

Abstract
Inflammasomes are cytosolic multi-molecular complexes that sense intracellular microbial danger signals and metabolic perturbations. Inflammasome activation leads to the activation of caspase-1 and the release of pro-inflammatory cytokines IL-1β and IL-18 accompanied by cell death. An inflammasome-based surveillance machinery for Gram-negative bacterial infections has been recently discovered. This noncanonical inflammasome relies on sensing the cytosolic presence of lipopolysaccharide (LPS) of Gram-negative bacteria via inflammatory caspases such as caspase-4, -5, and -11. This Review discusses the recent findings related to the mechanism of activation of the noncanonical inflammasome and its biological functions.
Graphical Abstract
Noncanonical inflammasome sensing of intracellular LPS
The innate immune system recognizes pathogen invasion via pattern recognition receptors (PRRs) that detect pathogen associated molecular patterns (PAMPs) [1]. Inflammasomes are a subset of PRRs located in the cytosol that are responsible for sensing various PAMPs and endogenous danger signals (danger associated molecular patterns or DAMPs). Inflammasomes are multimeric protein complexes consisting of a sensor protein from the AIM2 (absent in melanoma 2)-like receptor (ALR) or the nucleotide-binding domain and leucine rich repeat containing protein (NLR) families, an adaptor protein ASC (apoptosis-associated speck like protein containing CARD), and the protease caspase-1 [2]. The inflammasome complex undergoes oligomerization leading to the auto cleavage of caspase-1 into active caspase-1, which subsequently cleaves the inflammatory cytokines pro-IL-1β and pro-IL-18 into their active forms [2]. In addition, inflammasome activation results in an inflammatory cell death called pyroptosis in most cases [3]. Inflammasomes are important for initiating the inflammatory response against a wide variety of pathogens including bacteria and viruses [4]. Inflammasomes also play crucial roles in sterile inflammatory diseases such as atherosclerosis [5].
Assembly and activation of inflammasomes is triggered by a wide variety of microbial products including nucleic acids, toxins, flagellin, and cell wall components [6–11]. Crystalline substances such as alum and silica and endogenous danger signals such as ATP and uric acid can also activate the inflammasomes [12]. In addition to the above-described canonical inflammasomes, a noncanonical inflammasome was recently discovered, which is activated in response to intracellular LPS [13,14]. It was previously thought that LPS was solely recognized by the innate immune system through toll-like receptor 4 (TLR-4) in conjunction with MD2 and CD14 at the cell surface [15]. However, these proteins are not responsible for the cytosolic recognition of LPS [13,14].
In the noncanonical inflammasome, the inflammatory protease, caspase-11, which is closely related to caspase-1, acts as a receptor for LPS that gains access to the cytosol (Fig. 1) [16]. Caspase-11 is activated by Gram-negative bacteria including but not limited to Escherichia coli, Salmonella typhimurium, Shigella flexneri, and Burkholderia thailandensis [16–18]. Upon recognizing LPS, caspase-11 undergoes activation resulting in pyroptotic cell death (Fig. 1). Active caspase-11 also facilitates the activation of NLRP3 leading to the activation of caspase-1 and the secretion of IL-1β and IL-18 [18]. Caspase-11 directly binds the lipid A moiety of LPS, the same structural component that activates TLR-4, via its CARD domain [16]. Specifically, hexa-acylated lipid A is required for the optimal activation of caspase-11, although penta-acylated LPS can weakly activate caspase-11 [13]. Gram-negative bacteria containing hexa-acylated LPS, such as E. coli and S. flexneri robustly activate caspase-11 [17]. However, some pathogenic bacteria can modify their LPS by changing the acylation state of lipid A to evade recognition by the immune system and to minimize their inflammatory potential. For example, Francisella sp and Heliobacter pylori contain tetra-acylated lipid A, and they do not activate caspase-11 [13,14].
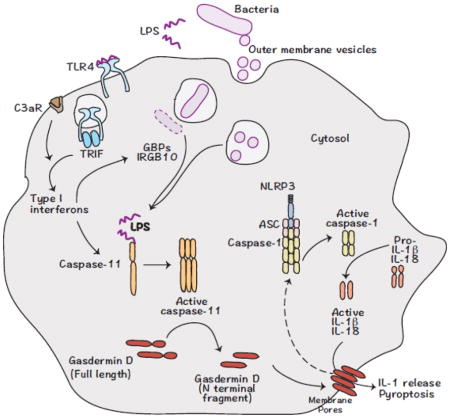
Figure 1. Noncanonical inflammasome recognition of bacterial pathogens.

LPS from Gram-negative bacterial pathogens trigger the expression of caspase-11 via TLR-4-TRIF-type I interferon signaling. C3aR signaling also contributes to the upregulation of caspase-11 expression. Extracellular vesicles released by bacteria, as well as GBPs and IRGB10 proteins induced by type I interferon signaling, play critical roles in the cytosolic localization of LPS. LPS that accumulates in the cytosol is recognized by caspase-11 in mice and caspase-4 and -5 in humans leading to their activation. Active caspase-4/-5/-11 cleaves gasdermin D to remove the inhibitory C terminal from the N terminal fragment, which migrates to the plasma membrane forming pores and eventually causing cell death. Gasdermin D is also involved in the activation of the NLRP3 inflammasome, presumably by inducing potassium efflux.
Caspase-11 can also be activated by endogenous DAMPs. Under the conditions of oxidative stress during infections and tissue damage, oxidized phospholipids such as 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphorylcholine (oxPAPC) that are generated by the oxidation of plasma membrane phospholipids accumulate in the cell. Damaged and dying cells release high concentrations of these oxidized phospholipids. It was recently discovered that oxPAPC, a LPS mimic, binds to and activates caspase-11 in dendritic cells (DCs) [19]. Interestingly, oxPAPC and LPS bind different domains of caspase-11; LPS binds to the CARD domain of caspase-11 but oxPAPC binds to the catalytic domain of caspase-11. Like LPS, oxPAPC induces IL-1β release following caspase-11 activation, but does not induce pyroptosis. The enzymatic activity of caspase-11 is not required for oxPAPC induced IL-1β release. Furthermore, oxPAPC elicits IL-1β release only from DCs, but not macrophages. DC recognition of oxPAPC via caspase-11 promotes adaptive T cell responses [19].
Transcriptional regulation of the non-canonical inflammasome
Caspase-11 is produced as two isoforms of 43 kDa and 38 kDa [20]. Unlike other caspases that are primarily regulated by proteolytic cleavage, caspase-11 is regulated at both transcriptional and post-translational levels. Caspase-11 is expressed broadly in immune and non-immune cells and its expression in resting cells is weak [17,18,21]. The promoter region of the caspase-11 gene has NFκB and STAT1 binding sites [22]. Accordingly, several pro-inflammatory stimuli that activate NFκB and STAT1 induce caspase-11 transcription [18]. The magnitude of caspase-11 expression driven by NFκB and STAT1 are different; while NFκB can induce caspase-11 expression weakly, STAT1 signaling robustly activates caspase-11 transcription [17].
The mechanism of caspase-11 transcription in Gram-negative bacterial infections has been extensively explored [17,23–25]. LPS from Gram-negative bacteria such as enterohemorrhagic Escherichia coli (EHEC) or Citrobacter rodentium is a strong inducer of caspase-11 expression (Fig. 1). LPS activation of TLR-4-TRIF signaling leads to the expression of type I interferons via the activation of interferon regulatory factor (IRF)-3. Autocrine or paracrine engagement of IFNAR1 receptor by type I interferons results in the activation of STAT1 and IRF9, which initiate the transcription of caspase-11. Accordingly, macrophages defective in any component of TRIF and type I interferon signaling are unable to robustly upregulate caspase-11 expression and activate inflammasome responses to Gram-negative bacterial infections [17].
Apart from the TRIF-type I interferon signaling axis, caspase-11 expression can also be induced by other immune pathways. IFN-γ acting via STAT1 can also trigger the expression of caspase-11. During infection with the invasive B. thailandensis in vivo, caspase-1-directed IL-18 was required for the production of IFN-γ that eventually primed the cells for caspase-11 expression [26]. Thus, IFN-γ might represent a potential link by which innate and adaptive lymphocytes such as NK and Th1 cells can potentiate noncanonical inflammasome activation. A recent study employing a whole genome CRISPR-Cas9 screen has demonstrated a role for the complement cascade in caspase-11 transcription [27]. It was shown that complement related carboxypeptidase B1 (Cbp1) processes C3a, which signals via the C3a receptor to promote caspase-11 expression. Cbp1-C3a-C3aR axis enhances the TLR-4-TRIF-IFNAR signaling by modulating P38 mitogen activated protein kinase (MAPK), thereby increasing caspase-11 expression (Fig. 1). Reactive oxygen species (ROS) accumulation can also augment caspase-11 expression by increasing c-Jun N-terminal kinase (JNK) signaling [28].
Noncanonical inflammasome in humans
Humans lack the caspase-11 gene, but do have the caspase-4 and caspase-5 genes, which are considered to be duplication products of an ancestral caspase-11 gene [29]. Unlike caspase-11, which is an inducible protein, caspase-4 is constitutively expressed in human cells [16]. On the other hand, caspase-5 expression is inducible [30]. Although caspase-5 transcription has not been extensively examined, type I and II interferons strongly induce caspase-5 expression [30]. Like caspase-11, both caspase-4 and caspase-5 are capable binding to hexa-acylated lipid A and undergo activation [16]. The presence of these two caspase-11-like proteins raised an obvious but important question of which one is responsible for the cytosolic detection of LPS in humans. Several reports have shown a requirement for caspase-4 in this process; siRNA knock down and CRISPR/Cas9-deletion of caspase-4 in HeLa cells, THP-1 cells and primary macrophages attenuated the pyroptotic and IL-1β responses to transfected LPS [16,31–33]. Whether caspase-5 is similarly required remains less clear. It appears that caspase-5 is minimally required when LPS is transfected into the cytosol, but becomes more important during actual infections [33]. The exact contribution of caspase-4 and caspase-5 to the cytosolic recognition of LPS during bacterial infections is yet to be completely understood. It is likely that caspase-4 and caspase-5 work together during infections to activate the noncanonical inflammasome. A recent study reported that LPS activates an alternative inflammasome pathway only in human monocytes, but not in macrophages and DCs [34]. This human monocyte-specific pathway does not require caspase-4/5, but is dependent on TLR-4-TRIF-RIPK1-FADD-CASP8 signaling, as well as the canonical NLRP3, ASC, and caspase-1 pathway. This alternative NLRP3 inflammasome activation occurs independent of potassium (K+) efflux and pyroptosome formation. An additional unique feature of this alternative inflammasome pathway is that it does not trigger pyroptosis [34]. Determining the relative contribution of the noncanonical inflammasome versus monocyte-specific alternative inflammasome to the overall IL-1β response in humans will be of future interest.
Cytosolic access of LPS
Bacterial vesicle-mediated delivery of LPS into the cytosol
Cytosolic presence of LPS is a requirement for the activation of caspase-11-dependent cell death and IL-1 responses in macrophages, DCs and several other cell types of human and murine origins [13,14,16]. However, free LPS fails to translocate across the plasma membrane of several immune and non-immune cells [16]. LPS of bacteria such as Burkholderia spp. can reach the cytosol when bacteria invade the cytosol [13]. Considering that caspase-11 activation is a feature common to both cytosolic and noncytosolic bacterial infections, a major question was how LPS gains access to the cytosol particularly during infections with noncytosolic and extracellular bacteria [17,18]. Many Gram-negative bacteria that activate caspase-11 have a type III secretion system (T3SS) to deliver bacterial effector proteins into the host cell, and it was a possibility that LPS was also delivered by T3SS. However, bacterial mutants lacking the T3SS are still able to activate the noncanonical inflammasome [17]. Additionally, non-pathogenic strains of E. coli that lack virulence factors robustly activate the noncanonical inflammasome [35]. Therefore, LPS entry into the cytosol during infection with extracellular bacteria is independent of the T3SS.
Gram-negative bacteria secrete LPS-laden outer membrane vesicles (OMVs), which range in size from 20 to 250 nm and are used by bacteria to deliver their contents into host cells [36]. It has been known for several years that OMVs are immunostimulatory, and studies have shown that the peptidoglycan associated with OMVs can activate NOD signaling in immune cells [37]. We recently demonstrated that OMVs function as a vehicle to deliver LPS into the host cytosol (Fig. 1) [35]. Purified OMVs from E. coli were sufficient to deliver LPS into the cytosol and activate capase-11 leading to pyroptosis and IL-1β release. In contrast, the membrane vesicles from Gram-positive bacterium, Listeria monocytogenes, did not induce appreciable cell death or IL-1β release. Remarkably, hypovesiculating bacterial strains that minimally produce OMVs failed to induce robust cell death and IL-1β responses [35]. OMVs enter the macrophages through clathrin-mediated endocytosis, and traffic to early endosomes, from where LPS is able to access the cytosol by an unknown mechanism [35]. It is possible that OMVs could fuse with the endosomal membrane and release LPS into the cytosol. Additional studies are needed to determine the precise mechanism of LPS entry into the cytosol from early endosomes.
Key roles for interferons-inducible proteins in cytosolic translocation of LPS
Host factors play critical roles in facilitating the cytosolic entry of LPS. Type I interferon signaling during Gram-negative bacterial infections upregulates the expression of a family of interferon-inducible proteins called guanylate-binding proteins (GBPs) [38]. GBPs are generally thought to play protective roles in infections with intracellular pathogens [39,40]. It was shown, during infection with L. monocytogenes and Mycobacterium bovis, that GBPs target phagocyte oxidase complexes and antimicrobial peptides to bacteria containing vacuoles and promote intracellular bacterial killing [40]. Most notably, GBPs play important roles in caspase-11 activation. GBPs have been shown to accumulate on phagosomal membranes and facilitate the release of bacterial products into the cytosol for recognition by caspase-11 [38]. GBPs also recruit an additional type I interferon inducible protein celled interferon response gene B10 (IRGB10) to the membranes of cytosolic bacteria. IRGB10 permeabilizes Gram-negative bacterial membranes to liberate bacterial products for recognition by the inflammasome sensors, AIM2 and caspase-11 [41]. It has also been demonstrated that macrophages deficient for these GBPs had impaired caspase-11 activation and diminished pyroptosis even after the transfection of LPS into the cytosol [42]. This suggests that GBPs play additional roles in the noncanonical inflammasome pathway. Overall, these studies highlight the critical contributions of the type I interferon signaling and interferon-inducible proteins to the cytosolic recognition of Gram-negative bacteria.
Effector functions of the noncanonical inflammasome
Pyroptosis
Activation of the noncanonical inflammasome by Gram-negative bacteria results in an inflammatory cell death called pyroptosis [18]. This type of programmed cell death is in general mediated by the inflammatory caspases including caspase-1 and caspase-4/-5/-11 and is characterized by plasma membrane perforation, cell swelling and osmotic lysis, and release of cytosolic contents to the extracellular space [16,18,43]. Pyroptosis mediated by the noncanonical inflammasome is a direct effector function of caspase-11. It was recently shown that active caspase-11 cleaves the effector protein gasdermin D liberating its N-terminal fragment, which executes the lytic cell death [44–46]. Additionally, caspase-4 and caspase-5 can also cleave gasdermin D to initiate pyroptosis in humans [44,45]. Cells that are gasdermin D-deficient are protected from pyroptosis following noncanonical inflammasome activation. Although caspase-1 also cleaves gasdermin D following canonical inflammasome activation, caspase-11 cleavage of gasdermin D and the subsequent pyroptosis in response to cytosolic LPS occur independent of caspase-1. The N-terminal peptide has a preference for binding to phospholipids, specifically phosphatidylinositol phosphates, highly abundant in the inner leaflet of the plasma membrane. As a result, the N-terminal fragment of gasdermin D accumulates on the plasma membrane, undergoes oligomerization, and forms pores that are on average 10–20 nm in diameter leading to osmotic swelling and cell lysis [47–50]. The affinity of gasdermin D for the lipid species on the inner leaflet of the plasma membrane ensures that gasdermin D kills cells only from within and not the bystander cells upon its release into the extracellular milieu. Furthermore, gasdermin D can also bind to cardiolipin found on the bacterial cell wall, allowing for direct bacterial killing [48].
Noncanonical NLRP3 activation
Activation of caspase-11 by Gram-negative bacterial pathogens results in the activation of the NLRP3 inflammasome leading to proteolytic activation of caspase-1, IL-1β, and IL-18 [18]. The link between caspase-11 and NLRP3 activation remained elusive, but the recent studies have shed some light on this aspect. It has been shown that gasdermin D cleavage by caspase-11 is required to promote NLRP3 activation of caspase-1 and IL-1β maturation and secretion during Gram-negative bacterial infection [45]. Gasdermin D-deficient cells are protected from pyroptosis following infection with Gram-negative bacteria and also have impaired IL-1β secretion. Furthermore, gasdermin D-deficient cells have impaired processing of capase-1 and IL-1β in cell lysates following LPS electroporation [43]. This indicates that caspase-11 activation of gasdermin D precedes NLRP3 activation of caspase-1 and IL-1β secretion. Plasma membrane pore formation by gasdermin D oligomers results in K+ efflux [51], which is an established trigger of NLRP3 activation [52,53]. Furthermore, it was recently shown that caspase-11 activation occurs upstream of NLRP3 inflammasome formation and results in a drop in intracellular K+ levels [54]. Therefore, it is most likely that gasdermin D activated by caspase-11, triggers NLRP3 inflammasome activation by inducing K+ efflux via the plasma membrane pores.
Unconventional protein secretion
Inflammasome activation is accompanied by unconventional protein secretion into the extracellular space [55]. Normally, proteins to be secreted follow the traditional secretory pathway from the endoplasmic reticulum (ER) to the Golgi apparatus to the plasma membrane. This pathway requires that proteins have a signal peptide sequence that directs them to the ER [56]. However, there are repertoires of secreted proteins that lack this signal sequence, and their route of secretion is still not well characterized [56]. These proteins follow an “unconventional secretory route”, and it is thought that the proteins released following inflammasome activation are secreted through the pores formed in the plasma membrane by gasdermin D [44]. Additionally, secretory lysosomes, multivesicular bodies, exosomes, microvesicles, and secretory autophagy also contribute to the release of leaderless proteins to the extracellular space [57]. Several of these secreted proteins such as IL-1α and HMBG1 are believed to act as alarmins or DAMPs, and regulate the inflammatory responses downstream of inflammasome activation [3,18]. The unconventional release of these endogenous alarmins following cytosolic LPS sensing is mediated by caspase-11 without requiring caspase-1 [18]. However, the full spectrum of proteins secreted in a caspase-11 dependent manner has yet to be determined.
The protein secretion from human macrophages following the activation of caspase-4 and caspase-5, human equivalents of murine caspase-11, has been recently characterized [58]. Lorey et al., described that caspase-4 and caspase-5 triggers the release of proteins from human macrophages via both extracellular vesicle- and non-vesicles-based mechanisms [58]. Expectedly, several danger signal proteins such as S100A8 and prothymosin-α were identified in the secretome. Some of these proteins including S100A8 and prothymosin-α have been shown to activate TLR-4 and thus, may enhance host’s inflammatory reaction. Furthermore, a set of translational machinery associated proteins including ribosomal proteins were secreted as well. The overall contribution of unconventionally secreted proteins to the host defense and sepsis pathogenesis remains to be determined.
Role of the noncanonical inflammasome in antimicrobial defense
Caspase-11 plays a protective role during infections with bacteria that invade the cytosol; caspase-11−/− mice are highly susceptible to infections with cytosolic bacteria B. thailandensis and B. pseudomallei [59]. Similarly, caspase-11-deficient mice were also susceptible to mutants of S. typhimurium and Legionella pneumophila that ruptures the pathogen-containing vacuoles and invade the cytosol. Another study showed that the noncanonical inflammasome is also functional in intestinal epithelial cells, where caspase-11 is required for IL-18 secretion and pathogen clearance during infection with S. typhimurium [60]. Here, caspase-11 was shown to be responsible for the shedding of infected epithelial cells, which provided an explanation for the higher bacterial burdens observed in caspase-11-deficient mice. Furthermore, caspase-11 has been shown to restrict the replication of L. pneumophila in murine lungs [61]. Recently, Choi et al., reported that caspase-11 is also required for effective bacterial clearance in a mouse model of uropathogenic Escherichia coli (UPEC)-induced cystitis [62]. Mechanistically, caspase-11-dependent secretion of IL-1β by bladder epithelial cells (BECs) was required for the recruitment of mast cells to the bladder epithelium, where they released chymase-loaded granules. Upon endocytic uptake by infected BECs, these granules induced caspase-1-dependent lysis and consequent shedding of BECs.
It has been thought for a while that the induction of pyroptosis exposes intracellular bacteria to the extracellular environment for subsequent clearance by secondary phagocytes, such as neutrophils. However, the precise steps governing this process remained unclear. Jorgensen et al., recently provided mechanistic insights into this phenomenon and demonstrated that intracellular bacteria are in fact retained within the pyroptotic cell corpses, which were termed as pore-induced intracellular traps (PITs) [63]. Interestingly, these PITs are believed to display potential find-me and eat-me signals that are recognized by complement and scavenger receptors on neutrophils for effective recruitment to and phagocytosis of PITs and the associated bacteria [63]. Furthermore, a combination of IL-1β, IL-18 and eicosanoids are also required for the recruitment of neutrophils to PITs [64]. Importantly, pyroptotic cell corpses injected into mice were also phagocytosed by neutrophils and macrophages in vivo.
A recent report has shown a new effector function for caspase-1 and caspase-11 in controlling bacterial growth in the cytosol of cells independently of pyroptosis [65]. Mouse embryonic fibroblasts (MEFs) and bone marrow-derived macrophages (BMDMs) lacking caspase-11 were more permissive for intracellular Salmonella growth relative to their caspase-11 sufficient counterparts. Interestingly, the ability of caspase-11 to inhibit Salmonella growth in the cytosol occurred prior to pyroptosis and was not dependent on gasdermin D and IL-1β. However, the enzymatic activity of caspase-11 was required for this process. These results indicate that the inflammatory caspases likely have additional effector mechanisms besides their typical pyroptotic functions to control intracellular infections. Further studies are needed to determine the mechanism by which caspase-11 controls cytosolic bacterial growth, and it also remains to be determined if caspase-11 functions similarly in other intracellular bacterial infections. Overall, these studies demonstrate that caspase-11 restricts bacterial replication in cell-intrinsic and -extrinsic fashions and that caspase-11 is essential for the antimicrobial defense.
Role of the noncanonical inflammasome in sepsis
While the studies described above clearly demonstrate the protective role of caspase-11 during Gram-negative bacterial infections, the pathological role of caspase-11 in vivo is also well documented. Excessive cytosolic localization of LPS leading to hyperactivation of caspase-11 is a major mechanism responsible for endotoxic shock in mice, and caspase-11−/− mice are resistant to lethal doses of LPS [13,14,18]. The mechanism(s) by which caspase-11 activation leads to lethality in endotoxemia is just beginning to be understood. It has been shown that mice lacking IL-1R and IL-18R are just as susceptible as wild type mice to endotoxic shock [18,66]. On the other hand, caspase-11-deficient mice and gasdermin D-deficient mice are protected from endotoxic shock. This argues that these IL-1 cytokines are less likely to play a major role in sepsis pathogenesis. This also implies that gasdermin D-mediated pyroptosis is more likely to be a critical factor in sepsis lethality. However, the precise molecular mechanism by which caspase-11 contributes to lethality in endotoxic shock is yet to be determined. Caspase-11 is considered to contribute to sepsis pathology by causing tissue damage and ultimately organ failure. Furthermore, the leaderless proteins released from cells undergoing pyroptosis can act as endogenous alarmins and contribute to the inflammation associated with sepsis. Therefore, characterizing the proteins secreted in a caspase-11 dependent manner will have important implications for understanding how the noncanonical inflammasome contributes to Gram-negative bacterial sepsis. Taken together, the noncanonical inflammasome pathway appears to function as a double-edged sword during Gram-negative bacterial infections; while it protects the host against certain cytosolic bacterial infections, its uncontrolled activation can be detrimental to the host.
Role of the noncanonical inflammasome in intestinal homeostasis
Caspase-11 is also emerging as an important player in regulating intestinal homeostasis. Multiple recent studies demonstrated that caspase-11 confers protection against dextran sodium sulfate (DSS)-induced colitis [67–69]. Under the conditions of DSS-induced damage to the epithelial layer, caspase-11 undergoes activation presumably in response to LPS from the microbiome and mediates intestinal barrier-protective effects. Relative to wild-type controls, mice lacking caspase-11, upon oral exposure to DSS, develop increased colonic inflammation and pathology and eventually succumb to death. Consistent with its expression and function in myeloid cells and intestinal epithelial cells, caspase-11’s role in both hematopoietic and nonhematopoietic compartments contribute to the protection against DSS colitis [60,68,69]. While the homeostatic function of caspase-11 in DSS-elicited colitis is evident from these studies, the underlying mechanisms are less clear. Specifically, there is some discrepancy in the requirement of IL-1β and IL-18 for the protective function. Demon et al., found that caspase-11-deficient mice produced normal or even increased levels of these cytokines depending on the DSS dose [68]. However, other studies reported that the protective role of caspase-11 was dependent, in part, on IL-1β and IL-18 as these cytokines were significantly reduced in caspase-11-deficient mice and their reconstitution attenuated disease severity [67,69]. This discrepancy might have arisen due to several technical and/or environmental variables. Nonetheless, these studies collectively establish the contribution of caspase-11 to epithelial cell repair, intestinal barrier integrity, and intestinal homeostasis.
Conclusions and Future directions
The noncanonical inflammasome has now been established as an integral part of intracellular innate surveillance mechanisms. The latest studies have provided new insights into the noncanonical inflammasome with regard to the cytosolic translocation of LPS and cell death induction (Fig. 1). Bacterial LPS that gains cytosolic access owing to the cytosolic invasion of bacteria or OMVs secreted by bacteria is sensed by caspase-11 triggering the activation of gasdermin D. The pore-forming ability of gasdermin D causes pyroptosis and NLRP3-dependent activation of IL-1β and IL-18. While the optimal activation of caspase-11 provides protection against bacterial infections, its excessive activation has the potential to cause extensive cell death and tissue damage of fatal consequence. While a substantial progress has been made in this area in the recent years, we still have a number of outstanding questions. The precise molecular mechanisms that govern the cytosolic translocation of LPS from the phagosomal compartments are not yet clear. Considering the broad expression of noncanonical inflammasome components, particularly caspase-11 and gasdermin D, it would be of interest to determine the cell type specific function of the noncanonical inflammasome in sepsis. Characterizing the cytosolic LPS sensing pathway in humans and determining the relative roles of caspase-4 and caspase-5 further could be helpful in treating septic patients.
Highlights.
Noncanonical inflammasome relies on sensing the cytosolic presence of lipopolysaccharide (LPS) of Gram-negative bacteria via inflammatory caspases such as caspase-4, -5, and -11.
This Review discusses the recent findings related to the mechanism of activation of the noncanonical inflammasome and its roles in anti-microbial defense.
Acknowledgments
We thank Sivapriya Kailasan Vanaja for critical review of the manuscript. Research in V.R. laboratory is supported by the Charles Hood Child Health Research Award by the Charles H. Hood Foundation and the NIH (AI119015).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nature Immunology. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 2.Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10:417–426. doi: 10.1016/s1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 3.Emerging inflammasome effector mechanisms. Nature Reviews Immunology. 2011;11:213–220. doi: 10.1038/nri2936. [DOI] [PubMed] [Google Scholar]
- 4.Vanaja SK, Rathinam VAK, Fitzgerald KA. Mechanisms of inflammasome activation: recent advances and novel insights. 2015 doi: 10.1016/j.tcb.2014.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–1361. doi: 10.1038/nature08938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. 2009;458:514–518. doi: 10.1038/nature07725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rathinam VAK, Jiang Z, Waggoner SN, Sharma S, Cole LE, Waggoner L, et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nature Immunology. 2010;11:395–402. doi: 10.1038/ni.1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhao Y, Yang J, Shi J, Gong YN, Lu Q, Xu H, et al. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature. 2011;477:596–600. doi: 10.1038/nature10510. [DOI] [PubMed] [Google Scholar]
- 9.Xu H, Yang J, Gao W, Li L, Li P, Zhang L, et al. Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature. 2014;513:237–241. doi: 10.1038/nature13449. [DOI] [PubMed] [Google Scholar]
- 10.Kofoed EM, Vance RE. Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature. 2011;477:592–595. doi: 10.1038/nature10394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wolf AJ, Reyes CN, Liang W, Becker C, Shimada K, Wheeler ML, et al. Hexokinase Is an Innate Immune Receptor for the Detection of Bacterial Peptidoglycan. Cell. n.d;0 doi: 10.1016/j.cell.2016.05.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nature Reviews Immunology. 2013;13:397–411. doi: 10.1038/nri3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA. Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science. 2013;341:1250–1253. doi: 10.1126/science.1240988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, et al. Noncanonical Inflammasome Activation by Intracellular LPS Independent of TLR4. Science. 2013;341:1246–1249. doi: 10.1126/science.1240248. [DOI] [PubMed] [Google Scholar]
- 15.Beutler B, Crozat K, Koziol JA, Georgel P. Genetic dissection of innate immunity to infection: the mouse cytomegalovirus model. 2005;17:36–43. doi: 10.1016/j.coi.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 16.Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. 2014;514:187–192. doi: 10.1038/nature13683. [DOI] [PubMed] [Google Scholar]
- 17.Rathinam VAK, Vanaja SK, Waggoner L, Sokolovska A, Becker C, Stuart LM, et al. TRIF Licenses Caspase-11-Dependent NLRP3 Inflammasome Activation by Gram-Negative Bacteria. Cell. 2012;150:606–619. doi: 10.1016/j.cell.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, et al. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479:117–121. doi: 10.1038/nature10558. [DOI] [PubMed] [Google Scholar]
- 19.Zanoni I, Tan Y, Di Gioia M, Broggi A, Ruan J, Shi J, et al. An endogenous caspase-11 ligand elicits interleukin-1 release from living dendritic cells. Science. 2016;352:1232–1236. doi: 10.1126/science.aaf3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang S, Miura M, Jung YK, Zhu H, Gagliardini V, Shi L, et al. Identification and characterization of Ich-3, a member of the interleukin-1beta converting enzyme (ICE)/Ced-3 family and an upstream regulator of ICE. J Biol Chem. 1996;271:20580–20587. doi: 10.1074/jbc.271.34.20580. [DOI] [PubMed] [Google Scholar]
- 21.Wang S, Miura M, Jung YK, Zhu H, Li E, Yuan J. Murine Caspase-11, an ICE-Interacting Protease, Is Essential for the Activation of ICE. Cell. 1998;92:501–509. doi: 10.1016/S0092-8674(00)80943-5. [DOI] [PubMed] [Google Scholar]
- 22.Schauvliege R, Vanrobaeys J, Schotte P, Beyaert R. Caspase-11 gene expression in response to lipopolysaccharide and interferon-gamma requires nuclear factor-kappa B and signal transducer and activator of transcription (STAT) 1. J Biol Chem. 2002;277:41624–41630. doi: 10.1074/jbc.M207852200. [DOI] [PubMed] [Google Scholar]
- 23.Broz P, Ruby T, Belhocine K, Bouley DM, Kayagaki N. Caspase-11 increases susceptibility to Salmonella infection in the absence of caspase-1. Nature. 2012;490:288–291. doi: 10.1038/nature11419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gurung P, Malireddi RKS, Anand PK, Demon D, Vande Walle L, Liu Z, et al. Toll or interleukin-1 receptor (TIR) domain-containing adaptor inducing interferon-β (TRIF)-mediated caspase-11 protease production integrates Toll-like receptor 4 (TLR4) protein- and Nlrp3 inflammasome-mediated host defense against enteropathogens. J Biol Chem. 2012;287:34474–34483. doi: 10.1074/jbc.M112.401406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Casson CN, Copenhaver AM, Zwack EE, Nguyen HT, Strowig T, Javdan B, et al. Caspase-11 activation in response to bacterial secretion systems that access the host cytosol. PLoS Pathog. 2013;9:e1003400. doi: 10.1371/journal.ppat.1003400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aachoui Y, Kajiwara Y, Leaf IA, Mao D, Ting JPY, Coers J, et al. Canonical Inflammasomes Drive IFN-γ to Prime Caspase-11 in Defense against a Cytosol-Invasive Bacterium. Cell Host & Microbe. 2015;18:320–332. doi: 10.1016/j.chom.2015.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Napier BA, Brubaker SW, Sweeney TE, Monette P, Rothmeier GH, Gertsvolf NA, et al. Complement pathway amplifies caspase-11-dependent cell death and endotoxin-induced sepsis severity. Journal of Experimental Medicine. 2016;213:2365–2382. doi: 10.1084/jem.20160027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lupfer CR, Anand PK, Liu Z, Stokes KL, Vogel P, Lamkanfi M, et al. Reactive Oxygen Species Regulate Caspase-11 Expression and Activation of the Non-canonical NLRP3 Inflammasome during. Enteric Pathogen Infection. 2014;10:e1004410. doi: 10.1371/journal.ppat.1004410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kersse K, Vanden Berghe T, Lamkanfi M, Vandenabeele P. A phylogenetic and functional overview of inflammatory caspases and caspase-1-related CARD-only proteins. Biochem Soc Trans. 2007;35:1508–1511. doi: 10.1042/BST0351508. [DOI] [PubMed] [Google Scholar]
- 30.Bian ZM, Elner SG, Khanna H, Murga-Zamalloa CA, Patil S, Elner VM. Expression and Functional Roles of Caspase-5 in Inflammatory Responses of Human Retinal Pigment Epithelial Cells. Investigative Ophthalmology & Visual Science. 2011;52:8646–8656. doi: 10.1167/iovs.11-7570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Casson CN, Yu J, Reyes VM, Taschuk FO, Yadav A, Copenhaver AM, et al. Human caspase-4 mediates noncanonical inflammasome activation against gram-negative bacterial pathogens. Proceedings of the National Academy of Sciences. 2015;112:6688–6693. doi: 10.1073/pnas.1421699112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schmid-Burgk JL, Gaidt MM, Schmidt T, Ebert TS, Bartok E, Hornung V. Caspase-4 mediates non-canonical activation of the NLRP3 inflammasome in human myeloid cells. European Journal of Immunology. 2015;45:2911–2917. doi: 10.1002/eji.201545523. [DOI] [PubMed] [Google Scholar]
- 33.Baker PJ, Boucher D, Bierschenk D, Tebartz C, Whitney PG, D’Silva DB, et al. NLRP3 inflammasome activation downstream of cytoplasmic LPS recognition by both caspase-4 and caspase-5. European Journal of Immunology. 2015;45:2918–2926. doi: 10.1002/eji.201545655. [DOI] [PubMed] [Google Scholar]
- 34.Gaidt MM, Ebert TS, Chauhan D, Schmidt T, Schmid-Burgk JL, Rapino F, et al. Human Monocytes Engage an Alternative Inflammasome Pathway. Immunity. 2016;44:833–846. doi: 10.1016/j.immuni.2016.01.012. [DOI] [PubMed] [Google Scholar]
- 35.Vanaja SK, Russo AJ, Behl B, Banerjee I, Yankova M, Deshmukh SD, et al. Bacterial Outer Membrane Vesicles Mediate Cytosolic Localization of LPS and Caspase-11 Activation. Cell. 2016;165:1106–1119. doi: 10.1016/j.cell.2016.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kulp A, Kuehn MJ. Biological functions and biogenesis of secreted bacterial outer membrane vesicles. Annu Rev Microbiol. 2010;64:163–184. doi: 10.1146/annurev.micro.091208.073413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Irving AT, Mimuro H, Kufer TA, Lo C, Wheeler R, Turner LJ, et al. The Immune Receptor NOD1 and Kinase RIP2 Interact with Bacterial Peptidoglycan on Early Endosomes to Promote Autophagy and Inflammatory Signaling. Cell Host & Microbe. 2014;15:623–635. doi: 10.1016/j.chom.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 38.Meunier E, Dick MS, Dreier RF, Schürmann N, Kenzelmann Broz D, Warming S, et al. Caspase-11 activation requires lysis of pathogen-containing vacuoles by IFN-induced GTPases. Nature. 2014;509:366–370. doi: 10.1038/nature13157. [DOI] [PubMed] [Google Scholar]
- 39.Shenoy AR, Wellington DA, Kumar P, Kassa H, Booth CJ, Cresswell P, et al. GBP5 Promotes NLRP3 Inflammasome Assembly and Immunity in Mammals. Science. 2012;336:481–485. doi: 10.1126/science.1217141. [DOI] [PubMed] [Google Scholar]
- 40.Kim BH, Shenoy AR, Kumar P, Das R, Tiwari S, MacMicking JD. A family of IFN-γinducible 65-kD GTPases protects against bacterial infection. Science. 2011;332:717–721. doi: 10.1126/science.1201711. [DOI] [PubMed] [Google Scholar]
- 41.Man SM, Karki R, Sasai M, Place DE, Kesavardhana S, Temirov J, et al. IRGB10 Liberates Bacterial Ligands for Sensing by the AIM2 and Caspase-11-NLRP3 Inflammasomes. Cell. 2016;167:382–396.e17. doi: 10.1016/j.cell.2016.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pilla DM, Hagar JA, Haldar AK, Mason AK, Degrandi D, Pfeffer K, et al. Guanylate binding proteins promote caspase-11-dependent pyroptosis in response to cytoplasmic LPS. Proceedings of the National Academy of Sciences. 2014;111:6046–6051. doi: 10.1073/pnas.1321700111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. 2009;7:99–109. doi: 10.1038/nrmicro2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660–665. doi: 10.1038/nature15514. [DOI] [PubMed] [Google Scholar]
- 45.Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signaling. Nature. 2015;526:666–671. doi: 10.1038/nature15541. [DOI] [PubMed] [Google Scholar]
- 46.He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015;25:1285–1298. doi: 10.1038/cr.2015.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 2016 doi: 10.1038/nature18590. [DOI] [PubMed] [Google Scholar]
- 48.Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;535:153–158. doi: 10.1038/nature18629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Aglietti RA, Estevez A, Gupta A, Ramirez MG, Liu PS, Kayagaki N, et al. GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proceedings of the National Academy of Sciences. 2016;113:7858–7863. doi: 10.1073/pnas.1607769113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sborgi L, Rühl S, Mulvihill E, Pipercevic J, Heilig R, Stahlberg H, et al. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. Embo J. 2016:e201694696. doi: 10.15252/embj.201694696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gutierrez KD, Davis MA, Daniels BP, Olsen TM, Ralli-Jain P, Tait SWG, et al. MLKL Activation Triggers NLRP3-Mediated Processing and Release of IL-1β Independently of Gasdermin-D. J Immunol. 2017;198:2156–2164. doi: 10.4049/jimmunol.1601757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Muñoz-Planillo R, Kuffa P, Martínez-Colón G, Smith BL, Rajendiran TM, Núñez G. K+ Efflux Is the Common Trigger of NLRP3 Inflammasome Activation by Bacterial Toxins and Particulate Matter. Immunity. 2013;38:1142–1153. doi: 10.1016/j.immuni.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pétrilli V, Papin S, Dostert C, Mayor A, Martinon F, Tschopp J. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ. 2007;14:1583–1589. doi: 10.1038/sj.cdd.4402195. [DOI] [PubMed] [Google Scholar]
- 54.Rühl S, Broz P. Caspase-11 activates a canonical NLRP3 inflammasome by promoting K(+) efflux. European Journal of Immunology. 2015;45:2927–2936. doi: 10.1002/eji.201545772. [DOI] [PubMed] [Google Scholar]
- 55.Keller M, Rüegg A, Werner S, Beer HD. Active Caspase-1 Is a Regulator of Unconventional Protein Secretion. Cell. 2008;132:818–831. doi: 10.1016/j.cell.2007.12.040. [DOI] [PubMed] [Google Scholar]
- 56.Nickel W, Rabouille C. Mechanisms of regulated unconventional protein secretion. Nat Rev Mol Cell Biol. 2009;10:148–155. doi: 10.1038/nrm2617. [DOI] [PubMed] [Google Scholar]
- 57.Monteleone M, Stow JL, Schroder K. Mechanisms of unconventional secretion of IL-1 family cytokines. Cytokine. 2015;74:213–218. doi: 10.1016/j.cyto.2015.03.022. [DOI] [PubMed] [Google Scholar]
- 58.Lorey MB, Rossi K, Eklund KK, Nyman TA, Matikainen S. Global Characterization of Protein Secretion from Human Macrophages Following Non-canonical Caspase-4/5 Inflammasome Activation. Mol Cell Proteomics. 2017;16:S187–S199. doi: 10.1074/mcp.M116.064840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Aachoui Y, Leaf IA, Hagar JA, Fontana MF, Campos CG, Zak DE, et al. Caspase-11 protects against bacteria that escape the vacuole. Science. 2013;339:975–978. doi: 10.1126/science.1230751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Knodler LA, Crowley SM, Sham HP, Yang H, Wrande M, Ma C, et al. Noncanonical inflammasome activation of caspase-4/caspase-11 mediates epithelial defenses against enteric bacterial pathogens. Cell Host & Microbe. 2014;16:249–256. doi: 10.1016/j.chom.2014.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Akhter A, Caution K, Abu Khweek A, Tazi M, Abdulrahman BA, Abdelaziz DHA, et al. Caspase-11 promotes the fusion of phagosomes harboring pathogenic bacteria with lysosomes by modulating actin polymerization. Immunity. 2012;37:35–47. doi: 10.1016/j.immuni.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Choi HW, Bowen SE, Miao Y, Chan CY, Miao EA, Abrink M, et al. Loss of Bladder Epithelium Induced by Cytolytic Mast Cell Granules. Immunity. 2016;45:1258–1269. doi: 10.1016/j.immuni.2016.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jorgensen I, Zhang Y, Krantz BA, Miao EA. Pyroptosis triggers pore-induced intracellular traps (PITs) that capture bacteria and lead to their clearance by efferocytosis. Journal of Experimental Medicine. 2016;213:2113–2128. doi: 10.1084/jem.20151613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jorgensen I, Lopez JP, Laufer SA, Miao EA. IL-1β, IL-18, and eicosanoids promote neutrophil recruitment to pore-induced intracellular traps following pyroptosis. European Journal of Immunology. 2016;46:2761–2766. doi: 10.1002/eji.201646647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Thurston TLM, Matthews SA, Jennings E, Alix E, Shao F, Shenoy AR, et al. Growth inhibition of cytosolic Salmonella by caspase-1 and caspase-11 precedes host cell death. Nature Communications. 2016;7:13292. doi: 10.1038/ncomms13292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sarkar A, Hall MW, Exline M, Hart J, Knatz N, Gatson NT, et al. Caspase-1 Regulates Escherichia coli Sepsis and Splenic B Cell Apoptosis Independently of Interleukin-1β and Interleukin-18. Am J Respir Crit Care Med. 2012;174:1003–1010. doi: 10.1164/rccm.200604-546OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Oficjalska K, Raverdeau M, Aviello G, Wade SC, Hickey A, Sheehan KM, et al. Protective role for caspase-11 during acute experimental murine colitis. The Journal of Immunology. 2015;194:1252–1260. doi: 10.4049/jimmunol.1400501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Demon D, Kuchmiy A, Fossoul A, Zhu Q, Kanneganti TD, Lamkanfi M. Caspase-11 is expressed in the colonic mucosa and protects against dextran sodium sulfate-induced colitis. 2014 doi: 10.1038/mi.2014.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Williams TM, Leeth RA, Rothschild DE, McDaniel DK, Coutermarsh-Ott SL, Simmons AE, et al. Caspase-11 attenuates gastrointestinal inflammation and experimental colitis pathogenesis. Am J Physiol Gastrointest Liver Physiol. 2015;308:G139–50. doi: 10.1152/ajpgi.00234.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]