

Abstract
Interferon regulatory factor 4 (IRF4) and IRF8 regulate B, T, macrophage, and dendritic cell differentiation. They are recruited to cis-regulatory Ets-IRF composite elements by PU.1 or Spi-B. How these IRFs target genes in most T cells is enigmatic given the absence of specific Ets partners. Chromatin immunoprecipitation sequencing in T helper 17 (TH17) cells reveals that IRF4 targets sequences enriched for activating protein 1 (AP-1)–IRF composite elements (AICEs) that are co-bound by BATF, an AP-1 factor required for TH17, B, and dendritic cell differentiation. IRF4 and BATF bind cooperatively to structurally divergent AICEs to promote gene activation and TH17 differentiation. The AICE motif directs assembly of IRF4 or IRF8 with BATF heterodimers and is also used in TH2, B, and dendritic cells. This genomic regulatory element and cognate factors appear to have evolved to integrate diverse immunomodulatory signals.
Interferon regulatory factor 4 (IRF4) and IRF8 are evolutionarily diverged members of the IRF family of transcription factors (1). Unlike other members, which are ubiquitously expressed, IRF4 and IRF8 are largely restricted to the immune system and play key roles in the differentiation and functioning of innate and adaptive immune cells (2, 3). IRF4 is required for B cells to undergo class switch recombination and plasma cell differentiation (4, 5). It additionally regulates the generation and/or functioning of various types of helper T (TH) cells, including TH17 (6), TH2 (7, 8), follicular TH (9), and induced regulatory T cells (10). A distinction from other members of the IRF family is that IRF4 and IRF8 bind with low affinity to interferon-stimulated response elements (ISREs) (11, 12) (supplementary text S1). IRF4 and IRF8 have evolved to interact with other transcription factors so as to facilitate their recruitment to distinct genomic regulatory elements. The Ets family transcription factors, PU.1 and Spi-B, that are expressed in B cells, macrophages, and dendritic cells represent interaction partners that are best characterized (13–15). They are able to recruit IRF4 or IRF8 to composite Ets-IRF elements (EICEs). Chromatin immunoprecipitation sequencing (ChIP-seq) analysis of IRF4 in interleukin 21 (IL-21)–stimulated T cells that do not express PU.1 or Spi-B has revealed co-binding with signal transducer and activator of transcription 3 (STAT3) (16). The mechanism underlying co-targeting of IRF4 and STAT3 to genomic regions remains to be defined but does not appear to involve DNA-dependent cooperative binding. Thus, in TH cells that do not express PU.1 or Spi-B, it remains to be determined how IRF4 is recruited to various genomic targets.
We used ChIP-seq analysis to identify the interaction partners for IRF4 in T cells, reasoning that the DNA binding site of such a transcription factor(s) should be juxtaposed with the IRF motif in a stereospecific manner. ChIP-seq with IRF4 in J558L B cells that express PU.1 was used to test the utility of this approach. As anticipated, the EICE motif occurred with highest frequency in the targeted regions (fig. S1A). In contrast, ChIP-seq with IRF4 that is induced during TH17 cell differentiation (fig. S1B) (supplementary text S2) revealed two types of composite IRF–AP-1 motifs, with either a 4- or 0-bp spacing (Fig. 1A). Enrichment of both motifs in IRF4 targeted regions was highly statistically significant relative to a background frequency estimated by MEME (materials and methods and supplementary text S3). These results suggested that IRF4 is recruited to the composite motifs by interactions with AP-1 family members. Because BATF is an activating protein 1 (AP-1) family transcription factor that is required for TH17 differentiation (17), we hypothesized that it was the sought-after interaction partner for IRF4 in TH17 cells. ChIP-seq analysis with BATF in TH17 cells also showed enrichment for AP-1–IRF composite motifs (4- and 0-bp spacing) that were present in 18% and 16% of the targeted sequences, respectively (fig. S1C). Analysis of coincident peaks (supplementary text S3) revealed that a majority of the IRF4-targeted sequences were co-bound by BATF (Fig. 1B) and delineated both types of composite motifs (Fig. 1C). These data implied that IRF4 is recruited to the distinctive composite elements by a BATF-containing AP-1 complex.
Fig. 1.

IRF4 and BATF cistromes in TH17 cells are enriched for composite AP-1–IRF motifs that direct cooperative binding. (A to C) ChIP-seq of in vitro differentiated TH17 cells (42 hours) with antibodies directed against IRF4 or BATF. (A) MEME analysis of highly represented motifs within the IRF4 cistrome (2333 peaks) along with their statistical significance (E) values. (B) Union analysis of the IRF4 and BATF cistromes. Numbers indicate unique or coincident peaks, the latter with peak maxima within 100 bp of each other. (C) MEME analysis of highly represented motifs within the coincident peak sequences of IRF4 and BATF (1936 peaks). (D to G) EMSAs using nuclear extracts from 293T cells overexpressing IRF4, BATF, or JunB. The AP-1 (red) and IRF (blue) sites are indicated. The sequences were derived from IRF4 and BATF co-targeted regions within the CTLA-4 or Bcl11b loci. (D) Protein-DNA complexes were supershifted with antibodies indicated above. (E to G) EMSAs using wild-type (WT) or mutant AICE motif probes. CTLA-4 or Bcl11b DNA probes containing base substitution mutations in either the IRF site (IRF mut), the AP-1 site (AP-1 mut), or both (IRF/AP-1 mut) (E); inverted IRF sites (F); or inserted nucleotides between motifs (G) were used in indicated binding reactions (probes in tables S6 and S7). Red and blue arrows mark the positions of the BATF-JunB-DNA and IRF4-BATF-JunB-DNA complexes, respectively. Data are representative of at least two independent experiments.
To test whether IRF4 and BATF assemble on the composite motifs, we performed electrophoretic mobility shift assays (EMSAs) by using TH17 nuclear extracts and DNA probes, representing the alternate configurations (fig. S2, A and B). Both prototype DNA probes gave rise to co-migrating protein-DNA complexes that contained IRF4 and BATF. These complexes also included JunB, a heterodimeric partner of BATF in TH17 cells (17). To demonstrate cooperative DNA-dependent assembly, we expressed each protein individually in 293T cells and then used these proteins for EMSAs. No specific protein-DNA complexes were detected with IRF4 alone (Fig. 1D). A complex was observed in reactions containing BATF and JunB. Addition of IRF4 to the BATF-JunB binding reactions resulted in a slower-migrating complex containing all three proteins. Mutational analysis of the IRF and AP-1 sites showed both to be required for the assembly of quaternary complexes with the two types of composite motifs (Fig. 1E). Fine specificity analysis revealed considerable degeneracy in the IRF recognition sequence in the context of IRF4 co-binding with BATF-JunB heterodimers (supplementary text S4). Thus, the AP-1 motif observed by IRF4 ChIP-seq analysis (Fig. 1A) may also be associated with degenerate IRF sites that are not readily revealed by MEME. An AP-1 site was not sufficient to mediate the recruitment of IRF4 to DNA. The latter conclusion was strongly reinforced by use of variant CTLA-4 or Bcl11b probes that differed in the orientation or spacing of the IRF site in relation to the AP-1 site (Fig. 1, F and G). Thus, binding of a BATF-JunB heterodimer to the AP-1 site functions to recruit IRF4 to DNA in alternate configurations via the latter’s recognition of an IRF site in the composite sequence. By analogy with the EICEs characterized in our earlier work (14), we designate the AP-1–IRF composite elements as AICE motifs.
To determine whether IRF4-BATF-JunB complexes bind to presumptive regulatory sequences in genes required by TH17 cells, we focused on Il17, Il21, Il22, Il23r, and Il12rb1. Each of these loci was found to contain one or more coincident binding peaks for IRF4 and BATF that were positioned in promoters and/or intronic regions (Fig. 2A, left). ChIP assays not only verified the binding of IRF4 and BATF to these regions but also demonstrated the co-binding of JunB (fig. S3A). Analysis of the DNA sequences underlying these peaks revealed that some had readily discernible AICE motifs whereas others contained AP-1 sites juxtaposed with degenerate IRF sites (Fig. 2A, right, and fig. S3). Nevertheless, all of these sequences promoted the assembly of IRF4-BATF-JunB complexes in TH17 nuclear extracts (Fig. 2A, right, and fig. S3C). We note that the sequences also varied in their affinity for BATF-JunB complexes (fig. S3, B and D, left). Importantly, IRF4 was seen to reciprocally enhance the binding of BATF-JunB (1.5- and 8-fold), more strongly on sequences that had variant lower affinity AP-1 sites (Fig. 2B and fig. S3, E and F). Mutational analysis confirmed the requirement of the AP-1 and IRF sites for DNA assembly (supplementary text S4). Thus, these complexes exhibit the hallmarks of DNA-dependent cooperative binding in spite of structurally divergent composite sites. We next tested whether loss of BATF or IRF4 resulted in impaired binding of the partner transcription factor to target sequences in vivo. IRF4 or BATF binding was reduced on co-targeted sequences in Batf−/− or Irf4−/− T cells, respectively, cultured under TH17 polarizing conditions (Fig. 2, C and D). ChIP-seq analyses of Batf−/− and Irf4−/− T cells revealed that co-targeting of IRF4 and BATF to the Ctla-4 and Il12rb1 loci was diminished or lost in the absence of the partner protein (fig. S4 and supplementary text S3). Loss of BATF or IRF4 did not affect the expression of the partner transcription factor. Therefore, IRF4-BATF-JunB complexes target a key set of TH17 genes and cooperatively assemble on structurally diverse AICE motifs in vitro as well as in vivo.
Fig. 2.
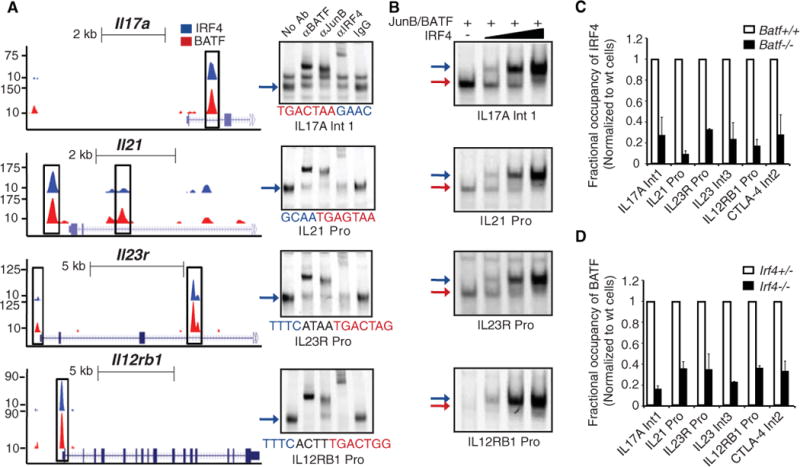
IRF4-BATF-JunB complexes assemble on presumptive regulatory sequences in key TH17 genes. (A) (Left) ChIP-seq tracks for IRF4 (blue) or BATF (red) at the Il17a, Il21, Il23r, and Il12rb1 loci. Coincident peaks containing AICE motifs are highlighted (boxed regions). (Right) EMSAs using nuclear extracts from TH17 cells and the indicated DNA probes and antibodies. (B) EMSAs using nuclear extracts from 293T cells overexpressing indicated proteins. Reactions were performed with increasing amounts (fivefold) of IRF4 protein. (C) ChIP analysis of IRF4 binding on indicated sequences in wild-type and Batf−/− T cells. (D) ChIP analysis of BATF binding on indicated sequences in Irf4+/− and Irf4−/− T cells. Purified CD4 cells were polarized for 42 hours under TH17 differentiating conditions. The average ± SD of two independent experiments is shown for each target sequence after normalization to control T cells.
Several complementary approaches were used to test functionality of molecular complexes between IRF4 and BATF. Given that the Il12rb1 promoter contains an AICE motif (Fig. 2), we tested whether its activity in TH17 cells is dependent on both the AP-1 and the IRF sites. The wild-type promoter displayed significant activity, which was abrogated by mutation of the AP-1 or IRF sites (Fig. 3A). Hence, both the AP-1 and IRF sites are required for the transcription-activating function of an AICE motif. We next performed gain-of-function experiments to determine whether coexpression of IRF4 and BATF promotes activation of target genes containing AICE motifs and the generation of TH17 cells. CTLA-4 induction was modestly elevated by increasing IRF4 or BATF levels in nonpolarized CD4+ T cells but more substantially by their combined expression (Fig. 3B and fig. S5A). Similarly, IRF4 or BATF overexpression did not appreciably increase the frequency of IL-17A–producing cells, whereas their coexpression resulted in a twofold enhancement (Fig. 3C and fig. S5B). Lastly, by mutational analysis of the BATF leucine zipper region, we were able to identify a residue H55 that participated in the recruitment of IRF4 to AICE motifs (18). Replacement of the histidine with a glutamine residue impaired the recruitment of IRF4 to both types of AICE motifs, with a more severe consequence on its binding to the abutting (Bcl11b) configuration (Fig. 3, D and E). Mutation of a residue Glu77 (E77) in the leucine zipper that is distal from the BATF DNA binding domain did not affect complex formation with IRF4. Whereas the wild-type and Glu77→Lys77 (E77K) BATF proteins robustly restored the generation of TH17 cells upon their expression in Batf−/− T cells, the His55→Gln55 (H55Q) protein exhibited reduced complementing activity in spite of comparable stability and DNA binding (Fig. 3, D, E, and F). Thus, by mutational analysis of the cis- and trans-acting components of the AICE motif as well as gain- and loss-of-function experiments, these results establish the functional importance of IRF4-BATF complexes in regulation of TH17 cells.
Fig. 3.
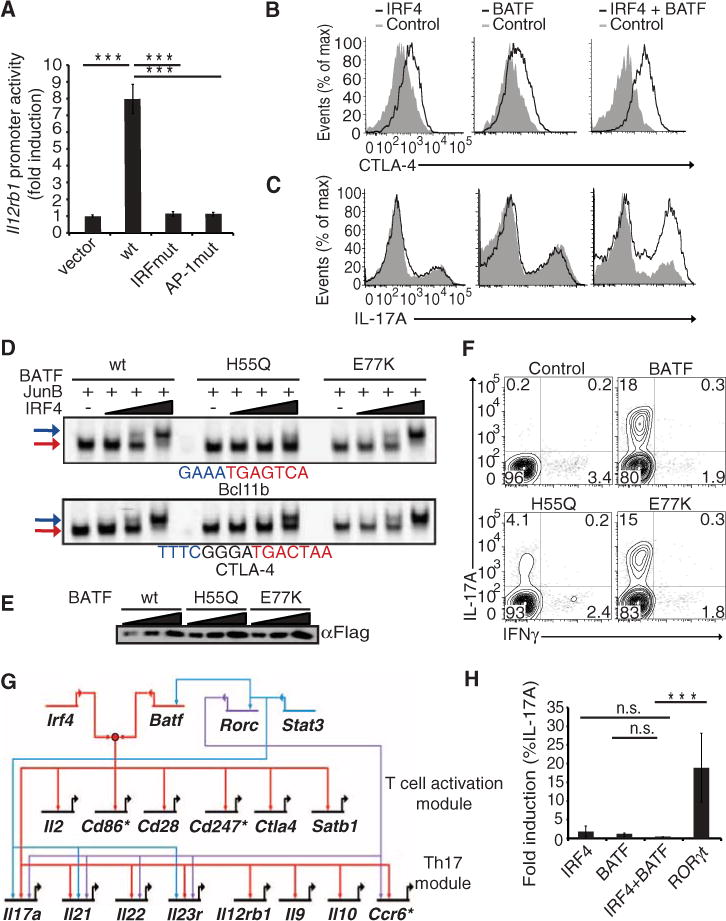
IRF4 and BATF complexes activate gene expression and TH17 differentiation. (A) TH17-polarized cells were transiently transfected with luciferase reporter plasmids containing the wild-type Il12rb1 promoter or mutant derivatives and a control Renilla luciferase reporter plasmid. Data are from five experiments (average ± SD) (B and C) Naïve CD4 cells activated under nonpolarizing conditions (TH0) or TH17-polarizing conditions, respectively, were transduced with the indicated retroviruses. Four days after infection, cells were stimulated with phorbol 12-myristate 13-acetate (PMA) and ionomycin for 4 hours and analyzed by flow cytometry for intracellular CTLA-4 or IL-17A after gating on transduced cells. (D) EMSAs using nuclear extracts from 293T cells overexpressing indicated proteins. Reactions used increasing amounts (fivefold) of IRF4. (E) Western blot analysis of wild-type and BATF mutant proteins used in (D). (F) Batf−/− T cells were activated under TH17-polarizing conditions and transduced with retroviruses encoding wild-type BATF or indicated mutants. Six days after infection, cells were restimulated under TH17 conditions and analyzed 5 days later for expression of IL17 and IFNγ by intracellular staining. (G) Network diagram of IRF4 and BATF co-targeted and regulated genes. Genes marked with * were differentially regulated in Batf−/− T cells based on genome-wide expression analysis but could not be confirmed via quantitative polymerase chain reaction (qPCR). RORγt and STAT3 inputs on these genes are indicated in blue (H). TH0 cells were transduced with indicated retroviruses and analyzed as described in (B). Data are representative of at least two experiments and where indicated include average values ± SD. n.s., not significant.
To identify IRF4-regulated genes among those that are co-targeted by IRF4 and BATF, we performed genome-wide expression analysis of Irf4−/− T cells polarized under TH17 conditions (fig. S6A). There was a profound defect in the expression of Il17a, Il21, Il22, and Il23r genes in Irf4−/− cells (fig. S6B). Notably, these cells were also defective in expression of 1l12rb1 and Ccr6 and genes involved in T cell activation, including Cd86, Cd247, Cd28, Ctla4, and Il2. Of the 362 genes that were positively regulated by IRF4 during TH17 differentiation, 155 (~42%) contained coincident IRF4 and BATF peaks (table S2). The hypergeometric P value for the overall enrichment of IRF4-regulated genes in the IRF4-BATF co-bound data set was <10−16. Ingenuity pathway analysis (IPA) revealed that 65 of the aforementioned 155 genes formed a highly interconnected network (fig. S6C). This network did not include genes encoding the other transcription factors required for TH17 differentiation, namely, Rorc, Rora, Ahr, and Stat3 (19–22). However, when these regulatory factors were introduced into the network (fig. S6C), they were seen to make a large number of connections that were statistically significant. Inspection of this network of genes revealed distinct regulatory modules (Fig. 3G). Analysis of this select set of co-targeted and IRF4-regulated genes in Batf−/− cells showed the majority of them to be dependent on BATF (17) (fig. S6D). Most of these genes contained identifiable AICE motifs in the DNA regions that were co-bound by IRF-4 and BATF (fig. S6E). Given that IRF4-BATF complexes appear to co-target and regulate a key set of genes in TH17 cells that is also controlled by Rorγt, we tested for their interdependency by gain-of-function experiments in TH0 cells. As expected, ectopic expression of Rorγt promoted the generation of TH17 cells (19) (Fig. 3H). In contrast, individual or coexpression of IRF4 and BATF in TH0 cells did not enhance TH17 cell generation. Because Rorγt is not induced under TH0 conditions (19), these results suggest that IRF4/BATF complexes function in a combinatorial manner with Rorγt to induce the expression of TH17 genes.
Expression profiling of AP-1 family genes in TH17 cells revealed the inducible expression of c-Jun and FosL2 in addition to BATF and JunB. Therefore, we tested whether alternate AP-1 complexes containing these subunits were capable of cooperatively assembling with IRF4 on AICE motifs. A c-Jun–BATF but not a JunB-FosL2 heterodimer was able to recruit IRF4 to the structurally distinct AICE motifs (Fig. 4, A and B, and fig. S7, A and B). Given a high degree of structural similarity of IRF4 with IRF8, we determined whether the latter factor could also be recruited to AICE motifs by BATF-Jun heterodimers. IRF8 was seen to cooperatively assemble on AICE motifs with BATF-JunB heterodimers (Fig. 4C and fig. S7C). However, this molecular property was not manifested by ubiquitously expressed IRFs, namely, IRF1 or IRF3 (Fig. 4D and fig. S7D). IRF1 and IRF3, unlike IRF4, could bind with high affinity to an ISRE DNA probe (fig. S7E). Structural modeling and biochemical analysis with the IRF4 DNA binding domain suggest an unusually pliant mode of cooperative DNA binding (fig. S8 and supplementary text S5). Thus, IRF4 and IRF8 are distinguished from other IRFs not only by their immune system–selective expression but also by their unique molecular properties of cooperative assembly with specific members of the Ets and AP-1 superfamilies on EICE and AICE motifs, respectively.
Fig. 4.

Specificity of AP-1–IRF complexes that cooperatively assemble on AICE motifs. (A to D) EMSAs using the Bcl11b probe and nuclear extracts from 293T cells overexpressing the indicated proteins: (A) c-Jun, BATF, IRF4; (B) FosL2, JunB, IRF4; (C) JunB, BATF, IRF8; or (D) JunB, BATF, IRF3, and IRF4. Complexes were supershifted with indicated antibodies. Red and blue arrows mark the ternary and quaternary protein-DNA complexes, respectively, as in Fig. 1. Data are representative of at least two independent experiments. (E to G) ChIPseq analysis of in vitro differentiated TH0 and TH2 cells (42 hours) and LPS-activated dendritic cells (6 hours) with antibodies directed against IRF4. Highly represented motifs and their respective E values within the IRF4 cistrome in TH0 (241 peaks), in TH2 (725 peaks), and in dendritic cells (10,364 peaks) are displayed.
Given the restricted expression and overlapping functions of IRF4 and BATF in T, B, and dendritic cells (5, 6, 17, 18, 23, 24), we used ChIPseq to determine whether IRF4 is able to target AICE motifs in alternate cellular contexts. We analyzed the IRF4 cistromes in activated but nonpolarized T cells (TH0), IL-4–polarized TH2 cells, and lipopolysaccharide (LPS)–activated dendritic cells. All three cellular contexts revealed statistically significant enrichment for the AICE motif in targeted sequences (Fig. 4, E to G, and table S3). Because dendritic cells express PU.1 and Spi-B whereas TH0 and TH2 cells do not, the EICE motif was observed within the IRF4 cistrome in the former context but was absent in the latter (Fig. 4G and table S3). As is the case for dendritic cells, the IRF4 cistrome in B cells was also seen to contain EICE as well as AICE motif targeted regions (supplementary text S6). A pairwise statistical analysis of the use of AICE and EICE motifs in the various immune cell contexts confirmed their distinctive distributions in IRF4-targeted sequences (table S4). These results suggest that, although IRF4 functions with BATF-containing AP-1 heterodimers on the AICE motif in both adaptive and innate immune cell contexts, TH17 and TH2 cells rely exclusively on such complexes.
We propose that the AICE motif is an immune-specific genomic regulatory element that is widely used in both innate and adaptive immune cells. Its functioning is predicted to be largely confined to cells of the immune system by virtue of the restricted expression of IRF4 and IRF8 as well as BATF or its paralogs (18). The involvement of the AICE motif in distinct innate or adaptive immune cell–specific programs of gene expression, as exemplified in TH17 cells, is likely to reflect the combinatorial functioning of AP-1–IRF4 or AP-1–IRF8 complexes with differentiation state–specific regulators such as Rorγt in TH17 cells or Gata3 in TH2 cells. In addition to its TH effector state–specific functions, the AICE motif also regulates key components within the T cell activation program. Given that AP-1 family members and IRF4 and IRF8 are signaling-induced transcription factors (2, 25), the AICE motif appears to have evolved to sense and integrate diverse signaling inputs in immune cells. It is capable of integrating such inputs from antigen receptors and their co-receptors as well as cytokine receptors and TLRs. Because AP-1 family members can cooperatively assemble on composite elements with nuclear factor of activated T cells (NFAT) family proteins (26) and juxtaposition of NFAT and IRF sites has been reported on the IL-4 and IL-10 genes (8, 27), it will be important to determine whether the AP-1–IRF4 or AP-1–IRF8 complexes described herein can cooperatively assemble with NFAT proteins, thereby enabling the integration of Ca signaling in the regulatory module. We propose that the acquisition of the AICE motif by simple variation of AP-1 sites in biologically important immune response genes may have provided a selective force for the evolution of the structurally divergent and specialized members of the IRF family, namely, IRF4 and IRF8.
Supplementary Material
Acknowledgments
We thank K. Dong for insightful discussions and suggestions and the fluorescence-activated cell sorting (FACS), antibody engineering, DNA sequencing, microarray, and oligonucleotide synthesis facilities at Genentech for providing valuable reagents and technical assistance. The data presented in the manuscript are tabulated in the main paper and the supplementary materials. Reagents will be provided upon request. D.R.L. and K.M.M. are Howard Hughes Medical Institute investigators. D.R.L. also acknowledges funding from NIH grants RC1 AI087266 and RC4 AI092765, and M.C. was supported by a fellowship from the Crohn’s and Colitis Foundation of America. Genentech, Incorporated, has filed a provisional patent application that is focused on targeting of IRF4-BATF complexes in the context of TH17-mediated inflammatory or autoimmune diseases. The complete gene expression and ChIPseq data sets can be accessed at www.ncbi.nlm.nih.gov/projects/geo/query/acc.cgi?acc=GSE40483.
Footnotes
Supplementary Materials
www.sciencemag.org/cgi/content/full/science.1228309/DC1
Materials and Methods
References (28–48)
References and Notes
- 1.Tamura T, Yanai H, Savitsky D, Taniguchi T. Annu Rev Immunol. 2008;26:535. doi: 10.1146/annurev.immunol.26.021607.090400. [DOI] [PubMed] [Google Scholar]
- 2.Lohoff M, Mak TW. Nat Rev Immunol. 2005;5:125. doi: 10.1038/nri1552. [DOI] [PubMed] [Google Scholar]
- 3.De Silva NS, Simonetti G, Heise N, Klein U. Immunol Rev. 2012;247:73. doi: 10.1111/j.1600-065X.2012.01113.x. [DOI] [PubMed] [Google Scholar]
- 4.Klein U, et al. Nat Immunol. 2006;7:773. doi: 10.1038/ni1357. [DOI] [PubMed] [Google Scholar]
- 5.Sciammas R, et al. Immunity. 2006;25:225. doi: 10.1016/j.immuni.2006.07.009. [DOI] [PubMed] [Google Scholar]
- 6.Brüstle A, et al. Nat Immunol. 2007;8:958. doi: 10.1038/ni1500. [DOI] [PubMed] [Google Scholar]
- 7.Lohoff M, et al. Proc Natl Acad Sci USA. 2002;99:11808. doi: 10.1073/pnas.182425099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rengarajan J, et al. J Exp Med. 2002;195:1003. doi: 10.1084/jem.20011128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bollig N, et al. Proc Natl Acad Sci USA. 2012;109:8664. doi: 10.1073/pnas.1205834109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zheng Y, et al. Nature. 2009;458:351. doi: 10.1038/nature07674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bovolenta C, et al. Proc Natl Acad Sci USA. 1994;91:5046. doi: 10.1073/pnas.91.11.5046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matsuyama T, et al. Nucleic Acids Res. 1995;23:2127. doi: 10.1093/nar/23.12.2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brass AL, Kehrli E, Eisenbeis CF, Storb U, Singh H. Genes Dev. 1996;10:2335. doi: 10.1101/gad.10.18.2335. [DOI] [PubMed] [Google Scholar]
- 14.Brass AL, Zhu AQ, Singh H. EMBO J. 1999;18:977. doi: 10.1093/emboj/18.4.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eisenbeis CF, Singh H, Storb U. Genes Dev. 1995;9:1377. doi: 10.1101/gad.9.11.1377. [DOI] [PubMed] [Google Scholar]
- 16.Kwon H, et al. Immunity. 2009;31:941. doi: 10.1016/j.immuni.2009.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schraml BU, et al. Nature. 2009;460:405. doi: 10.1038/nature08114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tussiwand R, et al. Nature. 2012;490:502. doi: 10.1038/nature11531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ivanov II, et al. Cell. 2006;126:1121. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 20.Yang XO, et al. Immunity. 2008;28:29. doi: 10.1016/j.immuni.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mathur AN, et al. J Immunol. 2007;178:4901. doi: 10.4049/jimmunol.178.8.4901. [DOI] [PubMed] [Google Scholar]
- 22.Kimura A, Naka T, Nohara K, Fujii-Kuriyama Y, Kishimoto T. Proc Natl Acad Sci USA. 2008;105:9721. doi: 10.1073/pnas.0804231105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tamura T, et al. J Immunol. 2005;174:2573. doi: 10.4049/jimmunol.174.5.2573. [DOI] [PubMed] [Google Scholar]
- 24.Ise W, et al. Nat Immunol. 2011;12:536. doi: 10.1038/ni.2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hess J, Angel P, Schorpp-Kistner M. J Cell Sci. 2004;117:5965. doi: 10.1242/jcs.01589. [DOI] [PubMed] [Google Scholar]
- 26.Macián F, García-Rodríguez C, Rao A. EMBO J. 2000;19:4783. doi: 10.1093/emboj/19.17.4783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee CG, et al. Mol Immunol. 2009;46:613. doi: 10.1016/j.molimm.2008.07.037. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.