

ABSTRACT
Relapsed T-cell malignancies have poor outcomes when treated with chemotherapy, but survival after allogeneic bone marrow transplantation (BMT) approaches 50%. A limitation to BMT is the difficulty of achieving remission prior to transplant. Chimeric antigen receptor (CAR) T-cell therapy has shown successes in B-cell malignancies. This approach is difficult to adapt for the treatment of T-cell disease due to lack of a T-lymphoblast specific antigen and the fratricide of CAR T cells that occurs with T-cell antigen targeting. To circumvent this problem two approaches were investigated. First, a natural killer (NK) cell line, which does not express CD5, was used for CAR expression. Second, CRISPR-Cas9 genome editing technology was used to knockout CD5 expression in CD5-positive Jurkat T cells and in primary T cells, allowing for the use of CD5-negative T cells for CAR expression. Two structurally distinct anti-CD5 sequences were also tested, i) a traditional immunoglobulin-based single chain variable fragment (scFv) and ii) a lamprey-derived variable lymphocyte receptor (VLR), which we previously showed can be used for CAR-based recognition. Our results show i) both CARs yield comparable T-cell activation and NK cell-based cytotoxicity when targeting CD5-positive cells, ii) CD5-edited CAR-modified Jurkat T cells have reduced self-activation compared to that of CD5-positive CAR-modified T cells, iii) CD5-edited CAR-modified Jurkat T cells have increased activation in the presence of CD5-positive target cells compared to that of CD5-positive CAR-modified T cells, and iv) although modest effects were seen, a mouse model using the CAR-expressing NK cell line showed the scFv-CAR was superior to the VLR-CAR in delaying disease progression.
KEYWORDS: anti-cds scFv, chimeric antigen receptor, immunotheraphy, lamprey variable lymphocyte receptor, T-cell lenkemia
Introduction
Chimeric antigen receptor (CAR) T-cell therapy is among the most promising anti-cancer therapeutics, with great success achieved in relapsed/refractory B-cell malignancies.1-4 This approach for the treatment of T-cell malignancies is complicated by the lack of a T-lymphoblast specific surface antigen. As a result, CAR T cells generated to target malignant T cells are at risk of fratricide and, therefore, their activation against targeted cancer T cells is compromised.5 We explore two alternative methods that can be used to apply this innovative therapy to T-cell disease; the first is through the use of a natural killer (NK) cell line as the CAR-expressing effector cell, and the second is by knocking-out surface expression of the target antigen in CAR T cells using CRISPR-Cas9 genome editing.
CD5 is a pan T-cell marker that is commonly over-expressed in most T-cell malignancies.6,7 Expression of CD5 by normal cells is restricted to thymocytes, peripheral T cells, and a minor subpopulation of B lymphocytes, called B-1 cells.8-10 Additionally, CD5 is a negative regulator of T-cell receptor (TCR) signaling and has a role in protecting against autoimmunity.11-13 As a result, we have chosen CD5 as the target antigen for our CARs. Clinical trials have previously studied CD5 as the tumor target antigen using immunotoxin-conjugated CD5 monoclonal antibodies, with responses documented in patients with cutaneous T-cell lymphoma and T-ALL.14,15 A pre-clinical study using anti-CD5 CAR T cells had favorable results, but did demonstrate some evidence of fratricide among the engineered CAR T cells due to inherent CD5 expression.5 CD5-negative cells, such as NK cells or CD5-CRISPR-Cas9-edited T cells, may be a more suited option for CAR-modified effector cells for the targeting of T-cell malignancies.16 The CRISPR system17-20 has been adapted to function in eukaryotes and can be used to induce genetic modifications, such as highly specific and permanent gene knockout.21-23 We hypothesized that CD5-CRISPR-edited T cells would have decreased self-activation when expressing a CD5-CAR compared to that of CD5-positive T cells.
We have previously shown that a variable lymphocyte receptor (VLR) can be used for CAR-mediated antigen recognition instead of the more traditional immunoglobulin-based single chain variable fragment (scFv).24 VLRs represent the functional unit of the adaptive immune system in jawless vertebrates (lamprey and hagfish), and are analogous, but not homologous to immunoglobulins.25,26 VLRs have a fundamentally different structure and geometry than immunoglobulin-based antibodies, while still demonstrating high degrees of specificity and avidity. Importantly for the production of CAR-based therapeutics, they exist naturally as single chain crescent-shaped proteins with their variable region consisting of multiple assembled repeating sequences, termed leucine rich repeats (LRRs).25-28 VLRs function as avidity-based antibodies with the individual monomeric VLR units exhibiting lower affinity towards their target compared to their multimeric form.29,30 The unique single chain structure of VLRs allows for rapid insertion into a CAR scaffold, compared to the corresponding use of an immunoglobulin, in which the variable heavy and light chains need further engineering for adapting to CAR technologies. We hypothesized that a CD5-directed VLR-CAR would have equal or superior efficacy compared to a corresponding scFv-CAR.
We tested both the CD5-VLR-CAR and CD5-scFv-CAR in NK-92 cells, non-edited and CD5-edited Jurkat T cells, and non-edited and CD5-edited primary T cells. Our in vitro studies demonstrate that both CD5-CARs have comparable outcomes in terms of T-cell activation and NK-92 cell mediated CAR cytotoxicity, and that CD5-edited CD5-CAR T cells have increased CD5-CAR expression and exhibit decreased self-activation while maintaining their ability to activate in the presence of CD5-positive target cells. However, in vivo the scFv-CAR had an advantage over the VLR-CAR when tested in a T-cell leukemia mouse model using NK-92 cells.
Results
Construction of CD5-directed CARs
The CD5-VLR-CAR (previously described).24 was generated using a VLR protein sequence shown to be specific for the CD5 antigen.29 The sequence for the CD5-scFv was generated using a published humanized murine immunoglobulin protein sequence,31 and the cDNA sequence designed to express the scFv was codon optimized for human cell expression. The C-terminus of VH was joined with the N-terminus of VL using a 15 bp linker encoding a glycine and serine pentapeptide repeat (G4 S)332 The entire CD5-scFv sequence totaled 720 bp compared to the shorter 570 bp CD5-VLR sequence. The two CD5 sequences were cloned into the CAR cassette, which is a second generation CAR composed of an N-terminal IL-2 signal peptide followed by the CD5-VLR or -scFV antigen binding domain, the transmembrane and intracellular domains of CD28, and the intracellular signaling domain of CD3ζ (Figure 1A). A bicistronic vector co-expressing eGFP and the CD5-CAR via a self-cleaving 2 A peptide sequence (P2 A) was used to enable selection of positively transduced cells by flow sorting (Figure 1B).
Figure 1.
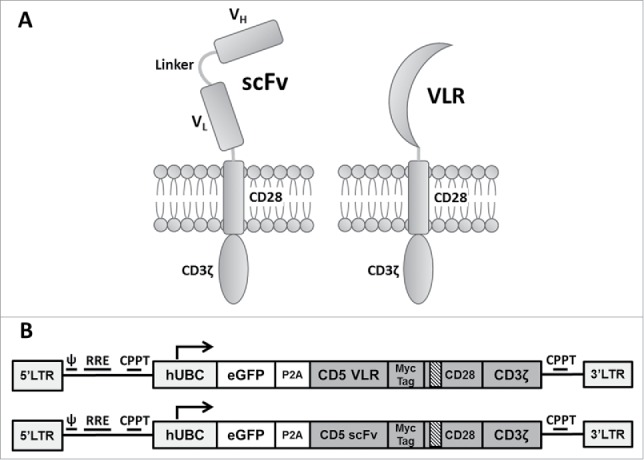
Schematic of CAR structures containing the CD5-directed variable lymphocyte receptor (VLR) or single chain variable fragment (scFv). (A) Second generation CAR structures with CD28 containing a scFv (left) or VLR (right) as the antigen recognition domain. (B) Schematics of the bicistronic transgene sequences used for expressing enhanced green fluorescent protein (eGFP) and the CD5-CARs using a P2 A sequence. It includes a 5′ long terminal repeat (LTR), human ubiquitin C promoter (hUBC), eGFP sequence, P2 A sequence, an interleukin-2 signal peptide (IL-2 SP), the CD5-VLR (top) or CD5-scFv (bottom), a myc epitope tag, the CD28 region, the CD3ζ intracellular domain and a 3′ LTR.
CD5-CAR NK-cell mediated cytotoxicity
To demonstrate CAR-directed cytotoxicity, the well-characterized cytotoxic human NK cell line, NK-92, was used, which is an interleukin-2 (IL-2) dependent immortalized cell line that has maintained its cytotoxic capabilities.33 NK-92 cells do not display CD5 on their surface, and this allows for expression of the CD5-CAR without self-activation and fratricidal killing of transduced cells. Generation of the CD5-VLR-CAR-expressing NK-92 cell line has been previously described.24 To generate CD5-scFv-CAR expressing NK-92 cells, they were transduced with the bicistronic construct expressing eGFP and the CD5-scFv-CAR. As expected, poor transduction efficiency (< 5%) was observed after the initial lentiviral vector transduction. As with the CD5-VLR-CAR-expressing NK-92 cells, flow sorting was used to generate a CD5-scFv-CAR expressing NK-92 cell line using eGFP as a selection marker for positively transduced cells. After two rounds of flow sorting for eGFP, a CD5-scFv-CAR expressing NK-92 population was generated with 99% eGFP expression (Figure 2A). qPCR analysis demonstrated an average of 1.0 transduced gene copy/cell in the sorted and expanded cells. To confirm CD5-CAR expression in the flow sorted NK-92 cell lines, western blot analysis was performed using a CD3ζ antibody. Bands of 48 and 55 kDa were visible corresponding to the CD5-VLR-CAR and CD5-scFv-CAR proteins respectively (Figure 2B). To assess their cytotoxic potential, CD5-CAR expressing NK-92 effector (E) cells were cultured with CD5-positive Jurkat and MOLT-4 T-cell leukemia target (T) cells at varying E:T ratios. The CD5-negative B-cell leukemia cell line, 697, was used as a negative control. The target cells were pre-labeled with the membrane dye PKH26, which allowed for easy distinction from the non-labeled effector cells using flow cytometry. Cytotoxicity was measured via uptake of 7-AAD, a marker for cell death, into target cells.34 A significant increase in cytotoxicity was observed with the CD5-CAR expressing NK-92 cells compared to naïve NK-92 cells, even at the lowest E:T ratios (p < 0.01 for all cell groups) (Figure 2C and Figure 2D). Greater cytotoxicity was observed in the CD5-scFv-CAR group at the higher E:T ratios, however, the difference in cytotoxicity was not significant between the VLR-CAR and scFv-CAR at the lower 1:1 E:T ratio. No increase in cytotoxicity was seen when the CD5-CAR NK-92 cells were tested against the CD5-negative 697 cell line (Figure 2E).
Figure 2.
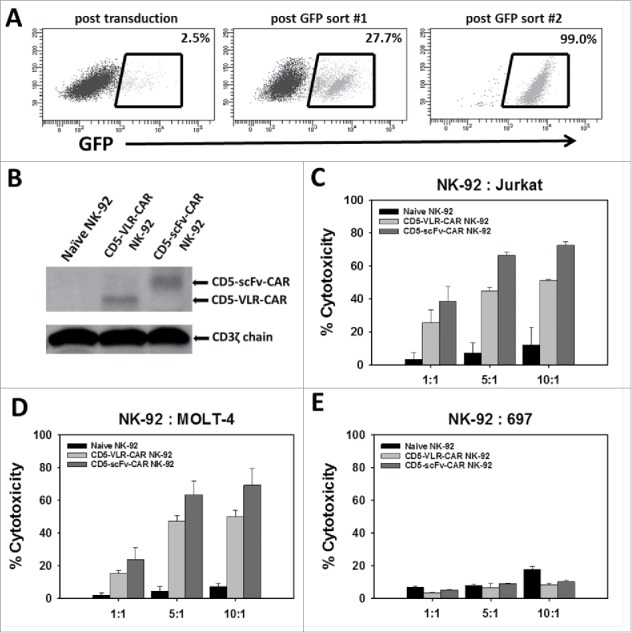
NK-92 cell mediated cytotoxicity against a CD5-positive T-ALL cell line using CD5-CARs. (A) NK-92 cells were transduced with the eGFP-P2 A-CD5-scFv-CAR lentiviral vector and sorted for GFP expressing cells. After two rounds of sorting an enriched population of CAR-expressing NK-92 cells was generated with 99% eGFP expression. (B) Western blot using anti-CD3ζ antibody on whole cell lysates of NK-92 cells shows the presence of CD5-VLR-CAR and CD5-scFv-CAR protein in the sorted and expanded cells. (C & D) Both CD5-CAR expressing NK-92 cells were mixed with CD5-positive target cells, Jurkat and MOLT-4, at various Effector: Target ratios and the percent cytotoxicity was measured by flow cytometry. CD5-CAR modified NK-92 cells showed a significantly greater cytotoxicity (p < 0.01) against the CD5-positive Jurkat and MOLT-4 cells when compared to unmodified NK-92 cells in a 4 hour assay. (E) No increase in cytotoxicity is seen when CD5-CAR NK-92 cells are cultured with CD5-negative 697 cells. Errors bars represent standard deviations.
CD5 CAR-directed T-cell activation
In order to analyze the effect of CD5-CARs on T cells, the CD5-positive Jurkat T-cell leukemia line was transduced with the lentiviral vector encoding eGFP and a CD5-CAR at MOIs ranging from 1 to 20. To measure T-cell activation induced by engagement of CD5-CARs with CD5 on neighboring cells, surface expression of the T-cell activation marker, CD69, was measured by flow cytometry 4 and 12 d after transduction (Figure 3A). The degree of activation correlated with the transduction vector amount, with increasing activation in a dose dependent manner. Higher activation was observed in the CD5-VLR-CAR expressing Jurkat T cells compared to those expressing the CD5-scFv-CAR, and no activation was observed in eGFP negative cells (Figure 3B and S1). To confirm integration of the CD5-CAR transgene into the Jurkat T-cell genome, proviral vector copy number (VCN) was measured using quantitative PCR. Increases in VCN were correlated with increases in vector amount and increases in activation (Figure 3C). The CD5-VLR-CAR Jurkat T cells had a higher VCN compared to the CD5-scFv-CAR cells at corresponding MOIs (data not shown), which is likely the reason for the slightly higher activation observed in the CD5-VLR-CAR cells (Figure 3B). When comparing the activation between the two CD5-CAR-modified cell populations as a function of VCN, we found a linear correlation in both groups (R2 = 0.91 for CD5-VLR-CAR, R2 = 0.82 for CD5-scFv-CAR) and the CD5-scFv-CAR cells exhibited higher activation compared to the CD5-VLR-CAR cells (Figure 3C). As a means of measuring CD5-CAR protein expression in the transduced T cells, Western blot analysis was performed on whole cell lysates 9 d after transduction. CD5-CAR proteins were detected using an anti-CD3ζ antibody. Proteins of approximately 48 and 55 kDa were observed, which corresponded to the predicted sizes of the CD5-VLR-CAR and CD5-scFv-CAR, respectively, as well as an 18 kDa band, which corresponded to the molecular weight of the endogenous CD3ζ protein known to be expressed in Jurkat T cells (Figure S2). CAR expression increased in a vector MOI-dependent manner. On day 12 post-transduction, activation and VCN were measured again in both CD5-CAR-expressing Jurkat T-cell populations. A decrease in VCN from day 4 to day 12 was observed, as was a corresponding decrease in CD69 expression (Figure 3D and S3). Although this decrease in Jurkat T-cell activation and VCN can, in part, be due to pseudo transduction, it also likely results from the faster proliferation rate of non-modified cells compared to CD5-CAR expressing cells, as well as from activation-induced cell death resulting from continuous activation of the transduced cell population through interactions with CD5 antigen on self and neighboring cells.
Figure 3.

Initial comparison of VLR- and scFv-based CD5-CARs. (A) Jurkat T cells were transduced with lentiviral vectors encoding either an scFv- or VLR-based CD5-CAR with co-expression of eGFP. Schematic of the Jurkat T cell activation assay shows time points for measurement of T-cell activation and Western blot analysis. (B) Activation measured by surface CD69 expression four days after transduction increased as the amount of viral vector increased. Greater activation was observed in the CD5-VLR-CAR Jurkat group. (C) The percentage of activated cells was compared to the vector copy number (VCN) obtained for each transduced population of cells. The inset to the figure defines each group. (D) CD69 expression was measured 4 and 12 d after transduction, which showed activation decreased over time in both CD5-CAR expressing Jurkat T cell groups. Errors bars represent standard deviations.
CD5 knockout in Jurkat T cells using CRISPR-Cas9 genome editing
To increase the effectiveness of anti-CD5-directed CAR T cells, we knocked out CD5 expression in Jurkat T cells using CRISPR-Cas9 genome editing. In T cells, only full-length CD5 protein is expressed. However, in CD5-positive B cells, alternative splicing of exon 1 results in an alternate exon, termed exon 1B, that encodes a truncated, cytosolic CD5 protein.35 We reasoned that targeting sequences early in the gene, upstream of the splice site, would generate a non-functional protein product and avoid the alternative splicing event. Although T cells do not express exon 1B naturally, a balance between the expression of exon 1A and exon 1B has been implicated in T cells, which may occur if exons downstream to 1A are edited.35 We generated three gRNAs with different targeting sequences within the first 100 bp of exon 1A to knockout CD5 expression. Each gRNA was expressed in conjunction with Cas9, derived from Streptococcus pyogenes, on a single plasmid. Using nucleoporation, we transfected naïve Jurkat T cells with each CRISPR-Cas9 construct and determined the percentage of CD5-negative cells five days after transfection. CD5-CRISPR gRNA #2 yielded the greatest increase in CD5-negative Jurkat T cells, resulting in 48% CD5-negative cells, compared to the mock transfected cells, which is a clone that is naturally 15% CD5-negative. gRNA #1 and gRNA #3 resulted in 38% and 24% CD5-negative cells, respectively (Figure 4A). Using COSMID (CRISPR Off-target Sites with Mismatches, Insertions, and Deletions), a public webtool, we were able to predict sites within the human genome that had the most likelihood of being targeted by our CRISPR system.36 Using the same search parameters, we identified potential off-target sites that could result from using gRNAs #1 and #2; gRNA #1 was predicted to have likely off-target sites in three genes, with one site being within the CD5 gene (separate location from the intended target site), and gRNA #2 was predicted to have likely off-target sites only within the CD5 gene. Given the more efficient CD5 knockout and decreased potential for off-target binding, gRNA #2 was used in subsequent experiments.
Figure 4.
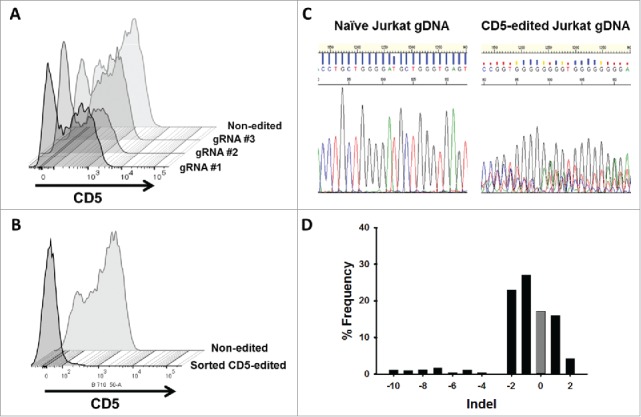
CD5 knockout in Jurkat T cells using CRISPR-Cas9 genome editing. (A) CD5 expression, measure by flow cytometry, in Jurkat T cells five days following mock transfection or transfection with plasmid encoding Cas9 and one of three different gRNA target sequences. Histogram plots for CD5 expression in mock transfected and transfected Jurkat T cells are shown along a single axis. (B) Overlay image of histogram plots of CD5 expression in naïve Jurkat T cells and flow-sorted CD5-negative Jurkat T cells that were transfected with the CD5-CRISPR gRNA #2. (C) Representative Sanger sequencing traces from naïve (top left) and sorted CD5-edited (top right) Jurkat T cell genomic DNA PCR amplified for CD5. TIDE analysis of the frequency of indels within the CD5 gene after the predicted break-site generated by Cas9 (D). Results show 77% CD5-negative cells were edited with 27% having a −1 deletion.
Flow sorting allowed for the isolation and expansion of the population of CD5-negative Jurkat T cells from the mixed population of cells edited with CD5-CRISPR gRNA #2. Only 2.1% of sorted cells expressed CD5 (Figure 4B). To characterize the frequency of mutations within the CD5-edited cells, we utilized TIDE (Tracking of Indels by DEcomposition) software. This analysis uses trace sequences generated by Sanger sequencing to identify the predominant insertions and deletions (indels) in CD5-edited Jurkat T-cell genomic DNA compared to naïve Jurkat T-cell genomic DNA. A representative section of the trace sequences is shown in Figure 4C. Using a decomposition algorithm, the software identifies the frequency of indels around the break site.37 TIDE analysis indicated that within our sorted population of CD5-negative Jurkat T cells, 77% of the population was edited. Within the edited population, 27% have a −1 deletion, compared to naïve Jurkat T cells, and approximately 17% of the population was not edited (Figure 4D). Although by flow cytometry we determined approximately 98% of the cells in this population are CD5-negative, some are not CRISPR-edited due to naturally occurring CD5-negative cells within our starting Jurkat T-cell population. Therefore, after sorting, approximately 17% of the collected cells, lack mutations in the genome but maintain their CD5-negative characteristics.
CD5-edited CAR-modified T cells have reduced self-activation and increased CD5-CAR expression
Naïve and sorted CD5-CRISPR-edited Jurkat T cells were transduced with the lentiviral vectors encoding eGFP and the CD5-CARs. Additionally, a third lentiviral vector encoding eGFP and BCL-VLR-CAR was used as a negative control. We previously showed the BCL-VLR-CAR expressed in Jurkat T cells does not stimulate T-cell activation in the absence of BCL cells.24 It was hypothesized that both CD5-CARs would activate naïve Jurkat T cells to a greater degree than CD5-edited Jurkat T cells, whereas the BCL-VLR-CAR would stimulate low and equivalent levels of T-cell activation in all Jurkat T cells. eGFP expression was used as a marker of transduced Jurkat T cells to identify the CAR-expressing population, and cells were transduced at MOIs of 1, 10 or 20. As expected, for all three vectors, there is an increase in eGFP-positive cells as the vector titer increases, and this increase is similar and consistent in both cell populations (Figure 5A and S4). As the vector amount of CD5-CAR increased, there is a decrease in CD5 expression on non-edited Jurkat T cells (Figure 5B). This decrease is most pronounced in CD5-scFv-CAR-modified Jurkat T cells. This effect was not observed in BCL-VLR-CAR-modified T cells, showing these results are a consequence of CD5-CAR expression, which is consistent with previously published results (5). Furthermore, we compared CD69 expression to eGFP expression in all cell groups. A positive correlation was observed between eGFP expression and activation in both the scFv- and VLR-based CARs, as well as in edited and non-edited cells (Figure 5C). The increase in activation was dramatically dampened in CD5-edited cells expressing either the CD5-VLR-CAR or the CD5-scFv-CAR. The BCL-VLR-CAR only stimulated very low levels of T-cell activation.
Figure 5.
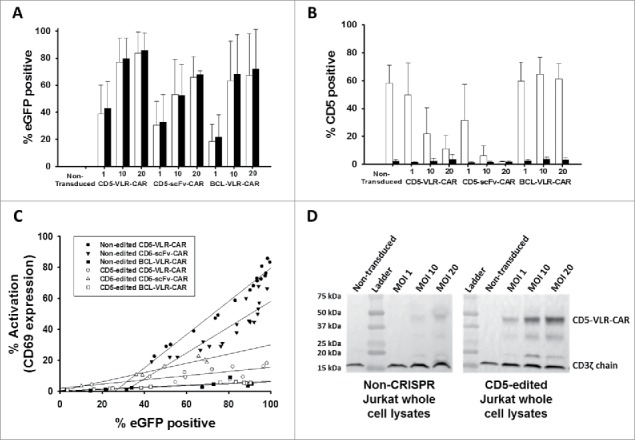
CD5-edited CD5-CAR-modified Jurkat T cells have reduced self-activation and increased CD5-CAR expression. Naïve (white) and CD5-edited Jurkat T cells (black) were transduced with eGFP-P2 A-CD5-VLR-CAR, eGFP-P2 A-CD5-scFv-CAR or control eGFP-P2 A-BCL-VLR-CAR lentiviral vectors at MOIs 1, 10 and 20. Polybrene was not used during transduction, which provided a greater separation in transduction efficiency between MOIs of 1 and 10. Experiments were performed with replicates of three or greater (error bars are generated using the standard deviation from the mean) except for CD5-scFv-CAR at MOI 10 and 20, which were done in duplicate, providing the difference of the mean. (A) Transduction efficiency, measured by eGFP-positive cells, of each CAR vector at MOIs 1, 10 and 20 in both populations of Jurkat T cells. (B) CD5 expression in both populations of Jurkat T cells transduced with each CAR vector at each MOI. (C) Activation was measured by monitoring CD69 expression and transduction efficiency was measured by eGFP expression. A correlation exists between activation and eGFP expression in CD5-CAR-transduced Jurkat T cells. Non-edited CD5-CAR-modified cells have increased T-cell activation compared to CD5-edited CD5-CAR-modified cells. (D) Western blots on whole cell lysates showing CD3ζ expression in non-edited Jurkat T cells (left) and CD5-edited Jurkat T cells (right) when transduced with the VLR-CAR vector. Endogenous CD3ζ is represented by the 18 kDa bands and CD3ζ in the CAR construct is represented by the 48, kDa band in the CD5-VLR-CAR construct. eGFP, CD5 and CD69 surface expression were measured by flow cytometry.
Western blot analysis using whole cell lysates collected 9 d after transduction confirmed the decrease in CD5 expression in CD5-CAR-modified Jurkat T cells compared to naïve Jurkat T cells and BCL-CAR-modified Jurkat T cells (Figure S5A). As expected, Western blot analysis showed CD5-edited Jurkat T cells have lower CD5 expression compared to non-edited cells for both transduced and non-transduced cells (Figure S5B). We hypothesized that if the decrease in CD5 levels is due to interactions between the CD5-CAR and the CD5 cell surface protein, then CD5-CAR levels may also be influenced by CD5 expression. Therefore, cells with lower CD5 expression levels will have increased CD5-CAR protein expression due to reduced interactions with the CD5 antigen. To test this, we ran flow cytometry using a CD5-Fc fusion protein consisting of the CD5 antigen fused to the Fc portion of an IgG. Jurkat T cells were stained with the CD5-Fc protein and then stained a second time using an anti-IgG Fc antibody conjugated to phycoerythrin (PE). As expected, the CD5-scFv-CAR-modified CD5-edited Jurkat T cells bind CD5-Fc to a greater degree than do CD5-scFv-CAR-modified non-edited Jurkat T cells. This data also shows evidence of potential pseudo-transduction at day 4, however, CD5-Fc binding decreases by day 8 and then appears to plateau. (Figure S6A). Significant differences are observed early after transduction, however they become less significant after CD5-CAR expression decreases in non-edited cells and normalizes. On day 8 post-transduction, 18.6% of non-edited Jurkat T cells were bound to CD5-Fc protein and eGFP, compared to 35.7% of CD5-edited Jurkat T cells (Figure S6B).
Figure 6.

CD5-edited CD5-CAR-modified effector cells in culture with naïve target T cells stimulates effector cell activation and target cell down-regulation of CD5. Naïve and CD5-edited Jurkat T cells were transduced with eGFP-P2A-CD5-scFv-CAR or eGFP-P2A-CD5-VLR-CAR lentiviral vectors at MOI 5. Polybrene was not used during transduction. Target naïve Jurkat T cells were labeled with VPD450. On day five post-transduction, effector cells were cultured with labeled target cells at E:T ratios 2:1, 1:1 and 1:5. The cells were analyzed by flow cytometry 24 hours later. White bars signify non-edited effector cells; black bars signify CD5-edited effector cells. Experiments were performed with three replicates and error bars represent standard deviation from the mean. (A and B) Percent of baseline CD5 expression in target Jurkat T cells cultured with non-edited and CD5-edited effector Jurkat T cells expressing the CD5-scFv-CAR (A) or the CD5-VLR-CAR (B). CD5 expression in target cells cultured alone (gray bar) was used as baseline and set at 100%. (C and D) T-cell activation of non-edited and CD5-edited effector Jurkat T cells expressing the CD5-scFv-CAR (C) or the CD5-VLR-CAR (D) when cultured alone and in culture with target Jurkat T cells.
Our experiments in Jurkat T cells serve as a basis for using primary T cells. We expanded primary T cells in media containing IL-2 and IL-7, and using the same CRISPR-Cas9 system used in Jurkat T cells, we knocked out CD5 expression in 38.6% of our primary T cells (Figure S7A and B). We transduced non-edited and CD5-edited primary T cells with CD5-scFv-CAR lentiviral vector and measured eGFP and CD5-Fc binding by flow cytometry on day 9 post-transduction. Similar to our Jurkat T cell data, we show increased percentage of CD5-edited cells bound to CD5-Fc protein compared to non-edited cells, with 64.4% CD5-Fc-bound CD5-edited cells, compared to 6.1% CD5-Fc-bound non-edited cells (Figure S7C and D).
Figure 7.
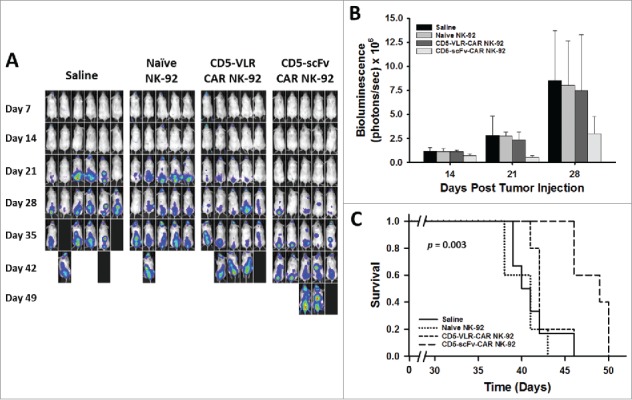
CD5-scFv-CAR NK-92 cells are superior to CD5-VLR-CAR NK-92 cells in delaying disease progression and improving survival in a T-ALL xenograft mouse model. (A) NSG mice were injected with 2 × 106 luciferase-expressing Jurkat T cells intravenously on day 0. Treatment was started 7 d after tumor injection with each mouse receiving a total of 4 treatments on days 7, 11, 14 and 18. Mice were assigned to 4 different treatment groups – saline, naïve NK-92, CD5-VLR-CAR NK-92 or CD5-scFv-CAR NK-92. A dose of 107 cells per mouse were administered at each treatment time. Bioluminescence imaging was performed every seven days to monitor tumor burden. (B) Total bioluminescence from Jurkat T cells on days 14, 21, and 28 post tumor injection. A significant decrease (p < 0.05) in tumor burden is observed in the CD5-scFv-CAR NK-92 group. Errors bars represent standard deviations. (C) Kaplan-Meier survival curves showing overall survival. Mice treated with CD5-scFv-CAR NK-92 cells showed significant increased survival (p < 0.05) when compared to all other treatment groups. Mice treated with CD5-VLR-CAR NK-92 cells did not have a significant advantage over saline and naïve NK-92 treatment groups.
The difference in CD5-Fc binding to edited compared to non-edited cells could be a result of steric hindrance from CD5 binding the CAR on non-edited cells, blocking CD5-Fc from binding the CAR, as opposed to reduced CAR expression on these cells. To test this, we performed Western blot analysis on Jurkat whole cell lysates using a CD3ζ antibody to detect endogenous CD3ζ (18 kDa) and CD3ζ in the CAR constructs (48, 55, and 47 kDa in the CD5-VLR-CAR, CD5-scFv-CAR and BCL-VLR-CAR constructs, respectively). Importantly, using endogenous CD3ζ as a reference, the CD5-edited Jurkat T cells express both CD5-CARs at greater levels compared to the non-edited Jurkat T cells (Figure 5D and S6C). Furthermore, there is not an effect on BCL-CAR expression when comparing transduced cells with or without CD5 editing (Figure S6C). We can conclude that non-edited Jurkat T cells have down-regulated CD5-CAR expression.
CD5-edited effector cells are efficiently stimulated by target T cells, which down-regulate CD5
We hypothesized that culturing CD5-CAR-modified effector cells with naïve Jurkat T cells would result in i) an increase in non-edited effector CD5 expression because of competition between CD5 expressed on the CAR-modified cells and CD5 expressed on the target cells, ii) target cell down-regulation of CD5 expression and iii) increased activation of CD5-edited effector cells compared to non-edited cells. Non-edited and CD5-edited Jurkat T cells were transduced with lentiviral vector encoding CD5-scFv-CAR or CD5-VLR-CAR at an MOI of 5. Flow cytometry five days after transduction confirmed eGFP expression, as well as an expected decrease in CD5 expression on the non-edited Jurkat T cells (as previously demonstrated in Figure S4 and S5). Naïve Jurkat T cells were labeled with Violet Proliferation Dye 450 (VPD450) to distinguish target cells from effector cells, and subsequently cultured with the CAR-modified effector cells at E:T ratios of 2:1, 1:1 and 1:5. After 24 hours, cells were collected and flow cytometry was used to measure CD5 expression on the effector and target cells, as well as CD69 expression on the effector cells. CD5 expression was low in effector cells in edited and non-edited transduced cells when co-cultured with target cells (Figure S8), showing there is little effect on CD5 expression on the effector cells during co-culture. To compare CD5 expression in the target cells, the level of CD5 expression in VPD450-labeled naïve Jurkat T cells cultured alone was set as the baseline CD5 expression in the target cells. When in culture with CD5-scFv-CAR- (Figure 6A) and CD5-VLR-CAR-modified effector cells (Figure 6B), CD5 expression decreased in the target cells, with a greater decrease observed in target cells cultured with the CD5-scFv-CAR-modified cells. At E:T ratios of 2:1 and 1:1, there is a significant difference in target cell CD5 expression between the groups cultured with CD5-edited CD5-scFv-CAR-effector cells (Figure 6A) and CD5-edited CD5-VLR-CAR-effector cells (Figure 6B) (2:1: p = 0.001, 1:1: p < 0.001). Additionally, significant differences in target cell CD5 expression were found at all E:T ratios comparing the non-edited effector cell group and the CD5-edited effector cell group (p < 0.05). However, at low E:T ratios (high percentage of target cells relative to effector cells), the decrease in CD5 expression was less pronounced (Figure 6A and Figure 6B, E:T ratio of 1:5 p = 0.028 and p = 0.045 in CD5-scFv-CAR-effector cell cultures and CD5-VLR-CAR-effector cell cultures, respectively). These results show CD5-edited CAR-modified effector T cells have increased association with the target cells compared to non-edited CAR-modified effector T cells, which results in the dramatic decrease in CD5 expression on the target cells. To determine if there are differences in effector cell activation, CD69 expression was measured. At all E:T ratios, CD5-edited CD5-scFv-CAR- (Figure 6C) and CD5-edited CD5-VLR-CAR-modified (Figure 6D) effector T cells had a significant increase in activation compared to their activation prior to culture with naïve target cells (Figure 6C and 6D, p < 0.05 for all ratios). A control experiment measuring the same parameters using non-CAR-modified, CD5-edited effector cells demonstrated the cells alone had no effect (data not shown). This data illustrates CD5-edited effector T cells have increased interactions with target cells compared to non-edited effector T cells, which results in an increase in effector cell activation.
CD5-scFv-CAR NK-92 cells are superior to CD5-VLR-CAR NK-92 cells in delaying disease progression in a xenograft T-cell leukemia mouse model
To further compare the cytotoxic potential of the two CD5-CAR structures, we tested the efficacy of the CD5-CAR expressing NK-92 cells in a T-cell leukemia xenograft mouse model. Luciferase-expressing Jurkat T cells were used to establish our leukemia model, which allowed for monitoring of tumor burden using bioluminescence imaging. Treatment was started seven days after tumor injection, as described in the Materials and Methods section. NK-92 cells were injected twice weekly for a total of 4 doses without IL-2 supplementation. The twice-weekly dosing regimen was based on our experiments showing non-irradiated NK-92 cells, in the absence of IL-2, do not persist in the peripheral blood beyond three days, and show no evidence of engraftment in the bone marrow (Figure S9). A significant decrease in tumor burden was evident in the CD5-scFv-CAR NK-92 treatment group at Day 21 (p = 0.02 using one-way ANOVA) (Figure 7A and 7B). Significance (p < 0.05) for multiple comparisons tests by Holm-Sidak method was shown for CD5-scFv-CAR vs saline, and CD5-scFv-CAR vs naïve NK-92 groups, but not for the CD5-scFv-CAR vs CD5-VLR-CAR group. A similar overall trend was observed at days 14 and 28 in terms of disease burden, however the one-way ANOVA test was underpowered to compare all groups. Although only modest effects were observed, as expected due to the cell dose and persistence of the NK-92 cells, the scFv-CAR-treated group had a significant advantage in survival compared to all three other groups (p = 0.003 by log-rank test, p < 0.05 for all multiple comparisons tests between CD5-scFv-CAR and other groups by Holm-Sidak method) with a median survival of 49 d compared to 40, 41, and 42 days for the saline, naïve NK-92 and CD5-VLR-CAR NK-92 groups, respectively (Figure 7C). In contrast, the CD5-VLR-CAR-NK-92 mice did not exhibit a significant survival advantage over the saline- and naïve NK-92-treated groups.
Discussion
Patients with relapsed T-cell acute lymphoblastic leukemia or lymphoblastic lymphoma (T-ALL/T-LLy) have dismal outcomes, with mortality rates greater than 80%, when treated with chemotherapy alone.38-40 Allogeneic hematopoietic stem cell transplantation (HSCT) offers the greatest chance of cure in these patients. A recent study by the Center for International Blood and Marrow Transplant Research showed the 3-year overall survival (OS) with HSCT is 48% for patients able to achieve complete second remission (CR2) prior to transplantation.41 For patients with first relapse of T-ALL/LLy, achieving CR2 is the most important step prior to HSCT, as disease status at the time of transplantation remains the most important factor associated with overall survival.42 However, attaining clinical remission after relapse remains the biggest therapeutic challenge in T-cell disease, and most patients are unable to receive transplantation given the aggressive nature of relapsed disease.43 Thus, in order to maximize and improve upon the benefits of an allogeneic HSCT, there remains a need to develop newer strategies to induce remission in these relapsed patients. CAR-based immunotherapy can play an important role by providing a sustained remission post-relapse, thereby acting as a bridge to stem cell transplantation. Unlike CAR therapy in B-cell malignancies, where sustained B-cell aplasia due to off-target toxicity can be managed with periodic intravenous immunoglobulin infusions, persistent T-cell aplasia caused by T-cell-directed CAR therapy would result in life threatening severe immunosuppression. Thus, HSCT to allow for immune reconstitution following CAR T cell therapy is a reasonable strategy.
CD5 was first studied as a target tumor antigen using a monoclonal CD5 antibody linked to an immunotoxin ricin A chain, and was tested in patients with T-cell leukemia and cutaneous T-cell lymphoma.14,15,44 A phase 1 clinical trial in patients with cutaneous T-cell lymphoma demonstrated partial responses in 4 patients with no significant side effects. Two additional studies have used the CD5-directed immunoconjugate to treat graft-versus-host disease by targeting normal T cells.45,46 Due to the success of these studies, the restricted expression of CD5, and the role of CD5 in the suppression of TCR signaling,11,47 we hypothesized CD5 would be a good target for CAR therapy. Two studies have previously demonstrated the effectiveness of using CD5 as a target antigen for a CAR construct. Mamonkin et al. showed that CD5-CAR expressing T cells were effective in targeting T-cell disease in an in vivo model, however, they also reported evidence of fratricide among the CAR-expressing T-cells.5 A clinical trial NCT03081910 has now been initiated based on this approach. Additionally, Chen et al. showed that NK-92 cells expressing an anti-CD5 CAR had potent anti-tumor activity against T-cell leukemia.16 While we have shown similar cytotoxicity results here using the anti-CD5-scFv-CAR, we have also demonstrated cytotoxicity and T-cell activation using an anti-CD5-VLR-CAR and have tested an alternative approach to using CD5-CAR T cells by CRISPR-Cas9 genome editing in an effort to prevent fratricide.
Overall, the studies of this report, with our previous studies and reports from others (5, 23), illustrate self-activation of CD5-positive CD5-CAR-modified effector cells due to interactions with self and neighboring CD5 antigens. We show when using both scFv- and VLR-based CD5-CARs that this effect diminished over time as the average number of transgene copies per cell decreased. This is likely due to a decreased proliferation rate and activation-induced cell death of the CAR-modified, activated cells.48 One approach to prevent effector cell activation in the absence of malignant cells is to use CD5-negative NK cells modified to express the anti-CD5 CARs. The use of NK cells as the CAR-expressing effector cells has been demonstrated in several previous preclinical CAR studies, in which either primary human NK cells or the IL-2 dependent NK cell line, NK-92, have been used as the effector cells.49-61 Primary human NK cells have been used in CAR-based clinical trials, with mRNA electroporation being the means of inducing CAR expression.61-63 Our in vitro and in vivo data show that NK-92 cells modified to express CD5-CARs are effective in targeting a CD5-positive T-cell leukemia cell line. While we only show a modest effect with NK-92 cells, our results are comparable to the NK-92 studies by Chen et al.16 The lack of persistence of NK-92 cells in the absence of IL-2, as we have demonstrated, is the likely reason for this low effectiveness. This can be overcome by repeated effector cell dosing or by transitioning to primary NK CD5-CAR cells to demonstrate enhanced anti-tumor efficacy in our T-cell leukemia mouse model.
Our second approach was to knock out the target antigen from the effector cells using genome-editing. The CRISPR-Cas9 system has been widely used since its discovery and characterization in prokaryotes and has been engineered for use in eukaryotes.18,20,22,64-66 Genome editing technologies are quickly progressing toward clinical uses, and the NIH recently approved the initiation of a clinical trial using CRISPR-Cas9 to treat refractory metastatic non-small cell lung cancer.67 We demonstrated CD5-edited CD5-CAR-modified Jurkat T cells exhibit decreased self-activation, yet increased activation when cultured with target cells. CD5-edited effector cells were significantly more activated when in culture with target T cells compared to their initial levels of activation in culture alone. We and others (5) have now shown that CD5-CAR expression in T cells results in down-regulation of CD5. Interestingly, our data also shows non-edited CAR-modified T cells have decreased CD5-CAR protein expression compared to CD5-edited CAR-modified T cells. This data is shown in both Jurkat T cells and primary T cells. CAR down-regulation is likely similar to the mechanism of CD5 down-regulation in that the interaction between the CAR and CD5 results in processing of both proteins from the cell membrane. This result can be important for the expression of other CARs on effector cells that express variable levels of the target antigen. Furthermore, in cultures with CD5-edited effector cells and target cells, effector cells interact more robustly with CD5 on target cells; whereas CD5-positive, non-edited effector T cells interact with CD5 antigen on both effector and target cells, reducing their potency. Overall, the data shows CD5-negative effector cells are advantageous compared to CD5-positive effector cells due to their decreased self-activation and increased CAR expression.
Interestingly, CD5-edited effector cells have a greater effect on target cell CD5 expression. This effect is much stronger with CD5-scFv-CAR effector cells compared to CD5-VLR-CAR effector cells. This is the first study comparing a VLR-based CAR to a more traditional scFv-based CAR. The CD5-VLR used in the CAR construct is an avidity-based antibody, with the multimeric form of the VLR antibody binding to human CD5 with a higher efficiency compared to the monomeric form.29 The scFv was derived from the murine H65 anti-human CD5 IgG antibody.31 It cannot be concluded from these in vitro studies which CD5-CAR would be most advantageous, as both demonstrate substantial target cell association and effector cell activation. However, our in vivo studies showed the VLR-CAR did not perform as well as the scFv-CAR. This result is possibly due to the avidity nature of this particular VLR. We cannot conclude that other VLRs would not provide adequate target cell killing, as our in vitro studies certainly show VLR sequences can efficiently activate T cells. There may be particular circumstances in which a VLR may be advantageous over an scFv. Previous CAR studies have shown that differing CAR affinities usually do not affect the maximum level of T-cell activation seen, however decreased selectivity can be seen with higher affinity CARs.68,69 Since the target cells in our study have high CD5 antigen expression, it is unlikely that affinity alone could account for the differences in efficacy between the VLR and scFv-CAR. It is possible that the limited persistence of NK-92 cells in the in vivo setting had a significantly higher negative effect on the VLR-CAR compared to the scFv-CAR.
Our results show both approaches, NK-cells as effector cells and CD5 knockout in effector T cells, modified with the CD5-CARs have the potential to overcome the barriers of self-activation and fratricide, which are issues that are hampering the use of CAR therapies from being applied to the treatment of T-cell malignancies. Although our goal is to provide a bridge to allogenic transplantation for relapsed patients, strategies using CAR-modified immunocompetent cells also are being developed as therapeutics to attain long-term remission in these patients. All approaches will require a functional anti-T-cell CAR construct, and this study advances our understanding of this possibility.
Materials and methods
Cell lines. The Jurkat and NK-92 cell lines were obtained from American Type Culture Collection (ATCC, Manassas, VA). The MOLT-4 and 697 cell lines were kindly provided to us by the laboratory of Dr. Douglas Graham (Emory Unisversity). The Jurkat T cell clone used is heterogeneous in CD5 expression. The primary culture media for the Jurkat cell line was RPMI (Corning, Manassas, VA) with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. For NK-92 cells, AIMV (Thermo Fisher Scientific, Waltham, MA) was used with 20% FBS, 1% penicillin/streptomycin and 1000 U/mL recombinant interleukin-2 (IL-2, PeproTech, Rocky Hill, NJ).
Primary Cells. Primary T cells were generously donated by the Chandrakasan laboratory at Emory University. These cells were isolated from PBMCs from healthy, consented donors. Cells were expanded in X-VIVO 15 media (Lonza, Switzerland) with 10% FBS, 1% penicillin/streptomycin/amphotericin B (Lonza, Switzerland), 50 ng/mL IL-2 (PeptroTech, Rocky Hill, NJ) and 5 ng/mL IL-7 (PeptroTech, Rocky Hill, NJ).
Cloning of the CD5-CAR sequences. The CD5-scFv cDNA sequence was derived from a humanized murine immunoglobulin protein sequence targeting CD5.31 The variable heavy and variable light sequences were joined by a (G4 S)3 peptide linker. The CD5-VLR cDNA sequence was generated from a published protein sequence of a VLR targeting CD5.29 Both cDNA sequences were then codon optimized for human cell expression and subcloned into a vector containing the remaining necessary components for CAR production, which were obtained by gene synthesis from Genewiz (South Plainfield, NJ). We used a bicistronic construct to allow for dual expression of enhanced green fluorescent protein (eGFP) and the CD5-CAR using a P2A peptide sequence. The BCL-VLR-CAR control was cloned by substituting the CD5-VLR for the BCL-VLR into the CAR cassette as previously described.24
Generation of CAR encoding lentiviral vector. High-titer, recombinant, self-inactivating (SIN) HIV lentiviral vector was produced using a four-plasmid system. Briefly, the expression plasmid encoding the CD5-CAR constructs and BCL-VLR-CAR construct, as well as packaging plasmids containing the gag, pol, and envelope (VSV-g) genes were transiently transfected into HEK-293 T cells by calcium phosphate transfection. Cells were cultured in DMEM (Thermo Fisher Scientific, Waltham, MA) supplemented with 10% FBS and 1% penicillin/streptomycin. Twenty-four hours after transfection, the cell culture medium was replaced with fresh medium. At 48 and 72 hours the vector supernatant was collected, filtered through a 0.22 μm filter and stored at −80 °C. After the final collection, the vector supernatant was pooled and concentrated overnight via centrifugation at 10,000 x g at 4 °C. Pelleted vector was then re-suspended in serum-free StemPro media (Thermo Fisher Scientific, Waltham, MA). Titering was performed on HEK-293 T cell genomic DNA using quantitative polymerase chain reaction (qPCR). Titers of the concentrated recombinant viral vectors were ∼1 × 107 TU/mL.
Lentiviral vector transduction of cell lines. Transduction of recombinant HIV-1-based lentiviral vector particles was carried out by incubating cells with vector in appropriate culture medium supplemented with 6 μg/mL polybrene (EMD Millipore, Billerica, MA), unless otherwise stated. Twenty-four hours after transduction, culture medium was replaced with fresh medium. The transduced cells were then cultured for at least 3 d before being used for downstream applications. Jurkat T cells were transduced at multiplicity of index (MOI) ranging from 1 to 20.
Lentiviral vector spinoculation of primary T cells. Transduction of recombinant HIV-1-based lentiviral vectors was carried out by incubating cells with vector in appropriate culture medium supplemented with 5 μg/mL polybrene (EMD Millipore, Billerica, MA) and then centrifuged at 3000 RPM for 2.5 hours. Twenty-four hours after spinoculation, culture medium was replaced with fresh medium. The transduced cells were then cultured for at least 3 d before being used for downstream applications.
Flow cytometry analysis and sorting. Analysis was done using a BD FACS Canto II Flow Cytometer and BD LSRII Flow Cytometer (BD Biosciences, San Jose, CA). Data was analyzed using the BD FACSDiva software and FlowJo, LLC. Antibodies used included anti-CD69 APC-Cy7 and anti-CD5 PerCP/Cy5.5 (BD Biosciences, San Jose, CA). Additionally, a CD5-Fc fusion protein was used to detect CD5-CAR surface expression through binding of the CAR to the CD5 portion of the protein (G&P Biosciences, Santa Clara, CA). A secondary anti-IgG-Fc conjugated to PE was used to detect the CD5-Fc (Jackson Immunoresearch Laboratories, West Grove, PA). For the cytotoxicity studies, target cells were stained with the membrane dye PKH26 and cell death was assessed using 7-AAD (described below). Flow sorting for CD5 and eGFP was performed using a SH800 S Cell Sorter (Sony Biotechnology Inc. U.S., San Jose, CA).
Western blotting with CD3ζ and CD5 antibodies. Cells were lysed using RIPA buffer (Sigma-Aldrich, St. Louis, MO). Cell lysates were clarified by centrifugation and protein was quantified using the Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA). Equal quantities of protein were loaded and cell lysates were separated by SDS-PAGE under reducing conditions and transferred to a nitrocellulose membrane (Bio-Rad Laboratories, Hercules, CA). The protein-loaded and blocked membrane was incubated with an anti–CD3ζ mAb (1:500) or anti-CD5 mAb (0.5 μg/mL) followed by a HRP-labeled goat anti-mouse IgG secondary Ab (1:2500) or HRP-labeled anti-goat IgG secondary Ab (1:1000), respectively.
Cytotoxicity assay. Target cells were labeled with membrane dye PKH26 using the manufacturer's protocol (Sigma-Aldrich, St. Louis, MO). Effector cells were left unstained. Effector (E) and target (T) cells were counted and viability assessed using trypan blue. Labeled target cells were mixed with effector cells in 12 × 75 mm FACS tubes at E:T ratios ranging from 0:1 to 10:1 in a total volume of 200 uL. Target cells (50,000) were added to 12 × 75 mm FACS tubes along with the corresponding number of effector cells. The cell mixture was incubated for 4 hours at 37 °C in 5% CO2. After incubation, cells were washed and stained with 7-AAD (BD Biosciences, San Jose, CA). Flow cytometry analysis was performed to assess 7-AAD positive cells. All experiments were performed in triplicate. To calculate specific cytotoxicity, the number of spontaneously lysed target cells in the absence of effector cells was subtracted from the number of dead target cells, which were identified as PKH26 and 7-AAD double positive in the measured sample.
Real time quantitative PCR. Genomic DNA was extracted using the Qiagen DNeasy Blood & Tissue Kit according to the manufacturer's recommended protocol (Qiagen, Germantown, MD). Oligonucleotide primers were designed for a 150bp amplicon of the Rev-response element (RRE). Real-time PCR was performed in an Applied Biosystems® StepOne™ System (Thermo Fisher Scientific, Waltham, MA) in 25 µl reaction volumes using 50 ng of template DNA, using the default thermocycler program for all genes: 10 minutes of pre-incubation at 95 °C followed by 40 cycles of 15 seconds at 95 °C and one minute at 60 °C.
Transfection of Jurkat T cells and primary T cells. Jurkat T cells and primary T cells were transfected using the Lonza Nucleofector 2b Device and the Amaxa Cell Line Nucleofector Kit V or the Amaxa Human T Cell Nucleofector kit, respectively, according to the manufacturer's protocol (Lonza, Switzerland). Cells were transfected with 6 µg of a single plasmid CRISPR-Cas9 system encoding both the guide RNA (gRNA) and Cas9. By day 5 post-transfection, the CD5 knockout was confirmed using BD LSRII Flow Cytometer (BD Biosciences, San Jose, CA).
Tracking of Indels by DEcomposition (TIDE) analysis of genome editing. Genomic DNA was isolated using the Qiagen DNeasy Blood & Tissue Kit according to the manufacturer's recommended protocol (Qiagen, Germantown, MD). Naïve Jurkat genomic DNA and CD5-edited Jurkat genomic DNA samples were sequenced by GeneWiz (South Plainfield, NJ) using primer sequences that flanked the predicted Cas9 cut site within the CD5 gene. The sequencing traces were uploaded into the TIDE software as well as the guide RNA sequence. Parameters were adjusted to fit the uploaded sequences and indels +/- 10 bp were analyzed. Sequencing and analysis of forward and reverse amplifications confirmed the results.
Co-culture assay using CAR-modified effector T cells and naïve target T cells. Naïve and CD5-edited Jurkat T cells were transduced by incubating with high titer, recombinant, SIN lentiviral vectors encoding eGFP-P2A-CD5-scFv-CAR or eGFP-P2A-CD5-VLR-CAR at MOI 5. After 24 hours, culture medium was replaced with fresh medium. On day 5 after transduction, flow cytometry using BD LSRII Flow Cytometer (BD Biosciences, San Jose, CA) confirmed eGFP expression. The same day, transduced cells were cultured with naïve Jurkat T cells labeled with Violet Proliferation Dye 450 (VPD450) at effector (E) to target (T) ratios of 2:1, 1:1 and 1:5. The final concentration of each culture was 5 × 105 cells/mL. Naïve Jurkat T cells were labeled according to the manufacturer's protocol (BD Biosciences, San Jose, CA). Flow cytometry was used to analyze changes in CD5 on the effector and target cells, as well as CD69 expression on effector cells at 24 hours after initiation of the co-culture. All experiments were performed in triplicate.
Generation of a T-cell leukemia murine xenograft model and treatment with CD5-CAR expressing NK-92 cells
NOD/SCID/IL2Rγnull (NSG) mice were purchased from The Jackson Laboratory (Bar Harbor, ME) and were maintained in a specific pathogen-free environment. Mice were cared for according to the established principles of the Institutional Animal Care and Use Committee (IACUC) and all animal protocols were approved by the IACUC. A luciferase-expressing Jurkat T-cell leukemia cell line was kindly provided to us by Dr. Douglas Graham (Atlanta, GA). To determine the treatment dosing regimen with NK-92 cells, we injected NSG mice with non-irradiated CD5-scFv-CAR NK-92 cells without supplementation of IL-2 and followed persistence of the NK-92 cells over time. Mice were evaluated for evidence of NK-92 cells by flow cytometry in peripheral blood, bone marrow and spleen 1, 3, and 18 d post injection. Based on results from this experiment, a twice-weekly dosing regimen for non-irradiated NK-92 cells without IL-2 supplementation was established.
Seven- to nine-week-old NSG mice were then intravenously injected with 2 × 106 luciferase-expressing Jurkat T cells on day 0 to establish disease. Cells were re-suspended in 100 uL phosphate buffered saline (PBS) prior to injection. Treatment was started on day 7 after tumor injection. There were four treatment groups; mice either received PBS (control), unmodified naïve NK-92 cells, CD5-VLR-CAR NK-92 cells or CD5-scFv-CAR NK-92 cells. For mice receiving cells, each treatment consisted of 107 NK-92 cells re-suspended in 100 uL PBS administered intravenously via a retro-orbital injection. Each mouse received 4 treatments on days 7, 11, 14 and 18. Mice underwent in vivo bioluminescence imaging every seven days to monitor tumor burden. Animals were monitored frequently and were euthanized upon signs of leukemia progression (weight loss >20%, decreased activity, and/or hind limb paralysis).
In vivo bioluminescence imaging. NSG mice were anesthetized with inhaled isoflurane and maintained with 2% isofluorane during imaging procedures. Bioluminescence imaging was performed with the IVIS® Spectrum imaging system (PerkinElmer, Boston, MA). Each mouse was given an intraperitoneal injection of 150 mg/kg D-luciferin (PerkinElmer, Boston, MA) dissolved in PBS. Images were captured 10–15 minutes after the D-luciferin injection. Bioluminescence intensity was quantified using the Living Image® advanced in vivo imaging software. Total flux values were determined by drawing regions of interest (ROI) of identical size over each mouse and are presented in photons/second.
Statistical analysis. Unpaired 2-tailed Student t test and One-way ANOVA were used to determine statistical significance. For in vivo survival data, Kaplan–Meier curves were plotted and compared using a log-rank test. A p-value of <0.05 is considered statistically significant for all studies unless otherwise stated. All statistics were calculated with SigmaPlot, version 13.0 (Systat Software, Chicago, IL).
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by grants from the National Institute of Health (NIH 1R43CA192710-01), Curing Kids Cancer, Hyundai Hope on Wheels and Winship Invests. Primary T cells from healthy, consented donors were generously gifted by the Chandrakasan laboratory at Emory University, and luc-modified Jurkat T cells, MOLT-4 and 697 cells were generously donated by the Graham laboratory at Emory University. All flow cytometry experiments were done with equipment in the Emory+Children's Pediatric Research Center Flow Cytometry Core.
References
- 1.Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, Fry TJ, Orentas R, Sabatino M, Shah NN, et al.. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385(9967):517–28. doi: 10.1016/S0140-6736(14)61403-3. PMID:25319501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF, et al.. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371(16):1507–17. doi: 10.1056/NEJMoa1407222. PMID:25317870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, Chung SS, Stefanski J, Borquez-Ojeda O, Olszewska M, et al.. Efficacy and toxicity management of 19–28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014. 6(224):224ra25. doi: 10.1126/scitranslmed.3008226. PMID:24553386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schubert ML, Hückelhoven A, Hoffmann JM, Schmitt A, Wuchter P, Sellner L, Hofmann S, Ho AD, Dreger P, Schmitt M. Chimeric Antigen Receptor T Cell Therapy Targeting CD19-Positive Leukemia and Lymphoma in the Context of Stem Cell Transplantation. Hum Gene Ther. 2016;10:758–771. doi: 10.1089/hum.2016.097. PMID:27479233. [DOI] [PubMed] [Google Scholar]
- 5.Mamonkin M, Rouce RH, Tashiro H, Brenner MK. A T-cell-directed chimeric antigen receptor for the selective treatment of T-cell malignancies. Blood. 2015;126(8):983–92. doi: 10.1182/blood-2015-02-629527. PMID:26056165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pui CH, Behm FG, Crist WM. Clinical and biologic relevance of immunologic marker studies in childhood acute lymphoblastic leukemia. Blood. 1993;82(2):343–62. PMID:8329694. [PubMed] [Google Scholar]
- 7.Campana D, van Dongen JJ, Mehta A, Coustan-Smith E, Wolvers-Tettero IL, Ganeshaguru K, Janossy G. Stages of T-cell receptor protein expression in T-cell acute lymphoblastic leukemia. Blood. 1991;77(7):1546–54. PMID:1826223. [PubMed] [Google Scholar]
- 8.Osman N, Ley SC, Crumpton MJ. Evidence for an association between the T cell receptor/CD3 antigen complex and the CD5 antigen in human T lymphocytes. Eur J Immunol. 1992;22(11):2995–3000. doi: 10.1002/eji.1830221135. PMID:1385158. [DOI] [PubMed] [Google Scholar]
- 9.Berland R, Wortis HH. Origins and functions of B-1 cells with notes on the role of CD5. Annu Rev Immunol. 2002;20:253–300. doi: 10.1146/annurev.immunol.20.100301.064833. PMID:11861604. [DOI] [PubMed] [Google Scholar]
- 10.Azzam HS, Grinberg A, Lui K, Shen H, Shores EW, Love PE. CD5 expression is developmentally regulated by T cell receptor (TCR) signals and TCR avidity. J Exp Med. 1998;188(12):2301–11. doi: 10.1084/jem.188.12.2301. PMID:9858516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sigal LH. Basic science for the clinician 54: CD5. J Clin Rheumatol, 2012;18(2):83–8. doi: 10.1097/RHU.0b013e318247bc64. PMID:22334268. [DOI] [PubMed] [Google Scholar]
- 12.Bikah G, Carey J, Ciallella JR, Tarakhovsky A, Bondada S. CD5-Mediated Negative Regulation of Antigen Receptor-Induced Growth Signals in B-1 B Cells. Science. 1996;274(5294):1906–1909. doi: 10.1126/science.274.5294.1906. PMID:8943203. [DOI] [PubMed] [Google Scholar]
- 13.Dalloul A. CD5: a safeguard against autoimmunity and a shield for cancer cells. Autoimmun Rev. 2009;8(4):349–53. doi: 10.1016/j.autrev.2008.11.007. PMID:19041428. [DOI] [PubMed] [Google Scholar]
- 14.Bertram JH, Gill PS, Levine AM, Boquiren D, Hoffman FM, Meyer P, Mitchell MS. Monoclonal antibody T101 in T cell malignancies: a clinical, pharmacokinetic, and immunologic correlation. Blood. 1986;68(3):752–61. PMID:3488778. [PubMed] [Google Scholar]
- 15.LeMaistre CF, Rosen S, Frankel A, Kornfeld S, Saria E, Meneghetti C, Drajesk J, Fishwild D, Scannon P, Byers V. Phase I trial of H65-RTA immunoconjugate in patients with cutaneous T-cell lymphoma. Blood. 1991;78(5):1173–82. PMID:1878584. [PubMed] [Google Scholar]
- 16.Chen KH, Wada M, Pinz KG, Liu H, Lin KW, Jares A, Firor AE, Shuai X, Salman H, Golightly M. Preclinical targeting of aggressive T-cell malignancies using anti-CD5 chimeric antigen receptor. Leukemia. 2017;31(10):2151–2160. doi: 10.1038/leu.2017.8. PMID:28074066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wiedenheft B, Sternberg SH, Doudna JA. RNA-guided genetic silencing systems in bacteria and archaea. Nature. 2012;482(7385):331–8. doi: 10.1038/nature10886. PMID:22337052. [DOI] [PubMed] [Google Scholar]
- 18.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337(6096):816–21. doi: 10.1126/science.1225829. PMID:22745249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mojica FJ, Díez-Villaseñor C, García-Martínez J, Almendros C. Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology. 2009;155(Pt 3):733–40. PMID:19246744. [DOI] [PubMed] [Google Scholar]
- 20.Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, Horvath P. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315(5819):1709–12. doi: 10.1126/science.1138140. PMID:17379808. [DOI] [PubMed] [Google Scholar]
- 21.Doench JG, Hartenian E, Graham DB, Tothova Z, Hegde M, Smith I, Sullender M, Ebert BL, Xavier RJ, Root DE. Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nat Biotechnol. 2014;32(12):1262–7. doi: 10.1038/nbt.3026. PMID:25184501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cong L, Zhang F. Genome engineering using CRISPR-Cas9 system. Methods Mol Biol. 2015;1239:197–217. doi: 10.1007/978-1-4939-1862-1_10. PMID:25408407. [DOI] [PubMed] [Google Scholar]
- 23.Kaya H, Mikami M, Endo A, Endo M, Toki S. Highly specific targeted mutagenesis in plants using Staphylococcus aureus Cas9. Sci Rep. 2016;6:26871. doi: 10.1038/srep26871. PMID:27226350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moot R, Raikar SS, Fleischer L, Querrey M, Tylawsky DE, Nakahara H, Doering CB, Spencer HT. Genetic engineering of chimeric antigen receptors using lamprey derived variable lymphocyte receptors. Mol Ther Oncolytics. 2016;3:16026. doi: 10.1038/mto.2016.26. PMID:27933313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Herrin BR, Cooper MD. Alternative adaptive immunity in jawless vertebrates. J Immunol. 2010;185(3):1367–74. doi: 10.4049/jimmunol.0903128. PMID:20660361. [DOI] [PubMed] [Google Scholar]
- 26.Mariuzza RA, Velikovsky CA, Deng L, Xu G, Pancer Z. Structural insights into the evolution of the adaptive immune system: the variable lymphocyte receptors of jawless vertebrates. Biol Chem. 2010;391(7):753–60. doi: 10.1515/bc.2010.091. PMID:20482318. [DOI] [PubMed] [Google Scholar]
- 27.Boehm T, McCurley N, Sutoh Y, Schorpp M, Kasahara M, Cooper MD. VLR-based adaptive immunity. Annu Rev Immunol. 2012;30:203–20. doi: 10.1146/annurev-immunol-020711-075038. PMID:22224775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kasahara M, Sutoh Y. Two forms of adaptive immunity in vertebrates: similarities and differences. Adv Immunol. 2014;122:59–90. doi: 10.1016/B978-0-12-800267-4.00002-X. PMID:24507155. [DOI] [PubMed] [Google Scholar]
- 29.Yu C, Ali S, St-Germain J, Liu Y, Yu X, Jaye DL, Moran MF, Cooper MD, Ehrhardt GR. Purification and identification of cell surface antigens using lamprey monoclonal antibodies. J Immunol Methods. 2012;386(1-2):43–9. doi: 10.1016/j.jim.2012.08.016. PMID:22964555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kirchdoerfer RN, Herrin BR, Han BW, Turnbough CL Jr., Cooper MD, Wilson IA. Variable lymphocyte receptor recognition of the immunodominant glycoprotein of Bacillus anthracis spores. Structure. 2012;20(3):479–86. doi: 10.1016/j.str.2012.01.009. PMID:22405006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Studnicka GM, Soares S, Better M, Williams RE, Nadell R, Horwitz AH. Human-engineered monoclonal antibodies retain full specific binding activity by preserving non-CDR complementarity-modulating residues. Protein Eng. 1994;7(6):805–14. doi: 10.1093/protein/7.6.805. PMID:7937712. [DOI] [PubMed] [Google Scholar]
- 32.Huston JS, McCartney J, Tai MS, Mottola-Hartshorn C, Jin D, Warren F, Keck P, Oppermann H. Medical applications of single-chain antibodies. Int Rev Immunol. 1993;10(2-3):195–217. doi: 10.3109/08830189309061696. PMID:8360586. [DOI] [PubMed] [Google Scholar]
- 33.Gong JH, Maki G, Klingemann HG. Characterization of a human cell line (NK-92) with phenotypical and functional characteristics of activated natural killer cells. Leukemia. 1994;8(4):652–8. PMID:8152260. [PubMed] [Google Scholar]
- 34.Lee-MacAry AE, Ross EL, Davies D, Laylor R, Honeychurch J, Glennie MJ, Snary D, Wilkinson RW. Development of a novel flow cytometric cell-mediated cytotoxicity assay using the fluorophores PKH-26 and TO-PRO-3 iodide. J Immunol Methods. 2001;252(1-2):83–92. doi: 10.1016/S0022-1759(01)00336-2. PMID:11334968. [DOI] [PubMed] [Google Scholar]
- 35.Renaudineau Y, Hillion S, Saraux A, Mageed RA, Youinou P. An alternative exon 1 of the CD5 gene regulates CD5 expression in human B lymphocytes. Blood. 2005;106(8):2781–9. doi: 10.1182/blood-2005-02-0597. PMID:15998834. [DOI] [PubMed] [Google Scholar]
- 36.Cradick TJ, Qiu P, Lee CM, Fine EJ, Bao G. COSMID: A Web-based Tool for Identifying and Validating CRISPR/Cas Off-target Sites. Mol Ther Nucleic Acids 2014;3:e214. doi: 10.1038/mtna.2014.64. PMID:25462530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brinkman EK, Chen T, Amendola M, van Steensel B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014;42(22):e168. doi: 10.1093/nar/gku936. PMID:25300484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Henze G, Fengler R, Hartmann R, Kornhuber B, Janka-Schaub G, Niethammer D, Riehm H. Six-year experience with a comprehensive approach to the treatment of recurrent childhood acute lymphoblastic leukemia (ALL-REZ BFM 85). A relapse study of the BFM group. Blood. 1991;78(5):1166–72. PMID:1878583. [PubMed] [Google Scholar]
- 39.Einsiedel HG, von Stackelberg A, Hartmann R, Fengler R, Schrappe M, Janka-Schaub G, Mann G, Hählen K, Göbel U, Klingebiel T, et al.. Long-term outcome in children with relapsed ALL by risk-stratified salvage therapy: results of trial acute lymphoblastic leukemia-relapse study of the Berlin-Frankfurt-Munster Group 87. J Clin Oncol. 2005;23(31):7942–50. doi: 10.1200/JCO.2005.01.1031. PMID:16258094. [DOI] [PubMed] [Google Scholar]
- 40.Reismuller B, Peters C, Dworzak MN, Pötschger U, Urban C, Meister B, Schmitt K, Dieckmann K, Gadner H, Attarbaschi A, et al.. Outcome of children and adolescents with a second or third relapse of acute lymphoblastic leukemia (ALL): a population-based analysis of the Austrian ALL-BFM (Berlin-Frankfurt-Munster) study group. J Pediatr Hematol Oncol. 2013;35(5):e200–4. doi: 10.1097/MPH.0b013e318290c3d6. PMID:23652878. [DOI] [PubMed] [Google Scholar]
- 41.Burke MJ, Verneris MR, Le Rademacher J, He W, Abdel-Azim H, Abraham AA, Auletta JJ, Ayas M, Brown VI, Cairo MS, et al.. Transplant Outcomes for Children with T Cell Acute Lymphoblastic Leukemia in Second Remission: A Report from the Center for International Blood and Marrow Transplant Research. Biol Blood Marrow Transplant. 2015;21(12):2154–9. doi: 10.1016/j.bbmt.2015.08.023. PMID:26327632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bakr M, Rasheed W, Mohamed SY, Al-Mohareb F, Chaudhri N, Al-Sharif F, Al-Zahrani H, Al-Dawsari G, Saleh AJ, Nassar A, et al.. Allogeneic hematopoietic stem cell transplantation in adolescent and adult patients with high-risk T cell acute lymphoblastic leukemia. Biol Blood Marrow Transplant. 2012;18(12):1897–904. doi: 10.1016/j.bbmt.2012.07.011. PMID:22824185. [DOI] [PubMed] [Google Scholar]
- 43.Raetz EA, Borowitz MJ, Devidas M, Linda SB, Hunger SP, Winick NJ, Camitta BM, Gaynon PS, Carroll WL. Reinduction platform for children with first marrow relapse of acute lymphoblastic Leukemia: A Children's Oncology Group Study[corrected]. J Clin Oncol. 2008;26(24):3971–8. doi: 10.1200/JCO.2008.16.1414. PMID:18711187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Laurent G, Pris J, Farcet JP, Carayon P, Blythman H, Casellas P, Poncelet P, Jansen FK. Effects of therapy with T101 ricin A-chain immunotoxin in two leukemia patients. Blood. 1986;67(6):1680–7. PMID:3085747. [PubMed] [Google Scholar]
- 45.Koehler M, Hurwitz CA, Krance RA, Coustan-Smith E, Williams LL, Santana V, Ribeiro RC, Brenner MK, Heslop HE. XomaZyme-CD5 immunotoxin in conjunction with partial T cell depletion for prevention of graft rejection and graft-versus-host disease after bone marrow transplantation from matched unrelated donors. Bone Marrow Transplant. 1994;13(5):571–5. PMID:7519937. [PubMed] [Google Scholar]
- 46.Byers VS, Henslee PJ, Kernan NA, Blazar BR, Gingrich R, Phillips GL, LeMaistre CF, Gilliland G, Antin JH, Martin P. Use of an anti-pan T-lymphocyte ricin a chain immunotoxin in steroid-resistant acute graft-versus-host disease. Blood. 1990;75(7):1426–32. PMID:2180494. [PubMed] [Google Scholar]
- 47.Tarakhovsky A, Kanner SB, Hombach J, Ledbetter JA, Müller W, Killeen N, Rajewsky K. A role for CD5 in TCR-mediated signal transduction and thymocyte selection. Science. 1995;269(5223):535–537. doi: 10.1126/science.7542801. PMID:7542801. [DOI] [PubMed] [Google Scholar]
- 48.Krammer PH, Arnold R, Lavrik IN. Life and death in peripheral T cells. Nat Rev Immunol. 2007;7(7):532–42. doi: 10.1038/nri2115. PMID:17589543. [DOI] [PubMed] [Google Scholar]
- 49.Hermanson DL, Kaufman DS. Utilizing chimeric antigen receptors to direct natural killer cell activity. Front Immunol. 2015;6:195. doi: 10.3389/fimmu.2015.00195. PMID:25972867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Glienke W, Esser R, Priesner C, Suerth JD, Schambach A, Wels WS, Grez M, Kloess S, Arseniev L, Koehl U. Advantages and applications of CAR-expressing natural killer cells. Front Pharmacol. 2015;6:21. doi: 10.3389/fphar.2015.00021. PMID:25729364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Klingemann H. Are natural killer cells superior CAR drivers? Oncoimmunology. 2014;3:e28147. doi: 10.4161/onci.28147. PMID:25340009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Suck G, Odendahl M, Nowakowska P, Seidl C, Wels WS, Klingemann HG, Tonn T. NK-92: an ‘off-the-shelf therapeutic’ for adoptive natural killer cell-based cancer immunotherapy. Cancer Immunol Immunother. 2015. PMID:26559813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chu J, Deng Y, Benson DM, He S, Hughes T, Zhang J, Peng Y, Mao H, Yi L, Ghoshal K, et al.. CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. Leukemia. 2014;28(4):917–27. doi: 10.1038/leu.2013.279. PMID:24067492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Romanski A, Uherek C, Bug G, Seifried E, Klingemann H, Wels WS, Ottmann OG, Tonn T. CD19-CAR engineered NK-92 cells are sufficient to overcome NK cell resistance in B-cell malignancies. J Cell Mol Med. 2016;20(7):1287–94. doi: 10.1111/jcmm.12810. PMID:27008316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Oelsner S, Friede ME, Zhang C, Wagner J, Badura S, Bader P, Ullrich E, Ottmann OG, Klingemann H, Tonn T, et al.. Continuously expanding CAR NK-92 cells display selective cytotoxicity against B-cell leukemia and lymphoma. Cytotherapy. 2017;19(2):235–249. doi: 10.1016/j.jcyt.2016.10.009. PMID:27887866. [DOI] [PubMed] [Google Scholar]
- 56.Boissel L, Betancur-Boissel M, Lu W, Krause DS, Van Etten RA, Wels WS, Klingemann H. Retargeting NK-92 cells by means of CD19- and CD20-specific chimeric antigen receptors compares favorably with antibody-dependent cellular cytotoxicity. Oncoimmunology. 2013;2(10):e26527. doi: 10.4161/onci.26527. PMID:24404423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schonfeld K, Sahm C, Zhang C, Naundorf S, Brendel C, Odendahl M, Nowakowska P, Bönig H, Köhl U, Kloess S, et al.. Selective inhibition of tumor growth by clonal NK cells expressing an ErbB2/HER2-specific chimeric antigen receptor. Mol Ther. 2015;23(2):330–8. doi: 10.1038/mt.2014.219. PMID:25373520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chu Y, Hochberg J, Yahr A, Ayello J, van de Ven C, Barth M, Czuczman M, Cairo MS. Targeting CD20+ Aggressive B-cell Non-Hodgkin Lymphoma by Anti-CD20 CAR mRNA-Modified Expanded Natural Killer Cells In Vitro and in NSG Mice. Cancer Immunol Res. 2015;3(4):333–44. doi: 10.1158/2326-6066.CIR-14-0114. PMID:25492700. [DOI] [PubMed] [Google Scholar]
- 59.Esser R, Müller T, Stefes D, Kloess S, Seidel D, Gillies SD, Aperlo-Iffland C, Huston JS, Uherek C, Schönfeld K, et al.. NK cells engineered to express a GD2 -specific antigen receptor display built-in ADCC-like activity against tumour cells of neuroectodermal origin. J Cell Mol Med. 2012;16(3):569–81. doi: 10.1111/j.1582-4934.2011.01343.x. PMID:21595822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jiang H, Zhang W, Shang P, Zhang H, Fu W, Ye F, Zeng T, Huang H, Zhang X, Sun W. Transfection of chimeric anti-CD138 gene enhances natural killer cell activation and killing of multiple myeloma cells. Mol Oncol. 2014;8(2): p. 297–310. doi: 10.1016/j.molonc.2013.12.001. PMID:24388357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shimasaki N, Coustan-Smith E, Kamiya T, Campana D. Expanded and armed natural killer cells for cancer treatment. Cytotherapy. 2016;18(11):1422–1434. doi: 10.1016/j.jcyt.2016.06.013. PMID:27497701. [DOI] [PubMed] [Google Scholar]
- 62.Li L, Allen C, Shivakumar R, Peshwa MV. Large volume flow electroporation of mRNA: clinical scale process. Methods Mol Biol 2013;969:127–38. doi: 10.1007/978-1-62703-260-5_9. PMID:23296932. [DOI] [PubMed] [Google Scholar]
- 63.Chu Y, Flower A, Cairo MS. Modification of Expanded NK Cells with Chimeric Antigen Receptor mRNA for Adoptive Cellular Therapy. Methods Mol Biol. 2016;1441:215–30. doi: 10.1007/978-1-4939-3684-7_18. PMID:27177669. [DOI] [PubMed] [Google Scholar]
- 64.Brouns SJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJ, Snijders AP, Dickman MJ, Makarova KS, Koonin EV, van der Oost J. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science. 2008;321(5891):960–4. doi: 10.1126/science.1159689. PMID:18703739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8(11):2281–308. doi: 10.1038/nprot.2013.143. PMID:24157548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Anders C, Niewoehner O, Duerst A, Jinek M. Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature. 2014;513(7519):569–73. doi: 10.1038/nature13579. PMID:25079318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cyranoski D. First trial of CRISPR in people. Nature. 2016;535(7613):476–477. doi: 10.1038/nature.2016.20302. PMID:27466105. [DOI] [PubMed] [Google Scholar]
- 68.Chmielewski M, Hombach A, Heuser C, Adams GP, Abken H. T cell activation by antibody-like immunoreceptors: increase in affinity of the single-chain fragment domain above threshold does not increase T cell activation against antigen-positive target cells but decreases selectivity. J Immunol. 2004;173(12):7647–53. doi: 10.4049/jimmunol.173.12.7647. PMID:15585893. [DOI] [PubMed] [Google Scholar]
- 69.Chmielewski M, Hombach AA, Abken H. CD28 cosignalling does not affect the activation threshold in a chimeric antigen receptor-redirected T-cell attack. Gene Ther. 2011;18(1):62–72. doi: 10.1038/gt.2010.127. PMID:20944680. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.