

ABSTRACT
Immunotherapy has expanded treatment options for cancers with historically poor outcomes, yet a significant proportion of patients still fail to achieve durable clinical benefit. We defined the contribution of β-adrenergic receptor (βAR) signaling, a component of the stress response, on success of immunotherapy for melanoma since the use of antagonists (β-blockers) is associated with improved clinical outcomes in some cancers. We show that metastatic melanoma patients who received immunotherapy had improved overall survival if they also received pan β-blockers. This retrospective analysis is reinforced by results showing that βAR blockade enhances the control of murine melanoma growth by anti-(α)PD-1 checkpoint blockade. However, this effect was most significant when β-blocker was combined with dual αPD-1 + high dose interleukin-2 therapy and was reproduced by selective blockade of β2ARs. These results identify a novel strategy that can be quickly introduced to potentially increase the number of patients who benefit from immune-based therapies.
KEYWORDS: Beta-adrenergic receptor, Beta-blockers, Immunotherapy, Metastatic melanoma patients, Mice
Introduction
Metastatic melanoma remains a significant clinical problem, with five-year survival rates of only 15–20%.1 Until recently, high dose interleukin-2 (IL-2) was the only FDA-approved immune based therapy available for the treatment of metastatic melanoma. However, overall response rates for metastatic melanoma patients treated with IL-2 are reported at less than 20%.2 Although complete responses from IL-2 therapy are rare, they are typically durable when achieved.3,4 These dismal survival rates are likely to improve over time following recent FDA approvals of checkpoint inhibitor therapies including antibodies that block cytotoxic T-lymphocyte-associated protein 4 (CTLA-4)5 or the programmed cell death protein 1 (PD-1)6,7 pathways. Although promising, the clinical response rate to these therapies remains low at less than 35% with a large percentage of patients experiencing no benefit or only a transient response to these treatment modalities.8,9 These observations highlight the need for novel therapeutic strategies which take advantage of the beneficial aspects of immunotherapy while improving clinical outcome by increasing the durable response rates.
The influence of β-adrenergic receptor (βAR) driven stress on the immune response has been long recognized and the role of stress in suppressing the anti-tumor immune response is well documented.10,11 However, the precise mechanism by which stress hinders anti-tumor immunity is still poorly understood. Recent pre-clinical studies have shown that under conditions of reduced physiological stress, the T cell dependent anti-tumor immune response is greatly enhanced12,13 and the efficacy of standard cancer therapies is improved.14,15 In contrast, stress driven βAR signaling is immunosuppressive, leading to increased numbers of immune suppressive cells,12 reduced expression of T cell growth-promoting cytokines16 and impeded T cell cytotoxicity.12 These findings suggest that targeting the βAR signaling pathway directly to reduce stress signaling may provide an innovative approach to improve cancer treatment.
βAR signaling can be inhibited pharmacologically by treatment with antagonists. Commonly known as β-blockers, βAR antagonists are recommended for all patients at risk of heart failure independent of age and sex.17,18 Clinically, β-blockers are widely used in multiple disease conditions including hypertension, acute myocardial infarction, congestive heart failure, essential tremor, migraines, glaucoma and anxiety disorders.17,19 The most commonly prescribed antagonists are pan β-blockers and β1-selective blockers which target both β1ARs and β2ARs or only β1ARs, respectively. Several studies demonstrate a positive correlation between β-blocker usage, as prescribed for non-cancer related indications, and outcome in various malignancies including breast20-24 and ovarian25,26 cancers. These pre-clinical and clinical observations suggest an important role for β-blockers in the anti-tumor immune response.
Combination therapy is an attractive strategy to overcome the resistance associated with single agent approaches. We hypothesized that βAR blockade delivered in combination with T cell dependent immunotherapy would improve the efficacy of immunotherapeutic approaches for melanoma. In this study, we performed a retrospective analysis to determine how β-blocker usage among metastatic melanoma patients that received immunotherapy impacts overall survival (OS) and investigated the feasibility of combined βAR blockade and immunotherapy in a mouse model of melanoma.
Results
β-blocker usage correlates with improved overall survival among metastatic melanoma patients that received immunotherapy
We performed a retrospective analysis of metastatic melanoma patients treated with immune-based therapies at the Penn State Cancer Institute between 2000 and 2015. Patients (N = 195) treated with IL-2, αCTLA-4 and/or αPD-1 were included in the analysis. We found that approximately 32% (N = 62) of these patients were prescribed some class of β-blockers. Previous studies suggest that the stress response most dramatically impacts immunity through β2AR.27,28 Since β2-selective blockers are not clinically prescribed, patients were stratified into those that were not prescribed β-blockers, those that were prescribed β1-selective blockers and those that were prescribed pan β-blockers that antagonize both β1ARs and β2ARs. Age at time of immunotherapy, gender, stage at initial diagnosis and type of immunotherapy received were similar between these cohorts (Table 1 and supplementary Table S1). We found no difference in OS following the initiation of immunotherapy between patients taking β1-selective blockers and those taking no β-blocker (Fig. 1). In contrast, we observed a significant survival benefit for patients prescribed pan β-blockers compared to either those taking no β-blocker or β1-selective blockers. The types of immunotherapies were broadly distributed among the patient groups stratified by β-blocker usage (Supplementary Table S2), suggesting an advantage for patients that received pan β-blocker independent of the specific types of immunotherapy received. These results suggest that pan β-blocker usage may improve clinical outcomes for patients that receive multiple types of immunotherapy in the setting of metastatic melanoma.
Table 1.
Characteristics of metastatic melanoma patients.
| All Patients | No β-blocker | β1-blocker | Pan β-blocker | p value | |
|---|---|---|---|---|---|
| N = 195 | N = 133 | N = 45 | N = 17 | ||
| Age at start of immunotherapy treatment | |||||
| Mean | 60.7 | 60.1 | 62.3 | 61.5 | |
| (SD) | (14.7) | (14.7) | (14.4) | (16.3) | 0.6294 |
| Median | 61.8 | 61.4 | 63.1 | 69.6 | |
| (Range) | (22–92) | (22–92) | (30–89) | (23–80) | |
| Sex | |||||
| Male | 124 (63.6%) | 83 (62.4%) | 31 (68.9%) | 10 (58.8%) | 0.6726 |
| Female | 71 (36.4%) | 50 (37.6%) | 14 (31.1%) | 7 (41.2%) | |
| Stage at Initial Diagnosis* | |||||
| 0 | 2 (1.2%) | 2 (1.2%) | 0 (0%) | 0 (0%) | |
| I | 31 (19.1%) | 22 (20.2%) | 7 (17.9%) | 2 (14.3%) | |
| II | 40 (24.7%) | 27 (24.8%) | 10 (25.6%) | 3 (21.4%) | |
| III | 57 (35.2%) | 38 (34.9%) | 13 (33.3%) | 6 (42.9%) | |
| IV | 32 (19.8%) | 20 (18.3%) | 9 (23.1%) | 3 (21.4%) | 0.9830 |
Descriptive characteristics of the patients in this study are summarized overall and by β-blocker usage. Where not indicated as SD or range, values represent the number of patients followed by the percentage of that column in parenthesis. The p-values were obtained using nonparametric Kruskal-Wallis tests or Fisher's exact tests with p<0.05 considered significant.
Note that all patients had metastatic disease at the time of immunotherapy regardless of staging at diagnosis.
Figure 1.
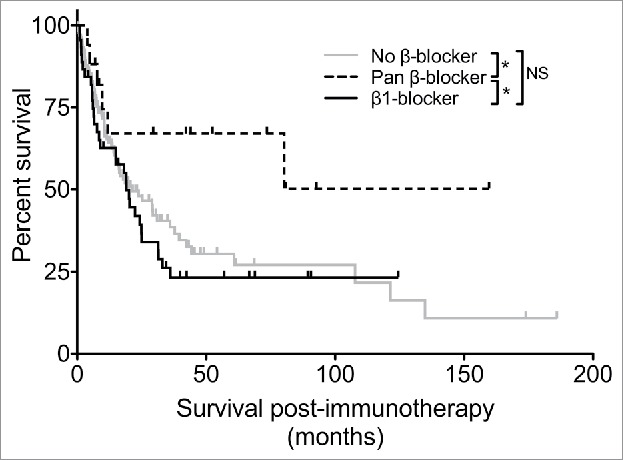
Malignant melanoma patients receiving pan β-blockers have prolonged survival following immunotherapy. Overall survival following the initiation of immunotherapy treatment was determined for melanoma patients treated with at least one immunotherapy (IL-2, αCTLA-4, αPD-1). Patients were stratified based on specific β1AR antagonist use, non-specific pan βAR antagonist use or no β-blocker use. N = 195; * = p <0.05 determined by log rank test; NS = not significant.
βAR blockade improves the anti-tumor efficacy of immune-based therapies in a preclinical model of melanoma
We developed a parallel murine model (Fig. 2A) in order to better understand the role βAR signaling could play in the enhanced efficacy of immunotherapies suggested by our clinical study. We utilized C57BL/6J mice bearing established (palpable, ∼1 week) subcutaneous wild type B16-F10-tumors lacking exogenous immunogenic T cell epitopes; a highly aggressive melanoma model.29,30 Tumor-bearing mice received IL-2, αPD-1, or their combination with and without a daily dose of the clinically available pan-β-blocker propranolol (Fig. 2A). Previous studies have shown only a minor or insignificant effect of αPD-1 therapy alone on the growth of B16-F10 tumors.31,32 Consistent with these previous results, we observed a statistically significant but not biologically relevant delay in tumor growth following administration of αPD-1 monotherapy (Fig. 2C) compared to control-treated (PBS) mice (Fig. 2B). In contrast, IL-2 administration produced a uniform delay in tumor growth (Fig. 2D) that was not further enhanced by combination with αPD-1 (Fig. 2E). Treatment with propranolol alone provided a minimal, although statistically significant delay in tumor growth (Fig. 2F). However, propranolol administration significantly delayed tumor growth when combined with αPD-1 (Fig. 2G) compared to either monotherapy. Propranolol with IL-2 (Fig. 2H) also produced a statistically significant delay in tumor growth compared to IL-2 alone (Fig. 2C), although the magnitude of improvement was less than observed in combination with αPD-1.
Figure 2.

Propranolol improves tumor control in mice treated with immunotherapy. (A) Experimental schema. When applicable, tumor-bearing mice received 10mg/kg propranolol (pan β-blocker) daily for three weeks, 200μg αPD-1 twice a week for three weeks and/or 120,000 IU IL-2 twice a day for two cycles of five days on, two days off. An equivalent volume of PBS was given daily to control mice. Mice were treated with (B) PBS control, (C) αPD-1, (D) IL-2, (E) αPD-1/IL-2, (F) Propranolol, (G) Propranolol + αPD-1, (H) Propranolol + IL-2, (I) Propranolol + αPD-1/IL-2. Days 15 and 30 are indicated with long- and short-dashed lines. N = 7–8/group; p values determined by mixed linear models; pairwise comparison to untreated: ** p < 0.01, **** p < 0.0001; pairwise comparison to propranolol: ˆˆ p < 0.01, ˆˆˆ p <0.001; pairwise comparison between immunotherapy and propranolol + immunotherapy: @ p < 0.05, @@ p < 0.01. (J) Survival analysis. N = 7/group; p values determined by log rank test. Data are representative of two experiments. MS = median survival.
Since several patients in our study received multiple immunotherapy regimens in sequence (Supplementary Table S2), we also evaluated the impact of combined αPD-1/IL-2 therapy in this mouse model. The combination of αPD-1/IL-2 did not substantially delay tumor progression beyond that observed with IL-2 alone (Fig. 2E). However, the addition of propranolol to this regimen significantly delayed tumor progression compared to αPD-1/IL-2 alone (Fig. 2I) although no complete regressions were observed. In addition, triple combination of propranolol + αPD-1/IL-2 (Fig. 2I) delayed tumor progression more effectively than propranolol + αPD-1 (Fig. 2G), suggesting that the addition of IL-2 optimally promotes the anti-tumor response.
Survival analysis of mice within these same cohorts revealed that β-blocker alone had no survival benefit (Fig. 2J; median 15 days) and propranolol did not improve the already significant increase induced by IL-2 therapy (median 23.5 versus 22.5 days). However, propranolol significantly prolonged the survival of αPD-1 treated mice (median 25 versus 18 days; Fig. 2J). Further, survival was most dramatically extended when propranolol was administered with combined αPD-1/IL-2 (median 39 days; Fig. 2J). Taken together, our data reveal that pan βAR blockade effectively enhanced the anti-tumor properties of αPD-1-based therapy but the combination of pan βAR blockade with αPD-1/IL-2 was most effective, suggesting that pan β-blockers may function through multiple mechanisms to improve this combined therapeutic approach.
β2AR blockade is sufficient to enhance the efficacy of immune-based therapies
We sought to determine the influence of β1AR and β2AR on the improved immunotherapy observed in this murine melanoma model. Although β2AR antagonists have minimal toxicity in healthy humans,33 there are currently no clinical indications for which these selective β-blockers are prescribed. However, these compounds have been used routinely in pre-clinical studies to dissect the roles of each βAR subset. We utilized selective β1AR (metoprolol) and β2AR (ICI 118,551) antagonists in combination with αPD-1/IL-2. Tumors in control treated mice (Fig. 3A) grew at a similar rate to those treated with either metoprolol (Fig. 3B) or ICI 118,551 (Fig. 3C). We again observed a significant delay in tumor growth when treating with αPD-1/IL-2 (Fig. 3D) compared to control treated mice (Fig. 3A). However, only β2AR blockade with ICI 118,551(Fig. 3F) demonstrated improved tumor control compared with αPD-1/IL-2 alone (Fig. 3D–F). Likewise, while survival was improved by combined αPD-1/IL-2 therapy (median 14 versus 23 days), only ICI 118,551 administration further improved survival in combination with αPD-1/IL-2 (Fig. 3G; median 35 days). Neither βAR-selective inhibitor alone improved survival of tumor-bearing mice compared to control treated mice. These data indicate that reduced signaling through β2AR plays an essential role in the improved tumor control observed following immunotherapy and suggest that the beneficial effect of pan β-blockers works primarily by modulating β2AR signaling.
Figure 3.
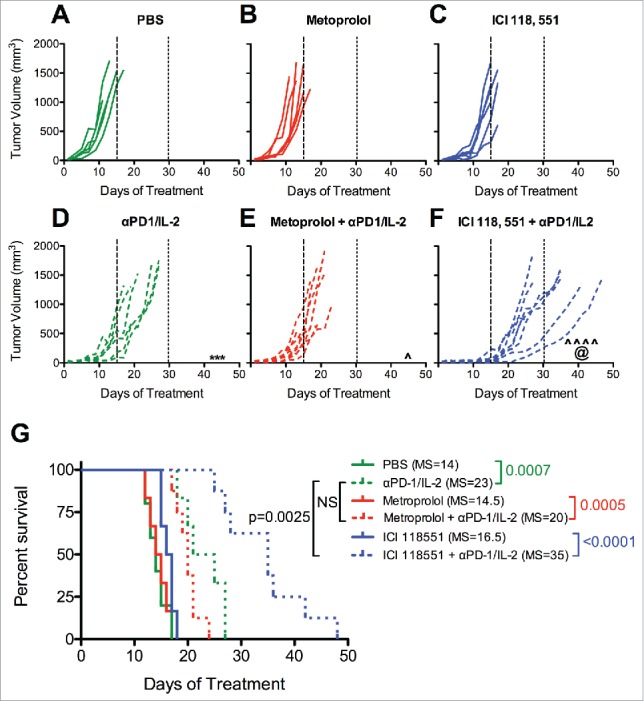
β2AR-selective blockade improves immunotherapy against melanoma. Mice were treated as indicated in Figure 2 with (A) PBS control, (B) Metoprolol (β1AR selective antagonist), (C) ICI 118, 551 (β2AR selective antagonist), (D) αPD-1/IL-2, (E) Metoprolol + αPD-1/IL-2, (F) ICI 118, 551 + αPD-1/IL-2. Days 15 and 30 are indicated with long- and short-dashed lines, respectively. N = 5–8/group; p values determined by mixed linear models; pairwise comparison to untreated: *** p < 0.001; pairwise comparison to same β-blocker: ˆ p < 0.05, ˆˆˆˆ p < 0.0001; pairwise comparison between immunotherapy alone and β-blocker + immunotherapy: @ p < 0.05. (G) Survival analysis. The median survival (days) is listed in parenthesis for each group. N = 5–7/group; p values determined by log rank test; NS = not significant; MS = median survival.
Discussion
Here we make the novel observation that metastatic melanoma patients who had received immunotherapy and were taking pan β-blockers experienced prolonged OS. This clinical observation is supported by our results demonstrating that combining a non-selective pan β-blocker or selective β2AR antagonist with either αPD-1 or combination αPD-1/IL-2 therapy significantly slowed tumor progression and extended survival in a murine melanoma model. Further, we have demonstrated that inhibition of signaling through β2AR plays the prominent role in the improved anti-tumor immunity seen following immunotherapy. Our data raise the possibility that reducing stress signaling, particularly through the β2AR signaling pathway, can improve the efficacy of immune based therapies for melanoma and perhaps other cancers as well. These results indicate that β-blockers and immunotherapy may synergize to enhance immune cell activity and/or function to a higher degree than either approach alone.
Given the positive impact on immunotherapy observed, βAR signaling could directly augment the anti-tumor immune response as β2ARs are expressed on most immune cell types. Among immune cell subsets, NK cells express the highest number of receptors followed by monocytes, B cells, CD8 T cells and CD4 T cells.34 Increased βAR signaling reduces proinflammatory cytokine secretion from monocytes and macrophages.35,36 Within the T cell compartment, CD4 T cells show decreased Th1 cytokine production37 and Treg function is enhanced38 following βAR stimulation; two potential mechanisms that could hinder anti-tumor immunity and immunotherapy efficacy. Additionally, β2AR signaling limited the IFNγ-driven CD8 T cell response to influenza infection39 and βAR stimulation resulted in reduced TNFα production by macrophages and T cells that was directly linked to the inability to generate anti-tumor CD8 T cells.40 Further, IFNγ production by and cytotoxicity of CD8+ T cells is inhibited by the β2AR agonist salmeterol.41 Relevant to the current study is the finding that βAR signaling can suppress T cell receptor-induced cytokine secretion and lytic activity exclusively through β2AR signaling.28 Previous work from some in our group demonstrates that environmental conditions that activate sympathetic signaling regulate immune responses. Increased necessity to thermoregulate body temperature due to cool housing temperature reduced tumor control and hindered CD8 T cells and dendritic cells in multiple murine tumor models.12,13 Thus, our results are consistent with a model in which β2AR blockade may directly improve immune cell functions, resulting in a prolonged control of tumor progression. Although in our study most patients prescribed β-blockers were receiving selective β1AR agents, it is of note that a recent meta-analysis concluded that pan β-blockers may in fact yield better clinical outcomes in patients with hypertension, diabetes, heart failure and acute myocardial infarction.19
While our results imply that the anti-melanoma immune response is improved with βAR blockade, direct effects on tumor growth may also be involved. Studies in murine xenograft models show that βAR blockade increases the efficacy of chemotherapies independent of adaptive immunity.14,15 However, the mechanistic relationship between βAR signaling, tumor growth and the anti-tumor immune response is not well understood in regards to any cancer, including melanoma. B16-F10 murine melanoma cells express a small proportion of functional βAR sites and thus may be responsive to manipulation of βAR signaling.42 Additionally, βAR expression has been detected in melanoma biopsies43 purporting that human melanoma may be directly sensitive to β-blocker treatment. Thus, the full impact of βAR signaling on melanoma progression and treatment remains incompletely defined.
Our data support recent results showing that the addition of βAR blockade significantly improved the impact of αPD-1 therapy across distinct murine tumor models, including B16-F10 melanoma expressing the model antigen ovalbumin and 4T1 mammary tumors.44 This recent study yielded similar results to those shown here in terms of the impact of propranolol on αPD-1 therapy of melanoma, yet used an altered treatment protocol, including more frequent αPD-1 administration. Here we have extended these findings using the parental B16-F10 melanoma model and determined that improved anti-tumor immunity is dependent on β2AR blockade. Additionally, we show in the current study that βAR blockade had a greater impact on dual αPD-1/IL-2 therapy than on αPD-1 therapy alone. Our finding that pan or selective β2AR antagonists improved αPD-1 therapy but only minimally impacted IL-2-based therapy may indicate that βAR blockade and IL-2 target overlapping mechanisms, such as T cell proliferation. Indeed, norepinephrine decreases T cell proliferation by reducing IL-2 production.16 Thus, in the presence of high dose IL-2, autologous IL-2 production gained from βAR blockade is unlikely to contribute to the overall response. In contrast, βAR blockade may improve T cell proliferation through autocrine IL-2 production and then the addition of αPD-1 therapy may further enhance T cell survival and function. βAR blockade may also promote T cell recruitment to the tumors by relieving their retention in the lymph nodes.45 Alternatively, βAR blockade may predominantly affect the sensitivity of the tumor to the immune system.
The basis for our finding that administration of β-blockers with combined αPD-1/IL-2 was more effective than βAR blockade with αPD-1 or αPD-1/IL-2 alone suggests that these three agents cooperate to improve the response to the individual agents. βAR blockade with αPD-1 may sensitize the responding immune cells to the effects of IL-2. Alternatively, βAR blockade with IL-2 may promote T cell exhaustion that masks any synergy between these two agents, but is revealed by the inclusion αPD-1. Further studies are required to dissect the cellular and molecular nature of these interactions and to define whether a more targeted approach can be identified.
Both our pre-clinical and clinical data implicate β2AR blockade in improving the impact of immunotherapy to melanoma. Most patients in our study were taking β1-selective blockers rather than pan β-blockers. These data suggest the presence of a sizeable population of metastatic melanoma patients who might benefit from combined pan βAR blockade and immunotherapy. Our retrospective analysis provides a strong rationale to further evaluate how pan βAR blockade may impact immunotherapy in the clinic. While we were unable to extract other patient outcomes associated with pan β-blocker use in this retrospective cohort of patients, future prospective studies are needed to define the relationship between pan βAR blockade and clinical outcomes such as time to metastasis or recurrence and the degree of response to immunotherapy (complete response, partial response, stable disease, progressive disease). Given our results implicating the role of β2AR signaling in resistance to immunotherapy, an additional retrospective analysis could be performed investigating whether use of β2AR agonists, commonly used for control of asthma, negatively impact the response to immunotherapy. Prospective clinical trials are needed to definitively test the benefit of this approach, but the combination of already FDA-approved drugs could be rapidly implemented for metastatic melanoma and potentially other cancers.
Patients and methods
Clinical Data Collection
The retrospective analysis of patient data was performed under a protocol approved by the Penn State Cancer Institute Scientific Review Committee and the Penn State Hershey Medical Center Institutional Review Board. Due to the retrospective nature of the study without collection of new data or specimens, this study was exempt from patient consent. Patients aged 18 or older with a history of melanoma who received IL-2, αPD-1 and/or αCTLA-4 immunotherapy were eligible. Some patients received more than one type of immunotherapy. Data were collected on eligible patients diagnosed between January 1, 2000 and March 31, 2015. Data including age at the start of immunotherapy treatment, gender, melanoma stage at diagnosis, cancer treatment and β-blocker usage were extracted from the Penn State Cancer Institute Tumor Registry and the Penn State Informatics for Integrating Biology and the Bedside (i2b2) System. Patients that were prescribed β1AR (Atenolol, Esmolol, and Metoprolol) and pan βAR (Carvedilol, Labetalol and Propranolol) antagonists were identified from these databases. Any patient who did not have data available in both the Tumor Registry and i2b2 were excluded. Patient medical records were also viewed to extract data missing from the Tumor Registry and i2b2.
Mice, cell lines and media
Female 6–8 week old C57BL/6J mice were purchased from The Jackson Laboratory and used within 2 weeks of receipt. All mice were maintained in specific pathogen-free conditions in a HEPA-filtered ventilated rack system at the Milton S. Hershey Medical Center animal facility. Mice were housed 5 or less per cage and fed and watered ad libitem. Mice were maintained in a 12-hour light/dark cycle. All experiments with animals were performed in accordance with institutional guidelines under protocol #46648 that was approved by the Institutional Animal Care and Use Committee at the Penn State College of Medicine. B16-F10 melanoma cells were obtained directly from the American Type Culture Collection (ATTC; CRL-6475) in February of 2015 and stored frozen in aliquots after 2 passages in vitro. Cells were cultured in RPMI-1640 with GlutaMAX™(Gibco) supplemented with 10% fetal bovine serum (HyClone), 100 U/mL penicillin, 100 μg/mL streptomycin, 2 mM L-glutamine, 50 μM 2-mercaptoethanol, 10 mM HEPES, and 25 μg/mL pyruvic acid. For in vivo experiments, cells were harvested at ∼90% confluency at less than 6 passages from the original stock. 1 × 105 freshly harvested tumor cells were suspended in PBS and injected subcutaneously into the left flank of C57BL/6J mice.
Murine tumor model and treatment regimens
Following tumor implantation, mice were monitored daily to identify palpable tumors at which time they were randomized into treatment groups. Treatments were administered to mice bearing palpable tumors at ∼6–9 days following inoculation (tumor volume ∼20 – 100mm3). All treatments were given intraperitoneally in a volume of 200 µl. Sterile PBS was used for control injections. IL-2 was obtained from Prometheus Inc. and administered at 120,000 IU twice a day for two five-day cycles separated by a two day break. αPD-1 (clone: RMP-1) was purchased from BioXcell and up to six 200 µg doses were administered (days 1, 4, 8, 11, 15, 18). All βAR antagonists were purchased from Sigma and given daily for 3 weeks at the following concentrations: ICI 118,551 (1 mg/kg), metoprolol (10 mg/kg) and propranolol (10 mg/kg) as previously described.27 Tumors were measured with digital calipers and tumor volume was calculated in mm3 using the formula (length*width*width)/2. Following the initiation of treatment, tumors were measured every two days until size reached 1000 mm3 at which point measurements were taken every day. The endpoint for survival studies included when tumor volume reached >1500 mm3, mice became lethargic or ascites developed prohibiting movement.
Statistical methods
Linear mixed models for longitudinal data were used to assess differences in tumor growth curves between different treatment groups. P-values less than 0.05 are reported and indicated on the figures. Kaplan-Meier survival plots and log-rank tests were used to display and analyze the time from the beginning of treatment to death or the date of sacrifice. All tests were two-sided. The statistical significance level used was 0.05, and it was not adjusted for multiple testing due to the exploratory nature of this study. For the analyses of clinical data, descriptive statistics were used to summarize patient's characteristics. The basic comparison of patient's characteristics between treatment groups was performed using Fisher's exact test or nonparametric Kruskal-Wallis test when appropriate. Overall survival was defined as the length of time from start of immunotherapy for the metastatic or recurrent disease (i.e. immunotherapy start date) to the date of death or last follow-up. Kaplan-Meier plots and log-rank tests were used to evaluate association of β-blocker usage and overall survival. All analyses were performed using statistical software SAS version 9.4, Graphpad Prism version 5.0f, and R programming language 3.1.2.
Supplementary Material
Funding Statement
This project was supported, in part, by the Pennsylvania Department of Health using Tobacco CURE Funds. The Department specifically disclaims responsibility for any analyses, interpretations or conclusions. KK was supported by National Cancer Institute/National Institutes of Health training grant T32 CA060395.
Disclosure of potential conflicts of interest
The authors declare no potential conflicts of interest
Acknowledgments
The authors thank Dr. Hong Zheng for providing editorial comments, Lisa Hand of the PSCI Cancer Registry for assistance with data collection, Matt Bolton and Masayo Mesler for assistance with data extraction from the i2b2 system, Jaenell Ditsious for data extraction support and Jeremy Haley for outstanding technical support. We also thank Prometheus Laboratories, Inc. for generously providing recombinant human IL-2 for this project.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 2.Bright R, Coventry BJ, Eardley-Harris N, Briggs N. Clinical response rates from interleukin-2 therapy for metastatic melanoma over 30 years' experience: a meta-analysis of 3312 patients. J Immunother. 2017;40:21–30. doi: 10.1097/CJI.0000000000000149. PMID:27875387 [DOI] [PubMed] [Google Scholar]
- 3.Atkins MB, Kunkel L, Sznol M, Rosenberg SA. High-dose recombinant interleukin-2 therapy in patients with metastatic melanoma: long-term survival update. Can J Sci Am. 2000;6 Suppl 1:S11–4. [PubMed] [Google Scholar]
- 4.Atkins MB, Lotze MT, Dutcher JP, Fisher RI, Weiss G, Margolin K, Abrams J, Sznol M, Parkinson D, Hawkins M, et al.. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol. 1999;17:2105–16. doi: 10.1200/JCO.1999.17.7.2105. PMID:10561265 [DOI] [PubMed] [Google Scholar]
- 5.Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, et al.. Improved survival with ipilimumab in patients with metastatic melanoma. New England J Med. 2010;363:711–23. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Topalian SL, Sznol M, McDermott DF, Kluger HM, Carvajal RD, Sharfman WH, Brahmer JR, Lawrence DP, Atkins MB, Powderly JD, et al.. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol. 2014;32:1020–30. doi: 10.1200/JCO.2013.53.0105. PMID:24590637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, Wolchok JD, Hersey P, Joseph RW, Weber JS, et al.. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369:134–44. doi: 10.1056/NEJMoa1305133. PMID:23724846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koller KM, Wang W, Schell TD, Cozza EM, Kokolus KM, Neves RI, Mackley HB, Pameijer C, Leung A, Anderson B, et al.. Malignant melanoma-The cradle of anti-neoplastic immunotherapy. Crit Rev Oncol Hematol. 2016;106:25–54. doi: 10.1016/j.critrevonc.2016.04.010. PMID:27637351 [DOI] [PubMed] [Google Scholar]
- 9.Hughes T, Klairmont M, Sharfman WH, Kaufman HL. Interleukin-2, ipilimumab, and anti-PD-1: clinical management and the evolving role of immunotherapy for the treatment of patients with metastatic melanoma. Cancer Biol Ther. 2015. doi: 10.1080/15384047.2015.1095401. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kin NW, Sanders VM. It takes nerve to tell T and B cells what to do. J Leukoc Biol. 2006;79:1093–104. doi: 10.1189/jlb.1105625. PMID:16531560 [DOI] [PubMed] [Google Scholar]
- 11.Bonneau RH, Kiecolt-Glaser JK, Glaser R. Stress-induced modulation of the immune response. Ann N Y Acad Sci. 1990;594:253–69. doi: 10.1111/j.1749-6632.1990.tb40485.x. PMID:2165759 [DOI] [PubMed] [Google Scholar]
- 12.Kokolus KM, Capitano ML, Lee CT, Eng JW, Waight JD, Hylander BL, Sexton S, Hong CC, Gordon CJ, Abrams SI, et al.. Baseline tumor growth and immune control in laboratory mice are significantly influenced by subthermoneutral housing temperature. Proc Natl Acad Sci U S A. 2013;110:20176–81. doi: 10.1073/pnas.1304291110. PMID:24248371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kokolus KM, Spangler HM, Povinelli BJ, Farren MR, Lee KP, Repasky EA. Stressful presentations:mild cold stress in laboratory mice influences phenotype of dendritic cells in naïve and tumor-bearing mice. Front Immunol. 2014;5:23. doi: 10.3389/fimmu.2014.00023. PMID:24575090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eng JWL, Reed CB, Kokolus KM, Pitoniak R, Utley A, Bucsek MJ, Ma WW, Repasky EA, Hylander BL, et al.. Housing temperature-induced stress drives therapeutic resistance in murine tumor models through β2-adrenergic receptor activation. Nat Commun. 2015;10:6426. doi: 10.1038/ncomms7426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pasquier E, Ciccolini J, Carre M, Giacometti S, Fanciullino R, Pouchy C, Montero MP, Serdjebi C, Kavallaris M, André N, et al.. Propranolol potentiates the anti-angiogenic effects and anti-tumor efficacy of chemotherapy agents: implication in breast cancer treatment. Oncotarget. 2011;2:797–809. doi: 10.18632/oncotarget.343. PMID:22006582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Slota C, Shi A, Chen G, Bevans M, Weng NP. Norepinephrine preferentially modulates memory CD8 T cell function inducing inflammatory cytokine production and reducing proliferation in response to activation. Brain Behav Immun. 2015;46:168–79. doi: 10.1016/j.bbi.2015.01.015. PMID:25653192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hoes AW. beta blockers for heart failure. Bmj. 2016;353:i2074. doi: 10.1136/bmj.i2074. PMID:27099266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE Jr., Drazner MH, Fonarow GC, Geraci SA, Horwich T, Januzzi JL, et al.. 2013 ACCF/AHA guideline for the management of heart failure: executive summary: a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation. 2013;128:1810–52. doi: 10.1161/CIR.0b013e31829e8807. PMID:23741057 [DOI] [PubMed] [Google Scholar]
- 19.DiNicolantonio JJ, Fares H, Niazi AK, Chatterjee S, D'Ascenzo F, Cerrato E, Biondi-Zoccai G, Lavie CJ, Bell DS, O'Keefe JH, et al.. beta-Blockers in hypertension, diabetes, heart failure and acute myocardial infarction: a review of the literature. Open heart. 2015;2:e000230. doi: 10.1136/openhrt-2014-000230. PMID:25821584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Powe DG, Voss MJ, Zanker KS, Habashy HO, Green AR, Ellis IO, Entschladen F.. Beta-blocker drug therapy reduces secondary cancer formation in breast cancer and improves cancer specific survival. Oncotarget. 2010;1:628–38. doi: 10.18632/oncotarget.197. PMID:21317458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barron TI, Connolly RM, Sharp L, Bennett K, Visvanathan K. Beta blockers and breast cancer mortality: a population- based study. J Clin Oncol. 2011;29:2635–44. doi: 10.1200/JCO.2010.33.5422. PMID:21632503 [DOI] [PubMed] [Google Scholar]
- 22.Ganz PA, Habel LA, Weltzien EK, Caan BJ, Cole SW. Examining the influence of beta blockers and ACE inhibitors on the risk for breast cancer recurrence: results from the LACE cohort. Breast Cancer Res Treat 2011;129:549–56. doi: 10.1007/s10549-011-1505-3. PMID:21479924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Melhem-Bertrandt A, Chavez-Macgregor M, Lei X, Brown EN, Lee RT, Meric-Bernstam F, Sood AK, Conzen SD, Hortobagyi GN, Gonzalez-Angulo AM. Beta-blocker use is associated with improved relapse-free survival in patients with triple-negative breast cancer. J Clin Oncol. 2011;29:2645–52. doi: 10.1200/JCO.2010.33.4441. PMID:21632501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Botteri E, Munzone E, Rotmensz N, Cipolla C, De Giorgi V, Santillo B, Zanelotti A, Adamoli L, Colleoni M, Viale G, et al.. Therapeutic effect of beta-blockers in triple-negative breast cancer postmenopausal women. Breast Cancer Res Treat. 2013;140:567–75. doi: 10.1007/s10549-013-2654-3. PMID:23912960 [DOI] [PubMed] [Google Scholar]
- 25.Diaz ES, Karlan BY, Li AJ. Impact of beta blockers on epithelial ovarian cancer survival. Gynecol Oncol. 2012;127:375–8. doi: 10.1016/j.ygyno.2012.07.102. PMID:22819786 [DOI] [PubMed] [Google Scholar]
- 26.Watkins JL, Thaker PH, Nick AM, Ramondetta LM, Kumar S, Urbauer DL, Matsuo K, Squires KC, Coleman RL, Lutgendorf SK, et al.. Clinical impact of selective and nonselective beta-blockers on survival in patients with ovarian cancer. Cancer. 2015;121:3444–51. doi: 10.1002/cncr.29392. PMID:26301456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leigh ND, Kokolus KM, O'Neill RE, Du W, Eng JW, Qiu J, Chen GL, McCarthy PL, Farrar JD, Cao X, et al.. Housing temperature-induced stress is suppressing murine graft-versus-host disease through beta2-adrenergic receptor signaling. J Immunol. 2015;195:5045–54. doi: 10.4049/jimmunol.1500700. PMID:26459348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Estrada LD, Agac D, Farrar JD. Sympathetic neural signaling via the beta2-adrenergic receptor suppresses T-cell receptor-mediated human and mouse CD8(+) T-cell effector function. Eur J Immunol. 2016;46:1948–58. doi: 10.1002/eji.201646395. PMID:27222010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kuzu OF, Nguyen FD, Noory MA, Sharma A. Current state of animal (mouse) modeling in melanoma research. Cancer Growth Metastasis. 2015;8:81–94. PMID:26483610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fidler IJ, Bucana C. Mechanism of tumor cell resistance to lysis by syngeneic lymphocytes. Cancer Res. 1977;37:3945–56. PMID:908034 [PubMed] [Google Scholar]
- 31.Kleffel S, Posch C, Barthel SR, Mueller H, Schlapbach C, Guenova E, Elco CP, Lee N, Juneja VR, Zhan Q, et al.. Melanoma cell-intrinsic PD-1 receptor functions promote tumor growth. Cell. 2015;162:1242–56. doi: 10.1016/j.cell.2015.08.052. PMID:26359984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen S, Lee LF, Fisher TS, Jessen B, Elliott M, Evering W, Logronio K, Tu GH, Tsaparikos K, Li X, et al.. Combination of 4-1BB agonist and PD-1 antagonist promotes antitumor effector/memory CD8 T cells in a poorly immunogenic tumor model. Cancer Immunol Res. 2015;3:149–60. doi: 10.1158/2326-6066.CIR-14-0118. PMID:25387892 [DOI] [PubMed] [Google Scholar]
- 33.Harry JD, Norris SC, Percival GC, Young J. The dose in humans at which ICI 118,551 (a selective beta 2-adrenoceptor blocking agent) demonstrates blockade of beta 1-adrenoceptors. Clin Pharmacol Ther. 1988;43:492–8. doi: 10.1038/clpt.1988.64. PMID:2896555 [DOI] [PubMed] [Google Scholar]
- 34.Maisel AS, Fowler P, Rearden A, Motulsky HJ, Michel MC. A new method for isolation of human lymphocyte subsets reveals differential regulation of beta-adrenergic receptors by terbutaline treatment. Clin Pharmacol Ther. 1989;46:429–39. doi: 10.1038/clpt.1989.161. PMID:2551559 [DOI] [PubMed] [Google Scholar]
- 35.Grailer JJ, Haggadone MD, Sarma JV, Zetoune FS, Ward PA. Induction of M2 regulatory macrophages through the beta2-adrenergic receptor with protection during endotoxemia and acute lung injury. J innate immun. 2014;6:607–18. doi: 10.1159/000358524. PMID:24642449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kizaki T, Shirato K, Sakurai T, Ogasawara JE, Oh-ishi S, Matsuoka T, Izawa T, Imaizumi K, Haga S, Ohno H. Beta2-adrenergic receptor regulate Toll-like receptor 4-induced late-phase NF-kappaB activation. Mol Immunol. 2009;46:1195–203. doi: 10.1016/j.molimm.2008.11.005. PMID:19167076 [DOI] [PubMed] [Google Scholar]
- 37.Sanders VM, Baker RA, Ramer-Quinn DS, Kasprowicz DJ, Fuchs BA, Street NE. Differential expression of the beta2-adrenergic receptor by Th1 and Th2 clones: implications for cytokine production and B cell help. J Immunol. 1997;158:4200–10. PMID:9126981 [PubMed] [Google Scholar]
- 38.Guereschi MG, Araujo LP, Maricato JT, Takenaka MC, Nascimento VM, Vivanco BC, Reis VO, Keller AC, Brum PC, Basso AS. Beta2-adrenergic receptor signaling in CD4+ Foxp3+ regulatory T cells enhances their suppressive function in a PKA-dependent manner. Eur J Immunol. 2013;43:1001–12. doi: 10.1002/eji.201243005. PMID:23436577 [DOI] [PubMed] [Google Scholar]
- 39.Grebe KM, Takeda K, Hickman HD, Bailey AL, Embry AC, Bennink JR, Yewdell JW. Cutting edge: Sympathetic nervous system increases proinflammatory cytokines and exacerbates influenza A virus pathogenesis. J Immunol. 2010;184:540–4. doi: 10.4049/jimmunol.0903395. PMID:20018617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kalinichenko VV, Mokyr MB, Graf LH Jr., Cohen RL, Chambers DA. Norepinephrine-mediated inhibition of antitumor cytotoxic T lymphocyte generation involves a beta-adrenergic receptor mechanism and decreased TNF-alpha gene expression. J Immunol. 1999;163:2492–9. PMID:10452985 [PubMed] [Google Scholar]
- 41.Zalli A, Bosch JA, Goodyear O, Riddell N, McGettrick HM, Moss P, Wallace GR. Targeting β2 adrenergic receptors regulate human T cell function directly and indirectly. Brain Behav Immun. 2015;45:211–8. doi: 10.1016/j.bbi.2014.12.001. PMID:25526818 [DOI] [PubMed] [Google Scholar]
- 42.Tsuji M., Kuno T., Tanaka C., Ichihashi M., Mishima Y.. Beta-adrenergic receptors of B16 melanoma cell. Arc Dermatol Res. 1983;275:415–416. doi: 10.1007/BF00417345. [DOI] [PubMed] [Google Scholar]
- 43.Yang EV, Kim SJ, Donovan EL, Chen M, Gross AC, Webster Marketon JI, Barsky SH, Glaser R. Norepinephrine upregulates VEGF, IL-8, and IL-6 expression in human melanoma tumor cell lines: implications for stress-related enhancement of tumor progression. Brain Behav Immun. 2009;23:267–75. doi: 10.1016/j.bbi.2008.10.005. PMID:18996182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bucsek MJ, Qiao G, MacDonald CR, Giridharan T, Evans L, Niedzwecki B, Liu H, Kokolus KM, Eng JW, Messmer MN, et al.. beta-adrenergic signaling in mice housed at standard temperatures suppresses an effector phenotype in CD8+ T cells and undermines checkpoint inhibitor therapy. Cancer Res. 2017;77:5639–51. doi: 10.1158/0008-5472.CAN-17-0546. PMID:28819022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nakai A, Hayano Y, Furuta F, Noda M, Suzuki K. Control of lymphocyte egress from lymph nodes through beta2-adrenergic receptors. J Exp Med. 2014;211:2583–98. doi: 10.1084/jem.20141132. PMID:25422496 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.