immun. 2009;10:641–6. doi: 10.1038/gene.2009.48. [DOI] [PubMed] [Google Scholar]
- 10.Toyoda H, Komurasaki T, Uchida D, Takayama Y, Isobe T, Okuyama T, et al. Epiregulin. A novel epidermal growth factor with mitogenic activity for rat primary hepatocytes. J Biol Chem. 1995;270:7495–500. doi: 10.1074/jbc.270.13.7495. [DOI] [PubMed] [Google Scholar]
- 11.Thuong NT, Dunstan SJ, Chau TT, Thorsson V, Simmons CP, Quyen NT, et al. Identification of tuberculosis susceptibility genes with human macrophage gene expression profiles. PLoS Pathog. 2008;4:e1000229. doi: 10.1371/journal.ppat.1000229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nalbandian A, Yan BS, Pichugin A, Bronson RT, Kramnik I. Lung carcinogenesis induced by chronic tuberculosis infection: the experimental model and genetic control. Oncogene. 2009;28:1928–38. doi: 10.1038/onc.2009.32. [DOI] [PubMed] [Google Scholar]
- 13.Toei M, Saum R, Forgac M. Regulation and isoform function of the V-ATPases. Biochemistry. 2010;49:4715–23. doi: 10.1021/bi100397s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hinton A, Bond S, Forgac M. V-ATPase functions in normal and disease processes. Pflugers Arch. 2009;457:589–98. doi: 10.1007/s00424-007-0382-4. [DOI] [PubMed] [Google Scholar]
- 15.Heinemann T, Bulwin GC, Randall J, Schnieders B, Sandhoff K, Volk HD, et al. Genomic organization of the gene coding for TIRC7, a novel membrane protein essential for T cell activation. Genomics. 1999;57:398–406. doi: 10.1006/geno.1999.5751. [DOI] [PubMed] [Google Scholar]
- 16.Li YP, Chen W, Stashenko P. Molecular cloning and characterization of a putative novel human osteoclast-specific 116-kDa vacuolar proton pump subunit. Biochem Biophys Res Commun. 1996;218:813–21. doi: 10.1006/bbrc.1996.0145. [DOI] [PubMed] [Google Scholar]
- 17.Scott BB, Chapman CG. The putative 116 kDa osteoclast specific vacuolar proton pump subunit has ubiquitous tissue distribution. Eur J Pharmacol. 1998;346:R3–R4. doi: 10.1016/s0014-2999(98)00163-0. [DOI] [PubMed] [Google Scholar]
- 18.Del Fattore A, Cappariello A, Teti A. Genetics, pathogenesis and complications of osteopetrosis. Bone. 2008;42:19–29. doi: 10.1016/j.bone.2007.08.029. [DOI] [PubMed] [Google Scholar]
- 19.Utku N, Heinemann T, Tullius SG, Bulwin GC, Beinke S, Blumberg RS, et al. Prevention of acute allograft rejection by antibody targeting of TIRC7, a novel T cell membrane protein. Immunity. 1998;9:509–18. doi: 10.1016/s1074-7613(00)80634-2. [DOI] [PubMed] [Google Scholar]
- 20.Singh CR, Moulton RA, Armitige LY, Bidani A, Snuggs M, Dhandayuthapani S, et al. Processing and presentation of a mycobacterial antigen 85B epitope by murine macrophages is dependent on the phagosomal acquisition of vacuolar proton ATPase and in situ activation of cathepsin D. J Immunol. 2006;177:3250–9. doi: 10.4049/jimmunol.177.5.3250. [DOI] [PubMed] [Google Scholar]
- 21.Sturgill-Koszycki S, Schlesinger PH, Chakraborty P, Haddix PL, Collins HL, Fok AK, et al. Lack of acidification in Mycobacterium phagosomes produced by exclusion of the vesicular proton-ATPase. Science. 1994;263:678–81. doi: 10.1126/science.8303277. [DOI] [PubMed] [Google Scholar]
- 22.Kolonko M, Geffken AC, Blumer T, Hagens K, Schaible UE, Hagedorn M. WASH-driven actin polymerization is required for efficient mycobacterial phagosome maturation arrest. Cell Microbiol. 2013 Sep 30; doi: 10.1111/cmi.12217. [DOI] [PubMed] [Google Scholar]
- 23.Wong D, Bach H, Sun J, Hmama Z, Av-Gay Y. Mycobacterium tuberculosis protein tyrosine phosphatase (PtpA) excludes host vacuolar-H+-ATPase to inhibit phagosome acidification. Proc Natl Acad Sci U S A. 2011;108:19371–6. doi: 10.1073/pnas.1109201108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Conboy IM, Manoli D, Mhaiskar V, Jones PP. Calcineurin and vacuolar-type H+-ATPase modulate macrophage effector functions. Proc Natl Acad Sci U S A. 1999;96:6324–9. doi: 10.1073/pnas.96.11.6324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ryckman KK, Jiang L, Li C, Bartlett J, Haines JL, Williams SM. A prevalence-based association test for case-control studies. Genet Epidemiol. 2008;32:600–5. doi: 10.1002/gepi.20342. [DOI] [PubMed] [Google Scholar]
- 26.Hu T, Sinnott-Armstrong NA, Kiralis JW, Andrew AS, Karagas MR, Moore JH. Characterizing genetic interactions in human disease association studies using statistical epistasis networks. BMC Bioinformatics. 2011;12:364. doi: 10.1186/1471-2105-12-364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hu T, Chen Y, Kiralis JW, Moore JH. ViSEN: methodology and software for visualization of statistical epistasis networks. Genet Epidemiol. 2013;37:283–5. doi: 10.1002/gepi.21718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Greene CS, Penrod NM, Williams SM, Moore JH. Failure to replicate a genetic association may provide important clues about genetic architecture. PLoS One. 2009;4:e5639. doi: 10.1371/journal.pone.0005639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Coussens AK, Wilkinson RJ, Nikolayevskyy V, Elkington PT, Hanifa Y, Islam K, et al. Ethnic variation in inflammatory profile in tuberculosis. PLoS Pathog. 2013;9:e1003468. doi: 10.1371/journal.ppat.1003468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pareek M, Evans J, Innes J, Smith G, Hingley-Wilson S, Lougheed KE, et al. Ethnicity and mycobacterial lineage as determinants of tuberculosis disease phenotype. Thorax. 2013;68:221–9. doi: 10.1136/thoraxjnl-2012-201824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thye T, Niemann S, Walter K, Homolka S, Intemann CD, Chinbuah MA, et al. Variant G57E of mannose binding lectin associated with protection against tuberculosis caused by Mycobacterium africanum but not by M. tuberculosis. PLoS One. 2011;6:e20908. doi: 10.1371/journal.pone.0020908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Olesen R, Wejse C, Velez DR, Bisseye C, Sodemann M, Aaby P, et al. DC-SIGN (CD209), pentraxin 3 and vitamin D receptor gene variants associate with pulmonary tuberculosis risk in West Africans. Genes Immun. 2007;8:456–67. doi: 10.1038/sj.gene.6364410. [DOI] [PubMed] [Google Scholar]
- 33.Morris GA, Edwards DR, Hill PC, Wejse C, Bisseye C, Olesen R, et al. Interleukin 12B (IL12B) genetic variation and pulmonary tuberculosis: a study of cohorts from The Gambia, Guinea-Bissau, United States and Argentina. PLoS One. 2011;6:e16656. doi: 10.1371/journal.pone.0016656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gustafson P, Gomes VF, Vieira CS, Rabna P, Seng R, Johansson P, et al. Tuberculosis in Bissau: incidence and risk factors in an urban community in sub-Saharan Africa. Int J Epidemiol. 2004;33:163–72. doi: 10.1093/ije/dyh026. [DOI] [PubMed] [Google Scholar]
- 35.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–5. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 37.StataCorp. Stata Statistical Software. [Release 11] College Station, TX: StataCorp LP; 2009. [Google Scholar]
- 38.Zamar D, McNeney B, Graham J. elrm: software implementation exact-like inference for logistic regression models. Journal of Statistical Software. 2007;21:21. [Google Scholar]
- 39.R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing; Vienna, Austria: 2013. [Google Scholar]
- 40.Grady BJ, Torstenson E, Dudek SM, Giles J, Sexton D, Ritchie MD. Finding unique filter sets in PLATO: a precursor to efficient interaction analysis in GWAS data. Pac Symp Biocomput. 2010:315–26. [PMC free article] [PubMed] [Google Scholar]
- 41.Fan R, Zhong M, Wang S, Zhang Y, Andrew A, Karagas M, et al. Entropy-based information gain approaches to detect and to characterize gene-gene and gene-environment interactions/correlations of complex diseases. Genet Epidemiol. 2011;35:706–21. doi: 10.1002/gepi.20621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moore JH, Gilbert JC, Tsai CT, Chiang FT, Holden T, Barney N, et al. A flexible computational framework for detecting, characterizing, and interpreting statistical patterns of epistasis in genetic studies of human disease susceptibility. J Theor Biol. 2006;241:252–61. doi: 10.1016/j.jtbi.2005.11.036. [DOI] [PubMed] [Google Scholar]
- 43.Anastassiou D. Computational analysis of the synergy among multiple interacting genes. Mol Syst Biol. 2007;3:83. doi: 10.1038/msb4100124. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.