

Abstract
Friedreich's Ataxia (FA) is an inherited neurologic disorder caused by an expanded GAA repeat within intron 1 of the frataxin (FXN) gene that reduces expression of FXN protein. Agents that increase expression of FXN have the potential to alleviate the disease. We previously reported that duplex RNAs (dsRNAs) and antisense oligonucleotides (ASOs) complementary to the GAA repeat could enhance expression of FXN protein. We now explore the potential of a diverse group of chemically modified dsRNAs and ASOs to define the breadth of repeat-targeted synthetic nucleic acids as a platform for therapeutic development for FA. ASOs and dsRNAs can activate FXN protein expression in FA patient-derived cell lines that possess varied numbers of GAA repeats. Increased FXN protein expression was achieved by ASOs incorporating diverse chemical modifications with low nanomolar potencies, suggesting substantial flexibility in choosing compounds for further chemical optimization and animal studies. Our data encourage further development of ASOs as agents to treat FA.
Keywords: : frataxin, Friedreich's ataxia, antisense oligonucleotide, gene activation
Introduction
Friedreich's Ataxia (FA) is an incurable disease caused by an expansion of a trinucleotide GAA repeat within intron 1 of the frataxin (FXN) gene [1,2]. This expansion does not change the coding region of FXN and does not result in expression of a mutant protein. Instead, the gene mutation reduces expression of FXN protein. Agents that increase expression of FXN protein to restore it to normal levels would have the potential to be a therapy for FA [3–11].
While the mechanism linking expansion of the trinucleotide repeat to reduced expression of FXN is not known with certainty, evidence suggests that the expanded intronic RNA forms an RNA loop (R-loop) with chromosomal DNA that induces histone and possibly DNA modification to act as a break on transcription [12,13]. Compounds that bind the expanded repeat would be expected to block the expanded RNA, inhibit recognition of DNA, prevent R-loop formation, and allow more expression of FXN. We have previously tested the hypothesis using duplex RNAs (dsRNAs) and antisense oligonucleotides (ASOs) complementary to the trinucleotide repeat [11]. Both types of synthetic oligonucleotide activated expression of FXN protein at or near the levels found in wild-type cells.
While FA has serious effects on cardiac muscle and other tissues, treatment of the central nervous system (CNS) is a major priority. Recently, an ASO, nusinersen (SPINRAZA), has been approved for treatment of spinal muscular atrophy [14]. Nusinersen is delivered to patients by intrathecal injection, suggesting that this route of administration might also be used to deliver ASOs to treat the neurological symptoms of FA.
In this study we focus on exploring gene activation by ASOs and dsRNAs in varied patient-derived cell lines (Table 1) using synthetic nucleic acids with diverse chemical modifications (Fig. 1). We find that increasing FXN protein levels through introduction of ASOs or dsRNAs can occur in different cell lines. Activation can be achieved by a wide variety of different chemically modified ASOs, providing a broad foundation for future drug development.
Table 1.
Friedreich's Ataxia Patient-Derived Fibroblast Cell Lines (Homozygous) and the Carrier Cell Line (Heterozygous)
| # GAA repeats | ||||
|---|---|---|---|---|
| Cell line no. | Gender | Age of onset | Allele 1 | Allele 2 |
| FA00130 | M | 41 | 136 | 540 |
| FA04111 | F | 12 | 380 | 454 |
| FA04676 | M | 10 | 678 | 678 |
| FA06327 | F | 16 | 397 | 507 |
| GM03816 | F | 36 | 330 | 380 |
| MC6353 | F | — | 27 | 1000 |
FIG. 1.

Chemical modifications of oligonucleotides.
Materials and Methods
Tissue culture and transfection
Fibroblast cells (GM03816, Coriell Institute; FA00130, FA04111, FA04676, FA06327 and MC6353) from Repository of FA Fibroblasts provided by Dr. Marek Napierala (University of Alabama at Birmingham) were cultured in minimum essential medium supplemented with 10% fetal bovine serum and 1% non-essential amino acid (NEAA), as described previously [11]. Briefly, all cells were grown at 37°C in 5% CO2. Lipofectamine RNAiMAX (Invitrogen) was used to transfect oligonucleotides following the manufacturer's recommended protocol in OptiMEM reduced serum medium (Invitrogen). Growth media was changed to full medium after 24 h. Transfected cells were harvested 72 h after transfection for RNA extraction and quantitative polymerase chain reaction (qPCR) analyses and 96 h after transfection for western blot analysis. The timing is based on a time course and its the earliest times at which maximal activation is observed.
Western blot analysis
Cell extracts were prepared using lysis buffer supplemented with 1% Protease Inhibitor Cocktail Set I (Calbiochem) as described previously [15]. Protein was separated on 4%–20% gradient (for 4F9 and ab110328 antibody) or 15% SDS-PAGE TGX precast gels (for ab110328 antibody; Bio-Rad). After gel electrophoresis, proteins were transferred to nitrocellulose membrane (Hybond-C Extra; GE Healthcare Life Sciences) at 100 V for 40–55 min. Membranes were blocked for 2 h at room temperature with 5% milk protein in 1× phosphate-buffered saline (PBS) containing 0.1% TWEEN-20 (PBST 0.1%). Blocked membranes were incubated with the primary antibodies at 4°C in PBST 0.1% and 1% milk with rocking overnight: anti-FXN (4F9, from Dr. Hélène Puccio at IGBMC, France) at 1:20,000, anti-tubulin at 1:10,000 (T5201; Sigma-Aldrich) (Supplementary Figure S1; Supplementary Data available online at www.liebertpub.nat). After primary antibody incubation, membranes were washed four times for 10 min at room temperature with PBS 1X 0.2% TWEEN-20 (PBST 0.2%) then incubated for 1 h at room temperature with horse radish peroxidase (HRP)-conjugated anti-mouse (715-035-150; Jackson Laboratories) in PBST 0.1%. Membranes were washed again 4× for 10 min in PBST 0.1% and 4× for 10 min in PBS at room temperature, then protein bands visualized using SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific). Alternatively, we used anti-FXN (ab110328; Abcam) as the primary antibody as described previously [11]. In our experience, antibody 4F9 produced results that were more consistent and were characterized by better signal to noise. For quantification of protein levels from western blots of cellular fractions, films were scanned and bands quantified using ImageJ.
Quantitative PCR
qPCR followed the protocol described in Li et al. [11]. In brief, equal amount of RNA (representing approximately the same number of cells and ranging from 1 to 2 μg of RNA) was treated with 2 units of DNase I (Worthington) in DNase I buffer (10 mM Tris–HCl, pH 7.0, 10 mM NaCl, 2 mM MgCl2, 0.5 mM CaCl2) for 20 min at room temperature to degrade genomic DNA contamination. Afterward, DNase I was heat inactivated at 75°C for 10 min. Treated RNAs were reverse transcribed using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). qPCR was performed using SYBR Supermix (Bio-Rad) with ∼50 ng of cDNA as template. Data were normalized relative to measured hypoxanthine guanine phosphoribosyl transferase (HPRT) levels.
Melting temperature determination
Thermal denaturation analysis of oligonucleotides to determine melting temperature Tm values was carried out using a CARY Varian 100 Bio UV-Vis spectrophotometer. Absorbance was monitored at 260 nm in a 1 cm quartz cuvette in buffer (0.25 M NaCl, 0.2 mM EDTA, 20 mM sodium phosphate, pH 7.0) to make the final volume 400 μL. The melting temperature was calculated and averaged from at least seven technical replicates.
Dose curve and EC50 calculations
The program GraphPad Prism 7.03 was used to draw the fitting curves for dose–response experiments. The Hill equations [16] were used for fitting curves in the following form: Y = Y0 + (Ymax − Y0)Xn/(Kn + Xn), where Y is the normalized fold activation, X is the oligo concentration, Y0 is baseline response (activation at an oligo concentration 0), Ymax is the maximum fold activation, K is the EC50 value, and n is the Hill coefficient. Data sets from at least three experiments were used for curve fitting. The error of EC50 is standard error of the mean, which is calculated from combining the data of each individual dose curve.
Results
Activating FXN expression with a dsRNA in diverse patient-derived cell lines
We had previously examined only one patient-derived fibroblast cell line (GM03816, 330/380 repeats) for RNA-induced activation of FXN [11]. In patients, the number of repeats varies. To test the hypothesis that FXN gene activation would be a general phenomenon shared by mutant cells possessing different numbers of GAA repeats, we tested four additional patient-derived fibroblast cell lines and one cell line from a donor who was a heterozygous carrier (Table 1).
We assayed the effect of adding unmodified dsRNA siGAA (Fig. 2a) to each cell line. We first measured FXN mRNA levels by qPCR (Fig. 2b) to provide a baseline comparison for the effect of ASO addition. The levels of RNA expression were similar, and the number of cell lines assayed was too few to observe any correlation with the mutant repeat number. We then transfected RNA into cells using cationic lipid. Addition of siGAA, but not a non-matched dsRNA control, increased expression of FXN mRNA (Fig. 2c). The increase in RNA levels was ∼2.5- to 6-fold. There was no clear correlation between increased expression and repeat number.
FIG. 2.

Effect of adding dsRNA siGAA to homozygous FA patient-derived cell lines or a cell line derived from a heterozygous donor. (a) Sequences of siGAA or noncomplementary dsRNA control CM. (b) Relative expression of FXN mRNA evaluated by qPCR in different FRDA cell lines. (c) Effect of transfecting siGAA (25 nM) on FXN expression (n = 3) in varied cell lines. CM is a noncomplementary negative control RNA. Numbers of GAA repeats in two alleles of patient FXN genes are shown in parentheses. dsRNA, duplex RNA; FA, Friedreich's ataxia; qPCR, quantitative polymerase chain reaction.
In parallel, we examined the increase in FXN protein by western analysis using antibodies that recognize endogenous protein (Fig. 3 and Supplementary Fig. S2). Consistent with our data on enhancement of mRNA expression, addition of siGAA increased expression in all cell lines tested relative to a noncomplementary dsRNA control (Fig. 3a) by three to sixfold (Fig. 3b). The addition of siGAA did not increase FXN protein expression in heterozygous carrier MC6353 cells, even though these cells have one mutant allele with 1,000 GAA repeats. We speculate that the normal allele may be endogenously upregulated to restore protein expression to counter any deficiency caused by the mutant allele.
FIG. 3.
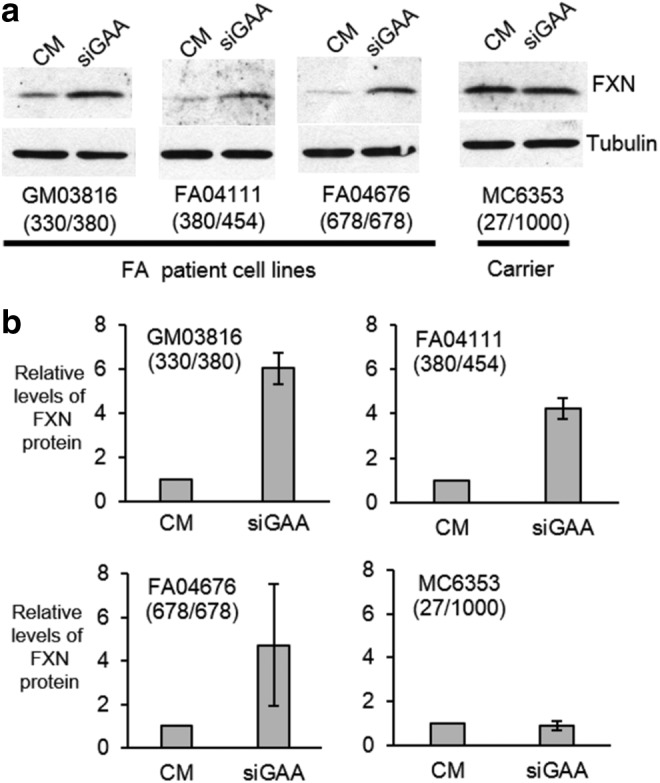
Effect on protein expression of adding dsRNA siGAA (25 nM) to homozygous FA patient-derived cell lines or a cell line derived from a heterozygous donor. (a) Western analysis of FXN protein expression in different FRDA cell lines after transfection with siGAA. (b) Quantification of data shown in (a), (n = 3). CM is a noncomplementary negative control RNA. Numbers of GAA repeats in two alleles of patient FXN genes are shown in parentheses.
Activating FXN expression with ASOs in diverse patient-derived cell lines
ASOs are demonstrating successful therapeutic effects upon administration into the CNS, and one ASO, nusinersen (SPINRAZA), has recently been approved for the treatment of spinal muscular atrophy [14]. Because of the success of ASOs upon delivery to the CNS we also examined their ability to activate FXN expression.
We tested two ASOs containing locked nucleic acid (LNA) substitutions. LNAs belong to the bridged nucleic acid (BNA) family of substitutions and contain a methylene bridge between the 2′ and 4′ positions [17–19] (Fig. 1). This bridge “locks” the ribose backbone in a conformation that is ideal for base pairing, reducing the entropic penalty paid during complementary hybridization. This constraint can increase the melting temperature of ASOs for complementary sequences by as much as 5°C per substitution, allowing the binding of ASOs to be tailored for optimal biological impact.
We observed that BNA-1 (Fig. 4a) activated FXN protein expression in all patient-derived cell lines tested (Fig. 4b). A noncomplementary control LNA was inactive and, as observed for siGAA, no activation was observed in MC6353 cells derived from a heterozygous carrier. Activation levels varied from approximately two to fourfold (Fig. 4c). We also tested a second LNA, BNA-2, that was modified with 2′4′-constrained ethyl (cEt) modifications rather than LNA. BNA-2 activated expression of FXN RNA and yielded similar levels of activation of FXN protein expression in four different patient-derived cell lines (Supplementary Fig. S3). Taken together, results from experiments with one dsRNA and two different BNA ASOs support the conclusion that upregulation of FXN protein expression can occur across a spectrum of FA genotypes.
FIG. 4.

Effect on protein expression of adding BNA-1 (12.5 nM) to homozygous FA patient-derived cell lines or a cell line derived from a heterozygous donor. (a) Sequences of BNA-1 and BNA-2. Data from parallel experiments using BNA-2 are shown in Supplementary Figure S3. (b) Western analysis of FXN protein expression in different FRDA cell lines after addition of BNA-1. (c) Quantification of data shown in (b), n = 3. Numbers of GAA repeats in two alleles of patient FXN genes are shown in parentheses.
Activation by dsRNAs and ASOs containing 2′-deoxy-2′-fluoro-beta-d-arabinonucleic acid
2′-Deoxy-2′-fluoro-beta-d-arabinonucleic acid (2′-F-ANA) (Fig. 1) mimics DNA in structure but can be successfully incorporated into dsRNAs to enhance stability in serum [20,21]. ASOs containing 2′-F-ANA substitutions possess enhanced resistance to nucleases and increased thermal stability for binding to complementary target sequences [22–25]. We tested both a modified dsRNA and several different ASOs containing 2′-F-ANA and 2′-Fluoro RNA (Fig. 5a). We observed 1.5- to 3-fold activation of RNA expression by fluorine-modified ASOs F-1 to F-6 (Fig. 5b) and fully modified fluoro dsRNA F-7 (Fig. 5c). We chose 2′Fluoro-derived ASO F-4 and 2′-FANA-derived ASO F7 for closer examination over a range of concentrations. ASO F-4 possessed an EC50 value of 2.5 nM (Fig. 6a). siRNA F-7 possessed an effective concentration for 50% maximal activation (EC50) of 5.1 nM (Fig. 6b).
FIG. 5.

Effect on FXN gene expression of adding ASOs or dsRNA modified with 2′F-ANA RNA or 2′F RNA in patient-derived GM03816 cells (330/380). (a) Sequences of fluorine-modified ASOs and dsRNA. (b) Activation of FXN mRNA expression upon addition of 12.5 nM ASOs (n = 3). (c) Activation of FXN mRNA expression upon addition of 25 nM dsRNA (n = 3). ASOs, antisense oligonucleotides.
FIG. 6.

Effect of ASO or dsRNA concentration on activation of FXN protein expression in GM03816 cells (330/380) by fluorine-containing (a) ASO F-4 (n = 3) and (b) dsRNA F-7 (n = 3).
Activation by ASOs containing methoxyethyl and BNA modifications
BNA modifications are powerful tools for improving recognition by ASOs and we designed a series of ASOs to more fully explore the potential of the modification for enhancing expression of FXN. The BNAs included LNA (sometimes modified with 5-methyl cytosine), constrained ethyl ((S)-cET), and triazole-linked LNA dimers [26]. The ASOs also included phosphorothioate linkages and 2′-methoxyethyl substitutions (Fig. 7a).
FIG. 7.

Activation of FXN expression by ASOs containing BNA modifications. (a) Sequences of BNA-modified ASOs. (b) Activation of FXN mRNA expression upon addition of 12.5 nM ASOs in patient-derived GM03816 cells (330/380) (n = 3).
We tested a large panel of ASOs and observed greater than twofold activation of FXN mRNA expression by most of the candidate compounds (Fig. 7b and Supplementary Fig. S4). ASOs with three mismatches at the central position 9, 10, 11 and ASOs containing scrambled sequences, MM-1, MM-2, and MM-3 did not activate FXN expression (Table 2 and Supplementary Fig. S5). These data further support the conclusion that sequence-specific gene activation of FXN expression is a general phenomenon.
Table 2.
Sequence of Control Antisense Oligonucleotides
| No. | Group | Chemistry | Sequence (5′-3′) | Tm (°C) |
|---|---|---|---|---|
| CM-PO | Controls | PO, LNA | GdCdTAdTdACdCdAGdCdGTdCdGTdCdATd | — |
| CM-PS | PS, LNA | GdCdTAdTdACdCdAGdCdGTdCdGTdCdATd | — | |
| MM-1 | MOE, 2′-FRNA, OMe, PS | P-TsTstCstTscTsaAsaTscTstsCstsTscsAsA | 56.1 | |
| MM-2 | MOE, 2′-FRNA, OMe, PS | P-TsCstTscTstCsaAsaTstCstsTscsTstsAsA | 57.8 | |
| MM-3 | MOE, 2′-FRNA, OMe, PS | P-TsTscTstCstTsaAsaCstTscsTstsCstsAsA | 56.3 |
All oligonucleotides used PS modification unless mentioned. MM1–3 have PO backbones except the labeled PS backbones.
Bold: LNA modification, bold italic: 2′-O-MOE, underline: 2′-F RNA, lowercase: 2′-O-Me, s: phosphorothioate, P: phosphate, d: deoxyribose.
LNA, locked nucleic acid; PS, phosphorothioate.
We characterized several compounds in more depth by calculating EC50 values for gene activation (Fig. 8). Compounds BNA-6, BNA-2, BNA-13, BNA-16, and BNA-17 were chosen for detailed evaluation based on possession of different design motifs and on their potency when screened at 12.5 nM. All five BNA ASOs showed dose-responsive increases in FXN protein expression. EC50 values ranged from 1.6 to 6 nM, demonstrating that BNA modifications are broadly effective when used to create ASOs for activating expression of FXN.
FIG. 8.

Effect of ASO concentration on activation of FXN protein expression by BNA-containing ASOs (a) BNA-6, (b) BNA-2, (c) BNA-13, (d) BNA-16, (e) BNA-17, (n = 3) in patient-derived GM03816 cells (330/380). The sequence for BNA-6 is found in Figure 6a.
Activation by ASOs containing 2′-MOE and PS modifications
2′-MOE is one of the most widely used modifications in clinical trials of therapeutic ASOs (27). Individual 2′-MOE substitutions do not increase the affinity of complementary binding as much as BNA substitutions but taken together they can be used to create tight-binding ASOs. We tested a series of ASOs that were either entirely substituted with 2′-MOE or that were partially substituted (Fig. 9a). All ASOs contained PS substitutions to enhance stability. Most ASOs activated expression of FXN mRNA (Fig. 9b). For more detailed analysis, compounds M-1, M-5, and M-8 were chosen because they possessed distinct design motifs and appeared potent when screened at 12.5 nM. The EC50 values of M-1, M-5, and M-8 were 4.4, 2.0, and 3.2 nM, respectively (Fig. 10).
FIG. 9.

Activation of FXN expression by ASOs containing 2′-methoxyethyl modifications in patient-derived GM03816 cells (330/380). (a) Sequences of 2′-methoxyethyl-modified ASOs. (b) Activation of FXN mRNA expression upon addition of 12.5 nM ASOs (n = 3).
FIG. 10.

Effect of concentration on activation by ASOs containing 2′-methoxyethyl modifications (a) M-1, (b) M-5, (c) M-8 (n = 3) in patient-derived GM03816 cells (330/380).
Discussion
The expanded trinucleotide GAA causes FA by reducing expression of FXN protein. Compounds that bind to the repeat region may have the potential to block the mutant repeat and enhance gene expression [11]. In combination with our previous study, the results presented here reinforce the conclusion that dsRNAs and ASOs provide a general approach increasing FXN gene expression. Activation was observed in all patient-derived cell lines tested, suggesting that a drug might have the potential to treat many FA patients with the expanded GAA mutation.
EC50 values in the low nanomolar range were routinely achieved. Cell culture potencies vary depending on how assays are performed. For our laboratory, the observed EC50 values are equal to the best we have achieved for ASOs. The variation between the “most” potent compound at 1.6 nM and the “least” potent compound at 6.0 nM was relatively small. This less than fourfold difference may have some significance, but the clear lesson is that the compounds behave similarly in cultured fibroblast cells. More definitive ranking will require animal testing or development of more realistic human cell models. We had previously shown that levels of frataxin activation after RNA addition were like levels in wild-type cells [11], so the absolute level of increased expression is promising.
There are currently no curative treatments for FA, and drugs that treat the underlying molecular cause of the disease are urgently needed. Both dsRNAs and ASOs have achieved recent successes in clinical trials [27]. These trials have demonstrated that nucleic acids can treat a range of diseases. Effects are long lasting, with infrequent dosing required to maintain effective gene silencing. For example, nusinersen, an FDA-approved drug designed to be administered to the CNS by intrathecal injection, is being administered just 3–4 times per year [14]. Nusinersen contains 2′-methoxyethyl modifications like those within many of the compounds we tested, and we anticipate that the ASOs evaluated in this study would also have good stabilities inside cells. While every drug and disease target will be different, the clinical experience with other targets suggests that the use of nucleic acids to upregulate FXN expression is a feasible approach for drug development.
Unlike ASOs, dsRNAs have not been successfully administered in the human CNS. While approaches are being developed to improve the delivery of dsRNAs to the CNS [28], ASOs are currently a more established platform for treating CNS diseases. For drug development, every ASO and every target present different challenges. As with any medicinal chemistry project, chemical properties will need to be tailored to achieve the best chance of success in clinical trials. Our data suggest that many different chemical modifications can contribute to potent EC50 values. This chemical diversity provides a broad platform for additional testing aimed at identifying compounds that will have optimal in vivo properties and the greatest likelihood of succeeding in clinical trials.
Promising compounds should be evaluated in animal models to guide in vivo use in humans. Animal models can test the uptake of ASOs by cells in the CNS and phenotypic changes may also be observable. It is important to recognize, however, that animal models for FA may not perfectly mimic the molecular mechanisms of the human mutant FXN gene. Specifically, the likely R-loop mechanism is complex and ASOs that are active in human cells may not be as active in mouse cells. Negative results, therefore, should be treated with caution and testing in ex vivo human cell models of FA will be a useful adjunct to animal testing. A compound that is a potent activator of FXN protein expression in cell culture and ex vivo testing, that has demonstrated low toxicity in animals, and is chemically closely related to compounds demonstrated to be safe in other clinical trials might be a viable clinical candidate given the high level of unmet need for FA.
Conclusion
Our data demonstrate the robustness of ASO and dsRNA-mediated activation of FXN protein expression. Activation occurs in patient-derived cell lines with varied numbers of repeats and using a chemically diverse collection of ASOs and dsRNAs. ASOs and dsRNAs are currently experiencing a remarkable period of clinical success for other diseases, with a particularly relevant example being nusinersen for treatment of SMA in the CNS. Successful gene activation by different chemically modified ASOs and dsRNAs lays a firm foundation for vigorously evaluating candidates for clinical development.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (GM R35118103) and the Robert A. Welch Foundation (I-1244). The authors thank Dr. Helen Puccio for providing anti-FXN antibody 4F9 and Marek Napierala for supplying us with patient-derived cell lines. The authors thank the Friedreich's Ataxia Research Alliance for generously funding this research.
Author Disclosure Statement
T.P.P., M.N., and F.R. are employees of Ionis Pharmaceuticals. L.L. and D.R.C. have filed a patent related to this research.
References
- 1.Pandolfo M. (2009). Friedreich ataxia: the clinical picture. J Neurol 256:3–8 [DOI] [PubMed] [Google Scholar]
- 2.Burk K. (2017). Friedreich ataxia: current status and future prospects. Cerebellum Ataxias 4:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Herman D, Jenssen K, Burnett R, Soragni E, Perlman SL. and Gottesfeld JM. (2006). Histone deacetylase inhibitors reverse gene silencing in Friedreich's ataxia. Nat Chem Biol 2:551–558 [DOI] [PubMed] [Google Scholar]
- 4.Sandi C, Mouro Pinto R, Al-Mahdawi S, Ezzatizadeh V, Barnes G, Jones S, Rusche JR, Gottesfeld JM. and Pook MA. (2011). Prolonged treatment with pimelic o-aminobenzamide HDAC inhibitors ameliorates the disease phenotype of a Friedreich ataxia mouse model. Neurobiol Dis 42:496–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chan PK, Torres R, Yandim C, Law PP, Khadayate S, Mauri M, Grosan C, Chapman-Rothe N, Giunte P, Pook M. and Festenstein R. (2013). Heterochromatinization induced by GAA-repeat hyperexpansion in Friedreich's ataxia can be reduced upon HDAC inhibition by vitamin B3. Hum Mol Genet 22:2662–2675 [DOI] [PubMed] [Google Scholar]
- 6.Libri V, Yandim C, Athanasopoulos S, Loyse N, Natisvili T, Pik Law P, Mohammad T, Mauri M, Tung Tam K, et al. (2014). Epigenetic and neurological effects and safety of high-dose nicotinamide in patients with Friedreich's ataxia: an exploratory, open-label, dose-escalation study. Lancet 384:504–513 [DOI] [PubMed] [Google Scholar]
- 7.Soragni E, Miao W, Iudicello M, Jacoby D, De Mercanti S, Clerico M, Longo F, Piga A, Ku S, et al. (2014). Epigenetic therapy for Friedreich ataxia. Ann Neurol 76:489–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sahdeo S, Scott D, McMackin MZ, Jasoliya M, Brown B, Wulff H, Perlman SL, Pook MA. and Cortopassi GA. (2014). Dyclonine rescues frataxin deficiency in animal models and buccal cells of patients with Friedreich's ataxia. Hum Mol Genet 23:6848–6862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Perdomini M, Belbellaa B, Monassier L, Reutenauer L, Messaddeq N, Cartier N, Crystal RG, Aubourg P. and Puccio H. (2014). Prevention and reversal of severe mitochondrial cardiomyopathy by gene therapy in a mouse model of Friedreich's ataxia. Nat Med 20:542–547 [DOI] [PubMed] [Google Scholar]
- 10.Soragni E. and Gottesfeld JM. (2016). Translating HDAC inhibitors in Friedeich's ataxia. Expert Opin Orphan Drugs 4:691–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li L, Matsui M. and Corey. DR. (2016). Activating frataxin expression by repeat-targeted nucleic acids. Nat Commun 7:10606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Groh M, Lufino MMP, Wade-Martins R. and Gromak. N. (2014). R-loops associated with triplet repeat expansions promote gene silencing in Friedreich ataxia and fragile X syndrome. PLoS Genet 10:e1004318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Groh M, Sliva LM. and Gromak N. (2014). Mechanisms of transcriptional dysregulation in repeat expansion disorders. Biochem Soc Trans 42:1123–1128 [DOI] [PubMed] [Google Scholar]
- 14.Corey DR. (2017). Nusinersen, an antisense oligonucleotide drug for spinal muscular atrophy. Nat Neurosci 20:497–499 [DOI] [PubMed] [Google Scholar]
- 15.Watts JK, Yu D, Charisse K, Montaillier C, Potier P, Manoharan M. and Corey DR. (2010). Effect of chemical modifications on modulation of gene expression by duplex antigene RNAs that are complementary to non-coding transcripts at gene promoters. Nucleic Acids Res 38:5242–5259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goutelle S, Maurin M, Rougier F, Barbaut X, Bourguignon L, Ducher M, and Maire P. (2008). The Hill equation: a review of its capabilities in pharmacological modelling. Fundam Clin Pharmacol 22:633–648 [DOI] [PubMed] [Google Scholar]
- 17.Koshkin AA, Singh SK, Nielsen P, et al. (1998). LNA (locked nucleic acids): synthesis of the adenine, cytosine, guanine, 5-methylcytosine, thymine and uracil bicyclonucleoside monomers, oligomerisation, and unprecedented nucleic acid recognition. Tetrahedron 54:3607–3630 [Google Scholar]
- 18.Obika S, Nanbu D, Hari Y, et al. (1998). Stability and structural features of the duplexes containing nucleoside analogues with a fixed N-type conformation, 2'-O,4'-C-methyleneribonucleosides. Tetrahedron Lett 39:5401–5404 [Google Scholar]
- 19.Braasch DA. and Corey DR. (2001). Locked nucleic acid (LNA): fine-tuning the recognition of DNA and RNA. Chem Biol 8:1–7 [DOI] [PubMed] [Google Scholar]
- 20.Deleavey GF. and Damha MJ. (2012). Designing chemically modified oligonucleotides for targeted gene silencing. Chem Biol 19:937–954 [DOI] [PubMed] [Google Scholar]
- 21.Dowler T, Bergeron D, Tedeschi AL, Paquet L, Ferrari N. and Damha MJ. (2006). Improvements in siRNA properties mediated by 2′-deoxy-2′-fluoro-beta-D-arabionucleic acid (FANA). Nucleic Acids Res 34:1669–1675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deleavey GF, Watts JK, Alain T, Robert F, Kalota A, Aishwarya V, Pelletier J, Gewirtz AM, Sonenberg N. and Damha MG. (2010). Synergystic effects between analogs of DNA an dRNA improve the potency of siRNA-mediated gene silencing. Nucleic Acids Res 38:4547–4557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kalota A, Karabon L, Swider CR, Viazovkina E, Elzagheid M, Damha MJ. and Gerwirtz AM. (2006). 2′-Deoxy-2′-fluoro-ß-D-arinonucleic (2′F-ANA) modified oligonucleotides (ON) effect highly efficient and persistent gene silencing. Nucleic Acids Res 34:4451–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Souleimanian N, Deleavey GF, Soifer H, Wang S, Tiemann K, Damha MJ, and Stein. CA. (2012). Antisense 2′-deoxy, 2′fluorarabino nucleic acid (2′F-ANA) oligonucleotides: in vitro gymnotic silences of gene expression whose potency is enhanced by fatty acids. Mol Ther Nucl Acids 1:e43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mangos MM, Min KL, Viazovkina E, Galarneau A, Elzagheid MI, Parniak MA, and Damha MJ. (2003). Efficient RNase H-directed cleavage of RNA promoted by antisense DNA or 2′F-ANA constructs containing acyclic nucleotide inserts. J Am Chem Soc 125:654–661 [DOI] [PubMed] [Google Scholar]
- 26.Sharma VK, Singh SK, Krishnamurthy PM, Alterman JF, Haraszti RA, Khvorova A, Prasad AK. and Watts JK. (2017). Synthesis and biological properties of triazole-linked locked nucleic acid. Chem Commun 53:8906–8909 [DOI] [PubMed] [Google Scholar]
- 27.Shen X. and Corey. DR. (2017). Chemistry, mechanism, and clinical status of antisense oligonucleotides and duplex RNAs. Nucl Acids Res [Epub ahead of print; DOI: 10.1093/nar/gkx1239] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nikan M, Osborn MF, Coles AH, Godinho BM, Hall LM, Haraszti MR, Echeverria D, Aronin N. and Khvorova. A. (2016). Docosahexaenoic acid conjugation enhances distribution and safety of siRNA upon local administration in mouse brain. Mol Ther Nucl Acids 5:e344. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.