

Gene variants tied to human facial features
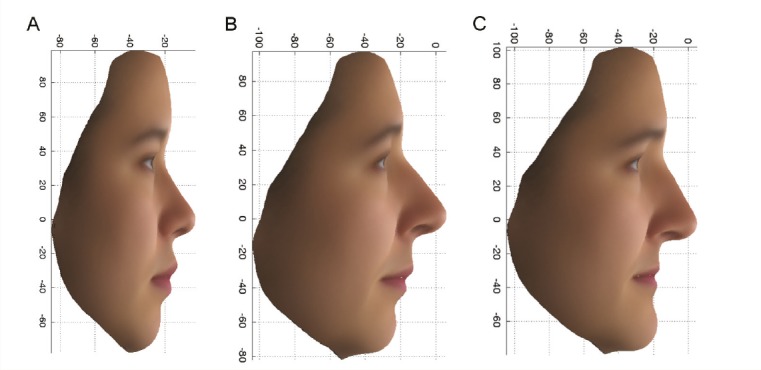
Average faces for 14 East Asian females (A); upper 10% (more East Asian) (B) and lower 10% (more European) (C) extremes.
Facial similarities tend to run in families, and genetically identical twins raised together or apart exhibit striking facial resemblance, suggesting strong genetic control of human facial features. Yet studies have largely failed to identify single gene variants with large effects on facial features. Daniel Crouch et. al (pp. E676–E685) used an analysis of images of identical and nonidentical twins to weight each point on the face by the extent to which it is genetically influenced. Next, the authors used the weights in a principal components analysis of 3D camera images of faces from 1,832 volunteers from the People of the British Isles project, 1,567 participants in the TwinsUK cohort, and 33 East Asian volunteers. Focusing on the upper and lower 10% of extremes of three composite facial phenotypes, which were derived from facial profiles and surface features of the region around the eyes, the authors identified two genetic variants tied to facial profiles in females and one genetic variant tied to surface features around the eyes in both males and females. One variant exhibited linkage with a gene implicated in Usher syndrome, which is marked by vision, hearing, and balance defects, whereas another was located in a gene involved in regulating steroid biosynthesis. The third variant was located in a zinc transporter with a potential role in mucolipidosis type IV, a condition occasionally accompanied by facial dysmorphia, particularly around the eyelids. According to the authors, identification of gene variants with large and specific effects on facial features paves the way toward unraveling the molecular mechanisms by which genes shape the extraordinary variability in human facial appearance. — P.N.
Reawakening inactivated X chromosomes to treat Rett syndrome

Mixed-mode approach reactivates healthy X chromosome in Rett syndrome mouse model.
Females inherit an X chromosome from each parent, one of which is inactivated during embryonic development. As such, female somatic cells express either the maternal or paternal X chromosome in roughly equal numbers, a critical distinction in how disease-related mutations on the X chromosome affect men and women. Lieselot Carrette et al. (pp. E668–E675) exploit the distinction to devise a mixed-mode treatment strategy for Rett syndrome, a neurodevelopmental disorder caused by a mutation in methyl-CpG binding protein 2 (MECP2) that largely afflicts young girls, causing severe seizures, motor abnormalities, speech deficits, and autism. The authors’ therapeutic strategy reawakens the inactivated but healthy X allele with a two-pronged approach: an antisense oligonucleotide to target the noncoding RNA responsible for X-inactivation, or Xist, and a small-molecule inhibitor of DNA methylation. Acting synergistically, the two approaches upregulate MECP2 from cultured inactivated X chromosomes approximately 30,000-fold. Furthermore, an in vivo model reveals that X chromosome reactivation occurs in the brains of mice with no apparent toxicity. According to the authors, the approach can be applied to similar neurodevelopmental disorders, such as fragile X syndrome and CDKL5 syndrome. — T.J.
Gender gap in mortality during famines and epidemics

Memorial to the Irish famine of 1845–1849, Dublin, Ireland. Image courtesy of Wikimedia Commons/AlanMc.
Life expectancy for women currently exceeds that for men nearly worldwide. Whether this gender gap persists under conditions of extremely high mortality risk remains unclear. Virginia Zarulli et al. (pp. E832–E840) analyzed mortality data for seven historic populations in which life expectancy for at least one of the sexes was 20 years or less. The populations include slaves in Trinidad and freed American slaves in Liberia in the early 19th century, the populations of Sweden, Ireland, and Ukraine during famines in 1772–1773, 1845–1849, and 1933, respectively, and the Icelandic population during the 1846 and 1882 measles epidemics. Life expectancy for women exceeded that for men in all populations by 0.5–3.5 years, with the possible exception of Trinidadian slaves. Women were more likely than men to survive to a given age, except in Trinidad, where male survival was greater early in life. In all populations, most of the difference in life expectancy between men and women was due to large sex differences in infant mortality. The results suggest that women’s survival advantage over men has a biological basis but is modulated by social and environmental factors, according to the authors. — B.D.
How amyloid plaques spread in the brain
Early-onset familial Alzheimer’s disease (AD) and hereditary cerebral amyloid angiopathy are neurological disorders caused by various mutations that produce a combination of normal and mutant forms of the amyloid-beta peptide in the brain. The amyloid-beta peptide can change its shape to form prions, which accumulate to form clumps called amyloid plaques. To examine the distinct conformational strains of amyloid-beta prions and the structural variability of plaque deposits, Carlo Condello et al. (pp. E782–E791) applied multiple structure-sensitive fluorescent amyloid-binding dyes and high-resolution confocal spectral imaging analysis to brain slices from patients with AD and patients with cerebral amyloid angiopathy. The authors found that the different disease types were associated with distinct amyloid-beta prion strains. To examine how amyloid plaques form, the authors next injected the mutant amyloid-beta peptide into the brains of mice expressing only the normal version of the peptide. The mutant amyloid-beta formed prions that induced the widespread deposition of amyloid plaques in the brain. Moreover, the molecular conformation of the induced amyloid deposits resembled those found in the patient samples. The findings suggest that mutant amyloid-beta adopts the disease-causing, self-propagating structure of the prion, which imparts its pathological conformation on previously normal amyloid-beta to form more prions. As more and more prions become sequestered into amyloid plaques, the plaques spread throughout the brain, according to the authors. — J.W.