

ABSTRACT
We recently identified CXCR4 as a novel vascular marker for vessel sprouting in hepatocellular carcinoma (HCC) tissues. Thus, CXCR4+ endothelial cells (ECs) could serve as a potential predictor for patients who may benefit from sorafenib treatment; however, the mechanism that regulates vascular CXCR4 expression in HCC remains largely unknown. Here, we revealed a large number of monocytes/macrophages (Mo/Mϕ) to be selectively enriched in the perivascular areas of CXCR4+ vessels in HCC samples. The depletion of Mo/Mϕ with gadolinium chloride (GdCl3) or zoledronic acid (ZA) treatment significantly reduced vascular CXCR4 expression in HCC tumors. This phenomenon was also confirmed in CCR2-KO mice, which exhibited reduced infiltration of inflammatory Mo/Mϕ in tumor tissues. Mechanistic studies revealed that inflammatory cytokines derived from tumor conditioned Mo/Mϕ, especially TNF-α, could up-regulate CXCR4 expression on ECs. TNF-α-induced activation of the Raf-ERK pathway, but not Notch signaling, was responsible for the expression of CXCR4. Moreover, the combination treatment of sorafenib with ZA was associated with improved anti-tumor efficacy by significantly reducing vascular CXCR4 expression. These findings revealed that Mo/Mϕ could regulate CXCR4 expression in the tumor vasculature. Thus, the inhibition of Mo/Mϕ inflammation might enhance the treatment efficacy of sorafenib in HCC.
KEYWORDS: CXCR4, tumor endothelial cell, monocyte/macrophage, TNF-α, HCC
Introduction
Hepatocellular carcinoma (HCC) is one of the most commonly diagnosed cancers and leading causes of cancer-related mortality worldwide.1 The dismal outcome has been attributed primarily to the high degree of vascularity, which increases early blood-borne metastasis and the frequency of recurrence.2 Sorafenib is a molecular-targeted drug that inhibits tumor angiogenesis and remains the only approved systemic therapy for advanced HCC patients; however, the clinical improvement associated with overall patient survival is limited.3,4
During the rapid growth of HCC, tumor angiogenesis often presents with abnormal cues to coordinate vessel growth and remodeling.5,6 The two main cellular elements of angiogenesis include endothelial tip cells and stalk cells, which functionally work together and comprise the main portion of the expanding vessels.7–9 Recently, we and other groups revealed a distinct vascular pattern: vessels that encapsulate tumor clusters (VETC), which could provide an epithelial-mesenchymal transition (EMT)-independent mode of HCC metastasis.10–12 We have also identified CXCR4 as a novel vascular marker for tumor endothelial cells (TECs) with tip cell characteristics and thus for VETC+ HCC. This marker could be a potential predictor for patients who may benefit from sorafenib treatment.13 However, the underlying mechanism of vascular CXCR4 expression remains largely unknown.
There is substantial evidence that the pro-inflammatory response within the tumor microenvironment could be rerouted into a tumor-promoting direction by stimulating angiogenesis and tissue remodeling.14–18 Monocytes/macrophages (Mo/Mϕ) constitute a major component of the leukocyte infiltrate in the tumor microenvironment.19 Tumor-associated Mo/Mϕ with an altered phenotype and the ability to secrete paracrine angiogenesis modulating factors, including vascular endothelial growth factor (VEGF), basic fibroblast growth factor (bFGF), tumor necrosis factor α (TNF-α), interleukin 1β (IL-1β), and matrix metalloproteinase (MMPs), have a profound influence on tumor angiogenesis.20 Mϕ can also directly modulate endothelial cell (EC) behaviors by promoting tip cell branching into a close apposition for fusion.21 Although these studies indicate that the interaction between Mo/Mϕ and EC plays a critical role in fostering angiogenesis, the capability, mechanism and significance of Mo/Mϕ in regulating CXCR4 expression on ECs remains largely unknown, especially in human tumors.
In the present study, we investigated the influence of Mo/Mϕ on tumor ECs in HCC, and revealed that the TNF-α derived from Mo/Mϕ was responsible for the increased CXCR4 expression on ECs via activation of the Raf-Erk pathway. Using two drugs that specifically target Mo/Mϕ, we found that depleting Mo/Mϕ reduced the CXCR4 expression on TECs, as well as the tumor growth and metastasis in a murine model of HCC, which potentiated a therapeutic collaboration with sorafenib treatment. These findings suggest that tumor-conditioned Mo/Mϕ may play a pivotal role in regulating CXCR4 expression on TECs in HCC. Furthermore, we provide evidence supporting the therapeutic effect of Mo/Mϕ depletion in combination with sorafenib for HCC treatment.
Results
Tumor-infiltrating Mo/Mϕ regulate vascular CXCR4 expression in HCC tumor tissues
To investigate the potential role of Mo/Mϕ in the expression of CXCR4 on human HCC vessels, we first examined the distribution of CD68+ Mo/Mϕ and CXCR4-expressing vessels by immunofluorescence staining in the paraffin-embedded tissues derived from 30 HCC patients. Confocal microscopic analysis revealed that Mo/Mϕ were prone to enriched in the perivascular areas of CXCR4+ vessels compared to CXCR4− vessels (Fig. 1A). We divided the HCC patients into two groups in accordance with the median value of CXCR4+ vascular density in the tumors. The statistical analysis showed that more Mo/Mϕ were distributed in the samples with CXCR4High vessels compared to those with the CXCR4Low vessels (Fig. 1B).
Figure 1.
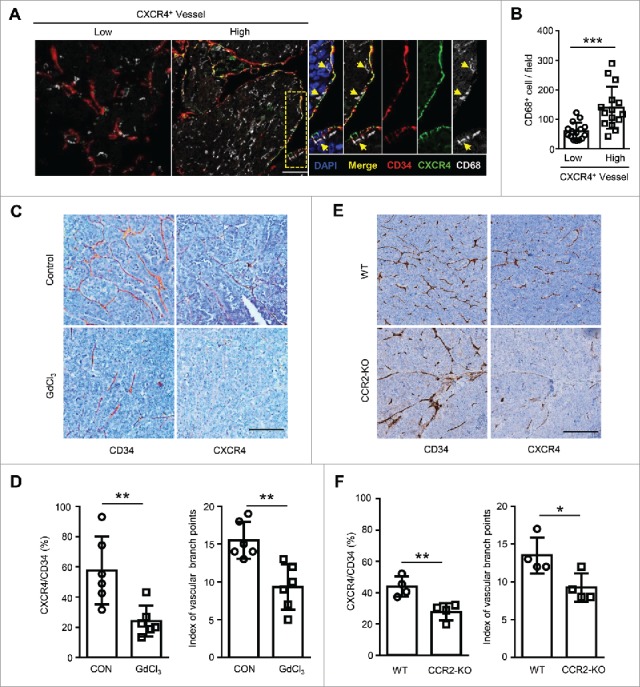
Tumor-infiltrating Mo/Mϕ regulate vascular CXCR4 expression in HCC tumor tissues. (A) The staining of CD34+ vascular (red), CXCR4+ (green) and CD68+ monocytes/macrophages (gray) in HCC tissues was examined by confocal microscopy. DAPI staining appears blue. Scale bar = 50 μm. (B) Patients were divided into two groups according to the median value of the intra-tumoral CXCR4+ vascular densities (n = 30). The infiltration of Mo/Mϕ between groups are shown. (C−F) Orthotopic HCC models derived from Hepa1-6 cells were established in C57 BL/6 (C and D) or CCR2-KO (E and F) mice. Immunohistochemical staining for CXCR4 and CD34 was performed on serial tissue sections of the mouse tumor allografts. Scale bar = 100 μm. The vascular CXCR4 density and the index of the vascular branch points were assessed (D and F). WT, wild type; CCR2-KO, CCR2 knock-out mice. Values given in B, D, and F represent the means ± SEM. * P < 0.05; ** P < 0.01; *** P < 0.001.
To determine the relationship between Mo/Mϕ and vascular CXCR4 expression, an orthotopic HCC model was established using the Hepa1-6 murine cell line. One week after the tumor cell inoculation, mice were randomly divided into two groups, and then subjected to a two-week treatment of PBS or gadolinium chloride (GdCl3), an inhibitor of Mo/Mϕ inflammation.22 Strikingly, with the marked reduction of the Mo/Mϕ in the tumor tissue of the GdCl3 treatment group (Fig. S1A), the vascular CXCR4 expression was significantly reduced (Fig. 1C-D). To exclude the direct effect of GdCl3 on ECs, we treated human umbilical vein endothelial cells (HUVECs) with GdCl3 for 24 hrs in vitro, in which condition over 95% of cultured alveolar macrophage underwent cell death.23 As shown in Figure S2, GdCl3 treatment had no effect on the apoptotic rate, the morphology or the CXCR4 expression of HUVECs, compared to the control condition. Since a CCR2 blockade reduces blood inflammatory monocytes and the infiltration of intra-tumorous macrophages in murine HCC tissues,24 CCR2 knock-out (CCR2-KO) mice were used to confirm the influence of Mo/Mϕ (Fig. S1B). As shown in Fig. 1E-F, the vascular CXCR4 expression was significantly reduced in CCR2-KO mice compared with the wild type (WT) mice. Moreover, the index of vascular branch points used to reflect the effect of CXCR4 on vessel sprouting in vivo13 was also significantly reduced following treatment with GdCl3 (Fig. 1D) and in CCR2-KO mice (Fig. 1F). Taken together, these data indicate that tumor-associated Mo/Mϕ can regulate the vascular expression of CXCR4 in HCC tumors.
Conditioned medium from tumor-activated Mo/Mϕ induce CXCR4 expression on ECs
To investigate the potential mechanism responsible for Mo/Mϕ-induced CXCR4 expression in HCC, HUVECs were cultured for 24 hrs in conditioned medium from human tumor cells (TSN), control human monocytes (CCM) or TSN-exposed human monocytes (TCM), which mimicked the tumor milieu.25,26 We found that TCM, but not TSN or CCM, effectively induced CXCR4 expression on HUVECs (Fig. 2A-B). Inflammatory cytokines derived from Mo/Mϕ play an important role in tumor angiogenesis.19 Therefore, we examined the cytokine profiles of CCM and TCM, and found that TSN-exposed Mo/Mϕ secreted significant amounts of TNF-α, IL-1β, IL-6, and IL-10 (Fig. 2C). To determine the specific cytokine(s) that induced the expression of CXCR4 on ECs, we tested the effect of recombinant human (rh) cytokines in the culture system. As shown in Fig. 2D, rhTNF-α and IL-1β, but not IL-6 or IL-10, significantly induced the expression of CXCR4 on HUVECs in a dose-dependent manner. To further confirm the role of TNF-α and IL-1β secreted by Mo/Mϕ, specific neutralizing antibodies were used to antagonize the effects of TNF-α and IL-1β. The results revealed that blocking TNF-α but not IL-1β, completely abrogated the up-regulation of CXCR4 induced by TCM (Fig. 2E-F). In addition, we found that TCM treatment induced thick actin stress fibers to traverse the HUVECs over nucleus, and thus elonged the cells (Fig. S2C).
Figure 2.
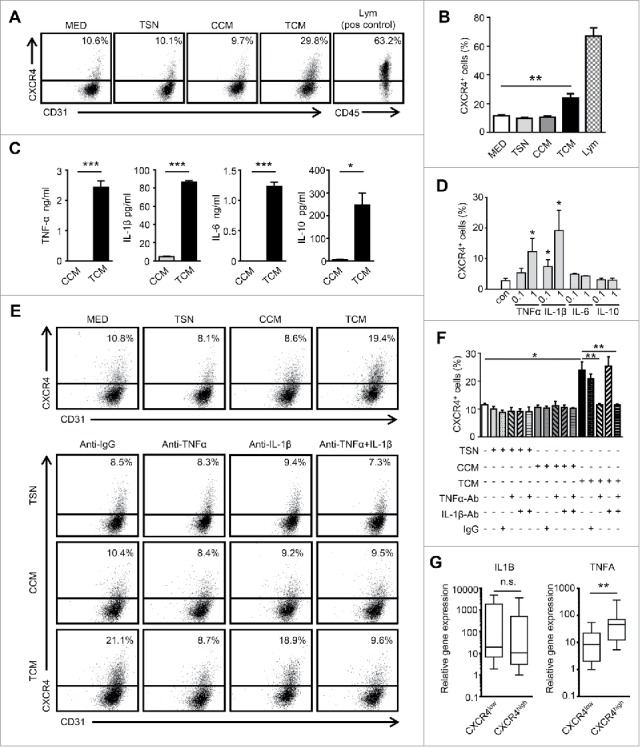
Conditioned medium from tumor-activated Mo/Mϕ induce CXCR4 expression on ECs. (A and B) HUVECs were cultured for 24 hrs in conditioned medium from tumor cells (TSN), control monocytes (CCM) or medium from TSN-exposed monocytes (TCM). Peripheral blood lymphocytes were used as the positive control for CXCR4 staining. (C) The levels of cytokines in CCM or TCM were determined by ELISA. (D) HUVECs were incubated for 24 hrs with rhTNF-α, rhIL-1β, rhIL-6, or rhIL-10 (ng/ml) at the indicated concentrations. (E and F) The HUVECs were cultured in TSN, CCM or TCM and mAb against TNF-α (1 μg/mL) or IL-1β (10 μg/mL), combined, or the control Ab (IgG1, 10 μg/mL). The percentages of HUVECs expressing CXCR4 were determined by FACS. (G) The relative expression of the indicated gene mRNA levels in the tumor tissues from HCC patients (n = 25) were analyzed by qPCR. All data shown are representative of at least three independent experiments. Lym, Peripheral blood lymphocytes. Error bar, SEM; n.s., not significant; * P < 0.05; ** P < 0.01; *** P < 0.001.
Consistent with the in vitro results, we observed that samples from HCC patients with CXCR4High vessels exhibited significantly higher levels of TNF-α compared with the samples with CXCR4Low vessels (Fig. 2G). However, there was no correlation between the IL-1β levels and vascular CXCR4 expression. These findings suggest that TNF-α derived from Mo/Mϕ in the tumor microenvironment are responsible for the CXCR4 up-regulation on TECs.
The essential role of the ERK pathway on the TNF-α-induced CXCR4 expression on ECs
We next sought to determine the underlying mechanism of TNF-α-induced CXCR4 expression on ECs. Our previous studies have suggested that the inhibition of the Raf-Erk pathway could reduce CXCR4 expression on ECs; moreover, NF-κB has been well characterized as a downstream target of TNF signaling.13,27 Therefore, we tested these potential pathways that might regulate TNF-α-induced CXCR4 expression on ECs. An irreversible inhibitor of IκBα phosphorylation (Bay11-7082), and a highly selective inhibitor of MEK1/2 (U0126) were used in our culture system. As expected, the activation of the NF-κB and Raf-Erk signaling pathways induced by TNF-α were completely inhibited by Bay11-7082 and U0126, respectively (Fig. 3A-B). However, blocking the Raf-Erk pathway, but not NF-κB pathway, effectively inhibited TCM and TNF-α-induced CXCR4 up-regulation on ECs (Fig. 3C-D).
Figure 3.
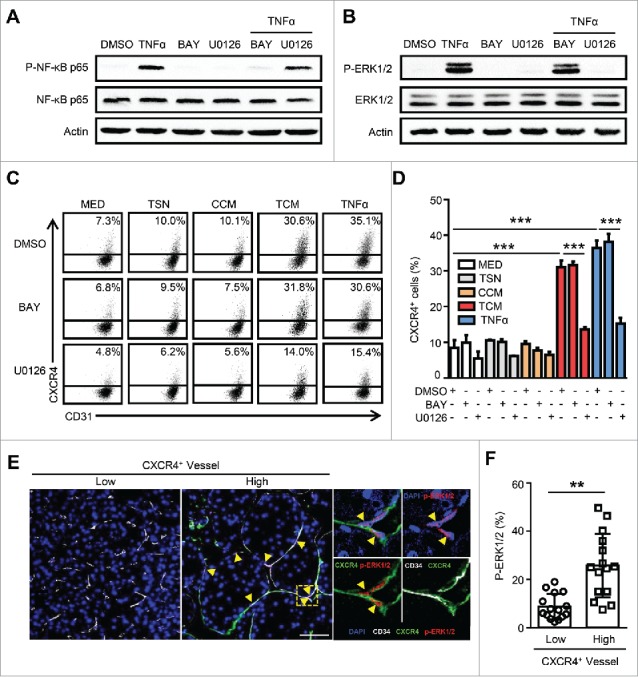
The essential role of the ERK pathway in TNF-α-induced CXCR4 expression in ECs. HUVECs were treated with the IκB inhibitor, Bay11-7082 (5 μg/mL), or the Erk1/2 inhibitor U0126 (10 μg/mL) for 1 hr followed by the administration of TNF-α (10 ng/mL) for 24 hrs. TNF-α induced activation of NF-κB at 30 min (A) and Erk1/2 at 15 min (B) are shown. (C and D) The percentages of CXCR4+ HUVECs were analyzed by FACS. (E and F) The expression of vascular CD34 (gray), CXCR4 (green), and p-ERK1/2 (red) in HCC tumor tissues was determined by confocal microscopy. DAPI staining appears blue. Scale bar = 50 μm. TSN, conditioned medium from tumor cells; CCM, conditioned medium from control monocytes; TCM, conditioned medium from TSN exposed-monocytes. All data shown are representative of at least three independent experiments. Error bar, SEM; * P < 0.05; ** P < 0.01; *** P < 0.001.
To further confirm these findings, we examined the in situ expression of CXCR4 and p-ERK in human HCC tissues using immunofluorescence staining. As shown in Fig. 3E, p-ERK1/2 was selectively located in the nucleus of ECs, but was negatively or only weakly expressed on other stromal and hepatoma cells in the tumor. Moreover, the expression of p-ERK1/2 was highly enriched on the CXCR4+ vessels, while a significantly smaller proportion of CXCR4− vessels exhibited the activation of this signaling (Fig. 3F). Consistently, we found that the activation of the ERK pathway was correlated with the presence of micrometastasis (microvascular invasion) in the adjacent nontumor tissues (within 1 cm from the primary tumor) of human HCC (Fig. S3). The above data reveal that the Raf-Erk pathway is essential for TNF-α-induced CXCR4 expression on ECs in HCC.
Notch signaling is dispensable for TNF-α-induced CXCR4 expression in HCC
Our previous study identified that CXCR4+ ECs represent angiogenic tip cells in HCC tissues.13 Studies by other groups have shown that Notch pathways are essential for the regulation of EC behavior during blood vessel sprouting (angiogenesis) through regulating key tip cell-related genes;8,28,29 therefore, we further tested the role of Notch signaling in our culture system. The HUVECs were treated with TSN, CCM, TCM and TNF-α for 24 hrs and then analyzed for gene expression by RT-PCR. Despite the increased expression of CXCR4 and JAG1 in the HUVECs, the expression of the Notch ligand DLL4, and more importantly, the critical downstream signal molecules HES1 and HEY1 were significantly down-regulated by TCM and TNF-α treatments (Fig. 4A). This was consistent with the findings from another group that TNF-α was capable of inhibiting Notch signaling.27 To evaluate whether the inhibition of the Notch pathway could up-regulate CXCR4 expression in HUVECs, a Notch signaling inhibitor, DAPT (gamma-secretase inhibitor),30 was applied in our culture system. Compared with the untreated group, DAPT treatment induced CXCR4 expression on HUVECs (Fig. 4B), indicating that the Notch pathway could regulate CXCR4 expression on ECs in vitro.
Figure 4.
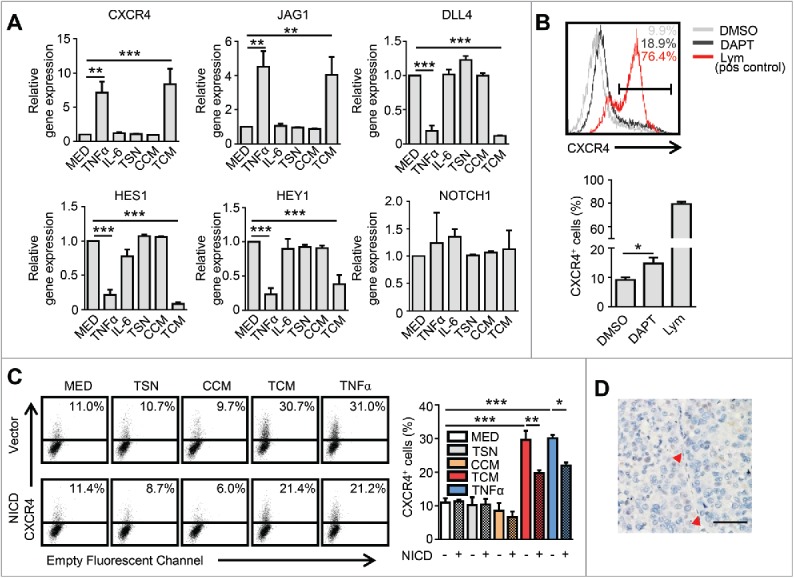
Notch signaling is dispensable for TNF-α-induced CXCR4 expression in HCC. (A) HUVECs were cultured in the indicated conditioned medium or TNF-α (10 ng/mL) for 24 hrs and subsequently harvested for analysis of gene expression by qPCR. (B) HUVECs were cultured in the presence or absence of the Notch signaling inhibitor, DAPT (10 μM), for 24 hrs and then harvested for an analysis of CXCR4 expression by FACS. Peripheral blood lymphocytes were used as the positive control for CXCR4 staining. (C) Cells transfected with lentiviral particles expressing activated Notch receptors (NICD) or the empty vector were harvested 24 hrs after exposure to indicated conditioned medium or TNF-α, and analyzed by FACS. (D) Analysis of NICD expression in the HCC tumor tissue by immunohistochemical staining. The red arrowhead indicates the endothelial cell nucleus. Scale bar = 50μm. All data shown are representative of at least three independent experiments. Lym, Peripheral blood lymphocytes; TSN, conditioned medium from tumor cells; CCM, conditioned medium from control monocytes; TCM, conditioned medium from TSN exposed-monocytes. Error bar, SEM; * P < 0.05; ** P < 0.01; *** P < 0.001.
To further verify whether Notch signaling was also responsible for the TNF-α-induced expression of CXCR4 on ECs, HUVECs were transfected with lentiviral particles expressing activated Notch receptors (NICD) or empty vectors (Fig. S4A-B). We observed that the up-regulation of CXCR4 induced by TCM and TNF-α was only partially attenuated following Notch activation by lentivirus-transfected NICD in HUVECs (Fig. 4C). We also examined the in situ expression of NICD on ECs in tumor tissues. Although NICD protein could be detected on the vasculature of colorectal carcinoma (Fig. S4C), which was consistent with another report;31 it was not expressed on CXCR4+ or CXCR4− vessels in the HCC tissues (Fig. 4D), suggesting that the Notch signaling is dispensable for the TNF-α-induced vascular CXCR4 expression in vivo.
Treatment with ZA plus sorafenib inhibits primary tumor growth and lung metastasis in an orthotopic HCC model
The above data indicate that Mo/Mϕ can induce CXCR4 expression on tumor vessels via the activation of Raf-ERK signaling. To further explore the potential clinical applications in HCC treatment, sorafenib (an inhibitor of Raf kinase32 which serves as a first-line treatment option for patients with advanced HCC),4 and zoledronic acid (ZA) (a bisphosphonate which can deplete phagocytes and has demonstrated efficacy in the reduction of osteoclast-induced bone loss in metastatic cancer patients),33,34 were applied in an orthotopic HCC model. One week following tumor implantation, mice were treated with either sorafenib or ZA or a combination of both treatments daily for another three weeks. As shown in Fig. 5A-B, both sorafenib and ZA monotherapies were able to reduce the tumor size compared with the control group. Moreover, the combination of sorafenib and ZA treatment displayed a trend towards the greatest reduction in tumor size. Lung metastasis was also significantly inhibited by sorafenib or ZA treatment compared with the control group, and the combination of these two drugs further inhibited lung metastasis (Fig. 5C and Fig. S5).
Figure 5.
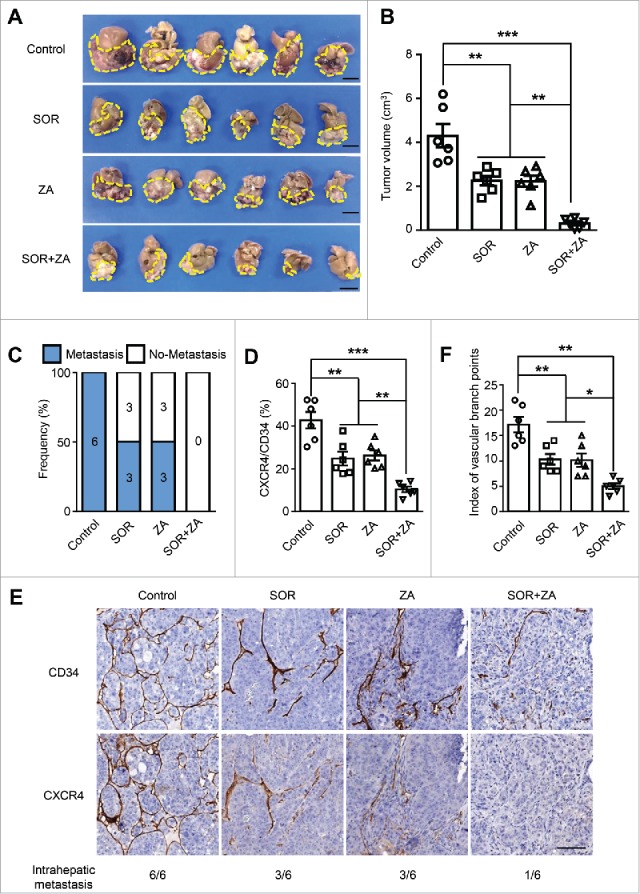
Treatment with ZA plus sorafenib in an orthotopic HCC model. In a Hepa1-6 allograft model, mice were treated daily with either sorafenib or ZA or a combination of both for three weeks (n = 6 per group). (A and B) The tumor volumes were measured using calipers. Scale bar = 1 cm. (C-F) Percentages of mice bearing macroscopic lung metastatic nodules in each group (C), the vascular CXCR4 densities (D), and the indexes of vascular branch points (F) in each group. (E) Immunohistochemical staining for CXCR4 and CD34 was performed on the serial tissue sections derived from the mouse tumor allografts derived from Hepa1-6 cells. The numbers of mice displaying intrahepatic metastasis relative to the total number of tumor-bearing mice in each group are indicated at the bottom. Scale bar = 100 μm. SOR, sorafenib; ZA, zoledronic acid. Error bar, SEM; * P < 0.05; ** P < 0.01; *** P < 0.001.
To test the effects of the above treatments on tumor angiogenesis, we further examined the CXCR4+ vascular density and vessel branch points in the tumors. Treatment with sorafenib or ZA resulted in a pronounced decrease in the CXCR4+ vascular density compared with that of mice in the control group. Furthermore, the combined treatment of sorafenib and ZA could further decrease CXCR4+ vascular density, compared with sorafenib or ZA treatment alone (Fig. 5D and E). Moreover, the index of the vascular branch points was also decreased in the combined treatment group (Fig. 5F). Collectively, these findings suggest that treatment with ZA and sorafenib inhibited primary tumor growth and lung metastasis in an orthotopic HCC model through the reduction of the CXCR4+ vascular density in the tumors.
Discussion
The inhibition of angiogenesis is considered to be a promising anti-tumor approach, particularly for tumors like HCC, which is characterized by a hypervascular nature and high propensity for early blood-borne metastases.7 In the present study, we found that tumor-conditioned Mo/Mϕ were pivotal inducers of CXCR4 vascular expression in both human HCC and mouse tissues. The inflammatory cytokines derived from tumor-activated Mo/Mϕ, especially TNF-α, were able to induce CXCR4 expression on ECs through the activation of the Raf-Erk pathway in HCC. Consistent with our previous study, the multikinase inhibitor sorafenib was able to decrease the CXCR4+ vascular density and tumor growth in a Hepa1-6 orthotopic HCC model. Moreover, the combined treatment with sorafenib and depletion of Mo/Mϕ further reduced the CXCR4+ vascular density, tumor growth, and lung metastasis compared with sorafenib treatment alone. These data revealed an important role of Mo/Mϕ in regulating the vascular expression of CXCR4 in tumors.
Previous studies involving various murine models and human tumor tissues have demonstrated a profound influence of Mo/Mϕ on the regulation of tumor angiogenesis.19,35 Our results showed that tumor-infiltrating Mo/Mϕ could promote tumor angiogenesis via the up-regulation of CXCR4 expression on tumor vessels. First, a greater number of Mo/Mϕ were located in the perivascular areas of vascular CXCR4High tumors compared to the CXCR4Low tumors in HCC. Secondly, using GdCl3 or ZA, inhibitors of Mo/Mϕ inflammation, and CCR2-KO mice, we found that the CXCR4+ vascular density was significantly reduced in mouse models of HCC. Thirdly, inflammatory cytokines derived from Mo/Mϕ, especially TNF-α, could induce CXCR4 expression on ECs in vitro. Taken together, our findings provide evidence for Mo/Mϕ-related tumor angiogenesis in HCC.
A typical pro-angiogenic phenotype of Mϕ is the up-regulation of various pro-angiogenic factors, such as VEGF, bFGF, IL-8, and MMPs.20 In this study, we showed that tumor-activated Mo/Mϕ produced significant levels of pro-inflammatory cytokines (e.g., TNF-α, IL-1β, and IL-6) and anti-inflammatory cytokines (e.g., IL-10). Experiments using recombinant human cytokines and inhibitors showed that TNF-α played a pivotal role in Mo/Mϕ-regulated tumor angiogenesis by inducing CXCR4 expression on ECs. Previous studies by other groups have demonstrated that inflammatory factors (e.g., IL-1α/β and TNF-α) could indirectly induce angiogenesis by stimulating both ECs and tumor cells to produce pro-angiogenic factors, such as VEGF-A, FGF-2, and IL-8.36 Our results provide evidence that inflammatory factors, especially TNF-α, may also directly induce tumor angiogenesis via the regulation of CXCR4+ angiogenic tip cells, which might serve as a link between the pro-inflammatory response and angiogenesis in the tumor milieu.
It is well-established that the activation of ERK pathway can promote tumor cell survival.37 In this study, we demonstrated that the Raf-Erk pathway was responsible for TNF-α induced CXCR4 expression on ECs. We also found that p-ERK1/2 was selectively located in the nucleus of ECs, but was weakly expressed on other stromal and hepatoma cells within HCC tumors. Moreover, patients exhibiting elevated p-ERK1/2 expression in the tumor vasculature were associated with higher vascular CXCR4 expression. Although studies from other groups have demonstrated that tumor cells containing higher levels of pERK might be more sensitive or responsive to sorafenib in HCC patients,38–40 our results suggest a novel role of the Raf-Erk pathway on TECs which may also influence the sensitivity to specific anti-angiogenesis treatment.
Previous studies have revealed a vital role of Notch signaling in tip-stalk cell selection.9 A recent study demonstrated that activated Notch1 receptors (NICD) were frequently observed in ECs of various human carcinomas, including colorectal carcinoma, lung adenocarcinoma, serous ovarian carcinoma, and breast carcinoma. Such sustained vascular Notch signaling further generated a senescent, pro-inflammatory endothelium and facilitated the transendothelial migration and metastasis of tumor cells.31 Although our data revealed that TNF-α could induce CXCR4 expression via the suppression of Notch signaling in ECs in vitro, low levels of NICD expression were identified in HCC tissues, suggesting that other mechanisms might exist to induce a high propensity of blood-borne tumor metastasis. Our previous findings have revealed a novel vascular morphology (VETC) in HCC, which correlated with the density of micrometastasis.11 We also showed that vascular CXCR4 expression was an important regulator of vascular morphology.13 In this study, we further identified that TNF-α-induced activation of the ERK pathway was correlated with vascular CXCR4 expression and the presence of micrometastasis, which was independent of Notch signaling.
At present, the benefit from sorafenib treatment remains limited; therefore, there is an unmet need for effective HCC therapies.4 In this study, we revealed that the combination of ZA, a drug for inhibiting Mo/Mϕ inflammation, with sorafenib was able to efficiently reduce CXCR4+ vascular density and significantly decreased both tumor growth and lung metastasis compared with sorafenib treatment alone in a mouse model of HCC. A recent study found that hypoxia induced from sorafenib treatment could result in the recruitment of tumor-associated macrophages to the tumor.41 This may partially explain the effect of the combination treatment in our model. Although a previous study reported that combination treatment of sorafenib with ZA was associated with a better anti-tumor efficacy compared with sorafenib treatment alone in xenograft nude mouse models,42 we have provided additional evidence for the use of combination treatment in an immune competent mouse model. Furthermore, we explored the potential underlying mechanism of such combination treatment, which was found to be mediated through the regulation of vascular CXCR4 expression.
In conclusion, our findings indicate that tumor-infiltrating Mo/Mϕ can up-regulate CXCR4 expression on tumor vessels via the secretion of TNF-α. The activation of the Raf-Erk signaling pathway was found to be essential for TNF-α-induced CXCR4 expression in HCC. The combined treatment of sorafenib with ZA was associated with a better anti-tumor efficacy compared to either treatment alone by significantly reducing CXCR4 expression on ECs. Taken together, these data reveal a novel mechanism by which Mo/Mϕ regulate tumor angiogenesis, and provide evidence supporting the use of combination therapy involving a Mo/Mϕ depleting drug with sorafenib for the treatment of HCC.
Materials and methods
Patients and specimens
Clinical samples were obtained from patients with pathologically confirmed HCC from the Sun Yat-sen University Cancer Center. A total of 30 patients who underwent a curative resection from 2007 to 2012, defined as the complete macroscopic removal of the tumor were randomly enrolled for analysis. The clinical characteristics of these patients are summarized in Supplementary Table S1. All samples were anonymously coded in accordance with local ethical guidelines (as requested by the Declaration of Helsinki) with written informed consent and a protocol approved by the Review Board of the Sun Yat-sen University Cancer Center.
Isolation and culture of HUVECs
For the ECs used in this study, human umbilical vein endothelial cells (HUVECs) were isolated and cultured in serum-free medium (SFM) (Gibco, 11111-044), supplemented with 20% fetal bovine serum (FBS, Gibco 10270-106), 0.1 mg/mL heparin and 0.03 mg/mL EC growth supplement (ECGS, EMD Millipore, 02–102) as previously described.14 HUVECs from passages 2 – 7 were used in all experiments.
Tumor cell lines and preparation of tumor culture supernatants
Murine hepatoma cell lines (Hepa1-6) and a transformed human embryonic kidney (HEK293T) cell line were maintained in Dulbecco's modified Eagle's medium (DMEM, Life Technologies, C11995500BT) supplemented with 10% FBS. Hepatoma HepG2 cell lines were obtained from the American Type Culture Collection. All cells were tested for mycoplasma contamination using a single-step polymerase chain reaction (PCR) method,43 and maintained in complete medium composed of DMEM supplemented with 10% FBS. Tumor culture supernatants (TSNs) were prepared by plating 5 × 106 tumor cells in 10 mL complete medium in 100-mm dishes for 24 h. Thereafter, the medium was changed to complete DMEM supplemented with 10% FBS. After two to three days, the supernatants were harvested, centrifuged, and stored in aliquots at −80°C.44
Isolation of monocytes and preparation of conditioned media
Monocytes were selected from PBMCs using anti-CD14 magnetic beads (Miltenyi Biotec, Bergisch Gladbach, Germany).44 To generate conditioned media, peripheral monocytes were cultured for 1 hrs with or without 15% TSN, and subsequently washed and cultured in DMEM containing 10% FBS for 16 hrs, which was defined as conditioned medium from the control monocytes (CCM) or conditioned medium from TSN-exposed monocytes (TCM), respectively. After the incubation, the media were harvested, centrifuged, and stored in aliquots at −80°C.25
Flow cytometric analysis
The cells were stained with surface markers and the data were acquired using the Gallios system (Beckman Coulter). AF488-conjugated monoclonal anti-CD31 antibodies (303110), APC-conjugated monoclonal anti-CXCR4 antibodies (306510), were purchased from Biolegend. Krome Orange-conjugated monoclonal anti-CD45 antibodies (A96416) were purchased from Beckman Coulter.
Regulation of CXCR4 expression
HUVECs were cultured for the indicated time with 20% conditioned medium or different concentrations of rhTNF-α and rhIL-1β (R&D Systems). In some experiments, before stimulating the HUVECs, the conditioned medium was pretreated with neutralizing mAbs against TNF-α or IL-1β (R&D Systems) for 15 min at room temperature. The cells were subjected to FACS analysis to detect the surface expression of CXCR4 protein, and the level of IL-6, IL-10, IL-1β, and TNF-α in the conditioned media of the monocytes was determined using commercial ELISA kits, in accordance with the manufacturer's instructions (eBioscience, San Diego, CA).
To explore the signaling pathways, HUVECs in SFM without ECGS were plated at a density of 4 × 104 cells per well and treated with the Erk1/2 inhibitor, U0126 (Selleck, S1102), or the IκB inhibitor, Bay11-7082 (Selleck, S2913), for 1 hrs followed by the administration of TNF-α (10 ng/mL) for 24 h.
Immunohistochemical and immunofluorescent staining
Formalin-fixed, paraffin-embedded tissues were cut into 4-µm sections and subjected to immunohistochemical or immunofluorescent staining as previously described.45 Antibodies used for immunohistochemical and immunofluorescent staining included: mouse monoclonal anti-CD34 antibody against human cell surface glycoprotein (hCD34, Santa Cruz Biotechnology, clone QBEnd/10); rabbit monoclonal anti-CXCR4 antibody against human or mouse (Abcam, clone UMB2); rat monoclonal anti-mouse CD34 antibody (mCD34, BioLegend, clone MEC14.7); phospho-Erk1/2 (Cell Signaling, 4370); Cleaved Notch1 (NICD) (Cell Signaling, 4147T); and rabbit monoclonal anti-CD11b antibody against human or mouse (Abcam, ab133357); Rhodamine Phalloidin (Cytoskeleton, PHDR1). For the immunofluorescence analysis, Alexa Fluor-conjugated secondary antibodies (Invitrogen Molecular Probes) were used to detect human antigens, and the sections were counterstained with 4′-6′-diamidino-2-phenylindole (DAPI, Sigma-Aldrich, D8417).
The stained tissue sections were scanned using the Vectra imaging system and analyzed using Inform image analysis software (Perkin-Elmer Applied Biosystems).
Evaluation of immunohistochemical and immunofluorescent staining signals
Assessment of the immunohistochemical and immunofluorescent staining signals was performed by two independent observers who were blinded to the tissue information as previously described.13 Occasional disagreements were discussed to reach a consensus or referred to a third observer. All slides were first screened at low power (× 100 magnification) and five representative spot images were captured at × 200 magnification using the Vectra imaging system.
To quantify the density of CXCR4+ vessels in HCC, the Nuance 2.0 image analysis software was used. The CD34+ vessel density was calculated as: (area of CD34+ vessel) / tissue area. The CXCR4+ vessel density was calculated as: (area of CXCR4+ CD34+ vessel) / (area of CD34+ vessel) (Figure. S1C). The density of CXCR4+ vessels in implanted tumor model was determined as follows: serial tissue sections of each mouse allograft were stained with an anti-CXCR4 or an anti- CD34 antibody, respectively. The density of CXCR4+ vessels in the tumor was defined as the CXCR4 staining area relative to the CD34 staining area. To determine the number of vascular branch points in mouse tumor allografts, the tissue sections were stained for CD34. We screened the five most intensely vascularized fields and counted the total number of vascular branch points manually. The CD34 staining area was then quantified using Inform software. The average number of vascular branch points per vessel area (magepixels) was presented as the index of vascular branch points (Figure. S1D).
Vector construction
Lentiviral vectors encoding human NOTCH-ICD were obtained from Addgene (TetO-FUW-NICD, Addgene plasmid 61540). The procedures for virus production were as described previously.13
Quantitative real-time PCR (qPCR)
The total RNA was extracted from sorted cells or frozen tissue using Trizol reagent (Life Technologies, AM9738). Aliquots (2 µg) of total RNA were reverse transcribed into cDNA using 5 × All-In-One RT MasterMix (Abm, G492), followed by quantitative real-time RT-PCR (qPCR) using SYBR Green real-time PCR Master Mix (TOYOBO QPS-201(-)). All reactions were run in triplicate and performed on a Roche Light Cycler 480 System (Roche Diagnostics). The cycle threshold (Ct) values did not differ by more than 0.5 among the triplicates. The level of target genes was normalized to that of β-actin (internal control) to permit the calculation of the 2−ΔCt value. The primers used for qPCR are provided in Supplementary Table S2.
Immunoblotting
Immunoblotting was performed as previously described.46 An equal amount of cellular proteins were separated using 12% SDS-PAGE, immunoblotted with Abs against β-Actin (Boster, BM0627), Erk1/2 (Cell Signaling, 4695), phospho-Erk1/2 (Cell Signaling, 4370), NF-kappaB p65 (Cell Signaling, 8242), phospho-NF-kappaB p65 (Cell Signaling, S536), and cleaved Notch1(NICD) (Cell Signaling, 4147T). The membranes were visualized using a commercial ECL kit (Thermo Scientific, 32106).
Animal experiments
C57 BL/6 (B6) mice were purchased from Guangdong Medical Laboratory Animal Center (China). B6.129S4-Ccr2tm1Ifc/J (referred to as CCR2-KO) homozygote mice were purchased from The Jackson Laboratory. All procedures for animal experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals (National Institutes of Health publication no. 80-23, revised 1996) and the Sun Yat-sen University Institutional Ethical Guidelines for animal experiments.
Orthotopic hepatic tumor was established by a subcapsular intrahepatic injection of Hepa1-6 hepatoma cells. Briefly, male B6 mice (five weeks of age) were anesthetized, and the abdominal cavity was opened. 1 × 106 Hepa1-6 tumor cells suspended in 25 μl of 50% Basement Membrane Extract (Trevigen) was inoculated under the left hepatic lobe capsule of the anesthetized mice.
Treatment was initiated one week after liver implantation with HCC cells. To inhibit Mo/Mϕ-induced inflammation, gadolinium chloride (GdCl3) (Sigma, 439770) or zoledronic acid (ZA) (Selleck, S1314) was used. One week after the orthotopic injection of Hepa1-6 cells, tumor-bearing mice were administered a tail vein injection of GdCl3 (10 mg/kg) twice a week or a subcutaneous injection of ZA (100 μg/kg) daily for two or three weeks. To evaluate the effect of sorafenib on allograft tumors, the mice were administered a daily oral gavage with 25 mg/kg sorafenib or a vehicle-only solution for three weeks. Mice were sacrificed four weeks after the tumor implantation.
The length (L) and width (W) of the dissected tumors was measured using calipers, and the tumor volume (V) was calculated using the formula: V = (L × W2) × 0.5.
Statistical analysis
Differences in the means of the continuous variables were compared using a Student's t-test, and the nonparametric exact Wilcoxon signed-rank test was used to compare data not normally distributed. The differences in proportions were tested using a χ2 test. IBM SPSS statistics software was used for all statistical analyses. All data were analyzed using two-tailed tests unless otherwise specified, and P < 0.05 was considered statistically significant.
Supplementary Material
Funding Statement
This work was supported by grants from National Key R&D Program of China (No. 2017YFA0505803), National Natural Science Foundation of China (No. 91442205, 81702409 and 81772589), The Natural Science Foundation of Guangdong Province (No. 2017A030310062), The Fundamental Research Funds for the Central Universities (No. 14ykpy42 and 16ykjc41) and Medical Scientific Research Foundation of Guangdong Province, China (No. A2017336).
Abbreviations
- CXCR4
C-X-C chemokine receptor type 4
- EC
endothelial cell
- GdCl3
gadolinium chloride
- HCC
hepatocellular carcinoma
- HUVEC
human umbilical vein endothelial cell
- IHC
immunohistochemistry
- mo
monocyte
- Mφ
macrophage
- ZA
zoledronic acid
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- 1.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA. 2015;65:87–108. PMID:25651787. [DOI] [PubMed] [Google Scholar]
- 2.Faivre S, Raymond E, Boucher E, Douillard J, Lim HY, Kim JS, Zappa M, Lanzalone S, Lin X, Deprimo S, et al.. Safety and efficacy of sunitinib in patients with advanced hepatocellular carcinoma: An open-label, multicentre, phase II study. Lancet Oncol. 2009;10:794–800. doi: 10.1016/S1470-2045(09)70171-8. PMID:19586800. [DOI] [PubMed] [Google Scholar]
- 3.Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A, et al.. Sorafenib in advanced hepatocellular carcinoma. N Eng J Med. 2008;359:378–90. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 4.Bruix J, Qin S, Merle P, Granito A, Huang Y-H, Bodoky G, Pracht M, Yokosuka O, Rosmorduc O, Breder V, et al.. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (resorce): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;389:56–66. doi: 10.1016/S0140-6736(16)32453-9. PMID:27932229. [DOI] [PubMed] [Google Scholar]
- 5.Ferrara N, Adamis AP. Ten years of anti-vascular endothelial growth factor therapy. Nat Rev Drug Discovery. 2016;15:385–403. doi: 10.1038/nrd.2015.17. PMID:26775688. [DOI] [PubMed] [Google Scholar]
- 6.Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. PMID:21376230. [DOI] [PubMed] [Google Scholar]
- 7.Jayson GC, Kerbel R, Ellis LM, Harris AL. Antiangiogenic therapy in oncology: Current status and future directions. Lancet. 2016;388:518–29. doi: 10.1016/S0140-6736(15)01088-0. PMID:26853587. [DOI] [PubMed] [Google Scholar]
- 8.Herbert SP, Stainier DY. Molecular control of endothelial cell behaviour during blood vessel morphogenesis. Nat Rev Mol Cell Biol. 2011;12:551–64. doi: 10.1038/nrm3176. PMID:21860391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ribatti D, Crivellato E. “Sprouting angiogenesis”, a reappraisal. Dev Biol. 2012;372:157–65. doi: 10.1016/j.ydbio.2012.09.018. PMID:23031691. [DOI] [PubMed] [Google Scholar]
- 10.Fang JH, Zhou HC, Zhang C, Shang LR, Zhang L, Xu J, Zheng L, Yuan Y, Guo RP, Jia WH, et al.. A novel vascular pattern promotes metastasis of hepatocellular carcinoma in an epithelial-mesenchymal transition-independent manner. Hepatology. 2015;62:452–65. doi: 10.1002/hep.27760. PMID:25711742. [DOI] [PubMed] [Google Scholar]
- 11.Ding T, Xu J, Zhang Y, Guo RP, Wu WC, Zhang SD, Qian CN, Zheng L. Endothelium-coated tumor clusters are associated with poor prognosis and micrometastasis of hepatocellular carcinoma after resection. Cancer-Am Cancer Soc. 2011;117:4878–89 [DOI] [PubMed] [Google Scholar]
- 12.Sugino T, Yamaguchi T, Ogura G, Saito A, Hashimoto T, Hoshi N, Yoshida S, Goodison S, Suzuki T. Morphological evidence for an invasion-independent metastasis pathway exists in multiple human cancers. BMC Med. 2004;2:9. doi: 10.1186/1741-7015-2-9. PMID:15066199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu J, Liang J, Meng YM, Yan J, Yu XJ, Liu CQ, Xu L, Zhuang SM, Zheng L. Vascular CXCR4 expression promotes vessel sprouting and sensitivity to sorafenib treatment in hepatocellular carcinoma. Clin Cancer Res. 2017;23:4482–92. doi: 10.1158/1078-0432.CCR-16-2131. PMID:28223275. [DOI] [PubMed] [Google Scholar]
- 14.Kuang DM, Zhao Q, Wu Y, Peng C, Wang J, Xu Z, Yin XY, Zheng L. Peritumoral neutrophils link inflammatory response to disease progression by fostering angiogenesis in hepatocellular carcinoma. J Hepatol. 2011;54:948–55. doi: 10.1016/j.jhep.2010.08.041. PMID:21145847. [DOI] [PubMed] [Google Scholar]
- 15.Budhu A, Forgues M, Ye QH, Jia HL, He P, Zanetti KA, Kammula US, Chen Y, Qin LX, Tang ZY, et al.. Prediction of venous metastases, recurrence, and prognosis in hepatocellular carcinoma based on a unique immune response signature of the liver microenvironment. Cancer Cell. 2006;10:99–111. doi: 10.1016/j.ccr.2006.06.016. PMID:16904609. [DOI] [PubMed] [Google Scholar]
- 16.Kuang DM, Xiao X, Zhao Q, Chen MM, Li XF, Liu RX, Wei Y, Ouyang FZ, Chen DP, Wu Y, et al.. B7-H1-expressing antigen-presenting cells mediate polarization of protumorigenic Th22 subsets. J Clin Invest. 2014;124:4657–67. doi: 10.1172/JCI74381. PMID:25244097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ju C, Tacke F. Hepatic macrophages in homeostasis and liver diseases: From pathogenesis to novel therapeutic strategies. Cell Mol Immunol. 2016;13:316–27. doi: 10.1038/cmi.2015.104. PMID:26908374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bron S, Henry L, Faes-Van't Hull E, Turrini R, Vanhecke D, Guex N, Ifticene-Treboux A, Marina Iancu E, Semilietof A, Rufer N, et al.. Tie-2-expressing monocytes are lymphangiogenic and associate specifically with lymphatics of human breast cancer. OncoImmunology. 2016;5:e1073882. doi: 10.1080/2162402X.2015.1073882. PMID:27057438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Murdoch C, Muthana M, Coffelt SB, Lewis CE. The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer. 2008;8:618–31. doi: 10.1038/nrc2444. PMID:18633355. [DOI] [PubMed] [Google Scholar]
- 20.Dirkx AE, Oude Egbrink MG, Wagstaff J, Griffioen AW. Monocyte/macrophage infiltration in tumors: Modulators of angiogenesis. J Leukoc Biol. 2006;80:1183–96. doi: 10.1189/jlb.0905495. PMID:16997855. [DOI] [PubMed] [Google Scholar]
- 21.Fantin A, Vieira JM, Gestri G, Denti L, Schwarz Q, Prykhozhij S, Peri F, Wilson SW, Ruhrberg C. Tissue macrophages act as cellular chaperones for vascular anastomosis downstream of vegf-mediated endothelial tip cell induction. Blood. 2010;116:829–40. doi: 10.1182/blood-2009-12-257832. PMID:20404134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pendino KJ, Meidhof TM, Heck DE, Laskin JD, Laskin DL. Inhibition of macrophages with gadolinium chloride abrogates ozone-induced pulmonary injury and inflammatory mediator production. Am J Respiratory Cell Mol Biol 1995;13:125–32. doi: 10.1165/ajrcmb.13.2.7542894. [DOI] [PubMed] [Google Scholar]
- 23.Mizgerd JP, Molina RM, Stearns RC, Brain JD, Warner AE. Gadolinium induces macrophage apoptosis. J Leukoc Biol. 1996;59:189–95. PMID:8603991. [PubMed] [Google Scholar]
- 24.Li X, Yao W, Yuan Y, Chen P, Li B, Li J, Chu R, Song H, Xie D, Jiang X, et al.. Targeting of tumour-infiltrating macrophages via CCL2/CCR2 signalling as a therapeutic strategy against hepatocellular carcinoma. Gut. 2017;66:157–67. doi: 10.1136/gutjnl-2015-310514. PMID:26452628. [DOI] [PubMed] [Google Scholar]
- 25.Kuang DM, Peng C, Zhao Q, Wu Y, Chen MS, Zheng L. Activated monocytes in peritumoral stroma of hepatocellular carcinoma promote expansion of memory T helper 17 cells. Hepatology. 2010;51:154–64. doi: 10.1002/hep.23291. PMID:19902483. [DOI] [PubMed] [Google Scholar]
- 26.Chen MM, Xiao X, Lao XM, Wei Y, Liu RX, Zeng QH, Wang JC, Ouyang FZ, Chen DP, Chan KW, et al.. Polarization of tissue-resident TFH-like cells in human hepatoma bridges innate monocyte inflammation and M2b macrophage polarization. Cancer Discovery. 2016;6:1182–95. doi: 10.1158/2159-8290.CD-16-0329. PMID:27531854. [DOI] [PubMed] [Google Scholar]
- 27.Sainson RC, Johnston DA, Chu HC, Holderfield MT, Nakatsu MN, Crampton SP, Davis J, Conn E, Hughes CC. TNF primes endothelial cells for angiogenic sprouting by inducing a tip cell phenotype. Blood. 2008;111:4997–5007. doi: 10.1182/blood-2007-08-108597. PMID:18337563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Potente M, Gerhardt H, Carmeliet P. Basic and therapeutic aspects of angiogenesis. Cell. 2011;146:873–87. doi: 10.1016/j.cell.2011.08.039. PMID:21925313. [DOI] [PubMed] [Google Scholar]
- 29.Williams CK, Segarra M, Sierra Mde L, Sainson RC, Tosato G, Harris AL. Regulation of CXCR4 by the Notch ligand delta-like 4 in endothelial cells. Cancer Res. 2008;68:1889–95. doi: 10.1158/0008-5472.CAN-07-2181. PMID:18339870. [DOI] [PubMed] [Google Scholar]
- 30.Dovey HF, John V, Anderson JP, Chen LZ, de Saint Andrieu P, Fang LY, Freedman SB, Folmer B, Goldbach E, Holsztynska EJ, et al.. Functional gamma-secretase inhibitors reduce beta-amyloid peptide levels in brain. J Neurochem 2001;76:173–81. doi: 10.1046/j.1471-4159.2001.00012.x. PMID:11145990. [DOI] [PubMed] [Google Scholar]
- 31.Wieland E, Rodriguez-Vita J, Liebler SS, Mogler C, Moll I, Herberich SE, Espinet E, Herpel E, Menuchin A, Chang-Claude J, et al.. Endothelial Notch1 activity facilitates metastasis. Cancer Cell. 2017;31:355–67. doi: 10.1016/j.ccell.2017.01.007. PMID:28238683. [DOI] [PubMed] [Google Scholar]
- 32.Liu L, Cao YC, Chen C, Zhang XM, McNabola A, Wilkie D, Wilhelm S, Lynch M, Carter C. Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer Res. 2006;66:11851–8. doi: 10.1158/0008-5472.CAN-06-1377. PMID:17178882. [DOI] [PubMed] [Google Scholar]
- 33.Ottewell PD, Monkkonen H, Jones M, Lefley DV, Coleman RE, Holen I. Antitumor effects of doxorubicin followed by zoledronic acid in a mouse model of breast cancer. J Natl Cancer Inst. 2008;100:1167–78. doi: 10.1093/jnci/djn240. PMID:18695136. [DOI] [PubMed] [Google Scholar]
- 34.Thompson K, Rogers MJ, Coxon FP, Crockett JC. Cytosolic entry of bisphosphonate drugs requires acidification of vesicles after fluid-phase endocytosis. Mol Pharmacol. 2006;69:1624–32. doi: 10.1124/mol.105.020776. PMID:16501031. [DOI] [PubMed] [Google Scholar]
- 35.Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141:39–51. doi: 10.1016/j.cell.2010.03.014. PMID:20371344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hagemann T, Wilson J, Burke F, Kulbe H, Li NF, Pluddemann A, Charles K, Gordon S, Balkwill FR. Ovarian cancer cells polarize macrophages toward a tumor-associated phenotype. J Immunol. 2006;176:5023–32. doi: 10.4049/jimmunol.176.8.5023. PMID:16585599. [DOI] [PubMed] [Google Scholar]
- 37.Han T, Xiang DM, Sun W, Liu N, Sun HL, Wen W, Shen WF, Wang RY, Chen C, Wang X, et al.. PTPN11/Shp2 overexpression enhances liver cancer progression and predicts poor prognosis of patients. J Hepatol. 2015;63:651–60. doi: 10.1016/j.jhep.2015.03.036. PMID:25865556. [DOI] [PubMed] [Google Scholar]
- 38.Lee FA, Zee BC, Cheung FY, Kwong P, Chiang CL, Leung KC, Siu SW, Lee C, Lai M, Kwok C, et al.. Randomized phase II study of the X-linked inhibitor of apoptosis (XIAP) antisense AEG35156 in combination with sorafenib in patients with advanced hepatocellular carcinoma (HCC). Am J Clin Oncol. 2016;39:609–13. doi: 10.1097/COC.0000000000000099. PMID:24977690. [DOI] [PubMed] [Google Scholar]
- 39.Chen D, Zhao P, Li SQ, Xiao WK, Yin XY, Peng BG, Liang LJ. Prognostic impact of pERK in advanced hepatocellular carcinoma patients treated with sorafenib. Eur J Surg Oncol. 2013;39:974–80. doi: 10.1016/j.ejso.2013.06.018. PMID:23845703. [DOI] [PubMed] [Google Scholar]
- 40.Kalathil SG, Lugade AA, Iyer R, Miller A, Thanavala Y. Endothelial progenitor cell number and ERK phosphorylation serve as predictive and prognostic biomarkers in advanced hepatocellular carcinoma patients treated with sorafenib. OncoImmunology. 2016;5:e1226718. doi: 10.1080/2162402X.2016.1226718. PMID:27853648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen Y, Ramjiawan RR, Reiberger T, Ng MR, Hato T, Huang Y, Ochiai H, Kitahara S, Unan EC, Reddy TP, et al.. CXCR4 inhibition in tumor microenvironment facilitates anti-programmed death receptor-1 immunotherapy in sorafenib-treated hepatocellular carcinoma in mice. Hepatology. 2015;61:1591–602. doi: 10.1002/hep.27665. PMID:25529917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang W, Zhu XD, Sun HC, Xiong YQ, Zhuang PY, Xu HX, Kong LQ, Wang L, Wu WZ, Tang ZY. Depletion of tumor-associated macrophages enhances the effect of sorafenib in metastatic liver cancer models by antimetastatic and antiangiogenic effects. Clin Cancer Res. 2010;16:3420–30. doi: 10.1158/1078-0432.CCR-09-2904. PMID:20570927. [DOI] [PubMed] [Google Scholar]
- 43.Uphoff CC, Drexler HG. Detection of mycoplasma in leukemia-lymphoma cell lines using polymerase chain reaction. Leukemia. 2002;16:289–93. doi: 10.1038/sj.leu.2402365. PMID:11840297. [DOI] [PubMed] [Google Scholar]
- 44.Kuang DM, Wu Y, Chen N, Cheng J, Zhuang SM, Zheng L. Tumor-derived hyaluronan induces formation of immunosuppressive macrophages through transient early activation of monocytes. Blood. 2007;110:587–95. doi: 10.1182/blood-2007-01-068031. PMID:17395778. [DOI] [PubMed] [Google Scholar]
- 45.Wu Y, Kuang DM, Pan WD, Wan YL, Lao XM, Wang D, Li XF, Zheng L. Monocyte/macrophage-elicited natural killer cell dysfunction in hepatocellular carcinoma is mediated by CD48/2B4 interactions. Hepatology. 2013;57:1107–16. doi: 10.1002/hep.26192. PMID:23225218. [DOI] [PubMed] [Google Scholar]
- 46.Li XF, Chen DP, Ouyang FZ, Chen MM, Wu Y, Kuang DM, Zheng L. Increased autophagy sustains the survival and pro-tumourigenic effects of neutrophils in human hepatocellular carcinoma. J Hepatol. 2015;62:131–9. doi: 10.1016/j.jhep.2014.08.023. PMID:25152203. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.