

Abstract
Objective
Chronic immune activation and elevated numbers of circulating activated monocytes (CD16+) are implicated in HIV-associated neuroinflammation. The objective was to compare the level of circulating CD16+ monocytes and interferon-γ-inducible protein 10 (IP-10) between HIV-infected cannabis users (HIV+MJ+) and non-cannabis users (HIV+MJ−), and determine whether in vitro Δ9-Tetrahydrocannabinol (THC), a constituent of cannabis, affected CD16 expression as well as IP-10 production by monocytes.
Design
The levels of circulating CD16+ monocytes and IP-10 from HIV+MJ− and HIV+MJ+ donors were examined. In vitro experimentation using THC was performed on primary leukocytes isolated from HIV-MJ-, HIV+MJ− and HIV+MJ+ donors to determine if THC has an impact on CD16+ monocyte and IP-10 levels.
Methods
Flow cytometry was used to measure the number of blood CD16+ monocytes and serum IP-10 from HIV+MJ− and HIV+MJ+ donors. Peripheral blood mononuclear cells (PBMC) were isolated from HIV-MJ− and HIV+ (MJ− and MJ+) donors for in vitro THC and IFNα treatment, and CD16+ monocytes and supernatant IP-10 were quantified.
Results
HIV+MJ+ donors possessed a lower level of circulating CD16+ monocytes and serum IP-10, compared to HIV+MJ− donors. Further, monocytes from HIV+MJ+ donors were unable to induce CD16 expression when treated with in vitro IFNα, while HIV-MJ− and HIV+MJ− donors displayed pronounced CD16 induction, suggesting anti-inflammatory effects by cannabis. Lastly, in vitro THC treatment impaired CD16− monocyte transition to CD16+ and monocyte-derived IP-10.
Conclusions
Components of cannabis, including THC, may decelerate peripheral monocyte processes that are implicated in HIV-associated neuroinflammation.
Keywords: HIV, cannabis, CD16+ monocyte, IP-10, THC
Introduction
Antiretroviral therapy (ART) has shifted HIV prognosis to a controllable disease, however, health complications remain and include cognitive decline, cardiovascular disease and malignancies [1]. Cognitive decline impacts 30–50% of HIV-infected (HIV+) individuals and is termed HIV-associated neurocognitive disorder (HAND) [2, 3]. The etiology of HAND is not fully understood but is due, in part, to dysfunction, damage and ultimately death of neurons, in the absence of productive HIV infection of neurons [4, 5]. Chronic immune activation and central nervous system (CNS) inflammation are mechanisms underlying neuronal damage [6, 7]. A growing body of evidence implicates CD16+ monocytes as a contributor to this neuroinflammation [6, 8, 9]. Specifically, increased levels of CD16+ monocytes in circulation have been observed in patients with chronic HIV infection and HIV-associated dementia [10–12]. Studies involving animal models and post-mortem HAND patients have identified an increased level of CD16+ monocytes in the CNS, and these cells stained positive for the HIV viral capsid protein, p24 [13–16].
The majority (85–95%) of circulating monocytes of healthy individuals are of the classical phenotype (CD14+CD16−), with CD14+CD16+ monocytes composing the remaining 5–15% [17]. CD16+ monocytes, which consist of intermediate (CD14hiCD16+) and non-classical (CD14loCD16+) monocyte populations, are often termed “inflammatory” monocytes due to their ability to secrete proinflammatory cytokines and promote T cell activation [17, 18]. Elevated levels of CD16+ monocytes in circulation have been observed in chronic inflammatory diseases including: viremic HIV infection, multiple sclerosis and systemic lupus erythematosus [10, 19, 20].
CD16+ monocytes have been identified as the major monocyte population infected with HIV in circulation [21]. In addition, CD16+ monocytes of HIV-infected individuals have increased expression of cell adhesion molecules and the chemokine receptor, CCR2, in comparison to CD16− monocytes, resulting in enhanced migration across in vitro models of the blood brain-barrier [22]. CD16+ monocytes are thought to be a major transport mechanism for HIV into the brain while also being able to secrete neurotoxic factors and pro-inflammatory cytokines [7–9, 13, 16]. Interferon-γ-inducible protein 10 (IP-10/CXCL10) is a proinflammatory factor secreted by monocytes during HIV infection and may play a key role in HIV-associated neuroinflammation [23, 24]. IP-10 is elevated in the serum and cerebrospinal fluid (CSF) of patients with HAND and plasma levels inversely correlate with N-acetylaspartate, a marker of neuronal injury [25–27]. Furthermore, in vitro experiments have revealed neurotoxic effects by IP-10 as evidenced by apoptosis of neurons [28].
A surface marker expressed by CD16+ monocytes is CD163 [29, 30]. Co-expression of CD16 and CD163 on monocytes has been observed at an increased level in post-mortem brain tissue of HIV+ individuals with cognitive impairment [14, 15]. CD16+CD163+ monocytes are also elevated in circulation of HIV+ individuals with detectable viral loads [31], suggesting that CD163 is expressed on CD16+ monocytes before entry into the brain. In addition, CD163 has been shown to have an important role for monocyte adherence to endothelial cells [32], providing a functional role for its expression on CD16+ monocytes that are migrating to the brain.
CD16− monocytes transition into the CD16+ phenotype in circulation and this process is of interest due to the pathogenic nature of the CD16+ monocyte subset during HIV infection [8, 18]. However, the specific mechanism(s) of enhanced CD16− monocyte transition to CD16+ during HIV infection remains unclear. Here we considered interferon alpha (IFNα) as a potential inducer of CD16− to CD16+ monocyte transition. This is supported by previous studies in which monocytes isolated from HIV-infected individuals displayed a type I interferon gene signature, suggesting exposure to IFNα in vivo [25, 33]. Additionally, the use of IFNα as a vaccine adjuvant in humans increased the percentage of CD16+ monocytes [34]. IFNα is a central component of the innate anti-viral immune response against HIV infection, but elevated IFNα can persist during the chronic stages of infection [35]. Sustained presence of IFNα is thought to contribute to the chronic immune activation and neurocognitive dysfunction observed during HIV pathogenesis [35–37].
Cannabis use is common amongst HIV-infected individuals in the United States and Canada, with an estimated prevalence of 20–37% [38–40]. HIV+ individuals use cannabis to help alleviate symptoms of HIV infection [41, 42]. The major psychotropic cannabinoid in cannabis, Δ9-Tetrahydrocannabinol (THC), has been identified as an immune modulator in animal models and cell-based systems, with most of its effects characterized as being immune suppressive and anti-inflammatory [43, 44]. THC modulates immune cell activity, in part, by binding cannabinoid receptor 1 and 2 (CB1 and CB2) [43]. In human monocytes, CB2 mRNA expression is higher than CB1, with both CB1 and CB2 being expressed at the protein level [45].
The central observation for the development of this study is that HIV-infected individuals using cannabis (HIV+MJ+) have lower levels of circulating CD16+ monocytes and serum IP-10 in comparison to HIV-infected persons not using cannabis (HIV+MJ-). From this initial observation, the objective of this study was to use human primary leukocytes isolated from HIV-MJ-, HIV+MJ− and HIV+MJ+ donors to determine; (a) whether in vitro IFNα promotes monocyte expression of CD16 and CD163; (b) the effects of in vitro THC treatment on the percentage of monocytes expressing CD16 and/or CD163 in response to IFNα; and (c) the effect of THC on monocyte production of IP-10.
Materials and Methods
HIV-infected donors
HIV+ male donors were recruited for blood draw under the IRB protocol (IRB# 11–202) by Dr. Peter Gulick and enrolled into the Mid-Michigan HIV consortium (MMHC). Donors received the standard of care and donor information was electronically available through the Research Data Capture (REDcap) (Vanderbilt University), which supports 21 CFR Part 11 compliance for clinical research and trials data and HIPAA guidelines. All HIV+ donors are currently on ART and negative for hepatitis C. Cannabis use was determined by self-reporting and confirmed by serum detection of THC metabolites using THC ELISA (RTU) Forensic Kit (Neogen Corporation, Lansing, MI). In this study, 4 of the 42 HIV+ donors had a discrepancy between self-reported use and THC metabolite detection and were classified based on results from the THC ELISA (RTU) Forensic Kit for cannabis use. HIV-MJ− donors tested negative for THC metabolites.
Collection of serum and leukocytes from whole blood of HIV-MJ-, HIV+MJ− and HIV+MJ+ donors
Whole blood was collected from HIV-MJ− (Stanford Blood Center) and HIV+ donors in acid citrate dextrose (ACD) or heparin tubes and either shipped (HIV-MJ− donors) or stored (HIV+ donors) overnight at room temperature. The next day, the number of leukocytes per mL of blood was obtained using a coulter counter. An aliquot of cells from whole blood collected in ACD or heparin tubes was used for cell surface staining. Before surface staining, red blood cells were removed using ACK lysis buffer. For serum collection, whole blood was collected in heparin tubes only. Serum was collected and stored at −80°C.
Peripheral blood mononuclear cell (PBMC) and CD16− monocyte isolation for in vitro studies
PBMCs were isolated from human leukocyte packs (Gulf Coast Regional Blood Center, Houston, TX) of HIV-MJ− donors and whole blood of HIV+ (MJ− and MJ+) donors by density gradient centrifugation using Ficoll-Paque PLUS (GE Healthcare Life Sciences, Pittsburgh, PA). Purified CD16− monocytes were isolated by negative selection using Human Monocyte Isolation Kit II (Miltenyi Biotec, Bergisch Gladbach, Germany) per manufacturer’s direction. The mean (±SD) monocyte purity for donors (N=14) used in this report was 88.4±5.7%. The mean percent of CD16− cells within the monocyte population was 99.2±0.6% (<1% CD16+ monocytes).
Chemicals
Δ9-Tetrahydrocannabinol (THC) and cannabidiol (CBD) were dissolved in 100% ethanol (National Institute on Drug Abuse, Bethesda, MD). For cell culture experiments, THC and CBD were serially diluted in RPMI 1640. The vehicle concentration for each treatment was 0.03% ethanol.
Cell culture and activation
PBMCs (5×106 cells/mL) or purified CD16− monocytes (1×106 cells/mL) were cultured in media containing RPMI1640 (Gibco™) supplemented with 5% human AB serum (Sigma-Aldrich, St. Louis, MO) and 100 U/ml Penicillin/100 μg/mL streptomycin (Gibco™). Leukocytes were stimulated with Universal Type I Interferon Alpha (PBL Assay Science, Piscataway Township, NJ) and incubated at 37°C and 5% CO2. For experiments involving THC/CBD treatment, cells were incubated at 37°C and 5% CO2 with the corresponding concentration of THC/CBD for 30 min prior to IFNα addition.
Flow cytometry
FACS buffer (PBS, 1% BSA, 0.1% NaN3) was used to wash cells in between staining and fixing steps. First, cells were incubated with FACS containing 20% human AB serum to block Fc receptors. Cells were then incubated with antibodies and LIVE/DEAD™ Fixable Near-IR Dead Cell Stain (Thermo Fisher Scientific, Waltham, MA). BD Cytofix™ (BD Biosciences, San Jose, CA) was used to fix cells. For intracellular staining, cells were stained with antibody in PERM wash (BD Biosciences). Fixed cells were analyzed on a FACS BD Canto II™ (BD Biosciences). Antibodies included anti-CD14-Pe-Cy7 (clone: M5E2), anti-CD16-APC (3G8) and anti-CD163-BV421 (GHI/61) from BioLegend (San Diego, CA), and anti-IFNAR2-APC-Vio770 (REA124) from Miltenyi Biotec. Anti-IP-10-PerCP-eFluor710 (4NY8UN) antibody was purchased from eBioscience (San Diego, CA). For intracellular IP-10 staining, a protein transport inhibitor (eBioscience) was added to cell culture 5h prior to experiment takedown. Data analysis was performed using FLOWJO v10 software. The gating strategy for CD16+ monocytes is in Supplementary Figure 1 (see Figure, Supplemental Digital Content 1). A Boolean gate was used to determine the percent of CD16+CD163+ cells within the monocyte population. For experiments involving purified monocytes, viable monocytes were gated based on SSC and FSC-A and analyzed for CD16 and CD163 expression.
Serum and supernatant IP-10 analysis
Serum or supernatants were collected and stored at −80°C. Serum/supernatants were thawed and IP-10 protein levels were quantified using LEGENDplex™ or ELISAmax™ from BioLegend per manufacturer’s direction.
Statistical analysis
Statistical analysis was performed using Prism 7 (GraphPad, San Diego, CA). The experimental data was graphed as the mean +/− SEM. The statistical tests performed for each experiment are indicated in the figure legends.
Results
HIV+MJ+ donors possess lower levels of circulating CD16+ monocytes and serum IP-10 compared to HIV+MJ− donors
Monocyte expression of CD16 and CD163, and serum IP-10 was determined in whole blood collected from HIV-MJ-, HIV+MJ− and HIV+MJ+ donors. There were no significant differences in age, body mass index (BMI), CD4 count, CD4/CD8 ratio and years infected with HIV between HIV+MJ− and HIV+MJ+ donors. In addition, there was a similar profile between HIV+MJ− and HIV+MJ+ donors in terms of being on ART, having undetectable viral loads, cigarette use, alcohol and other drugs of abuse (see Table, Supplemental Digital Content 2). When the levels of CD16+ monocytes were compared, HIV+MJ+ donors had a significantly lower level compared to HIV+MJ− donors (p=0.026) (Fig. 1A). A lower number of CD16+CD163+ monocytes was also observed in HIV+MJ+ donors when compared to HIV+MJ− donors but not significant (p=0.052) (Fig. 1B). In addition, serum IP-10 was also significantly lower in HIV+MJ+ donors compared to HIV+MJ− donors (p=0.005) (Fig. 1C).
Figure 1. HIV+MJ+ donors display a lower level of circulating CD16+ monocytes, CD16+CD163+ monocytes and serum IP-10 compared to HIV+MJ− donors.
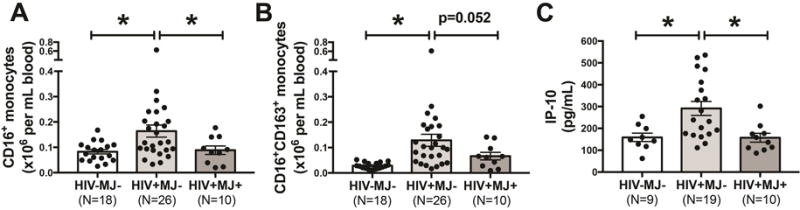
(A–C) Whole blood and serum was collected from HIV-MJ-, HIV+MJ− and HIV+MJ+ donors and the number (×106 per mL of blood) of CD16+ monocytes, CD16+CD163+ monocytes and serum IP-10 (pg/mL) was measured. For A–C, data was log transformed and a one-way ANOVA with a Dunnett’s post-hoc test was performed (p<0.05). All graphs are mean +/− SEM.
IFNα treatment of PBMCs and purified monocytes increases the expression of both CD16 and CD163 on monocytes in HIV-MJ− and HIV+MJ− donors but not HIV+MJ+ donors
Monocyte transition into the CD16+ phenotype in circulation is a key step prior to monocyte migration into the CNS during HIV infection [13, 46], but the specific mechanism(s) of this monocyte transition remains unclear. Since a type I IFN gene signature has been identified in monocytes from HIV+ individuals [25, 33], we sought to determine the effect of IFNα on monocyte expression of CD16 and CD163. Human PBMCs isolated from HIV-MJ− donors were stimulated with 50U/ml of IFNα and cells were harvested at 6, 16, 24 and 48h post stimulation. Supplementary Digital Content 3 illustrates the effect of IFNα on monocyte expression (within PBMCs) of CD16 at 48h. IFNα treatment for 48h led to a significant increase in the percent of monocytes expressing CD16 (p<0.0001) (Fig. 2A), while a significant increase in percent of CD163+ monocytes was observed only at 24h (p=0.01) (Fig. 2B). There was a notable increase in the percent of CD163+ monocytes in the non-stimulated monocytes at 48h, which may be due to adherence-mediated activation of monocytes [47]. A significant increase in the percent of monocytes co-expressing CD16 and CD163 (CD16+CD163+) was observed at 48h (p<0.0001) (Fig. 2C). PBMCs were then isolated from HIV+MJ− and HIV+MJ+ donors to determine if there was a difference in the IFNα-mediated induction of CD16 and CD163 on monocytes from HIV+MJ− and HIV+MJ+ donors. Interestingly, IFNα treatment significantly increased the percentage of monocytes expressing CD16 (p=0.027), CD163 (p=0.002) and CD16/CD163 (p=0.009) in HIV+MJ− donors (Figs. 2D–F), which was similar to that of HIV-MJ− donors. However, IFNα treatment only increased the percentage of CD163+ monocytes (p=0.005) and not CD16+ (p=0.621) or CD16+CD163+ (p=0.242) monocytes of HIV+MJ+ donors (Fig. 2D–F), suggesting that cannabis use may be suppressing monocyte induction of CD16.
Figure 2. IFNα treatment increases CD16 and CD163 expression on monocytes.
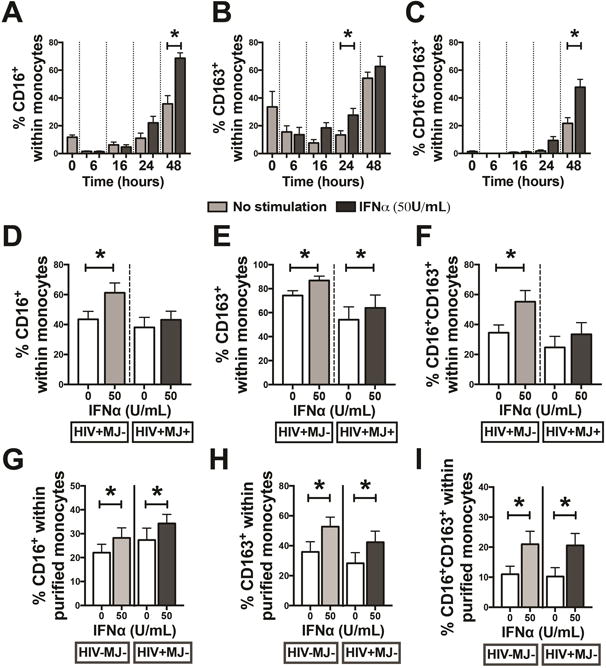
(A–C) HIV-MJ− PBMCs (N=7) were treated with IFNα (50U/mL) for 6, 16, 24 and 48h. (D–F) HIV+MJ− (N=6) and HIV+MJ+ (N=7) PBMCs were treated with IFNα (50U/mL) for 48h. (G–I) Purified CD16− monocytes from HIV-MJ− (N=7) and HIV+MJ− (N=7) donors were treated with IFNα (50U/mL) for 48h. Flow cytometry was used to measure the percentage of CD16+, CD163+ and CD16+CD163+ cells within the monocyte population. *Statistically different from non-stimulated controls (p<0.05, two-way RM ANOVA with a Tukey’s multiple comparisons post-test for A–C and two-way ANOVA with a Sidak’s multiple comparisons test for D–I). Graphs in A–I are mean +/− SEM.
To determine if IFNα is having a direct role on monocyte expression of CD16 and CD163, CD16− monocytes from HIV-MJ− and HIV+MJ− donors were purified prior to IFNα (50U/ml) treatment. As with PBMCs, IFNα treatment of purified monocytes led to an increased percentage of CD16+, CD163+ and CD16+CD163+ monocytes for both HIV-MJ− (p=0.032, 0.0001 and 0.0007, respectively) and HIV+MJ− (p=0.017, 0.0007 and 0.0005, respectively) donors (Figs. 2G–I).
In vitro THC treatment of HIV-MJ− PBMCs and purified monocytes impairs the IFNα-mediated induction of CD16 and CD163 expression on monocytes
Since HIV+MJ+ donors have lower levels of CD16+ and CD16+CD163+ monocytes (p=0.052) in whole blood compared to HIV+MJ− donors, we sought to determine whether in vitro THC treatment influenced monocyte expression of CD16 and CD163 in response to IFNα. PBMCs from HIV-MJ− donors were pre-treated with 1, 5 and 10μM of THC and stimulated with IFNα (50U/mL) for 48h. THC treatment markedly decreased the percentage of CD16+ monocytes in a concentration-dependent manner with significant suppression at 1, 5 and 10μM THC (p<0.0003 for all THC concentrations) (Fig. 3A). In addition, THC treatment significantly decreased the percentage of CD163+ (p<0.005 for 5 and 10μM) and CD16+CD163+ (p<0.0006 for all THC concentrations) monocytes (Figs. 3B and C). THC treatment had no significant effect on cell viability (>95% for each treatment group). As IFNα modulates cell function through the IFNα/β receptor (IFNAR) [48], we next sought to determine the effect of THC on monocyte expression of IFNAR using the same experimental approach as above. THC at 10μM modestly decreased the percentage of IFNAR+ monocytes after 48h of IFNα treatment (p=0.023) (Fig. 3D).
Figure 3. THC treatment decreased the percentage of CD16+, CD163+, CD16+CD163+ and IFNAR+ cells within the monocyte population.
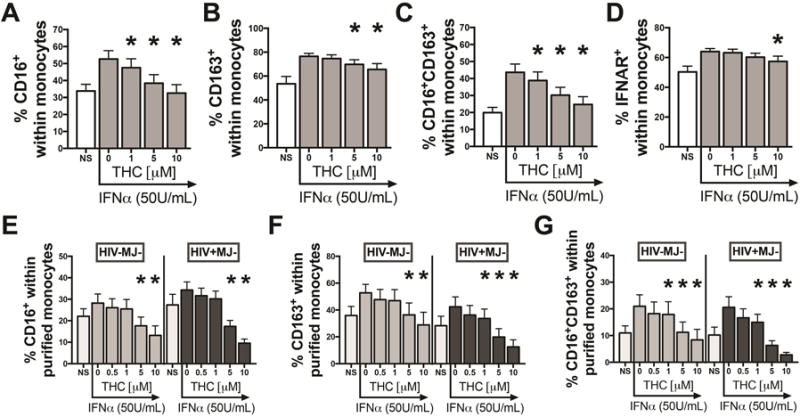
(A–D) PBMCs from HIV-MJ− donors (N=11 for A–C and N=5 for D) and (E–G) purified CD16− monocytes from HIV-MJ− (N=7) and HIV+MJ− (N=7) donors were pretreated with 0 (vehicle), 0.5 (purified only), 1, 5 and 10μM of THC for 0.5h and stimulated with IFNα (50U/ml) for 48h. *Statistically different from vehicle control (50 U/mL IFNα+vehicle) (p<0.05, RM one-way ANOVA with a Dunnett’s multiple comparisons post-test). NS represents vehicle without IFNα addition. Graphs in A–F are mean +/− SEM.
To determine if THC has a direct inhibitory effect on the monocyte population and not influencing monocyte activation via a bystander effect, CD16− monocytes from HIV-MJ− PBMCs were purified, pre-treated with 0.5, 1, 5 and 10μM of THC and stimulated with IFNα (50U/ml) for 48h. As seen in PBMCs, THC treatment decreased all three monocyte populations (CD16+, CD163+ and CD16+CD163+) in a concentration-dependent manner (grey bars in Fig. 3E–G). Next, we confirmed that THC treatment also directly impaired monocyte expression of CD16 and CD163 in purified monocytes of HIV+MJ− donors (black bars in Fig. 3E–G). THC treatment had no effect on cell viability (>95% for each treatment group).
Cannabidiol (CBD) does not impair CD16 or CD163 expression in IFNα-stimulated PBMCs from HIV-MJ− donors
THC has a binding affinity to both CB1 and CB2 with a Ki of 25.1nM and 35.2nM for CB1 and CB2, respectively [49]. By contrast, cannabidiol (CBD), another cannabinoid present in cannabis, displays high structure similarity to THC but has 80-fold lower binding affinity to CB1 and CB2 [49]. To better understand the role of CB1/CB2 in the THC-mediated impairment of CD16 and CD163 expression on monocytes, PBMCs from HIV-MJ− donors were pre-treated with THC or CBD at 1, 5 and 10μM and stimulated with 50U/mL of IFNα for 48h. As observed in Figure 3, THC significantly decreased the percentage of monocytes expressing of CD16 (p=0.0001 for 10μM THC), CD163 (p=0.004 for 10μM THC) and CD16/CD163 (p=0.0001 for 10μM THC), while CBD at the same concentrations elicited no significant effects on the percentage of monocytes expressing of CD16 (p=0.448 for 10μM THC), CD163 (p=0.626 for 10μM THC) and CD16/CD163 (p=0.219 for 10μM THC) (Fig. 4A–C).
Figure 4. THC but not CBD decreased the percentage of CD16+, CD163+ and CD16+CD163+ cells within the monocyte population of IFNα-treated PBMCs.
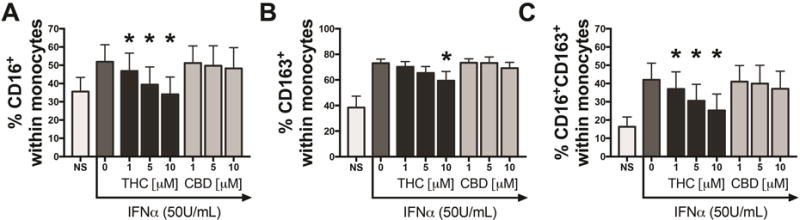
PBMCs from HIV-MJ− donors (N=5) were pretreated with 0 (vehicle), 1, 5 and 10μM of THC or 1, 5 and 10μM of CBD for 0.5h and stimulated with IFNα (50U/ml) for 48h. *Statistically different from vehicle control (50 U/mL IFNα+vehicle) group (p<0.05, RM one-way ANOVA with a Dunnett’s multiple comparisons post-test). NS represents vehicle without IFNα addition. Graphs in A–C are mean +/− SEM.
THC treatment of PBMCs and purified monocytes decreased supernatant IP-10 levels from HIV-MJ-, HIV+MJ− and HIV+MJ+ donors
After observing lower serum IP-10 in HIV+MJ+ donors, when compared to HIV+MJ− donors, we then examined the impact of in vitro THC on monocyte production of IP-10. First, to determine the cellular source of IP-10 in response to IFNα, PBMCs from HIV-MJ− donors were stimulated with IFNα (50U/mL) for 24h and intracellular IP-10 staining was performed. Figure 5A is one representative donor of three, which demonstrates IFNα treatment increases the percent of IP-10+ cells. Of the IP-10+ cells, >90% were CD14+ monocytes (Fig. 5A). Of the IP-10+ monocytes in the IFNα treatment group, 73% were CD16− and 27% were CD16+ (p=0.126) (data not shown), suggesting that CD16 expression isn’t a prerequisite for monocyte production of IP-10. Next, PBMCs from HIV-MJ−, HIV+MJ− and HIV+MJ+ donors were pre-treated with 1 and 5μM THC and stimulated with IFNα for 48h. IFNα triggered a significant increase in supernatant IP-10 levels in the three donor groups (p<0.009), with no significant differences detected across groups (left panel, Fig. 5B). IP-10 levels were suppressed by 1 and 5μM THC in HIV-MJ− donors (p=0.028 for 1μM and p=0.004 for 5μM THC), while significant suppression was only seen at 5μM THC in HIV+MJ− (p=0.009) and HIV+MJ+ (p=0.004) donors (right panel, Fig. 5B). To determine if THC directly impairs monocyte production of IP-10, monocytes were purified from HIV-MJ− and HIV+MJ− donors, treated with THC (0.5, 1, 5 and 10μM) and stimulated with IFNα for 48h. IFNα stimulation significantly increased supernatant IP-10 in purified monocytes from HIV-MJ− and HIV+MJ− donors (left panel, Fig. 5C). THC treatment decreased supernatant IP-10 in a concentration-dependent manner with significant differences observed at 0.5–10μM THC in both HIV-MJ− (p=0.019 for 0.5μM THC) and HIV+MJ− (p=0.007 for 0.5μM THC) donors (right panel, Fig. 5C).
Figure 5. THC decreased supernatant IP-10 levels in IFNα-stimulated PBMCs and purified monocytes from HIV-MJ−, HIV+MJ− and HIV+MJ+ donors.
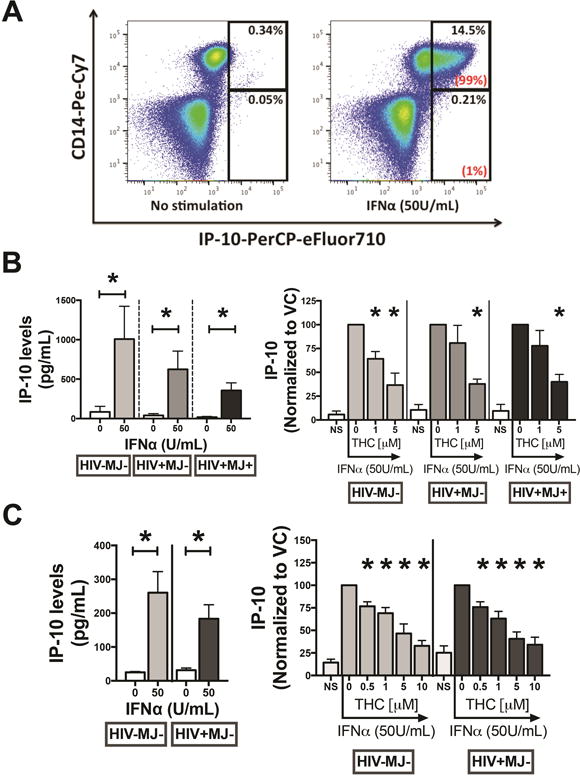
(A) PBMCs from HIV-MJ− donors (N=3) were stimulated with IFNα (50U/ml) for 24h and intracellular IP-10 staining was performed. Flow cytometry plots are from one representative donor. The percentages on the upper right of the gate refer to percent of CD14+IP-10+ or CD14−IP-10+ cells within total PBMCs. The percentages in parentheses display the portion of IP-10+ cells coming from the CD14+ and CD14− populations. (B) PBMCs from HIV-MJ− (N=3), HIV+MJ− (N=5) and HIV+MJ+ (N=5) donors were pre-treated with THC (1 and 5μM) for 0.5h and stimulated with IFNα (50U/ml) for 48h. Supernatants were harvested and LEGENDplex™ was used to quantify IP-10 levels (*p<0.05). (C) Purified CD16− monocytes from HIV-MJ− (N=7) and HIV+MJ− (N=7) donors were pre-treated with 0.5, 1 and 5 and 10μM of THC for 0.5h and stimulated with IFNα (50U/ml) for 48h. IFNα induction of IP-10 (pg/mL) is displayed on the left and the effect of THC on IP-10 levels is displayed on the right. For the right panels in B and C, IP-10 levels for each THC treatment group was normalized to the donors vehicle control (VC) response (e.g. 0μM THC), which served as 100%. For left panels in B and C, * denotes a statistical difference from non-stimulated controls (p<0.05, two-way ANOVA with a Sidak’s multiple comparisons test). For right panels in B and C, * denotes a statistical difference from vehicle control (50U/mL IFNα+vehicle) group (p<0.05) (RM one-way ANOVA with a Dunnett’s multiple comparisons post-test). Graphs in B and C are mean +/− SEM.
Discussion
In this study, we show that IFNα treatment of PBMCs and purified monocytes isolated from HIV-MJ− donors promotes monocyte transition into the CD16+ phenotype as well as increases the percent of CD163+ and CD16+CD163+ monocytes. These findings coincide with previous studies reporting that monocytes from HIV-infected individuals display a type I interferon gene signature [25, 33]. Similarly, in vivo IFNα therapy promoted an increase in the percent of CD16+ monocytes [34]. Taken together, these observations strongly support IFNα as an inflammatory factor that increases the frequency of CD16+ and CD16+CD163+ monocytes during HIV infection. These findings are noteworthy since circulating CD16+/CD16+CD163+ monocytes traffic to the brain during HIV infection promoting viral entry as well as secretion of inflammatory and neurotoxic factors [8, 9, 13, 16]. Interestingly, the IFNα-mediated monocyte transition to CD16+ was only observed in HIV+MJ− donors and not HIV+MJ+ donors, suggesting that cannabis use may impair the induction of CD16. Future studies investigating the differences in monocytes expression of IFNAR and key downstream signaling molecules between HIV+MJ− and HIV+MJ+ donors will provide insights into the lack of CD16 induction observed in HIV+MJ+ donors.
After the initial observation that HIV+MJ+ donors possess lower circulating CD16+ and CD16+CD163+ (p=0.052) monocytes compared to HIV+MJ− donors, we demonstrated that in vitro THC treatment decreased the percent of monocytes expressing CD16, CD163 and CD16/CD163 in IFNα-treated PBMCs isolated from HIV-MJ− donors. Further, THC treatment impaired monocyte expression of IFNAR in HIV-MJ− donors; however, the impairment was modest and only observed at the highest concentration of THC (10μM). With significant impairment in CD16 expression seen as low as 1μM THC, these findings suggest that THC is impairing IFNAR-mediated signaling. THC impairment of CD16 and CD163 expression on monocytes was also observed in purified CD16− monocytes demonstrating that THC acts directly on the monocyte population and not through a bystander mechanism. Further, treatment with the low affinity CB1/CB2 agonist, cannabidiol (CBD), yielded no significant effect on CD16 or CD163 expression, suggesting that THC is modulating monocyte activity through a cannabinoid receptor (CB1/CB2)-dependent mechanism. As HIV-infected individuals have chronic immune activation [50, 51] and CB1/CB2 expression may change with monocyte/macrophage activation status [43], the sensitivity of immune cells to THC treatment may vary between HIV-MJ− and HIV+MJ− donors. Therefore, we performed experiments using monocytes from HIV+MJ− donors, which demonstrated that monocytes isolated from HIV+MJ− donors displayed similar impairment by THC on CD16 and CD163 expression to that of HIV-MJ− donors. Overall, these findings suggest that the THC present in cannabis may be a significant contributor to the decreased levels of CD16+ monocytes observed in HIV+MJ+ donors.
Another interesting observation in this study is that serum IP-10 levels are lower in HIV+MJ+ donors compared to HIV+MJ− donors. IP-10 has been shown to be elevated in the CSF of patients with cognitive impairment and is thought to be an important contributor to neuroinflammation during HIV infection [24]. Furthermore, IP-10 has been shown to stimulate HIV replication in monocyte-derived macrophages and promote neuronal apoptosis in vitro [28, 52]. Using intracellular IP-10 staining, we report that the monocyte population is the primary cell type within the PBMCs of HIV-MJ− donors secreting IP-10 in response to IFNα and monocyte expression of CD16 isn’t necessary for IP-10 production. This is in agreement with a previous report showing the monocyte population is a major source of IP-10 when stimulated with TLR7/8 ligands [23]. When comparing the IFNα-mediated induction of IP-10 between HIV-MJ−, HIV+MJ− and HIV+MJ+ donors, similar induction profiles were observed. THC treatment was shown to decrease IP-10 in all three groups, with HIV-MJ− donors showing a slight increase in sensitivity to THC. Using purified monocytes from HIV-MJ− and HIV+MJ− donors, we demonstrate that THC has a direct effect on the monocytes resulting in decreased IP-10 levels. Furthermore, THC at a concentration of 0.5μM significantly decreased IP-10 levels, which is within the concentration range observed in blood of individuals smoking cannabis [53].
The results from this study show that in vitro THC treatment promotes anti-inflammatory effects on monocyte processes that are implicated in HIV-associated neuroinflammation, including monocyte transition into the CD16+ phenotype and secretion of IP-10. With these findings it is tempting to speculate that THC is one of the major components of cannabis that elicits the decrease in circulating CD16+ monocytes and serum IP-10 that was observed in HIV+MJ+ donors. However, the in vivo effects of THC when inhaled through cannabis use may be different than that observed in vitro due to the additional 60-plus cannabinoids that are present in cannabis as well as other plant-derived constituents [54]. Therefore cannabinoids in combination with other plant-associated compounds may contribute to the observed anti-inflammatory actions. In addition, cannabis use could indirectly have anti-inflammatory actions, such as through stress reduction, which can have an impact on inflammation [55, 56]
There were limitations in the cross-sectional design comparing blood CD16+ monocytes and serum IP-10 in HIV-MJ−, HIV+MJ− and HIV+MJ+ donors (Fig. 1). First, the absence of HIV-MJ+ donors hindered our ability to make comparisons between HIV-MJ+ and HIV+MJ+ donors. However, the HIV-MJ− donors served as a comparator to show the increased levels of inflammatory markers observed in HIV+MJ− donors. The central focus was to identify potential differences in the number of monocytes expressing CD16/CD163 and serum IP-10 between HIV+MJ− and HIV+MJ+ donors. Second, the exposure level of cannabis in the HIV+MJ+ population could not be quantified due to many variables. This remains a systemic limitation in studies investigating cannabis use, as exposure levels can be influenced by multiple variables [57]. However, we could confirm cannabis use and whether respondents were accurate in stating cannabis use in the patient questionnaire by assaying blood samples for the presence of THC metabolites. Lastly, the HIV-MJ− donors in this study were from different geographical locations compared to the HIV+MJ− and HIV+MJ+ donors. Importantly, all HIV+ donors, HIV+MJ− and HIV+MJ+, were from the Mid-Michigan area.
We conclude that within the context of HIV-associated neuroinflammation and cognitive decline, cannabinoid therapies may decelerate peripheral immune processes that are implicated in HIV-associated neuroinflammation.
Supplementary Material
Acknowledgments
We express our thanks to Linda Dale for coordinating blood collection from HIV+ donors. We would also like to thank Patrick O’Connell and Yuliya Pepelyayeva for their contribution in isolating peripheral blood mononuclear cells from HIV- and HIV+ donors.
Funding: The National Institutes of Drug Abuse Grant DA07908 and the National Institutes of Environmental Health Sciences Training Grant T32 ES07255 supported this work.
Footnotes
Conflict of Interest: None of the authors present on this paper report any conflict of interest.
Author contributions:
M.D.R. was central to the origination and development of this study. He performed the literature search, development and execution of the experimental design, and writing of the manuscript.
R.B.C. contributed to the development of the experimental design and manuscript. R.B.C. performed the whole blood cell analysis of CD16+ monocytes and was responsible for flow cytometric analysis of leukocyte samples. Mr. Crawford assisted with data analysis and graphical representation.
J.E.H. participated in discussions that were the basis for investigating the effects of cannabis use on monocytes in HIV patients. Furthermore, J.E.H. also contributed to formulating the experimental design for the study and in the final editing of the manuscript.
Y.A. assisted in the recruitment of the HIV donors used in this study. Y.A. also contributed to the experimental design and interpretation of results.
P.G.: The HIV donors recruited to this study were under the supervision of P.G. and he had a significant role in recruiting these patients for blood draw.
A.A. contributed to discussions that were the basis for investigating monocytes in HIV patients.
N.E.K. participated in discussions that were the basis for investigating the effects of cannabis use on monocytes in HIV patients. N.E.K. also contributed to the development of the experimental design, interpretation of results and writing of the manuscript.
References
- 1.Cardoso SW, Torres TS, Santini-Oliveira M, Marins LM, Veloso VG, Grinsztejn B. Aging with HIV: a practical review. Braz J Infect Dis. 2013;17(4):464–479. doi: 10.1016/j.bjid.2012.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heaton RK, Franklin DR, Ellis RJ, McCutchan JA, Letendre SL, Leblanc S, et al. HIV-associated neurocognitive disorders before and during the era of combination antiretroviral therapy: differences in rates, nature, and predictors. J Neurovirol. 2011;17(1):3–16. doi: 10.1007/s13365-010-0006-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rumbaugh JA, Tyor W. HIV-associated neurocognitive disorders: Five new things. Neurol Clin Pract. 2015;5(3):224–231. doi: 10.1212/CPJ.0000000000000117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wiley CA, Schrier RD, Nelson JA, Lampert PW, Oldstone MB. Cellular localization of human immunodeficiency virus infection within the brains of acquired immune deficiency syndrome patients. Proc Natl Acad Sci U S A. 1986;83(18):7089–7093. doi: 10.1073/pnas.83.18.7089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gonzalez-Scarano F, Martin-Garcia J. The neuropathogenesis of AIDS. Nat Rev Immunol. 2005;5(1):69–81. doi: 10.1038/nri1527. [DOI] [PubMed] [Google Scholar]
- 6.Hong S, Banks WA. Role of the immune system in HIV-associated neuroinflammation and neurocognitive implications. Brain Behav Immun. 2015;45:1–12. doi: 10.1016/j.bbi.2014.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Campbell JH, Ratai EM, Autissier P, Nolan DJ, Tse S, Miller AD, et al. Anti-alpha4 antibody treatment blocks virus traffic to the brain and gut early, and stabilizes CNS injury late in infection. PLoS Pathog. 2014;10(12):e1004533. doi: 10.1371/journal.ppat.1004533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Williams DW, Veenstra M, Gaskill PJ, Morgello S, Calderon TM, Berman JW. Monocytes mediate HIV neuropathogenesis: mechanisms that contribute to HIV associated neurocognitive disorders. Curr HIV Res. 2014;12(2):85–96. doi: 10.2174/1570162x12666140526114526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Campbell JH, Hearps AC, Martin GE, Williams KC, Crowe SM. The importance of monocytes and macrophages in HIV pathogenesis, treatment, and cure. AIDS. 2014;28(15):2175–2187. doi: 10.1097/QAD.0000000000000408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pulliam L, Gascon R, Stubblebine M, McGuire D, McGrath MS. Unique monocyte subset in patients with AIDS dementia. The Lancet. 1997;349(9053):692–695. doi: 10.1016/S0140-6736(96)10178-1. [DOI] [PubMed] [Google Scholar]
- 11.Thieblemont N, Weiss L, Sadeghi HM, Estcourt C, Haeffner-Cavaillon N. CD14lowCD16high: a cytokine-producing monocyte subset which expands during human immunodeficiency virus infection. Eur J Immunol. 1995;25(12):3418–3424. doi: 10.1002/eji.1830251232. [DOI] [PubMed] [Google Scholar]
- 12.Han J, Wang B, Han N, Zhao Y, Song C, Feng X, et al. CD14(high)CD16(+) rather than CD14(low)CD16(+) monocytes correlate with disease progression in chronic HIV-infected patients. J Acquir Immune Defic Syndr. 2009;52(5):553–559. doi: 10.1097/qai.0b013e3181c1d4fe. [DOI] [PubMed] [Google Scholar]
- 13.Fischer-Smith T, Croul S, Sverstiuk AE, Capini C, L’Heureux D, Regulier EG, et al. CNS invasion by CD14+/CD16+ peripheral blood-derived monocytes in HIV dementia: perivascular accumulation and reservoir of HIV infection. J Neurovirol. 2001;7(6):528–541. doi: 10.1080/135502801753248114. [DOI] [PubMed] [Google Scholar]
- 14.Tavazzi E, Morrison D, Sullivan P, Morgello S, Fischer T. Brain inflammation is a common feature of HIV-infected patients without HIV encephalitis or productive brain infection. Curr HIV Res. 2014;12(2):97–110. doi: 10.2174/1570162x12666140526114956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fischer-Smith T, Bell C, Croul S, Lewis M, Rappaport J. Monocyte/macrophage trafficking in acquired immunodeficiency syndrome encephalitis: lessons from human and nonhuman primate studies. J Neurovirol. 2008;14(4):318–326. doi: 10.1080/13550280802132857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clay CC, Rodrigues DS, Ho YS, Fallert BA, Janatpour K, Reinhart TA, et al. Neuroinvasion of fluorescein-positive monocytes in acute simian immunodeficiency virus infection. J Virol. 2007;81(21):12040–12048. doi: 10.1128/JVI.00133-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wong KL, Yeap WH, Tai JJ, Ong SM, Dang TM, Wong SC. The three human monocyte subsets: implications for health and disease. Immunol Res. 2012;53(1–3):41–57. doi: 10.1007/s12026-012-8297-3. [DOI] [PubMed] [Google Scholar]
- 18.Ziegler-Heitbrock L. The CD14+ CD16+ blood monocytes: their role in infection and inflammation. J Leukoc Biol. 2007;81(3):584–592. doi: 10.1189/jlb.0806510. [DOI] [PubMed] [Google Scholar]
- 19.Mukherjee R, Kanti Barman P, Kumar Thatoi P, Tripathy R, Kumar Das B, Ravindran B. Non-Classical monocytes display inflammatory features: Validation in Sepsis and Systemic Lupus Erythematous. Sci Rep. 2015;5:13886. doi: 10.1038/srep13886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chuluundorj D, Harding SA, Abernethy D, La Flamme AC. Expansion and preferential activation of the CD14(+)CD16(+) monocyte subset during multiple sclerosis. Immunol Cell Biol. 2014;92(6):509–517. doi: 10.1038/icb.2014.15. [DOI] [PubMed] [Google Scholar]
- 21.Ellery PJ, Tippett E, Chiu YL, Paukovics G, Cameron PU, Solomon A, et al. The CD16+ Monocyte Subset Is More Permissive to Infection and Preferentially Harbors HIV-1 In Vivo. The Journal of Immunology. 2007;178(10):6581–6589. doi: 10.4049/jimmunol.178.10.6581. [DOI] [PubMed] [Google Scholar]
- 22.Williams DW, Calderon TM, Lopez L, Carvallo-Torres L, Gaskill PJ, Eugenin EA, et al. Mechanisms of HIV entry into the CNS: increased sensitivity of HIV infected CD14+CD16+ monocytes to CCL2 and key roles of CCR2, JAM-A, and ALCAM in diapedesis. PLoS One. 2013;8(7):e69270. doi: 10.1371/journal.pone.0069270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Simmons RP, Scully EP, Groden EE, Arnold KB, Chang JJ, Lane K, et al. HIV-1 infection induces strong production of IP-10 through TLR7/9-dependent pathways. AIDS. 2013;27(16):2505–2517. doi: 10.1097/01.aids.0000432455.06476.bc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mehla R, Bivalkar-Mehla S, Nagarkatti M, Chauhan A. Programming of neurotoxic cofactor CXCL-10 in HIV-1-associated dementia: abrogation of CXCL-10-induced neuro-glial toxicity in vitro by PKC activator. J Neuroinflammation. 2012;9:239. doi: 10.1186/1742-2094-9-239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pulliam L, Rempel H, Sun B, Abadjian L, Calosing C, Meyerhoff DJ. A peripheral monocyte interferon phenotype in HIV infection correlates with a decrease in magnetic resonance spectroscopy metabolite concentrations. AIDS. 2011;25(14):1721–1726. doi: 10.1097/QAD.0b013e328349f022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ramirez LA, Arango TA, Thompson E, Naji M, Tebas P, Boyer JD. High IP-10 levels decrease T cell function in HIV-1-infected individuals on ART. J Leukoc Biol. 2014;96(6):1055–1063. doi: 10.1189/jlb.3A0414-232RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yuan L, Qiao L, Wei F, Yin J, Liu L, Ji Y, et al. Cytokines in CSF correlate with HIV-associated neurocognitive disorders in the post-HAART era in China. J Neurovirol. 2013;19(2):144–149. doi: 10.1007/s13365-013-0150-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sui Y, Stehno-Bittel L, Li S, Loganathan R, Dhillon NK, Pinson D, et al. CXCL10-induced cell death in neurons: role of calcium dysregulation. Eur J Neurosci. 2006;23(4):957–964. doi: 10.1111/j.1460-9568.2006.04631.x. [DOI] [PubMed] [Google Scholar]
- 29.Kim WK, Alvarez X, Fisher J, Bronfin B, Westmoreland S, McLaurin J, et al. CD163 identifies perivascular macrophages in normal and viral encephalitic brains and potential precursors to perivascular macrophages in blood. Am J Pathol. 2006;168(3):822–834. doi: 10.2353/ajpath.2006.050215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kristiansen M, Graversen JH, Jacobsen C, Sonne O, Hoffman HJ, Law SK, et al. Identification of the haemoglobin scavenger receptor. Nature. 2001;409(6817):198–201. doi: 10.1038/35051594. [DOI] [PubMed] [Google Scholar]
- 31.Fischer-Smith T, Tedaldi EM, Rappaport J. CD163/CD16 coexpression by circulating monocytes/macrophages in HIV: potential biomarkers for HIV infection and AIDS progression. AIDS Res Hum Retroviruses. 2008;24(3):417–421. doi: 10.1089/aid.2007.0193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wenzel I, Roth J, Sorg C. Identification of a novel surface molecule, RM3/1, that contributes to the adhesion of glucocorticoid-induced human monocytes to endothelial cells. Eur J Immunol. 1996;26(11):2758–2763. doi: 10.1002/eji.1830261131. [DOI] [PubMed] [Google Scholar]
- 33.Rempel H, Sun B, Calosing C, Pillai SK, Pulliam L. Interferon-alpha drives monocyte gene expression in chronic unsuppressed HIV-1 infection. AIDS. 2010;24(10):1415–1423. doi: 10.1097/QAD.0b013e32833ac623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arico E, Castiello L, Urbani F, Rizza P, Panelli MC, Wang E, et al. Concomitant detection of IFNalpha signature and activated monocyte/dendritic cell precursors in the peripheral blood of IFNalpha-treated subjects at early times after repeated local cytokine treatments. J Transl Med. 2011;9:67. doi: 10.1186/1479-5876-9-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cha L, Berry CM, Nolan D, Castley A, Fernandez S, French MA. Interferon-alpha, immune activation and immune dysfunction in treated HIV infection. Clin Transl Immunology. 2014;3(2):e10. doi: 10.1038/cti.2014.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rho MBWS, Glass JD, McArthur JC, Choi S, Griffin J, Tyor WR. A Potential Role for Interferon-α in the Pathogenesis of HIV-Associated Dementia. Brain Behav Immun. 1995;9:366–377. doi: 10.1006/brbi.1995.1034. [DOI] [PubMed] [Google Scholar]
- 37.Sas AR, Bimonte-Nelson H, Smothers CT, Woodward J, Tyor WR. Interferon-alpha causes neuronal dysfunction in encephalitis. J Neurosci. 2009;29(12):3948–3955. doi: 10.1523/JNEUROSCI.5595-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Okafor CN, Zhou Z, Burrell LE, 2nd, Kelso NE, Whitehead NE, Harman JS, et al. Marijuana use and viral suppression in persons receiving medical care for HIV-infection. Am J Drug Alcohol Abuse. 2017;43(1):103–110. doi: 10.1080/00952990.2016.1191505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ware MA, Rueda S, Singer J, Kilby D. Cannabis Use by Persons Living with HIV/AIDS: Patterns and Prevalence of Use. Journal of Cannabis Therapeutics. 2003;3(2):3–15. [Google Scholar]
- 40.Slawson G, Milloy MJ, Balneaves L, Simo A, Guillemi S, Hogg R, et al. High-intensity cannabis use and adherence to antiretroviral therapy among people who use illicit drugs in a Canadian setting. AIDS Behav. 2015;19(1):120–127. doi: 10.1007/s10461-014-0847-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Haney M, Gunderson EW, Rabkin J, Hart CL, Vosburg SK, Comer SD, et al. Dronabinol and marijuana in HIV-positive marijuana smokers: caloric intake, mood, and sleep. JAIDS Journal of Acquired Immune Deficiency Syndromes. 2007;45(5):545–554. doi: 10.1097/QAI.0b013e31811ed205. [DOI] [PubMed] [Google Scholar]
- 42.Abrams DI. Potential interventions for HIV/AIDS wasting: an overview. J Acquir Immune Defic Syndr. 2000;25(Suppl 1):S74–80. doi: 10.1097/00042560-200010001-00012. [DOI] [PubMed] [Google Scholar]
- 43.Cabral GA, Griffin-Thomas L. Emerging role of the cannabinoid receptor CB2 in immune regulation: therapeutic prospects for neuroinflammation. Expert Rev Mol Med. 2009;11:e3. doi: 10.1017/S1462399409000957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Karmaus PW, Chen W, Crawford R, Kaplan BL, Kaminski NE. Delta9-tetrahydrocannabinol impairs the inflammatory response to influenza infection: role of antigen-presenting cells and the cannabinoid receptors 1 and 2. Toxicol Sci. 2013;131(2):419–433. doi: 10.1093/toxsci/kfs315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roth MD, Castaneda JT, Kiertscher SM. Exposure to Delta9-Tetrahydrocannabinol Impairs the Differentiation of Human Monocyte-derived Dendritic Cells and their Capacity for T cell Activation. J Neuroimmune Pharmacol. 2015;10(2):333–343. doi: 10.1007/s11481-015-9587-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rappaport J, Volsky DJ. Role of the macrophage in HIV-associated neurocognitive disorders and other comorbidities in patients on effective antiretroviral treatment. J Neurovirol. 2015;21(3):235–241. doi: 10.1007/s13365-015-0346-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Coccia EM, Del Russo N, Stellacci E, Testa U, Marziali G, Battistini A. STAT1 activation during monocyte to macrophage maturation: role of adhesion molecules. Int Immunol. 1999;11(7):1075–1083. doi: 10.1093/intimm/11.7.1075. [DOI] [PubMed] [Google Scholar]
- 48.Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol. 2014;14(1):36–49. doi: 10.1038/nri3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McPartland JM, Glass M, Pertwee RG. Meta-analysis of cannabinoid ligand binding affinity and receptor distribution: interspecies differences. Br J Pharmacol. 2007;152(5):583–593. doi: 10.1038/sj.bjp.0707399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Younas M, Psomas C, Reynes J, Corbeau P. Immune activation in the course of HIV-1 infection: Causes, phenotypes and persistence under therapy. HIV Med. 2016;17(2):89–105. doi: 10.1111/hiv.12310. [DOI] [PubMed] [Google Scholar]
- 51.Jiang W, Lederman MM, Hunt P, Sieg SF, Haley K, Rodriguez B, et al. Plasma levels of bacterial DNA correlate with immune activation and the magnitude of immune restoration in persons with antiretroviral-treated HIV infection. J Infect Dis. 2009;199(8):1177–1185. doi: 10.1086/597476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lane BR, King SR, Bock PJ, Strieter RM, Coffey MJ, Markovitz DM. The C-X-C chemokine IP-10 stimulates HIV-1 replication. Virology. 2003;307(1):122–134. doi: 10.1016/s0042-6822(02)00045-4. [DOI] [PubMed] [Google Scholar]
- 53.Huestis MA. Human cannabinoid pharmacokinetics. Chem Biodivers. 2007;4(8):1770–1804. doi: 10.1002/cbdv.200790152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Croxford JL, Yamamura T. Cannabinoids and the immune system: potential for the treatment of inflammatory diseases? J Neuroimmunol. 2005;166(1–2):3–18. doi: 10.1016/j.jneuroim.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 55.Childs E, Lutz JA, de Wit H. Dose-related effects of delta-9-THC on emotional responses to acute psychosocial stress. Drug Alcohol Depend. 2017;177:136–144. doi: 10.1016/j.drugalcdep.2017.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Slavich GM, Irwin MR. From stress to inflammation and major depressive disorder: a social signal transduction theory of depression. Psychol Bull. 2014;140(3):774–815. doi: 10.1037/a0035302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.National Academies of Sciences E. Medicine The Health Effects of Cannabis and Cannabinoids: The Current State of Evidence and Recommendations for Research. Washington, DC: The National Academies Press; 2017. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.