

Abstract
Obsessive-compulsive disorder (OCD) is a leading cause of illness-related disability, but the neural mechanisms underlying OCD symptoms are unclear. One potential mechanism of OCD pathology is monoamine dysregulation. Because of the difficulty of studying monoamine signalling in patients, animal models offer a viable alternative to understanding this aspect of OCD pathophysiology. We used HPLC to characterize post-mortem monoamine levels in lateral orbitofrontal cortex (OFC), medial OFC, medial prefrontal cortex and dorsal and ventral striatum of SAPAP-3 knockout (KO) mice, a well-validated model of compulsive-like behaviours in OCD. As predicted from previous studies, excessive grooming was significantly increased in SAPAP-3 KO mice. Overall levels of the serotonin metabolite 5-hydroxyindoleacetic acid (HIAA) and the ratio of 5HIAA/serotonin (serotonin turnover) were increased in all cortical and striatal regions examined. In addition, dihydroxyphenylacetic acid/dopamine ratio was increased in lateral OFC, and HVA/dopamine ratio was increased in lateral and medial OFC. No baseline differences in serotonin or dopamine tissue content were observed. These data provide evidence of monoaminergic dysregulation in a translational model of OCD symptoms and are consistent with aberrant cortical and striatal serotonin and dopamine release/metabolism in SAPAP-3 KO mice. These results are guiding ongoing experiments using circuit and cell-type specific manipulations of dopamine and serotonin to determine the contributions of these monoaminergic systems to compulsive behaviours, and serve here as a touchstone for an expanded discussion of these techniques for precise circuit dissection.
This article is part of the discussion meeting issue ‘Of mice and mental health: facilitating dialogue between basic and clinical neuroscientists'.
Keywords: obsessive-compulsive disorder, orbitofrontal cortex, basal ganglia, monoamines, SAPAP3-KO, HPLC
1. Introduction
(a). Obsessive-compulsive disorder is a severe and chronic disorder with limited pharmacologic treatment options
Obsessive compulsive disorder (OCD) is a severe mental illness characterized by intrusive thoughts (obsessions) and repetitive acts (compulsions) [1,2]. These symptoms can be highly disruptive, leading to a significant impact on quality of life. In addition, the annual financial cost associated with OCD in the USA is estimated at $10 billion [3]. Accordingly, the World Health Organization has identified OCD as a major cause of illness-related disability, underscoring both the heavy burden this disorder places on patients and the steep cost to society at large [1,2,4]. Globally, OCD has a lifetime prevalence of 2–3% (i.e. two to three times more common than schizophrenia) [5,6]. Since illness onset often occurs during childhood or adolescence, and symptoms continue throughout life, this chronic disorder leads to significant morbidity [3]. OCD is therefore not only a devastating psychiatric illness, it is also extremely widespread.
Despite the high prevalence and severity of OCD, the underlying pathophysiologic mechanisms are still unclear. This lack of understanding of the neural basis of pathology limits the development of effective treatments. Current estimates indicate that approximately half of all patients have only partial responses to available pharmacologic treatments, and up to 10% are treatment resistant, meaning even our best treatments leave many patients without relief. Thus, in order to improve treatment of OCD, we need further research to identify the neural substrates of obsessions and compulsions.
In this manuscript, we use original research to highlight basic monoaminergic disruptions in a genetic knockout (KO) mouse model relevant to OCD. We then expand on how nuanced techniques for controlling and monitoring cellular activity could further elucidate the role of these monoaminergic abnormalities in future experiments. Thus, we provide a traditional, static view of dysregulation in the context of an OCD-related model, followed by a look towards cutting-edge approaches that will need to be implemented to expand our knowledge of neuronal circuit abnormalities relevant to OCD-related symptoms.
(b). Evidence from clinical obsessive-compulsive disorder studies suggests dysfunction in monoaminergic systems
While substantial research has focused on the role of cortico-striatal-thalamo-cortical circuits in OCD [1,2,7,8], particularly emphasizing the orbitofrontal cortex (OFC) and striatum, monoamine dysfunction served as a major focus of early human studies of OCD pathophysiology [9–19]. For over 30 years, it has been hypothesized that serotonergic dysfunction is central to OCD pathophysiology [18]. This hypothesis was initially grounded in the finding that serotonin reuptake inhibitors (SRIs), including both clomipramine and selective SRIs, were efficacious in treating a subset of OCD patients [17–19]. Furthermore, symptom exacerbation generally occurs when targeting receptors that negatively regulate presynaptic serotonin release [20,21], suggesting that deficient serotonin signalling contributes to OCD. In addition, associations of serotonin transporter gene polymorphisms with OCD [22–28] have led to speculation that OCD is associated with a hyposerotonergic state.
However, many inconsistencies have made it difficult to isolate specific abnormalities within the serotonergic system. For example, OCD patients and healthy controls show similar levels of plasma serotonin and cerebrospinal serotonin and metabolites [29–31], but recent work indicates that plasma serotonin concentration is positively correlated with OCD symptom reduction [30]. In addition, agonists can reduce or exacerbate OCD symptoms depending on the drug's receptor subtype affinity [28,31]. Because these approaches are unable to detect localized serotonergic disruptions in specific brain regions, they are unable to detect selective dysregulation of specific cortical and striatal serotonin inputs.
Similar challenges have been encountered during attempts to identify OCD-related pathologic changes in the dopaminergic system. Dopaminergic receptor antagonist augmentation of SRIs is efficacious in a subset of subjects with OCD [10,13,32–34]. Coupled with findings of reduced dopamine D1 and D2 receptor binding in the striatum [12,13,35,36] and exacerbation of OCD symptoms by indirect dopamine receptor agonists (cocaine, amphetamine, methylphenidate) [10], these clinical data would be consistent with a potential hyperdopaminergic state in people with OCD. However, these findings have not been consistently replicated [2,10], making the role of dopamine signalling in OCD unclear.
(c). Animal models are a powerful tool for investigating obsessive-compulsive disorder pathophysiology
The need for an improved understanding of the role of monoamines in the development, expression and treatment of OCD symptoms is clear. Although human studies are crucial for these efforts, they have inherent limitations due to their non-invasive nature. Animal studies of monoaminergic signalling are therefore essential to complement the human OCD literature by directly addressing questions of mechanism. One of the best currently available OCD-relevant genetic animal models is the SAPAP-3 KO mouse. SAPAP-3 is a post-synaptic density protein found at excitatory synapses, and is highly enriched in striatum relative to other SAPAP gene family members [37–40]. SAPAP-3 has been linked to OCD and related disorders including trichotillomania and skin picking via genetic studies in humans [41,42], strengthening its translational relevance. In addition to face validity (compulsive-like grooming behaviour accompanied by anxiety), SAPAP3-KOs have predictive validity, since chronic fluoxetine treatment ameliorates these behaviours [43]. Furthermore, SAPAP-3 KOs also show increased in vivo baseline striatal activity [44] and decreased fEPSPs in dorsolateral striatum slices [43], providing a potential link to human OCD functional imaging studies. This well-validated OCD-relevant animal model is now being used by numerous groups to investigate the neural mechanisms underlying compulsive-like behaviours [43–45].
Despite the growing use of SAPAP-3 KOs for translational OCD research, monoamine levels and turnover have not yet been examined in this model. To determine whether SAPAP3-KOs have monoaminergic abnormalities in prefrontal cortical and striatal subregions that have been consistently linked to OCD symptoms [1,8] and OCD-related phenotypes in animal models [2,43–47], we used HPLC to characterize dopamine, serotonin and their metabolites in post-mortem OFC (medial and lateral), mPFC, dorsal striatum (DS) and ventral striatum (VS) of SAPAP-3 KO mice and wild-type (WT) controls. This strategy allowed us to determine if global abnormalities in monoamine function are present in SAPAP-3 KOs, an important first step in investigating cortical and striatal neurochemical abnormalities in this OCD model.
2. Methods
(a). SAPAP3-knockout mice
All animal care and testing was approved by the University of Pittsburgh Institutional Animal Care and Use Committee and was in accordance with the National Institutes of Health's Guide for the Care and Use of Laboratory Animals. Data were collected from experimentally naive male and female homozygous SAPAP-3 KO (SAPAP-3−/−; n = 9 females and 8 males) and WT littermate control (SAPAP-3 +/+; n = 13 females and 8 males) mice on a C57BL/6 J background. Original SAPAP-3 KO breeders backcrossed for at least 10 generations were obtained from Dr Guoping Feng (Massachusetts Institute of Technology, Cambridge, MA). Breeding was then performed via heterozygote crosses (SAPAP3+/− × SAPAP3+/−), resulting in experimental groups comprised SAPAP3−/− mice and their WT littermate controls. All mice were group housed on 12 h light-dark schedule (lights on 7.00). Food (LabDiet 5001, LabDiet, St Louis, MO) and water were available ad libitum. Average age at tissue collection was approximately 7 months (207 ± 46 days).
(b). Grooming assessments
All testing took place in the same room during the light cycle. In each of two behavioural sessions (7 and 3 days prior to tissue collection), animals were allowed to acclimate to the testing room for 1 h in home cages. Each animal was then placed in one of eight clear acrylic chambers (20.32 × 20.32 × 30.48 cm, Collecting Warehouse, Fairfield, OH), which were cleaned with disinfectant spray (Virox, Oakville, ON, Canada) just prior to testing. Laminated white paper barriers were placed around the back and sides of the chambers to prevent mice from seeing each other. A video camera was placed in front of each chamber to capture behaviour for offline analysis. Collection of behavioural data was performed by an experimenter blinded to genotype. This procedure was repeated a second time for comparison with naive behaviour.
Video data were analysed offline by a pair of highly trained raters blinded to experimental conditions. Thirty minute videos were scored in 5 min bins for time spent grooming (defined as clearly visible cleaning or smoothing of the skin and fur with the mouth, front paws or hind paws). Each rater scored videos independently and without knowledge of the other rater's scores to ensure behavioural assessment was robust to errors in subjective ratings. Reported data are averaged between assessments and raters, except where otherwise noted.
(i). Tissue acquisition
Animals were sacrificed 3 days after grooming assessments during the light cycle using cervical dislocation, and brains were rapidly removed for slicing in ice-cold phosphate buffered saline. 0.7 mm thick coronal sections were cut at +2.77 mm, +1.97 mm and +1.21 mm anterior to Bregma [48]. Each section was then rapidly frozen on dry ice. 1.0 mm diameter tissue punches were then obtained from both hemispheres of lateral orbitofrontal cortex (lOFC) and medial orbitofrontal cortex (mOFC) (figure 1). A 1.75 mm diameter punch was taken from the midline of a medial prefrontal cortex (mPFC) section containing the prelimbic and infralimbic mPFC (figure 1). 1.0 mm diameter punches were obtained from both hemispheres of VS and DS. The DS section contained both dorsolateral and dorsomedial striatal subregions (figure 1). For a single subject, sections from both hemispheres were pooled together in a tube, and stored at −80°C prior to HPLC analysis.
Figure1.

Schematic diagram showing location of tissue punches for HPLC. Schematized depictions of tissue collection. In each coronal section, dashed circles depict the approximate location of tissue punches in post-mortem tissue. Sections are depicted anterior to posterior from left to right. (AP: +2.77 mm; +1.97 mm; +1.21 mm) For simplicity, lOFC, mOFC, VS and DS samples are depicted unilaterally, but all tissue was collected bilaterally. Note that there is minimal collection of non-targeted regions using this approach. (Online version in colour.)
(ii). Neurochemical detection via HPLC
All neurochemical assays were conducted at the University of Vanderbilt Neurochemistry Core (Biogenic Amines Assay); samples were shipped on dry ice. Brain sections were homogenized using a tissue dismembrator in 100–750 µl 0.1 M TCA (10–2 M sodium acetate, 10–4 M EDTA, 5 ng ml−1 isoproterenol (as internal standard); 10.5% methanol (pH 3.8)). Ten microlitres of homogenate were used for each protein assay. Samples were spun in a microcentrifuge at 10 000 g for 20 min and supernatant was removed for biogenic monoamine analysis with HPLC. Remaining samples were stored at –80°C for future use.
Biogenic amines were determined by a specific HPLC assay using an Antec Decade II (oxidation: 0.65) electrochemical detector operated at 33°C. 20 µl samples of the supernatant were injected using a Water 2707 autosampler onto a Phenomenex Kintex (2.6u, 100A) C18 HPLC column (100 × 4.60 mm). Biogenic amines were eluted with a mobile phase consisting of 89.5% 0.1 M TCA, 10–2 M sodium acetate, 10–4 M EDTA and 10.5% methanol (pH 3.8). Solvent was delivered at 0.6 ml min−1 using a Waters 515 HPLC pump. Using this HPLC solvent, biogenic amines elute in the following order: noradrenaline, adrenaline, 3,4-dihydroxyphenylacetic acid (DOPAC), dopamine, 5-hydroxyindoleacetic acid (5HIAA), homovanillic acid (HVA), serotonin 5-HT and 3-methoxytyramine (3-MT). HPLC control and data acquisition were managed by Empower software.
Protein concentration was determined by BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA). 10 µl of tissue homogenate from each sample was placed into a 96-well plate; 200 µl of mixed BCA reagent (25 ml of Protein Reagent A mixed with 500 µl of Protein Reagent B) was then added. The plate was incubated at room temperature for 2 h for colour development while a BSA standard curve was run. Absorbance was measured on a plate reader (POLARstar Omega; BMG LABTECH, Ortenberg, Germany).
(iii). Data analysis
All data analysis was conducted with custom Matlab scripts (ver. 2016a, MathWorks, Natick, MA) and SPSS statistical analysis software (IBM, Armonk, NY). Each animal's grooming data were averaged across both raters' assessments of both test sessions. Thus, each animal's grooming was an average of four ratings to minimize bias between raters and variability between test sessions. Time spent grooming was not normally distributed and non-parametric statistics were used as appropriate. Effects of sex and genotype on total grooming were assessed via a pair of Mann–Whitney tests with Bonferroni corrections for multiple comparisons. Data are reported as corrected p-values. Genotype effects on time spent grooming were also analysed for males and females separately using a Mann–Whitney procedure. Inter-rater reliability was calculated via the intraclass correlation (single measures; absolute agreement, one-way random raters model), and differences between rater's scores were assessed via Wilcox signed-rank test. Consistency across days within a rater was assessed with Spearman's correlation of each animal's total time grooming in assessment 1 and 2.
Differences in neurochemical content were assessed via analysis of variance (ANOVA) with sex, genotype and brain region as fixed factors (2 × 2 × 5 ANOVA). Main effects of brain region are not of central interest, as effects related to genotype were the main focus of the current study. For a given neurochemical, significant interactions between region and genotype were analysed with Bonferroni corrected post hoc independent samples t-tests on each region's data from SAPAP-3 KO and WT mice. All reported post hoc p-values were Bonferroni corrected.
We calculated the Spearman's rank correlation between neurochemical levels and grooming for samples that were significantly different according to genotype, pooling WT and SAPAP3−/− animals. All resulting p-values for this exploratory analysis are reported prior to Bonferroni correction.
3. Results
(a). SAPAP-3 knockouts demonstrate excessive grooming
Consistent with previous reports [43,44], time spent grooming was increased in SAPAP-3 KOs relative to WT controls (figure 2; U = 56, Z = −3.596, p < 0.001). These effects were similar when male (U = 2, Z = −3.151, p = 0.004) and female (U = 28, Z = −2.037, p = 0.042) animals were considered separately, and there was no effect of sex on time spent grooming (U = 130, Z = −1.360, p = 0.348). Total time grooming was highly correlated between both raters (intraclass correlation ICC37,38 = 0.994, p < 0.001), and grooming values did not significantly differ between them (Wilcox signed-rank test, Z1 = 1.617, signed rank = 482, p = 0.106, median rater 1: 300.40, median rater 2 = 284.43). For both raters, each animal's total time grooming was highly correlated across days (rater 1, r = 0.784, p < 0.001; rater 2, r = 0.823, p < 0.001).
Figure 2.
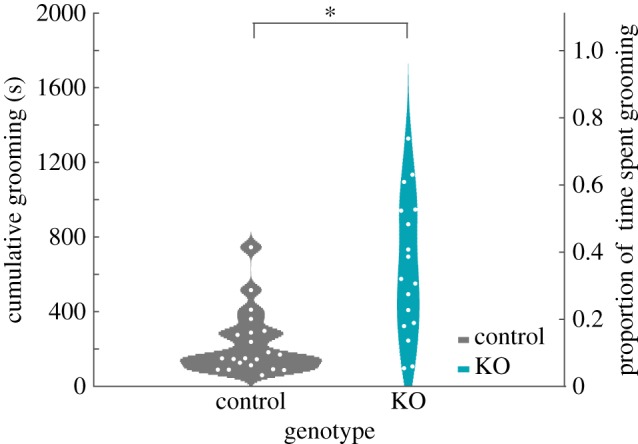
SAPAP-3 KOs display significantly increased grooming compared with WT mice. SAPAP-3 KO animals demonstrate increased grooming compared with WT littermate controls (Z = −3.596, p < 0.001). Data are depicted as violin plots of the cumulative time spent grooming (left Y axis) and proportion of time spent grooming (right Y axis) over a 30 min observation period. Each histogram represents the smoothed distribution of data points at each location on the Y axis. White dots represent each animal's grooming data plotted individually. Note that data are distributed non-normally. (Online version in colour.)
(b). Tissue samples
In order to assess post-mortem differences in monoamine neurochemistry between SAPAP-3 KO and WT mice, we collected bilateral lOFC, mOFC, mPFC, DS and VS from males and females of both genotypes. Total weight of protein recovered from each brain region did not differ according to sex or genotype (region, F4,169 = 36.068, p < 0.001; genotype, F1,169 = 0.163, p = 0.687; sex, F1,169 = 0.023, p = 0.879; all interactions non-significant), suggesting no gross differences in tissue quality or preparation between SAPAP-3 KO versus WT and male versus female.
(c). SAPAP-3 knockouts display broadly increased serotonin turnover in cortical and striatal regions
We observed that post-mortem basal 5-HT levels did not differ between SAPAP-3 KO and WT mice (figure 3; region, F4,169 = 20.179, p < 0.001; genotype, F1,169 = 2.103, p = 0.149; sex, F1,169 = 0.392, p = 0.532; all interactions non-significant). However, levels of the 5-HT metabolite, 5HIAA, were increased in SAPAP-3 KOs (figure 3; region, F4,169 = 16.173, p < 0.001; genotype, F1,169 = 37.591, p < 0.001; sex, F1,169 = 3.370, p = 0.68; all interactions non-significant), indicating dysregulation of 5-HT that is not packaged in vesicles and therefore is not shielded from catabolism by monoamine oxidase (MAO). The ratio of 5HIAA to 5-HT, commonly referred to as serotonin turnover, partially reflects serotonergic transmission and metabolism [49]. Using this measure, we detected increased serotonin turnover in SAPAP-3 KOs versus WT (figure 3; region, F4,168 = 11.993, p < 0.001; genotype, F1,168 = 14.304, p < 0.001; sex, F1,168 = 3.536, p = 0.62; all interactions non-significant).
Figure 3.
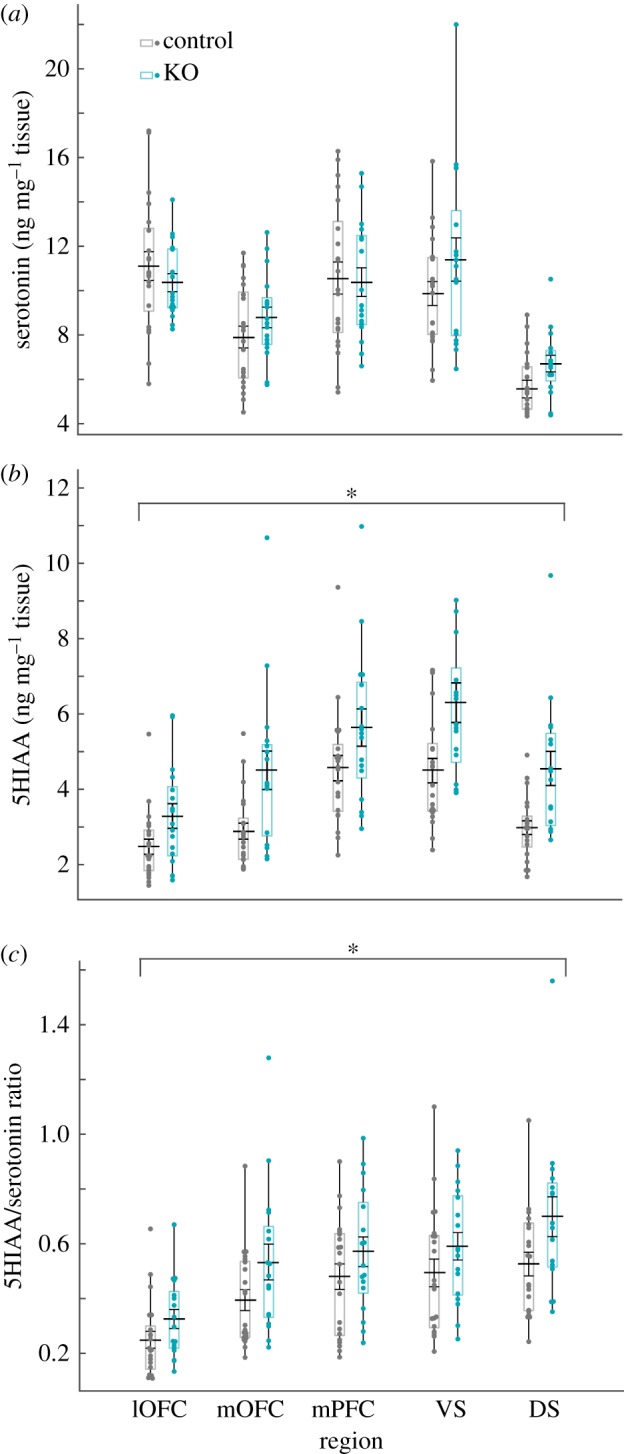
SAPAP-3 KOs demonstrate increased serotonin turnover in cortical and striatal regions. SAPAP-3 KO mice have elevated post-mortem 5HIAA and 5HIAA/serotonin ratios in frontal cortex and striatum. (a) Data depicted as box plot of ng of serotonin per mg of tissue in each region. Each box plot represents inner quartiles (25–75%) of distribution (bars indicate 9% and 91% of distribution). Black line and error bars inside box plots represent the mean ± s.e. Each data point is plotted in each distribution. (b) 5HIAA (serotonin metabolite) per mg of tissue in each region. Data plotted identically to (a). Main effect of genotype indicates that 5HIAA is increased in KO mice (F1,169 = 37.591, *p < 0.001). (c) The ratio of 5HIAA/serotonin, representing metabolized serotonin relative to serotonin content, is increased in SAPAP-3 KO mice (F1,168 = 14.304, *p < 0.001); this is potentially indicative of increased serotonin release and metabolism. Note different scales on Y axes are used throughout figure 3. (Online version in colour.)
(d). SAPAP-3 knockouts display dopaminergic abnormalities in orbitofrontal cortex
There were no differences in post-mortem levels of dopamine (figure 4; region, F4,169 = 503.545, p < 0.001; genotype, F1,169 = 1.046, p < 0.308; sex, F1,169 = 0.261, p = 0.610; all interactions non-significant) or dopamine metabolites DOPAC and 3-MT between SAPAP-3 KO and WT mice in any region (figure 4; DOPAC, region, F4,169 = 89.528, p < 0.001; genotype, F1,169 = 0.107, p = 0.744; sex, F1,169 = 0.592, p = 0.443; all interactions non-significant; 3-MT, region, F4,169 = 341.641, p < 0.001; genotype, F1,169 = 0.189, p = 0.664; sex, F1,169 = 0.848, p = 0.359; all interactions non-significant) (figure 5).
Figure 4.
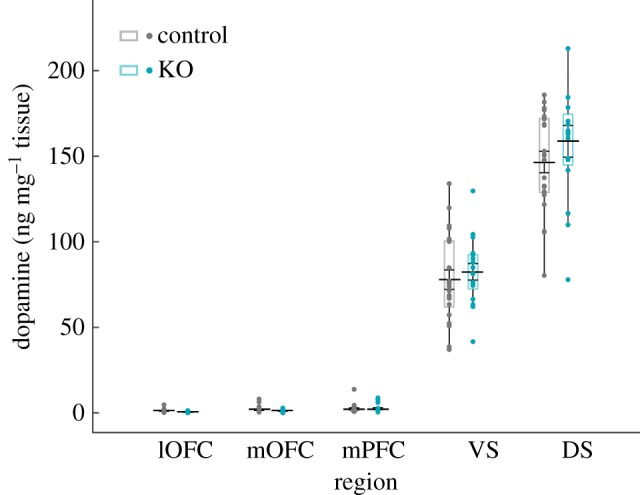
No differences in cortex or striatum dopamine levels in SAPAP-KOs. No significant differences are observed in post-mortem dopamine levels in frontal cortex and striatum between SAPAP-3 KO mice and littermate controls (genotype, F1,169 = 1.046, p = 0.308). Data depict ng of dopamine per mg of tissue in each region, and are plotted identically to figure 3. (Online version in colour.)
Figure 5.
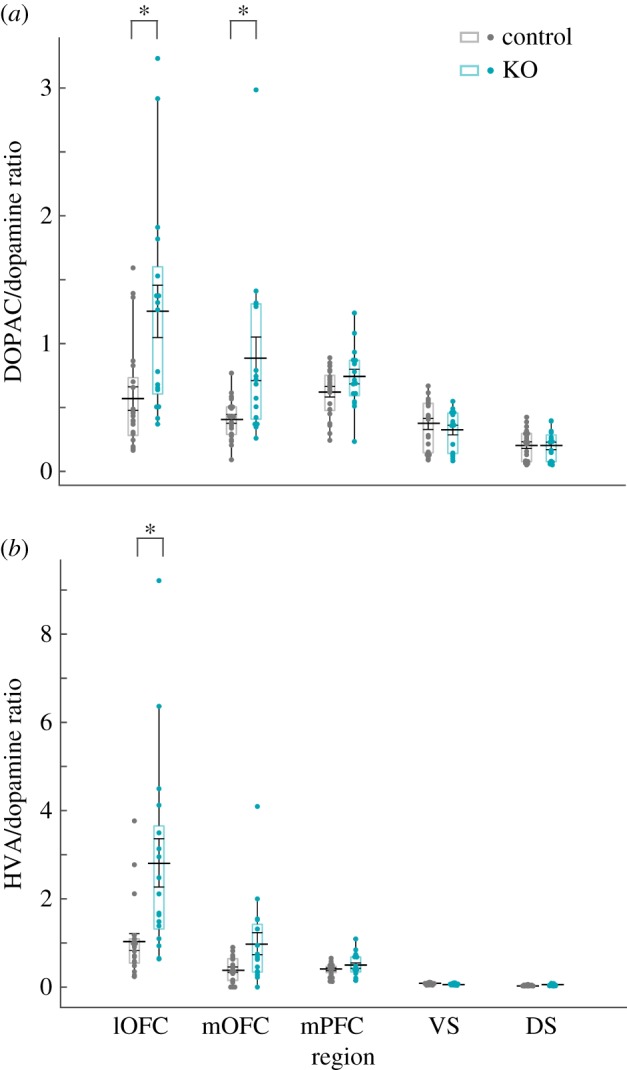
SAPAP-3 KO mice have elevated post-mortem metabolized dopamine ratios in OFC. (a) Data depict the ratio of the dopamine metabolite DOPAC to dopamine, and are plotted identically to figure 3. Interaction between genotype and region (F4,168 = 6.334, p < 0.001) that resulted in significantly greater ratios in both OFC regions in SAPAP-3 KOs (lOFC, t36 = −3.243, p = 0.015; mOFC, t35 = −3.061, p = 0.02). Asterisks represent within-region significant differences in DOPAC/dopamine ratio. (b) Data depict the ratio of HVA to dopamine, plotted identically to figure 2. A significant interaction between genotype and region (F4,168 = 8.207, p < 0.001) was due to increased HVA/dopamine ratios in lOFC of SAPAP-3 KOs (lOFC, t36 = −3.353, p = 0.01). Note different scales on Y axis are used throughout figure 5. (Online version in colour.)
Both DOPAC and 3-MT are precursors to further metabolic conversion into HVA by either catechol-O-methyl transferase or MAO, respectively. Therefore, we also examined HVA levels in all brain regions as a marker of dopaminergic release and metabolism [50,51]. HVA was weakly, but significantly, increased in SAPAP-3 KO versus WT mice (region, F4,169 = 198.169, p < 0.001; genotype, F1,169 = 5.012, p = 0.026; sex, F1,169 = 0.107, p = 0.744; all interactions non-significant). These results indicate there are modest differences in overall dopaminergic tone in post-mortem SAPAP-3 KO tissue.
There was a significant interaction between genotype and brain region on DOPAC relative to dopamine (region, F4,168 = 18.667, p < 0.001; genotype, F1,168 = 18.749, p < 0.001; sex, F1,168 = 1.897, p = 0.170; Genotype*Region, F4,168 = 6.334, p < 0.001). Post hoc analyses revealed that both lOFC and mOFC were associated with increased DOPAC turnover in SAPAP-3 KOs (lOFC, t36 = −3.243, p = 0.015; mOFC, t35 = −3.061, p = 0.02; VS, DS, and MPFC non-significant). Similarly, there was a significant interaction between genotype and brain region on HVA relative to dopamine (F4,168 = 32.217, p < 0.001; genotype, F1,168 = 16.277, p < 0.001; sex, F1,168 = 0.055, p = 0.815; Genotype*Region, F4,168 = 8.207, p < 0.001). Post hoc analyses indicated that lOFC HVA/dopamine was significantly greater in SAPAP-3 KO lOFC compared with WT mice (lOFC, t36 = −3.353, p = 0.01; mOFC, VS, DS, and mPFC: non-significant).
(e). Relationship between grooming levels and monoaminergic abnormalities in SAPAP3-knockouts
As a first attempt to elucidate the relationship between neurochemical alterations and behavioural abnormalities in SAPAP3-KOs, we performed an exploratory analysis to correlate grooming with each HPLC sample that differed significantly according to genotype. No neurochemical was significantly correlated with grooming following corrections for multiple comparisons (table 1). However, 5HIAA levels were correlated with grooming in all regions except mPFC prior to correction (table 1). Note that p-values displayed in table 1 are uncorrected for multiple comparisons.
Table 1.
Correlation between monoamines and grooming. Table displays the Spearman's rank order correlation between monoamine neurochemical levels and total time grooming in SAPAP-3 KO and WT mice. The p-values displayed are uncorrected. None of the correlations were significant following Bonferroni correction.
| lOFC | mOFC | mPFC | VS | DS | |
|---|---|---|---|---|---|
| 5HIAA |
r = 0.406 p = 0.011 |
r = 0.441 p = 0.006 |
r = 0.270 p = 0.101 |
r = 0.369 p = 0.023 |
r = 0.392 p = 0.017 |
| 5HIAA/5-HT |
r = 0.335 p = 0.040 |
r = 0.263 p = 0.111 |
r = 0.161 p = 0.334 |
r = 0.353 p = 0.030 |
r = 0.332 p = 0.048 |
| HVA |
r = 0.240 p = 0.146 |
r = 0.254 p = 0.124 |
r = 0.346 p = 0.033 |
r = 0.164 p = 0.324 |
r = 0.174 p = 0.303 |
| HVA/DA |
r = 0.352 p = 0.030 |
r = 0.012 p = 0.944 |
r = −0.135 p = 0.418 |
r = −0.160 p = 0.338 |
r = 0.009 p = 0.958 |
| DOPAC/DA |
r = 0.312 p = 0.056 |
r = 0.174 p = 0.304 |
r = −0.028 p = 0.867 |
r = −0.047 p = 0.78 |
r = −0.123 p = 0.469 |
4. Discussion
As expected based on previous studies, SAPAP-3 KO mice displayed excessive grooming [43]. In the same animals, we found evidence of serotonergic dysregulation (5HIAA and 5HIAA/serotonin ratio) in mOFC, lOFC, mPFC and dorsal and VS. We also observed evidence of dopamine abnormalities in OFC. Specifically, the ratio of HVA/dopamine was increased in lateral and medial OFC, and the ratio of DOPAC/dopamine was increased in lateral OFC. These findings are suggestive of increased cortical and striatal monoamine release in SAPAP-3 KO mice. They also align with a larger literature associating OCD with aberrant monoamine signalling [2,10,15,16,18] and documenting dysregulated neural activity in frontal cortex and striatum of both OCD patients and several animal models relevant to OCD [19,52–54].
(a). Serotonergic disruptions in SAPAP-3 knockout mice
These data are the first evidence of serotonergic abnormalities in SAPAP-3 KO mice, a validated animal model of compulsive-like behaviours [43,47,55]. To date most studies using SAPAP-3 KOs have focused on cortical and striatal systems [43–45,56]; our results therefore set the stage for more expansive investigation of the monoaminergic mechanisms underlying compulsive-like behaviours. Though SAPAP-3 is enriched in striatal regions relative to other SAPAP gene family members, it is also present in other brain regions [43,57]. Constitutive knock out of SAPAP-3 could therefore potentially impact monoaminergic signalling via multi-synaptic circuits either including or bypassing the striatum. For example, although this hypothesis has not yet been tested, SAPAP-3 deletion could generate dysregulated activity throughout cortico-striatal-thalamo-cortical circuits [58–61], leading to hyperactivity in descending projections from cortex to serotonin neurons in the raphe [62,63]. In turn, this could ultimately cause increased serotonin release in cortical and striatal regions. Further work is needed to directly elucidate how SAPAP-3 genetic deletion affects monoaminergic neural activity.
Our data suggest that serotonin release and metabolism are increased in cortex and striatum of SAPAP-3 KO mice. These preclinical data may seem somewhat at odds with some clinical data. For instance, the utility of SRIs in treating OCD symptoms [19,64], decreased serotonin transporter availability [28,31,65–69], and associations of polymorphisms of the genes encoding serotonin transporter [22–28] and serotonin 2A receptor [23] proteins with OCD have led to speculation that OCD is associated with a hypo-serotonergic state. However, it is important to note that the serotonin hypothesis has mixed support [1]. For instance, though the majority of the available clinical data suggest that OCD is associated with hypo-serotonergia, acute doses of metachlorophenylpiperazine (a serotonergic agonist) increase symptoms in OCD patients [70]. These data led to speculation that chronic treatment with SRIs induces adaptive hyposerotonergic states [71]. Further supporting the notion that OCD symptoms can be driven by increased serotonin release, SRI administration in OCD patients is correlated with decreased 5HIAA in cerebrospinal fluid [72] and decreased platelet serotonin concentrations [73].
There are several important considerations for interpreting the current data and comparing them to clinical observations. First, HPLC measurements from post-mortem tissue combine intracellular and extracellular serotonin, leading to challenges in making direct comparisons to human studies monitoring extracellular peripheral levels of neurotransmitter. HPLC also directly measures neurochemical levels, without inferring function through genetic expression or transporter availability. Second, increased serotonin turnover in SAPAP3-KOs could result from increased serotonin metabolism without changes in release. Third, assessment of these neurochemicals is often done in conjunction with a manipulation of neuronal activity (e.g. a drug or stimulation of a neuronal system), and it may therefore be more difficult to interpret the basal neurochemical levels obtained in our study. Fourth, it is unclear if these neurochemical abnormalities disrupt brain or behaviour function, and they could instead represent a compensatory process. This could be consistent with the absence of correlations between post-mortem monoamine content and the observed grooming phenotype that survived multiple comparisons. Finally, because mice cannot model all aspects of a human psychiatric disorder, it is possible that the SAPAP3-KO model does not accurately reflect serotonergic pathology in OCD patients. In vivo studies in SAPAP3-KOs examining the relationship between monoaminergic signalling and behaviour will yield more insight into these possibilities.
(b). Evidence of dopaminergic abnormalities in SAPAP-3 knockout orbitofrontal cortex
Our data are consistent with dysregulation of dopamine specifically in OFC of SAPAP3-KO mice, lending further support to an extensive literature associating abnormal dopamine transmission with dysregulated behavioural organization. For example, systemic administration of D1-receptor agonists or constitutive knockdown of the dopamine transporter can lead to compulsive-like grooming behaviour [74–77]. In addition, compulsive-like checking behaviours can be induced by D2-receptor agonists [78,79]. Our observation of increased dopamine turnover in OFC could contribute to compulsive-like behaviours in several ways. For example, OFC is essential for performing reward value comparisons and stimulus-outcome associations [80–85]. Dysregulated dopamine signalling in OFC could therefore lead to deficits in comparison of actual and expected rewards [86,87], ultimately leading to repeated perseverative performance of an action. In addition, OFC is strongly associated with behavioural flexibility in the context of reversal learning, and OCD patients have consistently demonstrated abnormal performance and/or neural activity on this task [52,88]. Thus, impaired dopamine signalling in OFC could impact the ability to learn new rules, contributing to cognitive inflexibility in OCD.
Although we selectively observed increased dopamine turnover in OFC, aberrant behavioural selection is less commonly associated with dopamine signalling within the OFC, and more commonly associated with striatal and mPFC dopamine signalling [86,89–92]. Our data may therefore point to a unique regional mechanism of monoaminergic dysfunction relevant to OCD. Of particular interest in this context is the fact that dopaminergic innervation of OFC is relatively weak compared with striatum and mPFC [93]; thus, modest abnormalities in OFC dopamine levels could represent a strong functional change in OFC. It will therefore be important to use in vivo approaches to precisely measure or control OFC dopamine signalling and examine the impact on performance of tasks impaired in OCD.
(c). Emerging technologies to further understand monoaminergic dysfunction in obsessive-compulsive disorder
More work is necessary to relate our findings to dynamic interactions between networks and resulting behaviours. For instance, monoaminergic abnormalities were not correlated with the compulsive grooming phenotype after corrections for multiple comparisons. However, this does not rule out a role for these changes in abnormal grooming, since a static and non-time-locked alteration in monoamine levels may not necessarily be directly related to a temporally fluctuating behavioural phenotype like grooming. The use of optogenetics to precisely control dopamine and serotonin signalling is an effective way to solve this problem, and this approach will be essential for elucidating the role of monoaminergic signalling in compulsive-like behaviour. However, it is also critical to develop strategies to optimize use of this technology by determining (1) when to optogenetically modulate these systems in relationship to behaviour, and (2) how to pattern optical stimulation, in order to elucidate the neural mechanisms that give rise to compulsive-like phenotypes.
Timing of light delivery: After over a decade of advances, neuroscientists are now poised to refine optogenetics to explore increasingly complex questions regarding the relationship between neural activity and behaviour. Behavioural state-dependent optogenetics is a nuanced approach to understanding when to optogenetically target monoaminergic activity relative to naturalistic behaviours. The strength of this technique was recently demonstrated in an important study from Costa and co-workers [94], in which the authors observed a complex relationship between movement initiation and execution in direct pathway dorsal striatum neurons [94]. This projection system is traditionally linked to movement initiation [60,95]. A clear prediction is that stimulating direct pathway neurons should therefore initiate and sustain behavioural sequences [96]. Stimulation of the direct pathway sustained ongoing behavioural sequences as predicted. However, stimulation also delayed action initiation when applied prior to sequence execution. The latter finding is contrary to theory and demonstrates that optogenetic effects of stimulation depended on the animal's ongoing behaviour. Simply stimulating without taking behavioural state into account would not have uncovered this important distinction. This demonstrates a more subtle relationship between action execution/movement and the direct pathway than previously theorized.
A similar approach has been used to reveal how the amygdala contributes to an opposing relationship between grooming and attack behaviours [97]. Specifically, optogenetic modulation of medial amygdala glutamatergic and GABAergic neurons during distinct behavioural states revealed that the circuits controlling self-grooming and attack behaviours are mutually antagonistic. These examples highlight the potential utility of state-dependent optogenetics to reveal complex relationships between compulsive-like grooming and monoaminergic activity that would be difficult to disentangle using standard optogenetic approaches.
State-dependent optogenetics can be further enhanced by ‘closed-loop’ approaches, which automatically detect ongoing neural activity or behaviours of interest to trigger optogenetic modulation [98–105]. Although this approach was first applied in an in vitro electrophysiological setting [100], more recent applications have focused on real-time in vivo modulation of seizure activity [98,101,102,105], memory consolidation [103,106] and somatosensation [99]. Building on this work, on-line detection of behavioural phenotypes, closed-loop optogenetics, and state-dependent modulations of neural activity will be important tools for elucidating the relationship between monoamine signalling and naturalistic compulsive-like phenotypes in SAPAP-3 KO mice.
Characteristics of light stimulation patterns: How optogenetic stimulation is patterned is another crucial parameter for optimization. While non-physiological modulation patterns have led to extremely important advances in neuroscience, it is clear that mimicking naturally occurring patterns of activity and determining how these patterns contribute to complex behaviours is an important next step. Step-function opsins offer one approach for studying how naturalistic patterns of activity impact behavioural phenotypes. These opsins create a stable step in membrane potential when activated by light, and therefore modulate excitability while preserving local circuit dynamics and the relative weight of excitatory and inhibitory inputs [107–111]. Thus, step-function opsins make targeted neurons more responsive to excitatory neurotransmitter release without directly causing neural firing. Frame-projected independent-fibre photometry is an alternative approach that allows naturally occurring levels of activity to be imaged and then optogenetically mimicked in the same population of neurons [112]. We propose that the use of these tools to naturalistically modulate the excitability of dopamine and serotonin neurons will be a powerful approach for elucidating the role of monoaminergic neurons in development and expression of compulsive-like phenotypes.
5. Conclusion
Our findings highlight disruptions in monoamine signalling in the OCD-relevant SAPAP3-KO mouse model, and serve as a starting point for future circuit-based investigation of the role of serotonin and dopamine signalling in compulsive-like behaviours. The current data suggest that dopamine signalling from ventral tegmental area to OFC may be a specific source of pathology in SAPAP-3 KO mice, and that serotonergic signalling may be impacted across cortical and striatal circuits, potentially pointing to global dysregulation in serotonergic tone. This study is therefore an important first step towards targeting distinct cortico-striatal circuits and monoamine systems with advanced optogenetic techniques to facilitate dynamic modulation. With this expanded technological toolbox and sophisticated behavioural paradigms, future studies will implement behaviour state-dependent and naturalistic optogenetic manipulations of dopaminergic and serotonergic neurons to assess how their dysfunction may play a role in compulsive-like behaviours.
Acknowledgements
The authors thank Victoria Corbit, Ruth K. Snyder and David Augustine for technical support and assistance with experiments, and Dr Alan Pehrson for helpful discussion of this manuscript. The authors also thank the Neurochemistry Core at Vanderbilt University for assistance with neurochemical analyses. We are grateful to the Royal Society for their support of the costs of attending the meeting ‘Of mice and mental health: facilitating dialogue between basic and clinical neuroscientists' convened by Amy Milton and Emily A. Holmes.
Data accessibility
Data available from the Dryad Digital Repository: doi:10.5061/dryad.h6qm7.
Authors' contributions
J.W. and S.E.A. designed experiments; J.W. collected and analysed data, and J.W., Z.F.L. and S.E.A. wrote the manuscript.
Competing interest
Dr Ahmari has no conflicts of interest to disclose.
Funding
Dr Wood: NIMH T32MH016804. Dr Ahmari: NIMH R01MH104255, Burroughs Wellcome Career Award for Medical Scientists.
References
- 1.Pauls DL, Abramovitch A, Rauch SL, Geller DA. 2014. Obsessive-compulsive disorder: an integrative genetic and neurobiological perspective. Nat. Rev. Neurosci. 15, 410–424. ( 10.1038/nrn3746) [DOI] [PubMed] [Google Scholar]
- 2.Wood J, Ahmari SE. 2015. A Framework for understanding the emerging role of corticolimbic-ventral striatal networks in OCD-associated repetitive behaviors. Front. Syst. Neurosci. 9, 171 ( 10.3389/fnsys.2015.00171) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rasmussen SA, Eisen JL. 1990. Epidemiology of obsessive compulsive disorder. J. Clin. Psychiatry 51 Suppl:10–3; discussion 4, 10–14. [PubMed] [Google Scholar]
- 4.Ahmari SE, Dougherty DD. 2015. Dissecting Ocd circuits: from animal models to targeted treatments. Depress. Anxiety 32, 550–562. ( 10.1002/da.22367) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Karno M, Golding JM, Sorenson SB, Burnam MA. 1988. The epidemiology of obsessive-compulsive disorder in five US communities. Arch. Gen. Psychiatry 45, 1094–1099. ( 10.1001/archpsyc.1988.01800360042006) [DOI] [PubMed] [Google Scholar]
- 6.Torres AR, et al. 2006. Obsessive-compulsive disorder: prevalence, comorbidity, impact, and help-seeking in the British national psychiatric morbidity survey of 2000. Am. J. Psychiatry 163, 1978–1985. ( 10.1176/ajp.2006.163.11.1978) [DOI] [PubMed] [Google Scholar]
- 7.Milad MR, Rauch SL. 2012. Obsessive-compulsive disorder: beyond segregated cortico-striatal pathways. Trends Cogn. Sci. 16, 43–51. ( 10.1016/j.tics.2011.11.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Graybiel AM, Rauch SL. 2000. Toward a neurobiology of obsessive-compulsive disorder. Neuron 28, 343–347. ( 10.1016/S0896-6273(00)00113-6) [DOI] [PubMed] [Google Scholar]
- 9.Denys D, et al. 2013. Dopaminergic activity in Tourette syndrome and obsessive-compulsive disorder. Eur. Neuropsychopharmacol. 23, 1423–1431. ( 10.1016/j.euroneuro.2013.05.012) [DOI] [PubMed] [Google Scholar]
- 10.Denys D, Zohar J, Westenberg HG. 2004. The role of dopamine in obsessive-compulsive disorder: preclinical and clinical evidence. J. Clin. Psychiatry 65(Suppl. 14), 11–17. [PubMed] [Google Scholar]
- 11.Denys D, Van Nieuwerburgh F, Deforce D, Westenberg H. 2006. Association between the dopamine D2 receptor TaqI A2 allele and low activity COMT allele with obsessive-compulsive disorder in males. Eur. Neuropsychopharmacol. 16, 446–450. ( 10.1016/j.euroneuro.2005.12.001) [DOI] [PubMed] [Google Scholar]
- 12.Denys D, van der Wee N, Janssen J, De Geus F, Westenberg HG. 2004. Low level of dopaminergic D2 receptor binding in obsessive-compulsive disorder. Biol. Psychiatry 55, 1041–1045. ( 10.1016/j.biopsych.2004.01.023) [DOI] [PubMed] [Google Scholar]
- 13.Klanker M, Feenstra M, Denys D. 2013. Dopaminergic control of cognitive flexibility in humans and animals. Front. Neurosci. 7, 201 ( 10.3389/fnins.2013.00201) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Figee M, et al. 2014. Deep brain stimulation induces striatal dopamine release in obsessive-compulsive disorder. Biol. Psychiatry 75, 647–652. ( 10.1016/j.biopsych.2013.06.021) [DOI] [PubMed] [Google Scholar]
- 15.Figee M, Pattij T, Willuhn I, Luigjes J, van den Brink W, Goudriaan A, Potenza MN, Robbins TW, Denys D. 2016. Compulsivity in obsessive-compulsive disorder and addictions. Eur. Neuropsychopharmacol. 26, 856–868. ( 10.1016/j.euroneuro.2015.12.003) [DOI] [PubMed] [Google Scholar]
- 16.Figee M, Vink M, de Geus F, Vulink N, Veltman DJ, Westenberg H, Denys D. 2011. Dysfunctional reward circuitry in obsessive-compulsive disorder. Biol. Psychiatry 69, 867–874. ( 10.1016/j.biopsych.2010.12.003) [DOI] [PubMed] [Google Scholar]
- 17.Insel TR. 1990. New pharmacologic approaches to obsessive compulsive disorder. J. Clin. Psychiatry 51 (Suppl. 47–51); discussion 6–8, 47–58. [DOI] [PubMed] [Google Scholar]
- 18.Insel TR, Mueller EA, Alterman I, Linnoila M, Murphy DL. 1985. Obsessive-compulsive disorder and serotonin: is there a connection? Biol. Psychiatry 20, 1174–1188. ( 10.1016/0006-3223(85)90176-3) [DOI] [PubMed] [Google Scholar]
- 19.Insel TR, Winslow JT. 1992. Neurobiology of obsessive compulsive disorder. Psychiatr. Clin. North Am. 15, 813–824. ( 10.1097/00004850-199206001-00008) [DOI] [PubMed] [Google Scholar]
- 20.Koran LM, Pallanti S, Quercioli L. 2001. Sumatriptan, 5-HT(1D) receptors and obsessive-compulsive disorder. Eur. Neuropsychopharmacol. 11, 169–172. ( 10.1016/S0924-977X(01)00082-7) [DOI] [PubMed] [Google Scholar]
- 21.Khanna S, John JP, Reddy LP. 2001. Neuroendocrine and behavioral responses to mCPP in obsessive-compulsive disorder. Psychoneuroendocrinology 26, 209–223. ( 10.1016/S0306-4530(00)00048-2) [DOI] [PubMed] [Google Scholar]
- 22.Murphy DL, Moya PR, Fox MA, Rubenstein LM, Wendland JR, Timpano KR. 2013. Anxiety and affective disorder comorbidity related to serotonin and other neurotransmitter systems: obsessive-compulsive disorder as an example of overlapping clinical and genetic heterogeneity. Phil. Trans. R. Soc. B 368, 20120435 ( 10.1098/rstb.2012.0435) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Taylor S. 2013. Molecular genetics of obsessive-compulsive disorder: a comprehensive meta-analysis of genetic association studies. Mol. Psychiatry 18, 799–805. ( 10.1038/mp.2012.76) [DOI] [PubMed] [Google Scholar]
- 24.Cengiz M, Okutan SN, Bayoglu B, Sakalli Kani A, Bayar R, Kocabasoglu N. 2015. Genetic polymorphism of the serotonin transporter gene, SLC6A4 rs16965628, is associated with obsessive compulsive disorder. Genet. Test. Mol. Biomarkers 19, 228–234. ( 10.1089/gtmb.2014.0319) [DOI] [PubMed] [Google Scholar]
- 25.Walitza S, Marinova Z, Grunblatt E, Lazic SE, Remschmidt H, Vloet TD, Wendland JR. 2014. Trio study and meta-analysis support the association of genetic variation at the serotonin transporter with early-onset obsessive-compulsive disorder. Neurosci. Lett. 580, 100–103. ( 10.1016/j.neulet.2014.07.038) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tukel R, Alkas E, Gurvit H, Aslantas Ertekin B, Ertekin E, Baran B, Kalem SA, Direskeneli GS. 2016. Serotonin transporter promoter polymorphism is associated with executive function impairments in patients with obsessive compulsive disorder. Clin. Neuropsychol. 30, 536–546. ( 10.1080/13854046.2016.1162329) [DOI] [PubMed] [Google Scholar]
- 27.Honda S, et al. 2017. A pilot study exploring the association of morphological changes with 5-HTTLPR polymorphism in OCD patients. Ann. Gen. Psychiatry 16, 2 ( 10.1186/s12991-017-0126-6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goddard AW, Shekhar A, Whiteman AF, McDougle CJ. 2008. Serotoninergic mechanisms in the treatment of obsessive-compulsive disorder. Drug Discov. Today 13, 325–332. ( 10.1016/j.drudis.2007.12.009) [DOI] [PubMed] [Google Scholar]
- 29.Baumgarten HG, Grozdanovic Z. 1998. Role of serotonin in obsessive-compulsive disorder. Br. J. Psychiatry Suppl. 35, 13–20. [PubMed] [Google Scholar]
- 30.Sampaio T, Lima C, Corregiari F, Bernik M. 2016. The putative catalytic role of higher serotonin bioavailability in the clinical response to exposure and response prevention in obsessive-compulsive disorder. Rev. Bras. Psiquiatr. 38, 287–293. ( 10.1590/1516-4446-2015-1721) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koo MS, Kim EJ, Roh D, Kim CH. 2010. Role of dopamine in the pathophysiology and treatment of obsessive-compulsive disorder. Expert Rev. Neurother. 10, 275–290. ( 10.1586/ern.09.148) [DOI] [PubMed] [Google Scholar]
- 32.Bloch MH, Landeros-Weisenberger A, Kelmendi B, Coric V, Bracken MB, Leckman JF. 2006. A systematic review: antipsychotic augmentation with treatment refractory obsessive-compulsive disorder. Mol. Psychiatry 11, 622–632. ( 10.1038/sj.mp.4001823) [DOI] [PubMed] [Google Scholar]
- 33.McDougle CJ, Epperson CN, Pelton GH, Wasylink S, Price LH. 2000. A double-blind, placebo-controlled study of risperidone addition in serotonin reuptake inhibitor-refractory obsessive-compulsive disorder. Arch. Gen. Psychiatry 57, 794–801. ( 10.1001/archpsyc.57.8.794) [DOI] [PubMed] [Google Scholar]
- 34.Denys D, de Geus F, van Megen HJ, Westenberg HG. 2004. A double-blind, randomized, placebo-controlled trial of quetiapine addition in patients with obsessive-compulsive disorder refractory to serotonin reuptake inhibitors. J. Clin. Psychiatry 65, 1040–1048. ( 10.4088/JCP.v65n0803) [DOI] [PubMed] [Google Scholar]
- 35.Hesse S, Muller U, Lincke T, Barthel H, Villmann T, Angermeyer MC, Sabri O, Stengler-Wenzke K. 2005. Serotonin and dopamine transporter imaging in patients with obsessive-compulsive disorder. Psychiatry Res. 140, 63–72. ( 10.1016/j.pscychresns.2005.07.002) [DOI] [PubMed] [Google Scholar]
- 36.Nikolaus S, Antke C, Beu M, Muller HW. 2010. Cortical GABA, striatal dopamine and midbrain serotonin as the key players in compulsive and anxiety disorders--results from in vivo imaging studies. Rev. Neurosci. 21, 119–139. ( 10.1515/REVNEURO.2010.21.2.119) [DOI] [PubMed] [Google Scholar]
- 37.Kim E, Naisbitt S, Hsueh YP, Rao A, Rothschild A, Craig AM, Sheng M. 1997. GKAP, a novel synaptic protein that interacts with the guanylate kinase-like domain of the PSD-95/SAP90 family of channel clustering molecules. J. Cell Biol. 136, 669–678. ( 10.1083/jcb.136.3.669) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Takeuchi M, Hata Y, Hirao K, Toyoda A, Irie M, Takai Y. 1997. SAPAPs. A family of PSD-95/SAP90-associated proteins localized at postsynaptic density. J. Biol. Chem. 272, 11 943–11 951. ( 10.1074/jbc.272.18.11943) [DOI] [PubMed] [Google Scholar]
- 39.Scannevin RH, Huganir RL. 2000. Postsynaptic organization and regulation of excitatory synapses. Nat. Rev. Neurosci. 1, 133–141. ( 10.1038/35039075) [DOI] [PubMed] [Google Scholar]
- 40.Kim E, Sheng M. 2004. PDZ domain proteins of synapses. Nat. Rev. Neurosci. 5, 771–781. ( 10.1038/nrn1517) [DOI] [PubMed] [Google Scholar]
- 41.Zuchner S, et al. 2009. Multiple rare SAPAP3 missense variants in trichotillomania and OCD. Mol. Psychiatry 14, 6–9. ( 10.1038/mp.2008.83) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bienvenu OJ, et al. 2009. Sapap3 and pathological grooming in humans: results from the OCD collaborative genetics study. Am. J. Med. Genet. B Neuropsychiatr. Genet. 150B, 710–720. ( 10.1002/ajmg.b.30897) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Welch JM, et al. 2007. Cortico-striatal synaptic defects and OCD-like behaviours in Sapap3-mutant mice. Nature 448, 894–900. ( 10.1038/nature06104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Burguiere E, Monteiro P, Feng G, Graybiel AM. 2013. Optogenetic stimulation of lateral orbitofronto-striatal pathway suppresses compulsive behaviors. Science 340, 1243–1246. ( 10.1126/science.1232380) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wan Y, Ade KK, Caffall Z, Ilcim Ozlu M, Eroglu C, Feng G, Calakos N. 2014. Circuit-selective striatal synaptic dysfunction in the Sapap3 knockout mouse model of obsessive-compulsive disorder. Biol. Psychiatry 75, 623–630. ( 10.1016/j.biopsych.2013.01.008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ahmari SE, Spellman T, Douglass NL, Kheirbek MA, Simpson HB, Deisseroth K, Gordon JA, Hen R. 2013. Repeated cortico-striatal stimulation generates persistent OCD-like behavior. Science 340, 1234–1239. ( 10.1126/science.1234733) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Burguiere E, Monteiro P, Mallet L, Feng G, Graybiel AM. 2015. Striatal circuits, habits, and implications for obsessive-compulsive disorder. Curr. Opin Neurobiol. 30, 59–65. ( 10.1016/j.conb.2014.08.008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Franklin KBJ, Paxinos G. 2013. Paxinos and franklin's The mouse brain in stereotaxic coordinates. 4th edn. Amsterdam, Netherlands: Academic Press, an imprint of Elsevier. 1 volume (unpaged) p. [Google Scholar]
- 49.Aghajanian GK, Rosecrans JA, Sheard MH. 1967. Serotonin: release in the forebrain by stimulation of midbrain raphe. Science 156, 402–403. ( 10.1126/science.156.3773.402) [DOI] [PubMed] [Google Scholar]
- 50.Roth RH, Murrin LC, Walters JR. 1976. Central dopaminergic neurons: effects of alterations in impulse flow on the accumulation of dihydroxyphenylacetic acid. Eur. J. Pharmacol. 36, 163–171. ( 10.1016/0014-2999(76)90268-5) [DOI] [PubMed] [Google Scholar]
- 51.Sharp T, Zetterstrom T, Ungerstedt U. 1986. An in vivo study of dopamine release and metabolism in rat brain regions using intracerebral dialysis. J. Neurochem. 47, 113–122. ( 10.1111/j.1471-4159.1986.tb02838.x) [DOI] [PubMed] [Google Scholar]
- 52.Remijnse PL, Nielen MM, van Balkom AJ, Cath DC, van Oppen P, Uylings HB, Veltman DJ. 2006. Reduced orbitofrontal-striatal activity on a reversal learning task in obsessive-compulsive disorder. Arch. Gen. Psychiatry 63, 1225–1236. ( 10.1001/archpsyc.63.11.1225) [DOI] [PubMed] [Google Scholar]
- 53.Chamberlain SR, et al. 2008. Orbitofrontal dysfunction in patients with obsessive-compulsive disorder and their unaffected relatives. Science 321, 421–422. ( 10.1126/science.1154433) [DOI] [PubMed] [Google Scholar]
- 54.Saxena S, Brody AL, Schwartz JM, Baxter LR. 1998. Neuroimaging and frontal-subcortical circuitry in obsessive-compulsive disorder. Br. J. Psychiatry Suppl. 1998, 26–37. [PubMed] [Google Scholar]
- 55.Albelda N, Joel D. 2012. Animal models of obsessive-compulsive disorder: exploring pharmacology and neural substrates. Neurosci. Biobehav. Rev. 36, 47–63. ( 10.1016/j.neubiorev.2011.04.006) [DOI] [PubMed] [Google Scholar]
- 56.Mahgoub M, Adachi M, Suzuki K, Liu X, Kavalali ET, Chahrour MH, Monteggia LM. 2016. MeCP2 and histone deacetylases 1 and 2 in dorsal striatum collectively suppress repetitive behaviors. Nat. Neurosci. 19, 1506–1512. ( 10.1038/nn.4395) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Welch JM, Wang D, Feng G. 2004. Differential mRNA expression and protein localization of the SAP90/PSD-95-associated proteins (SAPAPs) in the nervous system of the mouse. J. Comp. Neurol. 472, 24–39. ( 10.1002/cne.20060) [DOI] [PubMed] [Google Scholar]
- 58.Sesack SR, Grace AA. 2010. Cortico-basal ganglia reward network: microcircuitry. Neuropsychopharmacology 35, 27–47. ( 10.1038/npp.2009.93) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Crittenden JR, Graybiel AM. 2011. Basal Ganglia disorders associated with imbalances in the striatal striosome and matrix compartments. Front. Neuroanat. 5, 59 ( 10.3389/fnana.2011.00059) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Albin RL, Young AB, Penney JB. 1989. The functional anatomy of basal ganglia disorders. Trends Neurosci. 12, 366–375. ( 10.1016/0166-2236(89)90074-X) [DOI] [PubMed] [Google Scholar]
- 61.Graybiel AM. 2000. The basal ganglia. Curr. Biol. 10, R509–R511. ( 10.1016/S0960-9822(00)00593-5) [DOI] [PubMed] [Google Scholar]
- 62.Soiza-Reilly M, Commons KG. 2011. Glutamatergic drive of the dorsal raphe nucleus. J. Chem. Neuroanat. 41, 247–255. ( 10.1016/j.jchemneu.2011.04.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Commons KG, Beck SG, Bey VW. 2005. Two populations of glutamatergic axons in the rat dorsal raphe nucleus defined by the vesicular glutamate transporters 1 and 2. Eur. J. Neurosci. 21, 1577–1586. ( 10.1111/j.1460-9568.2005.03991.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McDougle CJ, Goodman WK, Leckman JF, Price LH. 1993. The psychopharmacology of obsessive compulsive disorder. Implications for treatment and pathogenesis. Psychiatr. Clin. North Am. 16, 749–766. [PubMed] [Google Scholar]
- 65.Zitterl W, et al. 2008. Changes in thalamus-hypothalamus serotonin transporter availability during clomipramine administration in patients with obsessive-compulsive disorder. Neuropsychopharmacology 33, 3126–3134. ( 10.1038/npp.2008.35) [DOI] [PubMed] [Google Scholar]
- 66.Zitterl W, et al. 2009. Diencephalic serotonin transporter availability predicts both transporter occupancy and treatment response to sertraline in obsessive-compulsive checkers. Biol. Psychiatry 66, 1115–1122. ( 10.1016/j.biopsych.2009.07.009) [DOI] [PubMed] [Google Scholar]
- 67.Hesse S, et al. 2011. The serotonin transporter availability in untreated early-onset and late-onset patients with obsessive-compulsive disorder. Int. J. Neuropsychopharmacol. 14, 606–617. ( 10.1017/S1461145710001604) [DOI] [PubMed] [Google Scholar]
- 68.Reimold M, et al. 2007. Reduced availability of serotonin transporters in obsessive-compulsive disorder correlates with symptom severity - a [11C]DASB PET study. J. Neural Transm. 114, 1603–1609. ( 10.1007/s00702-007-0785-6) [DOI] [PubMed] [Google Scholar]
- 69.Kim E, Howes OD, Park JW, Kim SN, Shin SA, Kim BH, Turkheimer FE, Lee Y-S, Kwon JS. 2016. Altered serotonin transporter binding potential in patients with obsessive-compulsive disorder under escitalopram treatment: [11C]DASB PET study. Psychol. Med. 46, 357–366. ( 10.1017/S0033291715001865) [DOI] [PubMed] [Google Scholar]
- 70.Zohar J, Mueller EA, Insel TR, Zohar-Kadouch RC, Murphy DL. 1987. Serotonergic responsivity in obsessive-compulsive disorder. Comparison of patients and healthy controls. Arch. Gen. Psychiatry 44, 946–951. ( 10.1001/archpsyc.1987.01800230026006) [DOI] [PubMed] [Google Scholar]
- 71.Zohar J, Insel TR, Zohar-Kadouch RC, Hill JL, Murphy DL. 1988. Serotonergic responsivity in obsessive-compulsive disorder. Effects of chronic clomipramine treatment. Arch. Gen. Psychiatry 45, 167–172. ( 10.1001/archpsyc.1988.01800260081011) [DOI] [PubMed] [Google Scholar]
- 72.Thoren P, Asberg M, Bertilsson L, Mellstrom B, Sjoqvist F, Traskman L. 1980. Clomipramine treatment of obsessive-compulsive disorder. II. Biochemical aspects. Arch. Gen. Psychiatry 37, 1289–1294. ( 10.1001/archpsyc.1980.01780240087010) [DOI] [PubMed] [Google Scholar]
- 73.Flament MF, Rapoport JL, Murphy DL, Berg CJ, Lake CR. 1987. Biochemical changes during clomipramine treatment of childhood obsessive-compulsive disorder. Arch. Gen. Psychiatry 44, 219–225. ( 10.1001/archpsyc.1987.01800150025004) [DOI] [PubMed] [Google Scholar]
- 74.Molloy AG, Waddington JL. 1984. Dopaminergic behaviour stereospecific promoted by the D1 agonist R-SK & F 38393 and selectively blocked by the D1 antagonist SCH 23390. Psychopharmacology 82, 409–410. ( 10.1007/BF00427697) [DOI] [PubMed] [Google Scholar]
- 75.Starr BS, Starr MS. 1986. Grooming in the mouse is stimulated by the dopamine D1 agonist SKF 38393 and by low doses of the D1 antagonist SCH 23390, but is inhibited by dopamine D2 agonists, D2 antagonists and high doses of SCH 23390. Pharmacol. Biochem. Behav. 24, 837–839. ( 10.1016/0091-3057(86)90421-1) [DOI] [PubMed] [Google Scholar]
- 76.White FJ, Bednarz LM, Wachtel SR, Hjorth S, Brooderson RJ. 1988. Is stimulation of both D1 and D2 receptors necessary for the expression of dopamine-mediated behaviors? Pharmacol. Biochem. Behav. 30, 189–193. ( 10.1016/0091-3057(88)90442-X) [DOI] [PubMed] [Google Scholar]
- 77.Berridge KC, Aldridge JW, Houchard KR, Zhuang X. 2005. Sequential super-stereotypy of an instinctive fixed action pattern in hyper-dopaminergic mutant mice: a model of obsessive compulsive disorder and Tourette's. BMC Biol. 3, 4 ( 10.1186/1741-7007-3-4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Einat H, Szechtman H. 1995. Perseveration without hyperlocomotion in a spontaneous alternation task in rats sensitized to the dopamine agonist quinpirole. Physiol. Behav. 57, 55–59. ( 10.1016/0031-9384(94)00209-N) [DOI] [PubMed] [Google Scholar]
- 79.Mundt A, et al. 2009. High-frequency stimulation of the nucleus accumbens core and shell reduces quinpirole-induced compulsive checking in rats. Eur. J. Neurosci. 29, 2401–2412. ( 10.1111/j.1460-9568.2009.06777.x) [DOI] [PubMed] [Google Scholar]
- 80.Rudebeck PH, Murray EA. 2011. Balkanizing the primate orbitofrontal cortex: distinct subregions for comparing and contrasting values. Ann. N. Y. Acad. Sci. 1239, 1–13. ( 10.1111/j.1749-6632.2011.06267.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Noonan MP, Kolling N, Walton ME, Rushworth MF. 2012. Re-evaluating the role of the orbitofrontal cortex in reward and reinforcement. Eur. J. Neurosci. 35, 997–1010. ( 10.1111/j.1460-9568.2012.08023.x) [DOI] [PubMed] [Google Scholar]
- 82.Schoenbaum G, Roesch MR, Stalnaker TA, Takahashi YK. 2009. A new perspective on the role of the orbitofrontal cortex in adaptive behaviour. Nat. Rev. Neurosci. 10, 885–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Schoenbaum G, Saddoris MP, Stalnaker TA. 2007. Reconciling the roles of orbitofrontal cortex in reversal learning and the encoding of outcome expectancies. Ann. N. Y. Acad. Sci. 1121, 320–335. ( 10.1196/annals.1401.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rolls E, Critchley H, Mason R, Wakeman E. 1996. Orbitofrontal cortex neurons: role in olfactory and visual association learning. J. Neurophysiol. 75, 1970–1981. [DOI] [PubMed] [Google Scholar]
- 85.Rolls ET. 2000. The orbitofrontal cortex and reward. Cereb. Cortex 10, 284–294. ( 10.1093/cercor/10.3.284) [DOI] [PubMed] [Google Scholar]
- 86.Schultz W. 1998. Predictive reward signal of dopamine neurons. J. Neurophysiol. 80, 1–27. [DOI] [PubMed] [Google Scholar]
- 87.Takahashi YK, Roesch MR, Stalnaker TA, Haney RZ, Calu DJ, Taylor AR, Burke KA, Schoenbaum G. 2009. The orbitofrontal cortex and ventral tegmental area are necessary for learning from unexpected outcomes. Neuron 62, 269–280. ( 10.1016/j.neuron.2009.03.005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chamberlain SR, Fineberg NA, Blackwell AD, Robbins TW, Sahakian BJ. 2006. Motor inhibition and cognitive flexibility in obsessive-compulsive disorder and trichotillomania. Am. J. Psychiatry 163, 1282–1284. ( 10.1176/ajp.2006.163.7.1282) [DOI] [PubMed] [Google Scholar]
- 89.Montague PR, Dayan P, Sejnowski TJ. 1996. A framework for mesencephalic dopamine systems based on predictive Hebbian learning. J. Neurosci. 16, 1936–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Glimcher PW. 2011. Understanding dopamine and reinforcement learning: the dopamine reward prediction error hypothesis. Proc. Natl. Acad. Sci. USA 108(Suppl. 3), 15 647–15 654. ( 10.1073/pnas.1014269108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Houk JC, Adams JL, Barto AG. 1995. A model of how the basal ganglia generate and use neural signals that predict reinforcement. Cambridge, MA: The MIT Press. [Google Scholar]
- 92.Joel D, Niv Y, Ruppin E. 2002. Actor-critic models of the basal ganglia: new anatomical and computational perspectives. Neural Netw. 15, 535–547. ( 10.1016/S0893-6080(02)00047-3) [DOI] [PubMed] [Google Scholar]
- 93.Bjorklund A, Dunnett SB. 2007. Dopamine neuron systems in the brain: an update. Trends Neurosci. 30, 194–202. ( 10.1016/j.tins.2007.03.006) [DOI] [PubMed] [Google Scholar]
- 94.Tecuapetla F, Jin X, Lima SQ, Costa RM. 2016. Complementary contributions of striatal projection pathways to action initiation and execution. Cell 166, 703–715. ( 10.1016/j.cell.2016.06.032) [DOI] [PubMed] [Google Scholar]
- 95.DeLong MR. 1990. Primate models of movement disorders of basal ganglia origin. Trends Neurosci. 13, 281–285. ( 10.1016/0166-2236(90)90110-V) [DOI] [PubMed] [Google Scholar]
- 96.Kravitz AV, Freeze BS, Parker PR, Kay K, Thwin MT, Deisseroth K, Kreitzer AC. 2010. Regulation of parkinsonian motor behaviours by optogenetic control of basal ganglia circuitry. Nature 466, 622–626. ( 10.1038/nature09159) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hong W, Kim DW, Anderson DJ. 2014. Antagonistic control of social versus repetitive self-grooming behaviors by separable amygdala neuronal subsets. Cell 158, 1348–1361. ( 10.1016/j.cell.2014.07.049) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sorokin JM, Davidson TJ, Frechette E, Abramian AM, Deisseroth K, Huguenard JR, Paz JT. 2017. Bidirectional control of generalized epilepsy networks via rapid real-time switching of firing mode. Neuron 93, 194–210. ( 10.1016/j.neuron.2016.11.026) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.O'Connor DH, Hires SA, Guo ZV, Li N, Yu J, Sun QQ, Huber D, Svoboda K. 2013. Neural coding during active somatosensation revealed using illusory touch. Nat. Neurosci. 16, 958–965. ( 10.1038/nn.3419) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sohal VS, Zhang F, Yizhar O, Deisseroth K. 2009. Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature 459, 698–702. ( 10.1038/nature07991) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Krook-Magnuson E, Armstrong C, Bui A, Lew S, Oijala M, Soltesz I. 2015. In vivo evaluation of the dentate gate theory in epilepsy. J. Physiol. 593, 2379–2388. ( 10.1113/JP270056) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Krook-Magnuson E, Szabo GG, Armstrong C, Oijala M, Soltesz I. 2014. Cerebellar directed optogenetic intervention inhibits spontaneous hippocampal seizures in a mouse model of temporal lobe epilepsy. eNeuro 1 ( 10.1523/ENEURO.0005-14.2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Siegle JH, Wilson MA. 2014. Enhancement of encoding and retrieval functions through theta phase-specific manipulation of hippocampus. eLife 3, e03061 ( 10.7554/eLife.03061) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Grosenick L, Marshel JH, Deisseroth K. 2015. Closed-loop and activity-guided optogenetic control. Neuron 86, 106–139. ( 10.1016/j.neuron.2015.03.034) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Paz JT, Davidson TJ, Frechette ES, Delord B, Parada I, Peng K, Deisseroth K, Huguenard JR. 2013. Closed-loop optogenetic control of thalamus as a tool for interrupting seizures after cortical injury. Nat. Neurosci. 16, 64–70. ( 10.1038/nn.3269) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Stark E, Roux L, Eichler R, Senzai Y, Royer S, Buzsaki G. 2014. Pyramidal cell-interneuron interactions underlie hippocampal ripple oscillations. Neuron 83, 467–480. ( 10.1016/j.neuron.2014.06.023) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Berndt A, Yizhar O, Gunaydin LA, Hegemann P, Deisseroth K. 2009. Bi-stable neural state switches. Nat. Neurosci. 12, 229–234. ( 10.1038/nn.2247) [DOI] [PubMed] [Google Scholar]
- 108.Liu Z, Wang Y, Cai L, Li Y, Chen B, Dong Y, Huang YH. 2016. Prefrontal cortex to accumbens projections in sleep regulation of reward. J. Neurosci. 36, 7897–7910. ( 10.1523/JNEUROSCI.0347-16.2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Rogerson T, Jayaprakash B, Cai DJ, Sano Y, Lee YS, Zhou Y, Bekal P, Deisseroth K, Silva AJ. 2016. Molecular and cellular mechanisms for trapping and activating emotional memories. PLoS ONE 11, e0161655 ( 10.1371/journal.pone.0161655) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Witten IB, et al. 2011. Recombinase-driver rat lines: tools, techniques, and optogenetic application to dopamine-mediated reinforcement. Neuron 72, 721–733. ( 10.1016/j.neuron.2011.10.028) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yamamoto K, et al. 2015. Chronic optogenetic activation augments abeta pathology in a mouse model of Alzheimer disease. Cell Rep. 11, 859–865. ( 10.1016/j.celrep.2015.04.017) [DOI] [PubMed] [Google Scholar]
- 112.Kim CK, et al. 2016. Simultaneous fast measurement of circuit dynamics at multiple sites across the mammalian brain. Nat. Methods 13, 325–328. ( 10.1038/nmeth.3770) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data available from the Dryad Digital Repository: doi:10.5061/dryad.h6qm7.