

ABSTRACT
Prognosis of glioblastoma remains dismal, underscoring the need for novel therapies. Immunotherapy is generating promising results, but requires combination strategies to unlock its full potential. We investigated the immunomodulatory capacities of poly(I:C) on primary human glioblastoma cells and its combinatorial potential with programmed death ligand (PD-L) blockade. In our experiments, poly(I:C) stimulated expression of both PD-L1 and PD-L2 on glioblastoma cells, and a pro-inflammatory secretome, including type I interferons (IFN) and chemokines CXCL9, CXCL10, CCL4 and CCL5. IFN-β was partially responsible for the elevated PD-1 ligand expression on these cells. Moreover, real-time PCR and chloroquine-mediated blocking experiments indicated that poly(I:C) triggered Toll-like receptor 3 to elicit its effect. Cocultures of poly(I:C)-treated glioblastoma cells with peripheral blood mononuclear cells enhanced lymphocytic activation (CD69, IFN-γ) and cytotoxic capacity (CD107a, granzyme B). Additional PD-L1 blockade further propagated immune activation. Besides activating immunity, poly(I:C)-treated glioblastoma cells also doubled the attraction of CD8+ T cells, and to a lesser extent CD4+ T cells, via a mechanism which included CXCR3 and CCR5 ligands. Our results indicate that by triggering glioblastoma cells, poly(I:C) primes the tumor microenvironment for an immune response. Secreted cytokines allow for immune activation while chemokines attract CD8+ T cells to the front, which are postulated as a prerequisite for effective PD-1/PD-L1 blockade. Accordingly, additional blockade of the concurrently elevated tumoral PD-L1 further reinforces the immune activation. In conclusion, our data proposes poly(I:C) treatment combined with PD-L1 blockade to invigorate the immune checkpoint inhibition response in glioblastoma.
KEYWORDS: glioblastoma (GBM), poly(I:C), Toll-like receptor 3 (TLR3), cancer immunotherapy, programmed death ligand 1 (PD-L1), programmed death ligand 2 (PD-L2), immune checkpoint blockade, primary patient-derived cells, brain tumor, glioma
Introduction
Glioblastoma is the most common malignant primary brain tumor and carries an extremely poor prognosis.1 The current standard of care consists of maximal surgical resection followed by chemoradiotherapy and eventually chemotherapy alone.2 Nevertheless, tumor recurrence is nearly inevitable, contributing to a median survival of only 14.6 months and a five-year survival of less than 5.5%.1,2 In order to control this devastating disease, new treatment modalities are urgently required.
Immunotherapy is being intensively investigated due to its ability to specifically target cancer cells and mediate long-term surveillance. The recent clinical success of immune checkpoint inhibitors in melanoma and lung cancer has accelerated research on employing these pathways to relief immunosuppression observed in cancer, including in glioblastoma.3,4 Immune checkpoints are molecules that dampen immune responses in order to prevent autoimmunity.5 However, they are commonly hijacked by tumors to escape immunity, which is a hallmark of cancer.6,7 Since tumor-infiltrating lymphocytes (TILs) are encountered in glioblastoma and expression of programmed death ligand 1 (PD-L1) has been observed,8,9 it has been proposed that this immune checkpoint pathway plays a role in controlling the antitumor immune response in glioblastoma.4,8 Preclinical glioblastoma mouse models have shown encouraging results with immune checkpoint blocking antibodies and several clinical trials investigating their therapeutic benefit in glioblastoma are ongoing.4,10–12 Nevertheless, combination therapy is key to improving cancer treatment efficacy and preclinical studies that help guide rational choices of combination strategies remain highly warranted.
Alleviation of existing immunosuppression in combination with boosting immune activation has been postulated as an immunotherapeutic strategy to pursue for eliciting effective immune responses at the tumor site. Polyriboinosinic-polyribocytidylic acid, or poly(I:C), is considered an immunostimulant with high potential to boost cancer immunotherapy.13 This synthetic mimetic of viral double-stranded RNA binds to either the endosomal Toll-like receptor 3 (TLR3), or the cytoplasmic receptors melanoma differentiation-associated protein 5 (MDA-5) or retinoic acid-inducible gene 1 (RIG-I).13,14 As such, poly(I:C) activates several immune cell types, including dendritic cells (DC) and natural killer (NK) cells, leading to antitumor responses.15 Furthermore, poly(I:C) also influences tumor cells, resulting in inhibition of proliferation and induction of apoptosis.15,16 Several clinical trials are/have been testing poly(I:C), including in glioblastoma.15
Since both immune checkpoint inhibitors and poly(I:C) are being investigated in a clinical setting, we examined in this study how poly(I:C) treatment of primary human glioblastoma cells affects their immunomodulatory capacity in order to assess its potential for combination therapy with immune checkpoint ligand blockade.
Results
Poly(I:C) modifies the immunological profile of primary glioblastoma cells
Primary glioblastoma cells were treated with increasing concentrations of poly(I:C) in order to assess its influence on the expression of immune checkpoint ligands PD-L1 and PD-L2 in glioblastoma. Poly(I:C) significantly upregulated the expression of both PD-1 ligands on glioblastoma cells (Fig. 1A). PD-L1 was upregulated to a greater extent than PD-L2 and this in a dose-dependent manner. The upregulation of PD-L1 was correlated with the upregulation of PD-L2 (R = 0.726, p < 0.05; Fig. 1B), but only for the latter the basal expression level was also correlated with expression after treatment with 10µg/ml poly(I:C) (R = 0.855, p < 0.01; Fig. 1B). Furthermore, poly(I:C) also significantly induced expression of both PD-1 ligands on PD-L1− and PD-L2− primary glioblastoma cells, resulting in 95% and 74% of the cells becoming PD-L1+ and PD-L2+, respectively (Fig. 1C). Of note, poly(I:C) appeared to be as potent as interferon (IFN)-γ in upregulating and inducing the PD-1 ligands. These results show that poly(I:C) is an efficient stimulator of PD-L1 and PD-L2 expression on glioblastoma cells.
Figure 1.
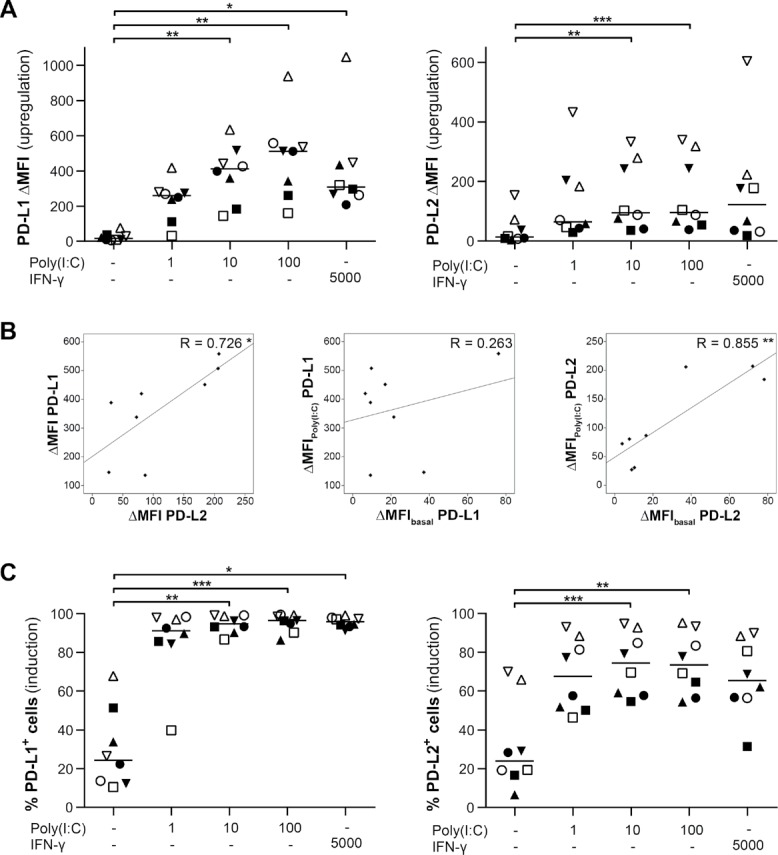
Poly(I:C) stimulates PD-L1 and PD-L2 expression on primary human glioblastoma cells. (A and C) Poly(I:C) upregulates (A; ΔMFI) and induces (C; % positive cells) expression of both PD-L1 and PD-L2 on primary glioblastoma cells; n = 8; Friedman test with Bonferroni correction; concentrations are denoted in µg/ml poly(I:C) and U/ml IFN-γ. (B) Plots showing correlations between PD-L1 and PD-L2 upregulation as well as between the expression levels at basal or poly(I:C) treatment conditions for each protein. Treatment with 10µg/ml poly(I:C) was selected for analysis; Pearson's correlation coefficient. Bar represents median. Symbols depict primary glioblastoma cells derived from different glioblastoma patients. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Poly(I:C) is recognized as a potent inducer of type I IFN.15,17 Hence, we investigated whether poly(I:C) could initiate a pro-inflammatory secretome in primary glioblastoma cells. poly(I:C) significantly induced the secretion of vast amounts of IFN-β, as well as IFN-α to a lesser extent (Fig. 2). On the other hand, IFN-γ was not secreted following poly(I:C) treatment (Fig. 2). In addition, we also observed a significant increase in the secretion of interleukin (IL)-15 (three-fold), while the secretion of transforming growth factor (TGF)-β was significantly decreased by 26% (Fig. 2). Finally, we assessed the secretion of poly(I:C)-inducible chemokines capable of attracting lymphocytes. The secretion of C-C motif chemokine ligand 4 (CCL4), CCL5 and C-X-C motif chemokine ligand 10 (CXCL10) was significantly invigorated upon poly(I:C) exposure, while also CXCL9 secretion was elevated (Fig. 2). These data show that exposure to poly(I:C) converts the secretome of glioblastoma cells towards an immunostimulatory profile.
Figure 2.
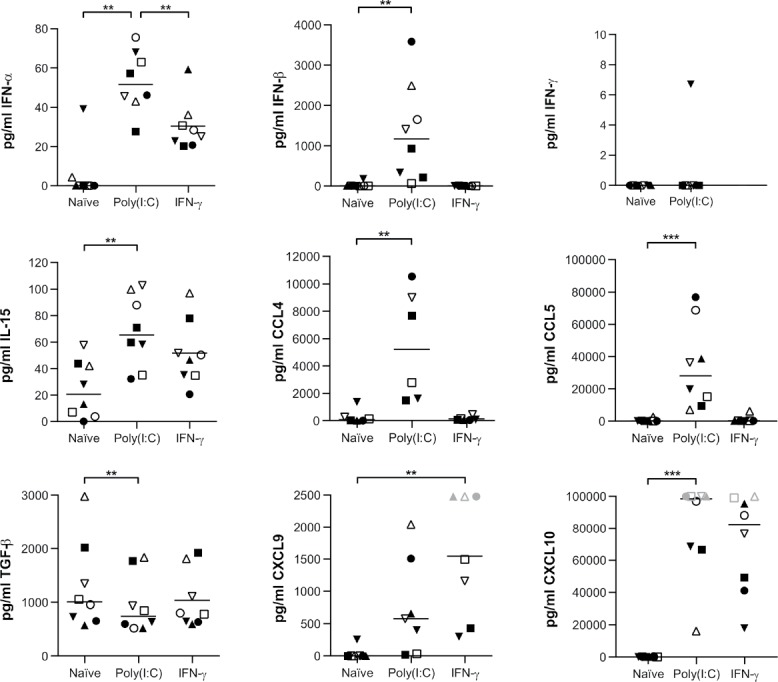
Poly(I:C) drives an immunostimulatory secretome of primary human glioblastoma cells. Poly(I:C) enhances the release of type I IFN, IL-15 and chemokines by primary glioblastoma cells, while TGF-β secretion is inhibited. IFN-γ secretion is not induced; n = 6–8; Friedman test with Bonferroni correction. 10µg/ml poly(I:C) and 5000IU/ml IFN-γ were used. Bar represents median. Symbols depict primary glioblastoma cells derived from different glioblastoma patients. Grey symbols represent data points above the detection limit; these were capped at the detection limit for visualization and analysis purposes since differences were apparent. **, p < 0.01; ***, p < 0.001.
Poly(I:C) mediates its effect via TLR3
Poly(I:C) can mediate its effect via the endosomal receptor TLR3 or the cytoplasmic receptors MDA-5/RIG-I (reviewed in reference 15). We assessed their adaptor molecules, TLR adaptor molecule 1 (TICAM1) for TLR3 and mitochondrial antiviral signaling protein (MAVS) for MDA-5 and RIG-I, in order to evaluate which receptor pathway in primary glioblastoma cells is activated by poly(I:C). RNA was isolated from glioblastoma cells following a 24-hour treatment with poly(I:C). The quantitative real-time PCR (qRT-PCR) data revealed a significant upregulation of TICAM1, but not of MAVS (Fig. 3A). The enhanced expression of the TLR3 downstream molecule TICAM1 indicates that poly(I:C) mediates its effect in glioblastoma cells via the endosomal TLR3-TICAM1 axis. Next, we validated the involvement of TLR3 by blocking its signaling using chloroquine, an inhibitor of TLR3 signaling.18 Indeed, the addition of chloroquine significantly reduced the poly(I:C)-mediated upregulation of both PD-L1 and PD-L2 (Fig. 3B–C). These data suggest that the effect of poly(I:C) is, at least primarily, mediated via TLR3-TICAM1 signaling.
Figure 3.
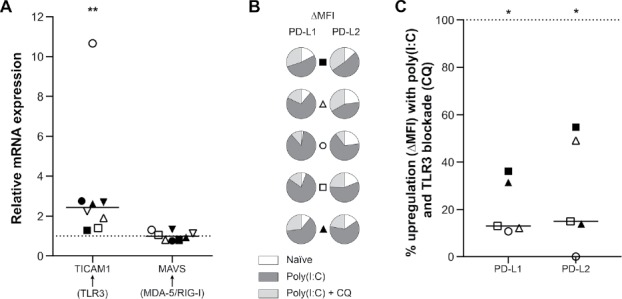
Poly(I:C) mediates its effect on PD-L1 and PD-L2 expression in primary human glioblastoma cells primarily via the TLR3-TICAM1 pathway. (A) Poly(I:C) upregulates mRNA expression of TLR3 adaptor molecule TICAM1 but not MAVS, the adaptor molecule for the cytosolic receptor pathway, in primary human glioblastoma cells. mRNA expression following poly(I:C) treatment is shown relative to the basal, naïve condition (dashed line at 1); n = 8; Wilcoxon Signed Ranks test. (B) Patient-specific presentation of the proportional PD-L1 and PD-L2 expression in the naïve setting (white) versus poly(I:C) treatment with (light grey) and without (dark grey) the TLR3 inhibitor chloroquine. TLR3 inhibition clearly diminishes the effect of poly(I:C) on both PD-1 ligands. (C) Relative upregulation (ΔMFI) of PD-L1 and PD-L2 by poly(I:C) when TLR3 signaling is blocked with chloroquine. Data are normalized to naïve (baseline) and poly(I:C) treatment (100%, dashed line) as to show the percentage of upregulation by poly(I:C) not accounted for by TLR3; n = 5; Wilcoxon Signed Ranks test. Bar represents median. Symbols depict primary glioblastoma cells derived from different glioblastoma patients. CQ: chloroquine; *, p < 0.05; **, p < 0.01.
Poly(I:C) triggers de novo production of PD-L1 and PD-L2 in primary glioblastoma cells
Since intracellular PD-L1 has been observed,8,9,19 we investigated whether poly(I:C) induced translocation of intracellular PD-1 ligands rather than their de novo synthesis. qRT-PCR data showed significantly elevated mRNA of both PD-L1 and PD-L2 following poly(I:C) treatment (Fig. 4A). In particular, PD-L1 mRNA levels were enormously elevated, which supports our flow cytometric data. Elevated transcription of PD-L1 was correlated with PD-L2 transcription (R = 0.707, p < 0.05; Fig. 4B). Next, we used immunohistochemistry (IHC) to reveal the cellular location of the PD-1 ligand proteins before and after poly(I:C) treatment. Whereas naïve glioblastoma cells were only faintly positive for PD-L1, poly(I:C)-treated glioblastoma cells presented a strong positive staining for PD-L1 protein (Fig. 4C). In contrast to naïve glioblastoma cells, poly(I:C)-treated glioblastoma cells displayed cytoplasmic PD-L1 with membrane accentuation. Moreover, the increase in PD-L1 protein observed following poly(I:C) treatment was greater than the amount of PD-L1 protein which was visually present in the cytoplasm of naïve glioblastoma cells. A similar observation regarding PD-L2 protein was made in naïve versus poly(I:C)-treated glioblastoma cells (Fig. 4D). These data indicate that the elevated membrane expression of PD-1 ligands on glioblastoma cells following treatment with poly(I:C) is mainly derived from de novo produced protein.
Figure 4.
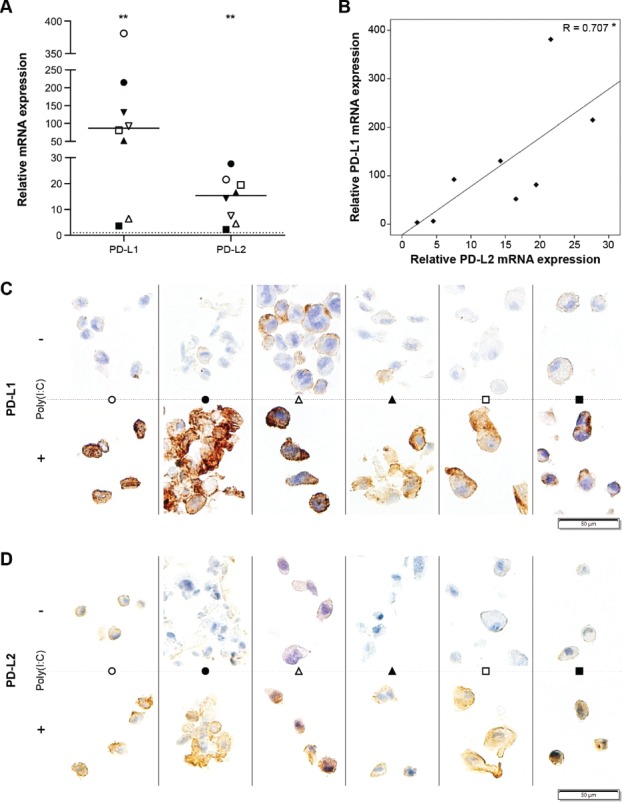
Poly(I:C) stimulates PD-L1 and PD-L2 expression on primary human glioblastoma cells via enhanced protein production. (A) Poly(I:C) upregulates mRNA transcription of PD-L1 and PD-L2 in primary human glioblastoma cells. mRNA expression following poly(I:C) treatment is shown relative to the basal, naïve condition (dashed line at 1); n = 8; Wilcoxon Signed Ranks test. (B) Plot showing the correlation between PD-L1 and PD-L2 mRNA expression following poly(I:C) treatment relative to naïve cells; Pearson's correlation coefficient. (C-D) Poly(I:C) increases both membrane and intracellular expression of PD-L1 (C) and PD-L2 (D) on primary human glioblastoma cells; n = 6; representative areas per specimen are shown. Bar represents median. Symbols depict primary glioblastoma cells derived from different glioblastoma patients. -, no poly(I:C); +, 10µg/ml poly(I:C); *, p < 0.05; **, p < 0.01.
The effect of poly(I:C) on the expression of PD-L1 and PD-L2 is partially mediated by IFN-β
We described the release of type I IFN (Fig. 2), which have been attributed the capacity to upregulate the expression of PD-L1.20 In a series of blocking experiments, we investigated whether IFN-α and IFN-β were involved in induction and upregulation of PD-L1 and PD-L2 following poly(I:C) treatment. IFN-β, in contrast to IFN-α, significantly contributed to the upregulation and induction of both PD-1 ligands (Fig. 5A–B). Whereas IFN-β contributed similarly to upregulation and induction of PD-L2 by poly(I:C), it was less involved in induction of PD-L1 compared to its upregulation. IFN-α was involved in the induction of neither PD-L1 nor PD-L2, but contributed to the upregulation of both ligands in three and four out of five primary glioblastoma cell lines, respectively, although not statistically significant (Fig. 5A–B). These results indicate that the effect of poly(I:C) on PD-L1 and PD-L2 is partially mediated via downstream-secreted IFN-β.
Figure 5.
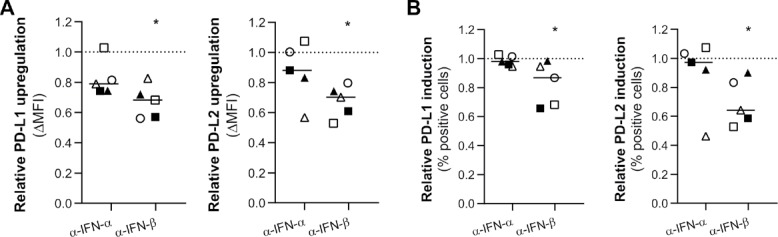
The poly(I:C)-mediated effect on PD-L1 and PD-L2 expression on primary human glioblastoma cells is in part via autocrine or paracrine signaling of downstream-secreted IFN-β. (A-B) Relative protein upregulation (A; ΔMFI) and induction (B; % positive cells) of blocking IFN-α or IFN-β additionally to poly(I:C) treatment, as determined by flow cytometry. Data are normalized to naïve (baseline) and poly(I:C) treatment (100%, dashed line) as to show the portion of upregulation or induction by poly(I:C) not accounted for by IFN-α or IFN-β. IFN-β, but not IFN-α, is significantly involved in the upregulation and induction of PD-L1 and PD-L2 on primary glioblastoma cells; n = 5; Wilcoxon Signed Ranks test. Bar represents median. Symbols depict primary glioblastoma cells derived from different glioblastoma patients. *, p < 0.05.
Poly(I:C) modifies the immunomodulatory capacity of primary glioblastoma cells
The observed poly(I:C)-induced pro-inflammatory secretome by and immunosuppressive PD-1 ligand expression on primary glioblastoma cells may have opposing effects on immune cells. Therefore, we investigated the overall immunomodulation of primary glioblastoma cells following treatment with poly(I:C) by setting up cocultures with peripheral blood mononuclear cells (PBMC), in which we focused on lymphocytes as effector immune cells. Based upon the activation marker CD69, the number of activated CD8+ T and CD4+ T cells as well as NK cells was significantly higher in cocultures with poly(I:C)-treated glioblastoma cells compared to naïve glioblastoma cells (Fig. 6A). A similar significant increase could be observed for the degranulation marker CD107a (Fig. 6A). These observations were validated by cytokine analyses, which showed a significant increase in IFN-γ as well as granzyme B (Fig. 6B). Secretion of TNF-α was not elevated in cocultures with poly(I:C)-treated glioblastoma cells (Fig. 6B), neither was IL-1β release (data not shown). Given primary glioblastoma cells did not release IFN-γ in response to poly(I:C) (Fig. 2) and granzyme B is restricted to lymphocytes,21 these results point towards both phenotypic and functional immune cell activation following poly(I:C) treatment of glioblastoma cells.
Figure 6.
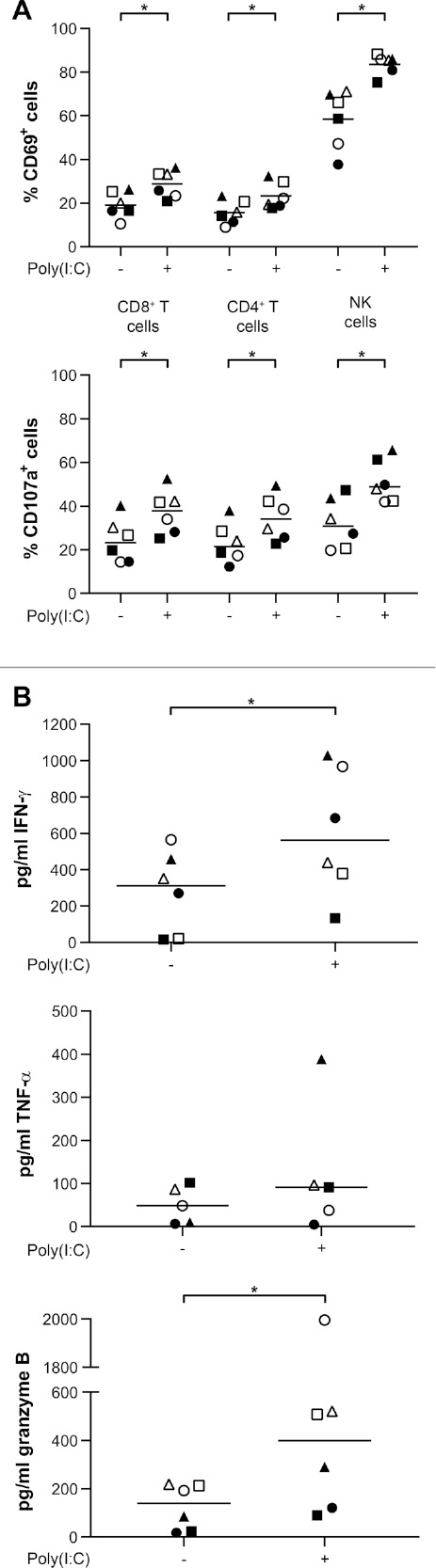
Poly(I:C)-treated primary human glioblastoma cells invigorate immune activation. (A) Poly(I:C)-treated primary human glioblastoma cells increase the number of activated (CD69+) lymphocytes and stimulate degranulation (CD107a+ lymphocytes); n = 6, Wilcoxon-Signed Ranks test. (B) Poly(I:C)-treated primary human glioblastoma cells increase immune activation and cytotoxic potential as shown by elevated release of IFN-γ and granzyme B, respectively. TNF-α secretion was not altered; n = 5–6, Wilcoxon-Signed Ranks test. Bar represents median. Symbols depict different PBMC donors each cocultured with primary glioblastoma cells derived from different glioblastoma patients. -, no poly(I:C); +, 10µg/ml poly(I:C); *, p < 0.05.
Poly(I:C)-treated primary glioblastoma cells attract effector lymphocytes
Using a transwell migration assay, we investigated whether lymphocytes are attracted to poly(I:C)-treated primary glioblastoma cells. CD8+ T cells were significantly more attracted to poly(I:C)-treated glioblastoma cells by nearly two-fold, whereas also CD4+ T cells migrated significantly more to this condition (Fig. 7A). On the other hand, chemoattraction of NK cells was not significantly altered, although two out of six PBMC donors presented a considerably enhanced migration (Fig. 7A). Of note, considering attraction of CD8+ T cells to poly(I:C)-treated glioblastoma cells was favored over CD4+ T cells, poly(I:C) treatment of primary glioblastoma cells also stimulated a more favorable CD8:CD4T cell ratio (Fig. 7B). Since T cells showed an enhanced migration, we next examined the chemokine receptor profile of the migrated T cells in both conditions. The receptors CXCR3 and CCR5 were significantly downregulated on both CD8+ and CD4+ T cells in the poly(I:C) condition (Fig. 7C). These data indicate that poly(I:C)-treated glioblastoma cells stimulate lymphocyte attraction via a mechanism that involves CXCR3 and CCR5.
Figure 7.
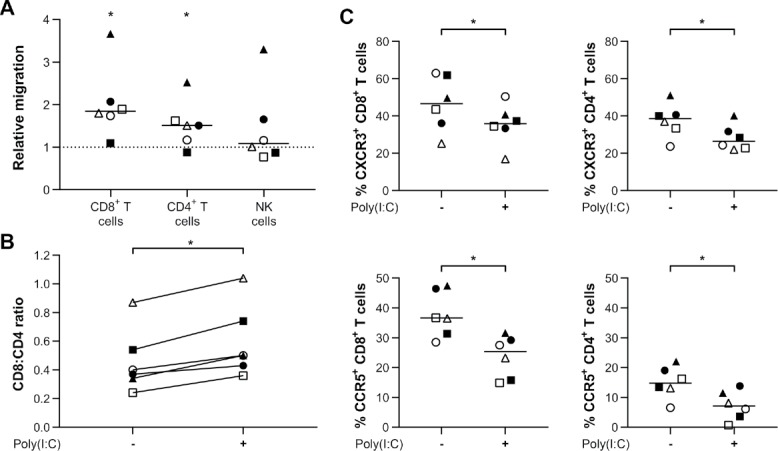
Poly(I:C)-treated primary human glioblastoma cells demonstrate elevated attraction of lymphocytes. (A) The migration of T cells towards primary human glioblastoma cells is enhanced by tumor priming with 10µg/ml poly(I:C). Data is normalized to and shown relatively to migration to naïve primary human glioblastoma cells (dashed line at 1); n = 6; Wilcoxon Signed Ranks test. (B) Preferential attraction of CD8+ T cells leads to increased CD8:CD4 T cell ratios following poly(I:C) treatment of primary glioblastoma cells; n = 6; Wilcoxon Signed Ranks test. (C) Expression of chemokine receptors CXCR3 and CCR5 is downregulated on T cells that migrated towards supernatant derived from poly(I:C)-treated primary human glioblastoma cells; n = 6; Wilcoxon Signed Ranks test. Bar represents median. Symbols depict different PBMC donors each cocultured with primary glioblastoma cells derived from different glioblastoma patients. -, no poly(I:C); +, 10µg/ml poly(I:C); *, p < 0.05.
PD-L1 blockade reinforces the immune activation triggered by poly(I:C)-treated primary glioblastoma cells
Poly(I:C)-treated primary glioblastoma cells activate immune cells, despite elevated tumoral PD-1 ligand expression. We examined the effect of additional PD-L1 or PD-L2 blockade on immune activation. PD-L1 blockade significantly propagated the immune activation established via poly(I:C) treatment of glioblastoma cells, as shown by IFN-γ secretion (Fig. 8A). Furthermore, additional PD-L1 blockade also significantly increased granzyme B release and CD8+ T cell proliferation (Fig. 8B–C). In contrast, additional blockade of PD-L2 had a more limited effect on enforcing immune activation. IFN-γ secretion and CD8+ T cell proliferation were only stimulated in up to half of the patients (Fig. 8A and 8C). Similar to PD-L1 blockade, also additional PD-L2 blockade resulted in a modest, but significant elevated granzyme B release (Fig. 8B). These data suggest that the combination of PD-L1 blockade with poly(I:C) can strengthen immune responses against glioblastoma.
Figure 8.
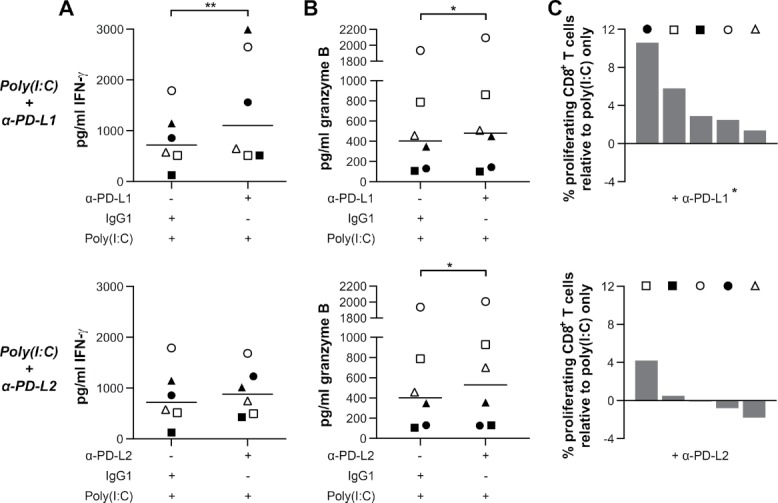
PD-L1 blocking invigorates the immunostimulatory effect of priming primary human glioblastoma cells with poly(I:C). (A and C) Additional blocking of PD-L1, but not PD-L2, further invigorates the immune response, as measured by IFN-γ secretion (A) and CD8+ T cell proliferation (C); n = 5–6; Wilcoxon Signed Ranks test. (B) Additional blockade of either PD-L1 or PD-L2 results in a modest but significant increase in granzyme B release; n = 6; Wilcoxon Signed Ranks test. Bar represents median. Symbols depict different PBMC donors each cocultured with primary glioblastoma cells derived from different glioblastoma patients. -, no compound; +, 10µg/ml blocking antibody, isotype antibody or poly(I:C); *, p < 0.05; **, p < 0.01.
Discussion
Inevitable progression and ultimate fatality underscore the urgency of new treatment modalities for glioblastoma. Recent breakthroughs in immunotherapy and improved understanding of tumor immunology have encouraged the belief for immunotherapeutic success in glioblastoma. The present study demonstrates that poly(I:C) induces an immunostimulatory profile in primary human glioblastoma cells, despite elevated expression of PD-L1 and PD-L2, which is in part due to secreted IFN-β. This situation enables immune activation which is invigorated by additional PD-L1 blockade. Moreover, attraction of lymphocytes, in particular CD8+ T cells, creates a tumor micro-environment (TME) primed for antitumor responses.
We show that both PD-L1 and PD-L2 are present at low levels on a subset of naïve primary human glioblastoma cells. Basal expression of PD-L1 on glioblastoma cells has been described.8,9,19 We are the first to show PD-L2 expression on human glioblastoma cells as well as induction and upregulation of PD-L1 and PD-L2 on glioblastoma cells due to poly(I:C). This is in concordance with poly(I:C)-mediated PD-L1 upregulation observed in neuroblastoma cells.22 Of note, another study demonstrated downregulation of PD-L1 on hepatoma cells in vivo,23 however, it was not determined whether poly(I:C) mediated this directly or via bystander cells. The positive correlation observed between the transcriptional activities of PD-L1 and PD-L2 is supported by a recent Genome Atlas analysis of glioblastoma samples.24
Next, we investigated how this effect of poly(I:C) on primary human glioblastoma cells was mechanistically driven. Since intracellular PD-L1 has been described,8,9,19 this could have served as a possible source of the elevated membrane expression observed following poly(I:C) treatment. However, IHC analysis revealed that the increase in membrane PD-L1 and PD-L2 expression originated foremost from de novo protein, as we could not detect sufficient intracellular amounts pre-exposure. Such de novo protein production is akin to the IFN-γ-mediated mechanism of PD-L1 upregulation in breast cancer cell lines25 and is supported by the increased transcription of PD-L1 and PD-L2 genes that we observed in poly(I:C)-treated primary glioblastoma cells.
Poly(I:C) is known to be an efficient inducer of pro-inflammatory cytokines, in particular of type I IFN.15 In this regard, we demonstrated that poly(I:C) indeed drastically modified the secretome of primary human glioblastoma cells. Our data show that poly(I:C) induces the release of both IFN-α and IFN-β by primary glioblastoma cells, which is in contrast to findings of Glas et al.16 The observed upregulation of TICAM1 but not MAVS suggests that this effect is mediated via the endosomal TLR3-TICAM1 pathway, as it has been shown that TLR3 and TICAM1 become upregulated following engagement with poly(I:C).26 Indeed, upon addition of chloroquine, a TLR3 inhibitor,18 the poly(I:C)-mediated upregulation of membrane PD-L1 and PD-L2 was drastically reduced. Moreover, it has been reported that the TLR3-TICAM1 pathway is required for IFN-β release by tumor cells in response to anthracyclines.27 Subsequent para-/autocrine activation of IFN-α and -β receptor subunit 1, IFNAR, led to tumor cell-mediated release of CXCL10 which ultimately recruited immune cells.27 These data support our findings that poly(I:C) induces the secretion of type I IFN by primary glioblastoma cells, but also of chemokines CXCL9, CXCL10, CCL4 and CCL5, either directly or via a para-/autocrine circuit. Furthermore, to our knowledge, we are the first to report on the receptor signaling pathway activated by poly(I:C) to augment PD-L1 and PD-L2 expression.
IFN-γ has most often been described to stimulate PD-L1 expression,19 however, also type I IFN have been attributed this capacity.17,20 Our data indicate that IFN-β contributes to both induction and upregulation of PD-1 ligands on primary human glioblastoma cells via the aforementioned para-/autocrine signaling, whereas IFN-α was not significantly involved in our experiments. This difference in contribution may possibly be a reflection of the difference in magnitude of secretion following poly(I:C) treatment, which was 23-fold higher for IFN-β. Nonetheless, others identified IFN-β as the most potent PD-L1 stimulant of both type I IFN in melanoma.20 Our data suggest that the effect of poly(I:C) on PD-1 ligand expression on primary glioblastoma cells is propagated via poly(I:C)-induced release of IFN-β.
Poly(I:C) generated a type I IFN response by primary human glioblastoma cells, which plays an important role in antitumor immunity,17 but also the release of other cytokines was modulated. Of note, we found elevated secretion of IL-15 and reduced secretion of TGF-β by poly(I:C)-treated primary human glioblastoma cells. IL-15 is a highly potent immune-activating cytokine, which potentiates the antitumor functions of several immune cell types, including NK cells, γδT cells, and DC.28–30 We are, to our knowledge, the first to report on IL-15 secretion by glioblastoma cells in particular and its production by human cancer cells following poly(I:C) treatment in general. Two murine studies demonstrated therapeutic efficacy of IL-15 in glioblastoma, dependent on activation and recruitment of lymphocytes.31,32 Moreover, type I IFN have been shown to induce IL-15 secretion as well as expression of IL-15 receptor-α, which allows transpresentation of IL-15.33 TGF-β is the most powerful tumor-promoting and immunosuppressive cytokine and actively plays a role in glioblastoma.34 We report a decreased secretion of TGF-β by primary glioblastoma cells following poly(I:C) treatment, which is in concordance with findings in neuroblastoma cells.22 Reduction of TGF-β release has been demonstrated by IFN-α in wound healing35 and IFN-β in adenoviral cancer therapy,36 which might provide a possible explanation for our observation. Altogether, the modified secretome of poly(I:C)-treated primary glioblastoma cells points towards an immunomodulatory shift towards activation.
We demonstrated that poly(I:C)-treated primary human glioblastoma cells indeed activate lymphocytic populations, i.e. CD8+ and CD4+ T cells as well as NK cells, and stimulate their degranulation. Overall, this resulted in an augmented IFN-γ and granzyme B secretion, that could be completely assigned to the PBMC. Boes et al. also observed increased activation of CD8+ and CD4+ T cells in coculture with poly(I:C)-treated neuroblastoma cells, based on membrane immunophenotyping.22 When in addition PD-L1 was blocked, we found immune activation was even more profound. Hence, these data indicate that poly(I:C) treatment of primary glioblastoma cells invigorates immune activation, despite elevated expression of immunosuppressive PD-1 ligands, and that this immune activation can further be propagated by additional PD-L1 blockade. This is in line with peripheral solid tumor murine models, where the combination of poly(I:C) with blockade of the PD-1/PD-L1 axis was more beneficial than either treatment individually.37,38 It will be interesting to investigate whether this also happens with brain tumors, given their unique localization. In this view, therapeutic efficacy of ipilimumab against brain metastases indicates the feasibility of antibody-mediated immune checkpoint blockade in the brain.8
Tumor-infiltrating effector lymphocytes are key to efficacious immunotherapy,39 which warrants strategies for their trafficking. We showed that poly(I:C)-treated primary human glioblastoma cells enhance attraction of CD8+ effector T cells, and to a lesser extent of CD4+ helper T cells. This is noteworthy, since tumor-infiltrating CD8+ T cells are inversely correlated with tumor grade40 and are positively associated with overall survival.41 Moreover, higher attraction of CD8+ T cells in comparison to CD4+ T cells also drives a more favorable CD8:CD4 T cell ratio, which has been shown to be lower in malignant compared to benign brain malignancies.42 In this context, dense CD8+ T cell infiltration has been shown to correlate with improved survival in brain metastases.43 We observed an increased CD8:CD4 T cell ratio, however, this remained rather low due to the use of healthy PBMC. This is in concordance with Kmiecik et al., who observed far more CD4+ than CD8+ T cells in the healthy donor peripheral blood in contrast to a more balanced CD8:CD4 TIL in glioblastoma.44 Hence, stimulating the attraction of CD8+ T cells may be indicative of improving the prognosis of glioblastoma patients. Furthermore, blockade of the PD-1/PD-L1 axis has been reported to primarily affect CD8+ T cells, whereas CTLA-4 blockade primarily acts on CD4+ T cells.45 Indeed, Nagato et al. identified CD8+ T cells as mediators of the therapeutic efficacy of poly(I:C) plus PD-1/PD-L1 blockade in peripheral solid tumor models,38 although Bald et al. ascribed prominent roles to CD4+ T cells and NK cells also.37 We observed a substantial release of CXCL9, CXCL10, CCL4 and CCL5, chemokines which strongly attract lymphocytes. Furthermore, lymphocytes migrating to supernatant derived from poly(I:C)-treated primary glioblastoma cells presented a downregulation of corresponding chemokine receptors CXCR3 and CCR5. This observation suggests that, based upon the process of receptor sequestration,46,47 ligands of both receptors are involved in the improved T cell chemotaxis. In this view, CXCL10-mediated intracranial infiltration of CD8+ T cells was observed following intramuscular injections with stabilized poly(I:C) in a glioblastoma mouse model,48 which suggests that poly(I:C) enables immune infiltration in glioblastoma, even when administered peripherally. Our data suggest that poly(I:C)-stimulated tumoral secretion of CXCL9, CXCL10, CCL4 and CCL5 could have participated in the enhanced T cell trafficking.
Immune checkpoint blockade prolongs survival in patients with pre-existing lymphocytic infiltrates and failure to PD-L1 treatment has been correlated with the absence of intratumoral CD8+ T cells.12,49 TILs are encountered in the majority of glioblastoma, however mostly sparse/moderate and rather at the perivascular and invasive areas than within the tumor tissue.8 Nduom et al. postulated that low tumoral PD-L1 expression in glioblastoma may be a reflection of a relatively low CD8+ T cell infiltrate, which might be an indication of non-responsiveness to PD-1 blockade in glioblastoma.9 Another indication may be the incapacity of TILs to readily invade the tissue, rendering the tumor “immune-excluded”.50 In this case, the tumor should advance in the cancer-immunity cycle for an optimal response to immune checkpoint blockade, e.g. by increasing immunological infiltration.50 Poly(I:C) is capable of activating cellular antitumor immunity in immune cell-poor tumors, as demonstrated by a beneficial outcome in conjunction with PD-1 blocking therapy in immune cell-poor melanoma.37 This supports data of us and others that poly(I:C) attracts effector lymphocytes, in particular CD8+ T cells.38,51 At the same time, poly(I:C) repolarizes tumor-resident microglia and macrophages towards an M1 active profile and matures DC.51,52 Hence, poly(I:C) primes the TME for battle, but it also stimulates the expression of PD-L1 and PD-L2. However, this provides an additional target and blockade of the PD-1/PD-L1 axis may relieve the brake in order to propagate immunity. Indeed, a murine glioblastoma DC vaccination study required combinatorial anti-PD-1 treatment to unleash an antitumor response.53
In this study, we focused on the lymphocytic compartment of immune cells. However, it is well known that the majority of the immune infiltrate in glioblastoma consists of tumor-associated microglia and macrophages (TAMs), which are polarized towards an immunosuppressive and tumor-supportive state.44 Pyonteck et al. showed that repolarization of TAMs via CSF-1R inhibition led to an antitumor response in glioma54 and this treatment option is currently under investigation in clinical trials in glioblastoma, alone or in combination with PD-1 blockade.55 Interestingly, also poly(I:C) is capable of reverting these tumor-residing microglia to an antitumor state.52 Moreover, poly(I:C) abrogated the immunosuppressive functions of myeloid-derived suppressor cells in a murine breast cancer model, leading to an enhanced T cell influx and enhanced survival.56 Recently, it has been shown that PD-1 on TAMs suppresses their phagocytic functions and antitumor immunity.57 On the other hand, PD-1 ligand expression by TAMs may contribute to immunosuppression.58 Hence, PD-L1 blockade could restore antitumor functions of and inhibit immunosuppression by TAMs. Therefore, we speculate that in vivo, treatment with poly(I:C), in particular in combination with PD-L1 blockade, may have a more profound effect than what we observed in vitro with lymphocytes.
The predictive value of PD-1, PD-L1 or PD-L2 as biomarker for blocking therapy is still under investigation,8,9 although tumoral PD-L1 expression has been confirmed as a significant, but not absolute, predictive marker in several solid malignancies.59 A robust assay for evaluation of the blocking efficacy in vitro has yet to be recommended. We and others have observed no evidence of enhanced cytotoxicity in vitro60 and only few papers in the enormous field of immune checkpoint blockade actually have. Furthermore, Beavis et al. have shown in vivo that the antitumor effect of combined PD-1 and adenosine receptor A2 blockade was largely independent on perforin, despite elevated granzyme B+ CD8+ T cells, but lost in IFN-γ knockout mice.60 Therefore, the effect of immune checkpoint blockade may be primarily IFN-γ-mediated, possibly amongst other cytokines. We propose analysis of IFN-γ secretion in in vitro tumor cell-immune cell cocultures as a read-out of immune activation mediated by immune checkpoint blockade. This outcome can be assessed in a quick and reliable manner using IFN-γ ELISA.
In conclusion, poly(I:C) shifts the immunomodulatory profile of primary human glioblastoma cells towards immune activation via the TLR3-TICAM1 pathway, despite a stimulated expression of immune checkpoint ligands PD-L1 and PD-L2, which is partially dependent on autocrine/paracrine secreted IFN-β. Poly(I:C)-treated primary glioblastoma cells stimulate the attraction of lymphocytes, in particular CD8+ T cells, while also enhancing lymphocytic activation. Immune activation is further invigorated by additional blockade of PD-L1. Both poly(I:C) and immune checkpoint blockade are being investigated in clinical trials. We propose the combinatorial treatment to strengthen their therapeutic efficacy and generate a profound immune response. Data in other murine cancer models look promising, but do not have to account for the blood-brain barrier as encountered in glioblastoma. Therefore, we plan to investigate this combination strategy in in vivo glioblastoma models as validation of this in vitro proof of concept.
Material and Methods
Ethics statement
Human tumor tissue was obtained from residual material following standard surgery of patients diagnosed with glioblastoma, performed in Antwerp University Hospital and AZ Nikolaas, and provided by Biobank@UZA (Belgium; ID: BE71030031000). The use of these tissues for this study was approved by the local Ethics Committee of the University of Antwerp (Belgium; reference number: 13/46/454). Characteristics of the primary tumor tissues are shown in Table 1. PBMC were isolated from adult volunteer whole blood donations (supplied by the Red Cross Flanders Blood Service, Belgium).
Table 1.
Characteristics of primary human glioblastoma tumor tissue specimens.
| Sex | Age | Diagnosis | Location tumor | MGMT† |
|---|---|---|---|---|
| ♂ | 64 | Recurrent‡ | Occipital (R) | - |
| ♀ | 74 | Newly diagnosed | Frontal (R) | + |
| ♀ | 85 | Newly diagnosed | Temporofrontal (R) | - |
| ♀ | 51 | Newly diagnosed | Frontal (R) | Unknown |
| ♀ | 62 | Newly diagnosed | Temporal (R) | - |
| ♀ | 59 | Newly diagnosed | Frontal (R) | - |
| ♂ | 50 | Newly diagnosed | Temporal (L) | - |
| ♂ | 62 | Newly diagnosed | Temporal (R) | + |
All patients were diagnosed with glioblastoma multiforme (World Health Organization grade IV astrocytoma). †, methylguanine methyltransferase (MGMT) promotor hypermethylation; ‡, patient received treatment prior to resection of the recurrent tumor. L, left; R, right.
Primary human cell culture
Primary human glioblastoma cell cultures were established from patient-derived tumor tissue samples.61 Tumor specimens were cut into small pieces and cultured at 37°C and 5% CO2 in Dulbecco's Modified Eagle Medium (DMEM; Gibco, 10938) supplemented with 10% fetal bovine serum (FBS; Gibco, 10270), 2mM L-glutamine (Gibco, 25030), 100U/ml penicillin and 100µg/ml streptomycin (Gibco, 15140). Following outgrowth, cells were harvested using 0.05% trypsin-EDTA (Gibco, 25300) and filtered over a 40µm cell strainer (Falcon, 352340) prior to subculturing. All cultures were confirmed as Mycoplasm free following MycoAlert analysis (Lonza, LT07–218).
PBMC were isolated from peripheral whole blood using Ficoll-Paque PLUS density gradient centrifugation (GE Healthcare, 17–1440) and frozen in FBS containing 10% dimethyl sulfoxide (Merck, 1.02931.0500) in liquid nitrogen (Air Liquide). For experiments, PBMC were thawed one day in advance in Roswell Park Memorial Institute medium (RPMI; Gibco, 52400) supplemented with 10% FBS, 2mM L-glutamine, 1mM sodium pyruvate (Gibco, 11360), 100 U/ml penicillin, 100µg/ml streptomycin and 1.5KU/ml DNase I (Sigma, D4263).
Flow cytometric tumor phenotyping
The response to poly(I:C) was analyzed by incubating the primary human glioblastoma cells with 1, 10 and 100µg/ml poly(I:C) (InvivoGen, tlrl-pic), or 5000IU/ml IFN-γ (ImmunoTools, 11343536) as positive control. After 24 hours, supernatant was collected for cytokine analysis. Primary glioblastoma cells were harvested and stained for 15 minutes with phycoerythrin-conjugated antibodies against PD-L1 (BD Biosciences, 557924), PD-L2 (BD Biosciences, 558066), or isotype control (BD Biosciences, 555749). Additionally, dead cells were excluded using propidium iodide (Life Technologies, P3566). Expression of PD-L1 and PD-L2 on viable primary glioblastoma cells was measured on a BD FACScan flow cytometer (Becton Dickinson). Data analysis was performed using FlowJo v10 software (TreeStar). Representative flow cytometry profiles are shown in Supplementary Figure S1. Difference in mean fluorescence intensity (ΔMFI) was calculated to evaluate target upregulation (ΔMFI = MFItarget – MFIisotype), while the overton subtraction tool in FlowJo was used to determine the percentage of positive cells (overton subtraction on: histogramtarget – histogramisotype).
Blocking experiments were performed similarly with one additional step. In order to investigate the involvement of TLR3, the primary glioblastoma cells were incubated for one hour with 50µM chloroquine (Abcam, ab142116) prior to the stimulation with 10µg/ml poly(I:C). For the assessment of the involvement of type I IFN, pre-incubation was performed with 10µg/ml blocking antibody against IFN-α (InvivoGen, maba-hifna-3), IFN-β (InvivoGen, mabg-hifnb-3), or corresponding isotype control (InvivoGen, maba2-ctrl and mabg2a-ctrlm). The effect of blocking on PD-L1 and PD-L2 upregulation by poly(I:C) was calculated using the following formula: relative upregulation = (MFIpoly(I:C)+blocking – MFIunstimulated+isotype) / (MFIpoly(I:C)+isotype – MFIunstimulated+isotype). The blocking effect on PD-L1 and PD-L2 induction by poly(I:C) was calculated accordingly using overton data.
Cytokine analysis
Secretion of cytokines and chemokines by primary human glioblastoma cells in response to stimulation with poly(I:C) was analyzed using electrochemiluminescent detection on a SECTOR3000 (MesoScale Discovery/MSD) using Discovery Workbench 4.0 software (MSD). The human cytokine panel included IFN-β (MSD, K151ADB-1), TGF-β (MSD, K151IUC-1), CXCL9 (MSD, N45IA-1), CCL5 (MSD, K151BFB-1) and IFN-α, IL-15, CXCL10 and CCL4 (MSD, K15067L-1). Additionally, IFN-γ secretion was analyzed using ELISA (eBioscience, 88–7316) with Nunc-Maxisorp flat-bottom 96-well plates (eBioscience, 44–2404). The ELISA was read on an iMark Microplate Absorbance Reader (Bio-Rad) using Microplate Manager 6 Software (Bio-Rad). Immune activation in cocultures of primary glioblastoma cells and PBMC was assessed using ELISA for secretion of IFN-γ (eBioscience) and granzyme B (R&D Systems, DY2906 with DY008), and using electrochemiluminescence for TNF-α secretion (MSD, K15067L-1). Standards and samples were measured in duplicate. All cytokine secretion assays were performed according to the manufacturer's instructions.
RNA isolation and quantitative real-time PCR
Primary human glioblastoma cells were incubated with 10µg/ml poly(I:C) for 24 hours. RNA was isolated from the cell pellet using the miRNeasy Mini Kit (Qiagen, 217004) according to the manufacturer's instructions, including a DNase treatment (Qiagen, 79254). A NanoDrop 1000 spectrophotometer (Thermo Fisher Scientific) was used to determine the RNA concentration.
RNA levels were analyzed by qRT-PCR on a LightCycler 480 (Roche) using the Power SYBR Green RNA-to-CT 1-Step Kit (Thermo Fisher Scientific, 4389986) according to the manufacturer's instructions. The custom-made primers (Biolegio) are shown in Table 2. The threshold for primer efficiency was set at [90–110%]. Determination of the number of housekeeping genes and data analysis were performed using qbase+ software (Biogazelle). A melt curve analysis was included in order to detect non-specific amplification.
Table 2.
Primers for quantitative real-time PCR.
| Gene | Forward primer | Reverse primer |
|---|---|---|
| GAPDH | 5′-TGCACCACCAACTGCTTAGC-3′ | 5′-GGCATGGACTGTGGTCATGAG-3′ |
| MAVS | 5′-CACAGCAAGAGACCAGGATCG-3′ | 5′-GCCGCTGAAGGGTATTGAAG-3′ |
| PD-L1 | 5′-CTGTCACGGTTCCCAAGGAC-3′ | 5′-GGTCTTCCTCTCCATGCACAA-3′ |
| PD-L2 | 5′-GTACATAATAGAGCATGGCAGCA-3′ | 5′-CCACCTTTTGCAAACTGGCTGT-3′ |
| RPL13A | 5′-CCTGGAGGAGAAGAGGAAAGAGA-3′ | 5′-TTGAGGACCTCTGTGTATTTGTCAA-3′ |
| SDHA | 5′-ACTCAGCATGCAGAAGTCAATGC-3′ | 5′-ACCTTCTTGCAACACGCTTCCC-3′ |
| TICAM1 | 5′-GGCTTTCAGGGTGGTCAGAA-3′ | 5′-GACCGTAGCAATACCCCCAC-3′ |
For the qRT-PCR reaction, 200mM of each primer and 20ng RNA were added to the reaction mix. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH), ribosomal protein L13a (RPL13A) and succinate dehydrogenase complex flavoprotein subunit A (SDHA) served as housekeeping genes as determined using qbase+ software.
Immunohistochemical tumor phenotyping
Primary human glioblastoma cells were incubated with 10µg/ml poly(I:C) for 24 hours. Next, the harvested cells were fixed for at least one hour in 4% buffered formaldehyde (Chem-Lab NV, CL02.0644.5000) and embedded in paraffin in order to acquire cell blocks, which were cut into 5µm-thick sections. The sections were baked in an oven for 0.5 to 1 hour at 60°C in order to facilitate attachment and to soften the paraffin. Staining was performed as previously described with adaptations.62 IHC for PD-L1 was performed on an Omnis instrument (Dako) with EnVision FLEX TRS Low pH antigen retrieval buffer at 95°C for 30 minutes (Dako Omnis, GV80511–2). The IHC staining protocol from the manufacturer's datasheet of PD-L1 (clone 28–8, Abcam, ab205921) was slightly adapted (20-minute incubation with 1:50 diluted antibody). The EnVision FLEX+ DAB detection kit (Dako Omnis, GV80011–2) was used according to the manufacturer's instructions, with the addition of the EnVision FLEX+ Rabbit LINKER (Dako Omnis, GV80911–2). Sections were counterstained with hematoxylin (Dako Omnis, GC808) for 5 minutes as part of the automated staining protocol. IHC for PD-L2 was performed on a Benchmark Ultra XT autostainer (Ventana Medical Systems Inc.) with CC1 antigen retrieval buffer at 97°C for 52 minutes (Ventana, #950–124). The UltraView DAB detection kit (Ventana, #760–051) was used according to the manufacturer's instructions. The IHC staining protocol from the manufacturer's datasheet of PD-L2 (clone # 176611, R&D Systems, MAB1224) was slightly adapted (12-minute incubation with 1:1500 diluted antibody). Sections were counterstained with hematoxylin II (Ventana, #79–2208) as part of the automated staining protocol. After the BenchMark staining, PD-L2 IHC slides were washed 1 minute in reagent buffer and 1 minute in distilled water. After the staining process with either antibody, the sections were dehydrated in graded alcohol, cleared in xylene, mounted with Quick-D Mounting Medium (Klinipath, 7280) and coverslipped. Images were taken using a BX51 microscope (Olympus) equipped with an DP71 digital camera (Olympus). CellSens Dimension Software (Olympus) was used for image acquisition and processing.
Coculture experiments
One day prior to coculturing, primary human glioblastoma cells were incubated with 10µg/ml poly(I:C), and frozen PBMC were thawed. After 24 hours, supernatant was collected and the primary glioblastoma cells were harvested, resuspended in their supernatant and seeded at 20×103 cells per tube. Next, 800×103 PBMC in DMEM were added to the respective tubes, and the cocultures were incubated for 24 or 96 hours. After 24 hours, lymphocytes were stained for immunophenotyping; after 24 and 96 hours, supernatant was collected for cytokine analysis of granzyme B and IFN-γ, respectively.
Blocking experiments were performed similarly with one additional step. Prior to PBMC addition, the primary glioblastoma cells were incubated for one hour with 3µg blocking antibody against PD-L1 (eBioscience, 16–5983), PD-L2 (eBioscience, 16–5888), or isotype control (eBioscience, 16–4714). Following the addition of PBMC, the coculture contained 10µg/ml blocking antibody and was maintained for 96 hours.
Flow cytometric immunophenotyping
Immunophenotyping of lymphocytes was performed on the cocultures in the immune activation, migration and proliferation assays. For the assessment of lymphocytic activation after 24 hours of coculture, the cells were stained for 15 minutes with an immune activation panel consisting of activation marker CD69 (BD Biosciences, 557756), degranulation marker CD107a (BD Biosciences, 555800), lineage markers CD3 (BD Biosciences, 560835), CD8 (Molecular Probes, MHCD0828) and NKp46 (BD Biosciences, 558051), and LIVE/DEAD Fixable Aqua Dead Cell Stain (Molecular Probes, L34957). CD107a and the corresponding isotype antibody were added to the cocultures from the beginning. For the assessment of lymphocyte migration and the chemokine receptor profile, the panel consisted of chemokine receptors CXCR3 (BD Biosciences, 550967) and CCR5 (BD Biosciences, 557752), lineage markers CD3 (BD Biosciences, 560176), CD8 (Molecular Probes) and CD56 (BD Biosciences, 345811), and the LIVE/DEAD viability stain (Molecular Probes). In these experiments, an isotype panel harvesting corresponding species- and isotype-matched antibodies at identical concentrations was included to serve as controls (BD Biosciences, 555748, 555749 and 560167 for immune activation, and 345818 and 557907 for chemokine receptors), along with the lineage markers and the viability stain. For the proliferation assay, the panel consisted of lineage markers CD3 (BD Biosciences, 560176) and CD8 (BD Biosciences, 345774), and the LIVE/DEAD viability stain (Moelcular Probes). Samples were analyzed on a BD FACSAria II flow cytometer (Becton Dickinson). Data were acquired using FACSDiva software (BD Biosciences) and analyzed using FlowJo v10 software. Representative flow cytometry profiles are shown in Supplementary Figure S1.
Chemotaxis assay
Transwell chemotaxis assays were performed using 24-well transwells with a 5µm pore size polycarbonate membrane (Corning, 3421). After 24 hours, supernatant of poly(I:C)-treated and naïve primary human glioblastoma cells was collected and transferred to the 24-well plate. Next, the transwell was placed in the well and 1×106 PBMC were added to the transwell insert. Migrated cells were collected from the lower compartment after three hours of incubation at 37°C with 5% CO2. Following staining, cell counting and phenotyping was performed on a BD FACSAria II using a fixed volume and a continuous flow rate. Migration due to poly(I:C) treatment of primary glioblastoma cells was calculated as: relative migration = (number of migrated cells towards supernatantpoly(I:C)-treated / number of migrated cells towards supernatantnaïve).
Proliferation assay
The proliferation assay was set up similarly to the coculture experiment described earlier. Three days prior to coculturing, PBMC were thawed, seeded at 1×106 PBMC/ml and stimulated with 2µg/ml phorbol 12-myristate 13-acetate (Sigma-Aldrich, P8139) and 50IU/ml IL-2 (Immunotools, 11340025). One day prior to coculturing, primary human glioblastoma cells were incubated with 10µg/ml poly(I:C). The next day, supernatant from the primary glioblastoma cell cultures was collected and the tumor cells were harvested, resuspended in their supernatant and seeded at 20×103 cells per tube. One hour prior to coculturing, PD-L1 or PD-L2 on the primary human glioblastoma cells was blocked as described earlier. On the same day, the PBMC were labelled with 5µM 5,6-carboxyfluorescein diacetate succinimidyl ester (CFSE; Molecular Probes, C34554) according to the manufacturer's instructions. Next, 200×103 PBMC in DMEM were added to the respective tubes, which were then incubated at 37°C with 5% CO2. After six days, CD8+ T lymphocytic proliferation was asses by quantifying the percentage of CFSE-diluted CD8+ T lymphocytes on a BD FACSAria II. The proliferation percentages were normalized to the isotype control, which was set at 0, and the effect of blocking antibodies was subsequently evaluated relative to this value.
Statistics
Prism 7.03 software (GraphPad) was used for data comparison and artwork. Statistical analysis was performed using SPSS Statistics 24 software (IBM). Paired-wise non-parametric tests were performed to compare medians between two (Wilcoxon signed ranks test) or multiple groups (Friedman test with Dunn-Bonferroni post-hoc testing). Correlations were investigated using the Pearson's correlation coefficient. Median values are depicted. Differences were predefined to be statistically significant when p < 0.05.
Supplementary Material
Funding Statement
Research Foundation Flanders, 1523715N, Research Foundation Flanders, 1121016N, Research Foundation Flanders, 1S32316N, University Foundation of Belgium, University of Antwerp (Research fund), Multidisciplinary Oncological Center Antwerp (Antwerp University Hospital), Flanders Innovation & Entrepreneurship, 141433. This work was performed with support of the Research Foundation Flanders (grant number: 1523715N), the Research Fund of the University of Antwerp and the Multidisciplinary Onco-logical Center (MOCA) of the Antwerp University Hospital (UZA). J. De Waele and J. Van Audenaerde are research fellows of the Research Foundation Flanders (fellowship numbers: 1121016N and 1S32316N), E. Marcq of Flanders Innovation & Entrepreneurship (fellowship number: 141433). We also thank the Vereycken family, Mr. Willy Floren and the University Foundation of Belgium for their financial support.
Disclosure of interest
The authors report no conflict of interest.
Acknowledgments
The authors express their gratitude to the members of the Departments of Neurosurgery of UZA and AZ Nikolaas, and the Biobank@UZA (Antwerp, Belgium; ID: BE71030031000; Belgian Virtual Tumorbank funded by the National Cancer Plan) for sample collection and provision; to Prof. Peter Ponsaerts for the use of the microscope; to Prof. Guy Van Camp and dr. Ken Op de Beeck for the use of the qRT-PCR machine; to Christophe Hermans, Céline Merlin, Hilde Lambrechts, Hans De Reu and Okke Geurts for technical assistance; to Tina Lauwers and Lesley De Backer (MOCA, UZA) for their help in collecting patient information; and to dr. Nicolas Goffart (University of Liège/Utrecht University) for advice on the primary cell culture.
References
- 1.Ostrom QT, Gittleman H, Xu J, Kromer C, Wolinsky Y, Kruchko C, Barnholtz-Sloan JS. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2009–2013. Neuro Oncol. 2016;18(suppl_5):v1–v75. doi: 10.1093/neuonc/now207. PMID:28475809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJB, Belanger K, Brandes AA, Marosi C, Bogdahn U, et al.. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. New Engl J Med. 2005;352(10):987–96. doi: 10.1056/NEJMoa043330. PMID:15758009. [DOI] [PubMed] [Google Scholar]
- 3.Postow MA, Callahan MK, Wolchok JD. Immune Checkpoint Blockade in Cancer Therapy. J Clin Oncol. 2015;33(17):1974–82. doi: 10.1200/JCO.2014.59.4358. PMID:25605845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tan AC, Heimberger AB, Khasraw M. Immune Checkpoint Inhibitors in Gliomas. Curr Oncol Rep. 2017;19(4):23. doi: 10.1007/s11912-017-0586-5. PMID:28303490. [DOI] [PubMed] [Google Scholar]
- 5.Marcq E, Pauwels P, van Meerbeeck JP, Smits EL. Targeting immune checkpoints: New opportunity for mesothelioma treatment? Cancer Treat Rev. 2015;41(10):914–24. doi: 10.1016/j.ctrv.2015.09.006. PMID:26433514. [DOI] [PubMed] [Google Scholar]
- 6.Drake CG, Jaffee E, Pardoll DM. Mechanisms of immune evasion by tumors. Adv Immunol. 2006;90:51−81. doi: 10.1016/S0065-2776(06)90002-9. PMID:16730261. [DOI] [PubMed] [Google Scholar]
- 7.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74. doi: 10.1016/j.cell.2011.02.013. PMID:21376230. [DOI] [PubMed] [Google Scholar]
- 8.Berghoff AS, Kiesel B, Widhalm G, Rajky O, Ricken G, Wohrer A, Dieckmann K, Filipits M, Brandstetter A, Weller M, et al.. Programmed death ligand 1 expression and tumor-infiltrating lymphocytes in glioblastoma. Neuro Oncol. 2015;17(8):1064–75. doi: 10.1093/neuonc/nou307. PMID:25355681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nduom EK, Wei J, Yaghi NK, Huang N, Kong LY, Gabrusiewicz K, Ling X, Zhou S, Ivan C, Chen JQ, et al.. PD-L1 expression and prognostic impact in glioblastoma. Neuro Oncol. 2016;18(2):195–205. doi: 10.1093/neuonc/nov172. PMID:26323609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reardon DA, Gokhale PC, Klein SR, Ligon KL, Rodig SJ, Ramkissoon SH, Jones KL, Conway AS, Liao X, Zhou J, et al.. Glioblastoma Eradication Following Immune Checkpoint Blockade in an Orthotopic, Immunocompetent Model. Cancer Immunol Res. 2016;4(2):124–35. doi: 10.1158/2326-6066.CIR-15-0151. PMID:26546453. [DOI] [PubMed] [Google Scholar]
- 11.Wainwright DA, Chang AL, Dey M, Balyasnikova IV, Kim CK, Tobias A, Cheng Y, Kim JW, Qiao J, Zhang L, et al.. Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA-4, and PD-L1 in mice with brain tumors. Clin Cancer Res. 2014;20(20):5290–301. doi: 10.1158/1078-0432.CCR-14-0514. PMID:24691018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zeng J, See AP, Phallen J, Jackson CM, Belcaid Z, Ruzevick J, Durham N, Meyer C, Harris TJ, Albesiano E, et al.. Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. Int J Radiat Oncol Biol Phys. 2013;86(2):343–9. doi: 10.1016/j.ijrobp.2012.12.025. PMID:23462419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cheever MAM. Twelve immunotherapy drugs that could cure cancers. Immunological Reviews. 2008;222:357–68. doi: 10.1111/j.1600-065X.2008.00604.x. PMID:18364014. [DOI] [PubMed] [Google Scholar]
- 14.Pichlmair A, Reis e Sousa C. Innate recognition of viruses. Immunity. 2007;27(3):370–83. doi: 10.1016/j.immuni.2007.08.012. PMID:17892846. [DOI] [PubMed] [Google Scholar]
- 15.Ammi R, De Waele J Willemen Y, Van Brussel I Schrijvers DM, Lion E, Smits ELJ. Poly(I:C) as cancer vaccine adjuvant: Knocking on the door of medical breakthroughs. Pharmacol Therapeut. 2015;146:120–31. doi: 10.1016/j.pharmthera.2014.09.010. PMID:25281915. [DOI] [PubMed] [Google Scholar]
- 16.Glas M, Coch C, Trageser D, Dassler J, Simon M, Koch P, Mertens J, Quandel T, Gorris R, Reinartz R, et al.. Targeting the cytosolic innate immune receptors RIG-I and MDA5 effectively counteracts cancer cell heterogeneity in glioblastoma. Stem Cells. 2013;31(6):1064–74. doi: 10.1002/stem.1350. PMID:23390110. [DOI] [PubMed] [Google Scholar]
- 17.Parker BS, Rautela J, Hertzog PJ. Antitumour actions of interferons: implications for cancer therapy. Nat Rev Cancer. 2016;16(3):131–44. doi: 10.1038/nrc.2016.14. PMID:26911188. [DOI] [PubMed] [Google Scholar]
- 18.Kuznik A, Bencina M, Svajger U, Jeras M, Rozman B, Jerala R. Mechanism of endosomal TLR inhibition by antimalarial drugs and imidazoquinolines. J Immunol. 2011;186(8):4794–804. doi: 10.4049/jimmunol.1000702. PMID:21398612. [DOI] [PubMed] [Google Scholar]
- 19.Wilmotte R, Burkhardt K, Kindler V, Belkouch MC, Dussex G, Tribolet N, Walker PR, Dietrich PY. B7-homolog 1 expression by human glioma: a new mechanism of immune evasion. Neuroreport. 2005;16(10):1081–5. doi: 10.1097/00001756-200507130-00010. PMID:15973152. [DOI] [PubMed] [Google Scholar]
- 20.Shin DS, Zaretsky JM, Escuin-Ordinas H, Garcia-Diaz A, Hu-Lieskovan S, Kalbasi A, Grasso CS, Hugo W, Sandoval S, Torrejon DY, et al.. Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discov. 2017;7(2):188–201. doi: 10.1158/2159-8290.CD-16-1223. PMID:27903500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trapani JA. Granzymes: a family of lymphocyte granule serine proteases. Genome Biol. 2001;2(12):REVIEWS3014. 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boes M, Meyer-Wentrup F. TLR3 triggering regulates PD-L1 (CD274) expression in human neuroblastoma cells. Cancer Lett. 2015;361(1):49–56. doi: 10.1016/j.canlet.2015.02.027. PMID:25697485. [DOI] [PubMed] [Google Scholar]
- 23.Ho V, Lim TS, Lee J, Steinberg J, Szmyd R, Tham M, Yaligar J, Kaldis P, Abastado JP, Chew V. TLR3 agonist and Sorafenib combinatorial therapy promotes immune activation and controls hepatocellular carcinoma progression. Oncotarget. 2015;6(29):27252–66. doi: 10.18632/oncotarget.4583. PMID:26287667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Z, Zhang C, Liu X, Wang Z, Sun L, Li G, Liang J, Hu H, Liu Y, Zhang W, et al.. Molecular and clinical characterization of PD-L1 expression at transcriptional level via 976 samples of brain glioma. Oncoimmunology. 2016;5(11):e1196310. doi: 10.1080/2162402X.2016.1196310. PMID:27999734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Soliman H, Khalil F, Antonia S. PD-L1 expression is increased in a subset of basal type breast cancer cells. PLoS One. 2014;9(2):e88557. doi: 10.1371/journal.pone.0088557. PMID:24551119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lin Q, Wang L, Lin Y, Liu X, Ren X, Wen S, Du X, Lu T, Su SY, Yang X, et al.. Toll-like receptor 3 ligand polyinosinic:polycytidylic acid promotes wound healing in human and murine skin. J Invest Dermatol. 2012;132(8):2085–92. doi: 10.1038/jid.2012.120. PMID:22572822. [DOI] [PubMed] [Google Scholar]
- 27.Sistigu A, Yamazaki T, Vacchelli E, Chaba K, Enot DP, Adam J, Vitale I, Goubar A, Baracco EE, Remedios C, et al.. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat Med. 2014;20(11):1301–9. doi: 10.1038/nm.3708. PMID:25344738. [DOI] [PubMed] [Google Scholar]
- 28.Anguille S, Van Acker HH, Van den Bergh J, Willemen Y, Goossens H, Van Tendeloo VF, Smits EL, Berneman ZN, Lion E. Interleukin-15 Dendritic Cells Harness NK Cell Cytotoxic Effector Function in a Contact- and IL-15-Dependent Manner. PLoS One. 2015;10(5):e0123340. doi: 10.1371/journal.pone.0123340. PMID:25951230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Van Acker HH, Anguille S, Willemen Y, Van den Bergh JM, Berneman ZN, Lion E, Smits EL, Van Tendeloo VF. Interleukin-15 enhances the proliferation, stimulatory phenotype, and antitumor effector functions of human gamma delta T cells. J Hematol Oncol. 2016;9(1):101. doi: 10.1186/s13045-016-0329-3. PMID:27686372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Van Audenaerde JRM, De Waele J, Marcq E, Van Loenhout J, Lion E, Van den Bergh JMJ, Jesenofsky R, Masamune A, Roeyen G, Pauwels P, et al.. Interleukin-15 stimulates natural killer cell-mediated killing of both human pancreatic cancer and stellate cells. Oncotarget. 2017;8(34):56968–79. doi: 10.18632/oncotarget.18185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Garofalo S, D'Alessandro G, Chece G, Brau F, Maggi L, Rosa A, Porzia A, Mainiero F, Esposito V, Lauro C, et al.. Enriched environment reduces glioma growth through immune and non-immune mechanisms in mice. Nat Commun. 2015;6:6623. doi: 10.1038/ncomms7623. PMID:25818172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mathios D, Park CK, Marcus WD, Alter S, Rhode PR, Jeng EK, Wong HC, Pardoll DM, Lim M. Therapeutic administration of IL-15 superagonist complex ALT-803 leads to long-term survival and durable antitumor immune response in a murine glioblastoma model. International Journal of Cancer. 2016;138(1):187–94. doi: 10.1002/ijc.29686. PMID:26174883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mattei F, Schiavoni G, Belardelli F, Tough DF. IL-15 Is Expressed by Dendritic Cells in Response to Type I IFN, Double-Stranded RNA, or Lipopolysaccharide and Promotes Dendritic Cell Activation. The Journal of Immunology. 2001;167(3):1179–87. doi: 10.4049/jimmunol.167.3.1179. PMID:11466332. [DOI] [PubMed] [Google Scholar]
- 34.Han JF, Alvarez-Breckenridge CA, Wang QE, Yu JH. TGF-beta signaling and its targeting for glioma treatment. Am J Cancer Res. 2015;5(3):945–55. PMID:26045979. [PMC free article] [PubMed] [Google Scholar]
- 35.Finnson KW, McLean S, Di Guglielmo GM, Philip A. Dynamics of Transforming Growth Factor Beta Signaling in Wound Healing and Scarring. Adv Wound Care (New Rochelle). 2013;2(5):195–214. doi: 10.1089/wound.2013.0429. PMID:24527343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cao GW, Su JD, Lu WX, Zhang FH, Zhao GL, Marteralli D, Dong ZY. Adenovirus-mediated interferon-ss gene therapy suppresses growth and metastasis of human prostate cancer in nude mice. Cancer Gene Ther. 2001;8(7):497–505. doi: 10.1038/sj.cgt.7700333. PMID:11498771. [DOI] [PubMed] [Google Scholar]
- 37.Bald T, Landsberg J, Lopez-Ramos D, Renn M, Glodde N, Jansen P, Gaffal E, Steitz J, Tolba R, Kalinke U, et al.. Immune cell-poor melanomas benefit from PD-1 blockade after targeted type I IFN activation. Cancer Discov. 2014;4(6):674–87. doi: 10.1158/2159-8290.CD-13-0458. PMID:24589924. [DOI] [PubMed] [Google Scholar]
- 38.Nagato T, Lee YR, Harabuchi Y, Celis E. Combinatorial immunotherapy of polyinosinic-polycytidylic acid and blockade of programmed death-ligand 1 induce effective CD8T-cell responses against established tumors. Clin Cancer Res. 2014;20(5):1223–34. doi: 10.1158/1078-0432.CCR-13-2781. PMID:24389326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Melero I, Rouzaut A, Motz GT, Coukos G. T-cell and NK-cell infiltration into solid tumors: a key limiting factor for efficacious cancer immunotherapy. Cancer Discov. 2014;4(5):522–6. doi: 10.1158/2159-8290.CD-13-0985. PMID:24795012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Han S, Zhang C, Li Q, Dong J, Liu Y, Huang Y, Jiang T, Wu A. Tumour-infiltrating CD4(+) and CD8(+) lymphocytes as predictors of clinical outcome in glioma. Br J Cancer. 2014;110(10):2560–8. doi: 10.1038/bjc.2014.162. PMID:24691423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang I, Tihan T, Han SJ, Wrensch MR, Wiencke J, Sughrue ME, Parsa AT. CD8+ T-cell infiltrate in newly diagnosed glioblastoma is associated with long-term survival. J Clin Neurosci. 2010;17(11):1381–5. doi: 10.1016/j.jocn.2010.03.031. PMID:20727764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu JS, Lee PK, Ehtesham M, Samoto K, Black KL, Wheeler CJ. Intratumoral T cell subset ratios and Fas ligand expression on brain tumor endothelium. J Neurooncol. 2003;64(1-2):55–61. doi: 10.1007/BF02700020. PMID:12952286. [DOI] [PubMed] [Google Scholar]
- 43.Erdag G, Schaefer JT, Smolkin ME, Deacon DH, Shea SM, Dengel LT, Patterson JW, Slingluff CL Jr.. Immunotype and immunohistologic characteristics of tumor-infiltrating immune cells are associated with clinical outcome in metastatic melanoma. Cancer Res. 2012;72(5):1070–80. doi: 10.1158/0008-5472.CAN-11-3218. PMID:22266112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kmiecik J, Poli A, Brons NH, Waha A, Eide GE, Enger PO, Zimmer J, Chekenya M. Elevated CD3+ and CD8+ tumor-infiltrating immune cells correlate with prolonged survival in glioblastoma patients despite integrated immunosuppressive mechanisms in the tumor microenvironment and at the systemic level. J Neuroimmunol. 2013;264(1-2):71–83. doi: 10.1016/j.jneuroim.2013.08.013. PMID:24045166. [DOI] [PubMed] [Google Scholar]
- 45.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–64. doi: 10.1038/nrc3239. PMID:22437870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Basu S, Broxmeyer HE. CCR5 ligands modulate CXCL12-induced chemotaxis, adhesion, and Akt phosphorylation of human cord blood CD34+ cells. J Immunol. 2009;183(11):7478–88. doi: 10.4049/jimmunol.0900542. PMID:19917679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Meiser A, Mueller A, Wise EL, McDonagh EM, Petit SJ, Saran N, Clark PC, Williams TJ, Pease JE. The Chemokine Receptor CXCR3 Is Degraded following Internalization and Is Replenished at the Cell Surface by De Novo Synthesis of Receptor. The Journal of Immunology. 2008;180(10):6713–24. doi: 10.4049/jimmunol.180.10.6713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhu X, Fallert-Junecko BA, Fujita M, Ueda R, Kohanbash G, Kastenhuber ER, McDonald HA, Liu Y, Kalinski P, Reinhart TA, et al.. Poly-ICLC promotes the infiltration of effector T cells into intracranial gliomas via induction of CXCL10 in IFN-alpha and IFN-gamma dependent manners. Cancer Immunol Immunother. 2010;59(9):1401–9. doi: 10.1007/s00262-010-0876-3. PMID:20549206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al.. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515(7528):568–71. doi: 10.1038/nature13954. PMID:25428505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. 2017;541(7637):321–30. doi: 10.1038/nature21349. PMID:28102259. [DOI] [PubMed] [Google Scholar]
- 51.Salmon H, Idoyaga J, Rahman A, Leboeuf M, Remark R, Jordan S, Casanova-Acebes M, Khudoynazarova M, Agudo J, Tung N, et al.. Expansion and Activation of CD103(+) Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. Immunity. 2016; 44(4):924–38. doi: 10.1016/j.immuni.2016.03.012. PMID:27096321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kees T, Lohr J, Noack J, Mora R, Gdynia G, Todt G, Ernst A, Radlwimmer B, Falk CS, Herold-Mende C, et al.. Microglia isolated from patients with glioma gain antitumor activities on poly (I:C) stimulation. Neuro Oncol. 2012;14(1):64–78. doi: 10.1093/neuonc/nor182. PMID:22015597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Antonios JP, Soto H, Everson RG, Orpilla J, Moughon D, Shin N, Sedighim S, Yong WH, Li G, Cloughesy TF, et al.. PD-1 blockade enhances the vaccination-induced immune response in glioma. JCI Insight. 2016;1(10):e87059. doi: 10.1172/jci.insight.87059. PMID:27453950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, Olson OC, Quick ML, Huse JT, Teijeiro V, et al.. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. 2013;19(10):1264–72. doi: 10.1038/nm.3337. PMID:24056773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Quail DF, Joyce JA. The Microenvironmental Landscape of Brain Tumors. Cancer Cell. 2017;31(3):326–41. doi: 10.1016/j.ccell.2017.02.009. PMID:28292436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Forghani P, Waller EK. Poly (I: C) modulates the immunosuppressive activity of myeloid-derived suppressor cells in a murine model of breast cancer. Breast Cancer Res Treat. 2015;153(1):21–30. doi: 10.1007/s10549-015-3508-y. PMID:26208484. [DOI] [PubMed] [Google Scholar]
- 57.Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, Gupta R, Tsai JM, Sinha R, Corey D, et al.. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature. 2017;545(7655):495–9. doi: 10.1038/nature22396. PMID:28514441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Noguchi T, Ward JP, Gubin MM, Arthur CD, Lee SH, Hundal J, Selby MJ, Graziano RF, Mardis ER, Korman AJ, et al.. Temporally Distinct PD-L1 Expression by Tumor and Host Cells Contributes to Immune Escape. Cancer Immunol Res. 2017;5(2):106–17. doi: 10.1158/2326-6066.CIR-16-0391. PMID:28073774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al.. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–54. doi: 10.1056/NEJMoa1200690. PMID:22658127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Beavis PA, Milenkovski N, Henderson MA, John LB, Allard B, Loi S, Kershaw MH, Stagg J, Darcy PK. Adenosine Receptor 2A Blockade Increases the Efficacy of Anti-PD-1 through Enhanced Antitumor T-cell Responses. Cancer Immunol Res. 2015;3(5):506–17. doi: 10.1158/2326-6066.CIR-14-0211. PMID:25672397. [DOI] [PubMed] [Google Scholar]
- 61.Goffart N, Kroonen J, Di Valentin E, Dedobbeleer M, Denne A, Martinive P, Rogister B. Adult mouse subventricular zones stimulate glioblastoma stem cells specific invasion through CXCL12/CXCR4 signaling. Neuro-Oncology. 2015;17(1):81–94. doi: 10.1093/neuonc/nou144. PMID:25085362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Marcq E, Siozopoulou V, De Waele J, van Audenaerde J, Zwaenepoel K, Santermans E, Hens N, Pauwels P, van Meerbeeck JP, Smits EL. Prognostic and predictive aspects of the tumor immune microenvironment and immune checkpoints in malignant pleural mesothelioma. Oncoimmunology. 2017;6(1):e1261241. doi: 10.1080/2162402X.2016.1261241. PMID:28197385. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.