

Abstract
The validity of rodent models for the study of psychiatric disorders is controversial. Despite great efforts from academic institutions and pharmaceutical companies, as of today, no major therapeutic intervention has been developed for the treatment of psychiatric disorders based on mechanistic insights from rodent models. Here, we argue that despite these historical shortcomings, rodent studies are nevertheless instrumental for identifying neuronal circuit mechanisms underlying behaviours that are affected in psychiatric disorders. Focusing on schizophrenia, we will give four examples of rodent models that were generated based on genetic and environmental risk factors or pathophysiological evidence as entry points. We will then discuss how circuit analysis in these specific examples can be used for testing hypotheses about neuronal mechanisms underlying symptoms of schizophrenia, which will then guide the development of new therapies.
This article is part of a discussion meeting issue ‘Of mice and mental health: facilitating dialogue between basic and clinical neuroscientists’.
Keywords: mice, behaviour, cognition, neuronal circuitry, schizophrenia
1. Introduction
This manuscript emerged from an inspiring meeting at the Royal Society in London organized by Emily Holmes and Amy Milton. The meeting ‘Of mice and mental health: facilitating dialogue between basic and clinical neuroscientists’ was intended to foster interactions between clinical and basic scientists with the goal of ultimately developing better treatment strategies for mental disorders. However, during the intensive discussions it became clear that a far more basic question had to be addressed first: how useful are rodent models, in general, for understanding human psychiatric disorders?
The reality is that the five most-prescribed pharmacological agents for treating mental disorders share mechanisms of action with compounds that were discovered serendipitously over half-a-century ago. One famous example is chlorpromazine, the first antipsychotic medication targeting dopamine D2 receptors, which was originally developed in the 1950s to stabilize patients in preparation for surgery, but was subsequently discovered to be effective for treating patients with psychosis. Similarly, chance rather than mechanistic insight, led to the identification of lithium for the treatment of manic disorders and selective serotonin reuptake inhibitors as anti-depressive compounds. Since then, and despite decades of research with preclinical models, rodent findings have not been the basis of any conceptually new intervention that has subsequently been found to cure or treat any of the major mental disorders [1]. Why has it been so difficult to make progress?
In contrast to other fields of medicine, no biological markers exist for psychiatric disorders and diagnosis is based solely on behavioural symptoms. Owing to the absence of such markers, animal models often aim to recapitulate behavioural symptoms. This approach has its limitations as some of the behavioural symptoms are distinctive for humans and are not measurable in animals. For example, it is hard to determine if mice are experiencing delusions or hallucinations, or to probe verbal memory in rats. In addition, even for seemingly homologous behaviours, the brain circuitry and anatomy underlying these behaviours may not necessarily be conserved between species. Consistent with both points, anatomy, especially at the microcircuit level, can differ between humans and rodents, and some brain areas, like the cortex, are comparatively more developed in humans. Finally, the neuronal mechanisms underlying a behavioural abnormality may have to be studied under the pathophysiological conditions of the disorder as conclusions drawn from studying ‘healthy’ rodent brains may not be sufficient for developing better treatments. In this context, animal models should be based not only on behavioural abnormalities but also on disease risk factors or pathophysiological changes, which are difficult to model in the absence of definitive biological markers.
Here, we would like to argue that despite these limitations there is much to be learned from rodent models. Technological advances in the ability to record or manipulate pre-defined neuronal circuits in the brain allow us to understand neuronal mechanisms of behaviour in an unprecedented way. In combination with precise genetic and molecular tools, neuronal mechanisms of rodent behaviour can now be identified with the potential to open up novel therapeutic entry points in humans. Although there are species differences in brain anatomy, the gross anatomy of the brain, including long range projections as well as many of the neuronal and molecular mechanisms underlying brain function, is evolutionarily conserved between rodents and humans. As changes in long range functional and structural connectivity have been identified in many psychiatric disorders, studying how these circuits regulate behaviour in rodents should give us insight about how they could be dysregulated in human disease states. Finally, although we may not be able to directly study symptoms like hallucinations in the mouse, we can assess other behavioural abnormalities—and we will give examples for these below—that rely on conserved circuitry between humans and rodents.
Aiding this approach, clinical psychiatry has also been refining the precision with which behavioural alterations are defined. Together, clinical and preclinical researchers are now developing comparable behavioural tasks for rodents and humans with the overarching goal to study truly homologous behaviours in both species. One example is the development of touch-screen-based cognitive tasks that can be used in rodents and humans as a way towards inter-species standardization of cognitive behaviours affected in schizophrenia and other disorders [2]. Also, the Royal Society meeting gave a great example of how exposure therapy combined with extinction training is now used in human subjects with post-traumatic stress disorder (PTSD). Notably, the initial concept and specific protocol that is now used in humans was developed from the insights originally gained with rats [3].
For the reasons outlined above, we are optimistic that rodent research will be instrumental for making progress on mental disorders. With a focus on schizophrenia as this is the area of our expertise, we will give examples of how the mouse has advanced our understanding of neurobiological mechanisms that may underlie positive, negative and cognitive symptoms of schizophrenia. While there are many excellent examples, we will focus mostly our own work that we are familiar with using four different entry points: a genetic risk factor, an environmental risk factor and two pathophysiological imaging findings.
2. Schizophrenia is diagnosed by positive, negative and cognitive symptoms
Like many other psychiatric disorders, schizophrenia is diagnosed by its symptoms, which fall into three main categories: positive, negative and cognitive. Positive symptoms include hallucinations, delusions and disorganized thinking. Negative symptoms include social withdrawal, blunted affect and a decrease in incentive motivation, while cognitive symptoms encompass deficits in processing speed, working memory, attentional set-shifting and verbal memory [4]. Although the positive symptoms are essential for diagnosis, the negative and cognitive symptoms are more predictive for the long-term prognosis of the disorder [5]. Moreover, the only currently available therapeutics for schizophrenia—dopamine D2 receptor blockers—decrease positive symptoms, but barely improve negative and cognitive symptoms [6]. Whereas positive symptoms are difficult to model in mice at the behavioural level, some of the negative symptoms (e.g. deficits in motivation) and cognitive symptoms (e.g. deficits in working memory and attentional set-shifting) can be studied in mice. Therefore, one major focus of basic research is to better understand the neurobiology underlying the cognitive and negative symptoms of schizophrenia in order to develop novel therapeutics that improve these behaviours.
(a). Schizophrenia has a strong genetic component
There is strong evidence that schizophrenia has a genetic component. Classical twin studies demonstrated a 50% concordance rate for schizophrenia among monozygotic twins and a rate of only 15% if the twins are dizygotic. Furthermore, whole genome studies have identified over 100 gene variants that are associated with the disorder [7]. While there have been promising examples, such as for the complement component 4 (C4) gene alleles [8], where the study of common variants has produced important insight into possible biological mechanisms of schizophrenia, most common variants only minimally increase the risk for the disorder. Moreover, many gene polymorphisms lie within non-coding regions, making it both difficult to infer their functional impact and challenging to generate appropriate mouse models.
In addition to common genetic variants, a few rare mutations have also been associated with schizophrenia. The best described rare mutation is the 22q11.2 deletion that leads to the 22q11.2 deletion syndrome (22q11DS), characterized by a 25-fold increased risk for developing schizophrenia as well as cardiac and facial anomalies [9]. This deletion syndrome also increases the risk of other psychiatric disorders, including schizoaffective disorder, attention-deficit hyperactivity disorder, bipolar disorder, anxiety and affective disorders [10,11]. Moreover, 22q11.2 microdeletion carriers show language delay, decreased full scale IQ, learning disabilities and mental retardation, in addition to deficits in attention and working memory [9,10]. Schizophrenia is the most prevalent diagnosis and it has been estimated that about 1–2% of patients with schizophrenia could be explained by 22q11DS [9]. As the individual genes comprising the 22q11.2 deletion are largely conserved between mice and humans, a homologous deletion has been introduced into the mouse to study its consequences on brain development, function and behaviour.
(i). Example 1: Modelling the 22q11.2 deletion in the mouse
A region of mouse chromosome 16 is homologous to the 22q11.2 region in humans, containing murine versions to all genes except CLTCL1 (clatherin, heavy polypeptide-like 1) with only minimal reorganization of gene order [12]. The 22q11.2 deletion present in the majority of patients (greater than 85%) spans approximately 3 Mb in humans [12]. A smaller, 1.5 Mb deletion is present in approximately 15% of 22q11DS patients, but their clinical presentation is similar, suggesting that this smaller region is sufficient for producing the deletion syndrome [12]. Several mouse models, including the Df(16)A+/− and the Df(16)1+/− models described below, have been generated with deletions that either fall within or encompass the 1.5 Mb microdeletion [13]. Although these rare microdeletions are present only in 1–2% of patients with schizophrenia, their high penetrance for the disease makes these aetiologically valid genetic models an excellent opportunity for an investigation of the pathogenic circuitry that may underlie behavioural abnormalities present in 22q11DS carriers and, probably schizophrenia more broadly.
One example of how deletion models can be exploited to uncover pathogenic mechanisms of cognitive symptoms is the use of the Df(16)A+/− model to study circuit-level alterations underlying deficits in spatial working memory. Schizophrenia patients with and without the 22q11DS show impairments in a number of cognitive tasks, including spatial working memory [14]. Similarly, Df(16)A+/− mice show impairments in the acquisition of a delayed non-match to sample T-maze task that relies on spatial working memory [15]. As spatial working memory is thought to depend on an intact hippocampal–prefrontal circuit, Sigurdsson et al. investigated whether hippocampal–prefrontal communication is impaired in Df(16)A+/− mice during the performance of the working memory task [16]. Recording neural activity from the dorsal hippocampus (dHPC) and prefrontal cortex (PFC), the authors found that synchronous activity between dHPC and the medial PFC increases during task acquisition in normal mice. By contrast, Df(16)A+/− mice take longer to acquire the task and exhibit reduced dHPC-PFC synchrony. Moreover, at the onset of training, the strength of hippocampal–prefrontal synchrony in a specific frequency (4–12 Hz; theta range) predicts the time it takes Df(16)A+/− mice to acquire the task. This work suggests that deficits in communication between the hippocampus and the prefrontal cortex may underlie working memory impairments in the 22q11DS. While this has not been directly tested in 22q11DS carriers, there is evidence for altered hippocampal–prefrontal connectivity in patients with schizophrenia (figure 1) based on functional coupling between these structures at rest and during working memory [17].
Figure 1.
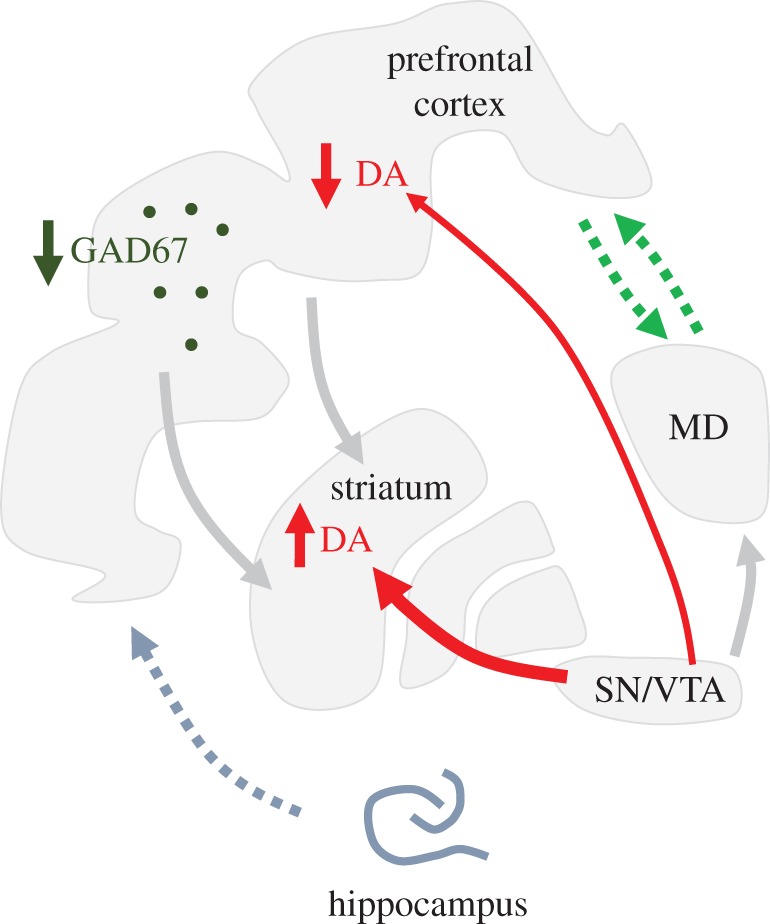
Brain circuit abnormalities in schizophrenia: Pathophysiological abnormalities observed in patients with schizophrenia include decreased expression of inhibitory neuron markers (e.g. GAD67: Glutamate Decarboxylase 67 in the cortex), disruption of thalamo–prefrontal communication (green dotted arrows) and disruption of hippocampal–prefrontal communication (grey dotted arrow) and increased nigrostriatal, but decreased mesocortical, dopamine transmission (red arrows). Animal models can determine whether the different alterations can coexist and are causally connected. They further identify potential circuit entry points for therapies targeting positive, negative and cognitive symptoms of schizophrenia that are potentially caused by these circuit abnormalities. MD, medio-dorsal thalamus; SN, substantia nigra; VTA, ventral tegmental area; DA, dopamine.
Another example of how the 22q11DS mouse model yielded insights into the pathophysiology of the deletion syndrome comes from a study examining the strength of thalamocortical (TC) connections into the auditory cortex, a region that is active during auditory hallucinations [18]. Studies in Df(16)1+/− mice found that auditory TC connections were weakened in post-adolescent, but not pre-adolescent animals, mirroring the developmental timeline of behavioural impairments in pre-pulse inhibition (PPI) in these mice [19]. This was intriguing given that PPI, a measure of sensory-motor gating, is impaired in 22q11DS carriers and patients with schizophrenia [13]. Furthermore, the authors found that this reduction in TC strength was specific for the auditory cortex, and was due to an unexpected increase in dopamine D2Rs in the medial geniculate nucleus of the thalamus—an area that normally contains very low levels of D2Rs. Given that TC strength and PPI were normalized following the acute administration of haloperidol, the authors proposed that the therapeutic effects of antipsychotic medications in schizophrenia could be due to targeting thalamic, rather than striatal, D2Rs.
(b). Epidemiological studies in humans point to the importance of environmental risk factors
While there is clearly a role for genetic risk factors in the aetiology of schizophrenia, the concordance rate between monozygotic twins is only 50%, suggesting that environmental factors contribute significantly to disease risk. Schizophrenia is considered a neurodevelopmental disorder and therefore the investigation of early environmental factors that could play a role in the aetiology of the disease has been the focus of many studies. One early life factor associated with schizophrenia is maternal infection during pregnancy (reviewed in [20]). Early epidemiological studies found an increased rate of schizophrenia among offspring who were in utero during major influenza epidemics compared to non-epidemic periods [20]. This association was replicated in several, but not all, geographical populations analysed, potentially reflecting the difficulty of accurately classifying exposure status in these studies [20].
Subsequent studies used large birth cohorts where exposure to infection was determined from medical records or based on seropositivity for infection during pregnancy. These studies found an increased risk of schizophrenia among offspring of mothers who received a diagnosis of influenza, toxoplasmosis, rubella or bacterial infection during pregnancy [20]. In addition, high levels of pro-inflammatory cytokines in the maternal serum during pregnancy were also found to increase the risk of schizophrenia in the in utero offspring [20,21]. The lack of pathogen specificity for this risk factor, combined with the risk conferred by elevated inflammatory molecules, led to the hypothesis that the detrimental effects of early infection on the developing offspring brain are due to pathological maternal immune activation (MIA).
Like the genetic risk factors described above, prenatal MIA is not specific to schizophrenia, but may also increase the risk of for autism, bipolar disorder and depression (e.g. [22]). And, unlike the 22q11.2 mutation, the effect size of the risk conferred by prenatal infection is low. But given the prevalence of this risk factor, the increased population-attributable risk conferred by exposure to prenatal infection has been estimated to be as high as 14% [23]. Therefore, it is of great interest to understand the neurobiological changes resulting from this common risk factor, as well as how they relate to disease-relevant behaviours.
(i). Example 2: Modelling maternal immune activation in the mouse
Several groups have studied prenatal infection in rodent models [24]. One of the key early observations in this field was that the behavioural impairments found in the adult offspring of mothers infected with live influenza virus at mid-gestation were phenocopied by simply activating the maternal immune system with a synthetic double-stranded RNA, polyinosinic–polycytidylic acid (PolyIC) [25]. This finding provided evidence that the negative effects of prenatal infection on the developing fetus could be largely explained by abnormal activation of the maternal immune system, rather than by direct viral infection of the fetus itself. This is supported by findings showing no evidence of viral presence in the fetal mouse brain 24 h following maternal intranasal infection with influenza [26] and by studies in humans, which failed to detect the presence of the influenza virus in the cord blood of offspring of mothers with documented influenza infection during pregnancy [27].
One of the best-replicated post mortem findings in the schizophrenia literature is histological alterations in prefrontal cortical interneurons (figure 1) [28,29]. Interneurons that express the molecular marker parvalbumin (PV) show decreases of GAD67 expression, an enzyme responsible for the production of GABA [28,29]. Alterations are also seen in other interneuron markers including the vesicular GABA transporter (vGAT), and the post-synaptic GABA receptor subunit, GABA-Aα2 [29]. At a functional level, patients with schizophrenia show deficits in GABAergic transmission assayed with radioligand studies [30] and an impairment in the induction of cortical gamma frequency oscillations, which are thought to depend upon normal functioning of PV interneurons [31]. Although these data imply that the observed histological abnormalities in subclasses of interneurons impair cortical inhibition, it remains unclear whether specific interneurons are affected not only at the histological but also at the functional level. Moreover, it is still unknown whether these functional changes affect behaviours that are altered in the disease.
The PolyIC model described above recapitulates the histological alterations in prefrontal PV interneurons observed in patients [32] and can thus be used to determine whether the histological abnormalities reflect functional abnormalities. Using slice electrophysiology, we recently demonstrated that adult MIA offspring show a reduction in GABAergic transmission onto pyramidal cells in the prefrontal cortex, which was due to a selective reduction in the strength of inputs from PV-containing interneurons, whereas another interneuron population expressing the marker calretinin (CR) was not affected. At the mechanistic level, decreased synaptic transmission was due to a decrease in pre-synaptic release probability, potentially caused by the reduced levels of GAD67 [33].
At the behavioural level, adult MIA offspring showed increased innate anxiety and impaired attentional set-shifting. To investigate whether the deficits in PV transmission could be responsible for these behavioural abnormalities, we inhibited prefrontal PV interneurons in normal mice while they were performing tests of anxiety and attentional set-shifting. Remarkably, we found that acutely decreasing PV GABAergic transmission was sufficient to increase anxiety and impair attentional set-shifting, phenocopying the behavioural impairments seen in the MIA offspring [33]. To the extent that the PolyIC MIA offspring recapitulate the landscape of neurobiological changes present in at least a subpopulation of patients with schizophrenia, this work suggests that deficits in GABAergic transmission from prefrontal PV interneurons may be responsible for the impairments in attentional set-shifting seen in some patients with schizophrenia. Strengthening the potential relevance of the animal findings for the human disease, a clinical study had previously demonstrated that patients with a documented history of infection during pregnancy had poorer performance on an attentional set-shifting task than patients without this exposure [34].
3. Using human brain imaging as a guide towards developing mouse models of schizophrenia
(a). Thalamo-cortical dysfunction
In addition to modelling genetic and environmental risk factors for schizophrenia, rodent models can also approximate the observations made in humans with brain imaging to determine whether specific physiological alterations give rise to abnormal behaviour. With regard to cognitive symptoms of schizophrenia, substantial evidence suggests that they are at least partially due to alterations in prefrontal cortical functioning [35,36]. In support of this, patients with frontal lobe damage exhibit impairments in many of the executive processes that are affected in patients with schizophrenia, including working memory and attentional set-shifting [37]. Moreover, functional brain imaging studies have consistently associated decreased performance in tasks of executive function with altered activation of the prefrontal cortex [35,36].
However, the prefrontal cortex receives strong innervation from the thalamus, and there is increasing evidence that the prefrontal cortex does not act in isolation from its main thalamic counterpart, the medio-dorsal nucleus (MD). Indeed, a recent meta-analysis of 41 imaging studies found altered MD activation in concert with altered prefrontal activation during executive functioning (figure 1) [38]. Even at rest, decreased functional connectivity between the thalamus and the PFC has been repeatedly measured in patients [39,40], and is already observed in individuals at high risk for the disorder, where it can predict conversion to schizophrenia [40].
(i). Example 3: Studying the role of thalamo-cortical circuitry in working memory
Human imaging studies are largely correlative. To determine whether a primary dysfunction of the MD can lead to deficits in cognitive processes associated with abnormalities in the prefrontal cortex, animal models are required.
In order to model the reductions in MD function observed in imaging studies, we generated a mouse model to selectively decrease neuronal activity in the MD while mice are performing a T-maze working memory task [41]. We expressed a designer receptor that can be activated by a designer drug (DREADD) selectively in the MD using a viral vector. We found that inhibiting only 30% of MD neurons was sufficient to induce deficits in a spatial working memory task, especially when the animals needed to retain information for longer delay intervals (greater than 30 s) [41]. Using in vivo physiology, we further observed that MD inhibition disrupted synchronous activity between the MD and the PFC during the task, suggesting that acutely inhibiting MD function impaired working memory by affecting thalamo-cortical information flow [41].
We tested this hypothesis by inhibiting the projections from the MD to the PFC using optogenetic tools [42]. As lesion studies indicated that the medial part of the mouse PFC (mPFC) is essential for working memory, we hypothesized that inhibiting projections from the MD to the mPFC would impair working memory performance, while inhibiting projections to the orbital frontal cortex (OFC), a brain area not thought to be involved in spatial working memory, would not. Inhibiting MD-mPFC projections impaired working memory performance during long delays similar to inhibiting the MD itself. By contrast, the inhibition of MD-OFC projections did not affect task performance. Optogenetic inhibition further revealed that the MD-mPFC projection is primarily required for the maintenance of task-relevant information during the delay phase of the task [42]. By contrast, ventral hippocampal input to the mPFC is preferentially involved in the encoding of spatial information earlier in the task [43]. MD-mPFC circuitry is known for its reciprocal connectivity, and inhibition of the reciprocal mPFC to MD projection during the delay also impaired subsequent task performance, suggesting that reciprocal activity between these structures is required for working memory maintenance [42].
The phase-specific inhibition experiments indicate that mPFC activity during the delay carries critical information for task performance that is modulated by the MD. Consistent with this hypothesis, neurophysiological recordings revealed elevated neuronal activity among a population of mPFC neurons whose temporally sparse but sequential activity spanned the entire delay. Importantly, this activity predicted task performance as elevated activity collapsed in incorrect trials. Moreover, elevated activity was dependent upon MD input for its sustained maintenance across the delay. These data indicate that thalamo-prefrontal projections are critical for sustaining prefrontal activity during working memory maintenance [42].
Strikingly, Schmitt et al. [44] made similar observations using a two-alternative forced-choice task (2AFC). In the 2AFC task, sequential elevated mPFC activity during the delay was dependent upon the MD and predicted task performance [44]. One of two distinct auditory stimuli presented at the start of a trial instructed the mouse whether to subsequently make a decision based on an auditory or a visual stimulus. This rule changed on a trial-by-trial basis and the authors observed that the elevated delay activity in mPFC tracked the task rules (e.g. whether to use the visual or the auditory cue). By contrast, MD activity during the delay could not distinguish between the rules. Based on these findings, the authors proposed that the MD is recruited by the PFC to stabilize cortical rule representations without directly relaying information about the rules themselves [44]. Sustaining persistent cortical activity during delay periods may be a function of not only the MD but also other higher order thalamic nuclei, as Guo et al. (2017) have described for thalamo-frontal circuits involving motor cortex [45].
If MD activity is required for sustaining cortical representations during the delay, enhancing MD function may enhance task performance. Indeed, in both the T-maze spatial working memory task and the 2AFC task, accuracy of performance was improved by increasing MD excitability [42,44]. This suggests that enhancing MD function could be a therapeutic strategy for improving working memory in schizophrenia and other mental disorders with similar cognitive impairments.
(b). The dopamine hypothesis of schizophrenia
The dopamine hypothesis is one of the oldest and most influential hypotheses about the biological basis of schizophrenia. In 1966, Jacques Van Rossum formally proposed that ‘overstimulation of dopamine receptors could be part of the etiology of schizophrenia’ [46]. This was based on the observation that amphetamine, which was thought to increase dopamine transmission, exaggerates the positive symptoms of schizophrenia [46]. Moreover, chlorpromazine, which had just been found by chance to have antipsychotic efficacy, blocked the effects of amphetamine in animals. Chlorpromazine was also known to have Parkinsonian side effects in humans, suggesting an antagonistic dopaminergic action. Even stronger support for the dopamine hypothesis surfaced in the 1970s when Philip Seeman and Solomon Snyder established that the therapeutic dose of antipsychotic medication is inversely related to its binding affinity at dopamine receptors [46].
Direct evidence in support of the dopamine hypothesis has come from brain imaging studies using ligands that can assess presynaptic dopamine uptake or postsynaptic occupancy of dopamine D2Rs by dopamine. At the presynaptic level, increased uptake of the dopamine precursor 18F-fluorodopa (or L-β-11C-DOPA) has consistently been measured in the striatum of patients with schizophrenia [47]. Importantly, enhanced uptake is already observed in subjects who are at high risk for schizophrenia, suggesting that it occurs early in the disease process [47]. Consistent with enhanced presynaptic function, amphetamine-induced dopamine release, as measured by ligand replacement at the dopamine D2R, is enhanced in patients with schizophrenia [48]. At the postsynaptic level, the enhanced occupancy of D2Rs by dopamine has been measured in drug naive patients with schizophrenia suggesting increased D2R signalling in the striatum of patients with schizophrenia. Strikingly, the enhanced striatal dopamine (figure 1) correlates with the severity of positive symptoms and predicts treatment response [48]. However, despite this strong relationship between striatal D2Rs and positive symptoms, it is important to note that 20–30% of psychotic patients do not respond to D2R blockers [49] and that not all patients with schizophrenia show enhanced precursor uptake or enhanced dopamine release.
(i). Example 4: Modelling enhanced D2R function in mice
Little is known about how enhanced D2R signalling affects striatal circuit function and behaviour, and whether striatal D2Rs are causally involved in the generation of negative and cognitive symptoms. To address these questions, we overexpressed D2Rs selectively in the mouse striatum from early development on (D2R-OE mice) [50,51]. Overexpression was achieved by using an artificial transcription factor system that allowed for both spatial and temporal control of transgene expression. We could thus distinguish effects of D2R upregulation during development versus adulthood.
An extensive behavioural analysis revealed two main outcomes. First, we observed deficits in cognitive processes that are dependent upon the PFC including spatial working memory and conditioned associative learning [50,51], two behaviours that are also affected in patients with schizophrenia. It was surprising that D2R upregulation in the striatum led to deficits in tasks that have traditionally been associated with mPFC function and pointed to alterations at the circuit level. Indeed, D2R-OE mice display a reduction in neuronal burst activity specifically in dopamine neurons projecting to the mPFC, which may be responsible for decreased prefrontal dopamine turnover and enhanced D1R sensitivity observed in these animals [50,51]. These findings recall classical studies in monkeys by Patricia Goldman-Rakic and others that point to an inverted U-relationship between prefrontal D1R activation and working memory [52]. A similar scenario may exist in patients with schizophrenia. A recent imaging finding has found attenuated amphetamine-induced dopamine release in the PFC of patients that was associated with decreased fMRI activation during a working memory task [53]. The findings obtained with the D2R-OE mice demonstrate that the enhanced striatal D2R function can lead to decreased cortical dopamine release and cognitive deficits. If this holds true in patients, increased striatal and decreased cortical dopamine should be measurable in individual patients, which now can be addressed using PET imaging.
Strikingly, the cognitive deficits and the decrease in dopamine neuron burst firing observed in D2R-OE mice are not reversed by switching off D2R overexpression in adulthood [51]. This suggests that developmental alterations in striatal D2R signalling may lead to persistent changes in brain function resulting in cognitive deficits. Although D2R blockers are effective for positive symptoms, it is now well established that they do not improve cognitive deficits. It is possible that persistent brain changes induced by an early hyperdopaminergic state in the striatum can no longer be reversed at the time of diagnosis, usually in late adolescence or early adulthood, when the treatment starts.
Findings from the D2R-OE mice also shed light on neurobiological changes contributing to negative symptoms such as motivational impairments [54]. In contrast to the cognitive deficits, the motivational impairments observed in D2R-OE mice were reversed after switching off the transgene, indicating that they require sustained D2R upregulation in the adult animal [51]. A careful analysis of the neurophysiological consequences of chronic D2R upregulation revealed that striatal neurons are hyper-excitable and tonic firing of ventral tegmental dopamine neurons is decreased in D2R-OE mice [51]. Both of these physiological alterations are reversed by switching off the transgene and could therefore contribute to the deficits in motivation seen in the D2R-OE mice.
D2R upregulation further alters the anatomy of the two functionally opposing striatal output pathways by inducing the growth of collaterals from the direct pathway into the external globus pallidus, which is the canonical output nucleus of the indirect pathway [55]. These anatomical changes were surprisingly plastic as they were reversed by switching off the transgene in the adult animal and by two weeks of treatment with the D2R blocker, haloperidol. Moreover, in vivo recordings revealed that the anatomical changes were associated with a functional imbalance between both pathways [55]. Owing to the tight association between D2R occupancy, positive symptom severity and treatment response to D2R blockers, these anatomical changes may be related to the positive symptoms of schizophrenia.
4. Conclusion
Here, we have described four different mouse models that were generated based on different biological entry points and were used to study neuronal circuitry and mechanisms of behaviours with relevance to schizophrenia. The six main findings of these studies are summarized as follows:
(1) The Df(16)A+/− mouse model, which recapitulates the 22q11DS deletion, displays impaired hippocampal–prefrontal communication and working memory
(2) The Df(16)1+/− mouse model shows increased thalamic D2R levels, thalamo-auditory cortex transmission and impaired sensory-motor gating. Altered transmission and sensory-motor gating are reversed by acute treatment with antipsychotic D2R blockers.
(3) Maternal immune activation leads to selective deficits in GABAergic transmission from prefrontal parvalbumin interneurons, possibly causing attentional set-shifting deficits.
(4) Medio-dorsal thalamic input to the prefrontal cortex is required for sustained cortical activity and working memory. Excitation of the medio-dorsal thalamus enhances working memory.
(5) Developmental upregulation of D2Rs induces persistent deficits in prefrontal-dependent cognitive tasks and a decreased functioning of the meso-cortical dopamine system.
(6) Striatal D2R upregulation alters the anatomical and functional balance of the striatal output pathways, which is reversed by chronic treatment with antipsychotic D2R blockers in the adult animal.
The main question arising from such findings in the mouse is how they will guide us towards a better understanding of schizophrenia and towards the development of new therapeutic strategies. We believe that findings like these could be helpful in several regards:
(1) Rodent findings provide testable hypotheses about pathophysiological mechanisms underlying symptoms of the disorder. Observations from the Df(16)1+/− model of the 22q11DS suggest that enhanced D2R binding in the auditory thalamus mediates auditory hallucinations. This hypothesis could be tested in human patients by quantifying D2R binding in the auditory thalamus (especially in 22q11DS carriers) and correlating D2R binding with positive symptoms. In a similar vein, results from the D2R-OE mice predict that striatal connectivity to the external globus pallidus could be important for positive symptoms. Again, this hypothesis can be tested in humans. Specifically, we anticipate that resting state connectivity between dorsal striatum and the external globus pallidus is decreased in drug naive patients and that this decrease is reversed by antipsychotic medication. Moreover, if aberrant connectivity is related to positive symptoms, the degree of functional connectivity should correlate with the severity of positive symptoms. Ideally, in such translational experiments imaging would be used in mice and humans to examine a phenomenon using the same methodology. Importantly, both examples demonstrate how animal models can provide testable hypotheses about pathophysiological mechanisms underlying symptoms of the disorder. Notably, they even may yield insight into human symptoms, such as psychosis, that cannot directly be studied in rodents at the behavioural level.
(2) Rodent findings provide potential biomarkers that will improve diagnostic categories and guide treatment. We described several plausible neuronal circuit mechanisms underlying deficits in working memory, a core cognitive symptom of schizophrenia. The 22q11DS model implicates hippocampal–prefrontal circuits, the thalamo-cortical silencing model implicates thalamo-prefrontal circuitry and the D2R-OE mice point to the meso-cortical dopamine system. Functional connectivity and PET imaging should be able to determine whether these circuit alterations exist in isolated patient subpopulations or co-occur in the same patients. Furthermore, using circuit alterations as biomarkers should help to identify patient subpopulations that may be particularly responsive to targeting pre-identified circuits for treating cognitive symptoms. This is important as cognitive deficits predict long-term prognosis, but are largely resistant to currently available drug treatments.
(3) Rodent findings emphasize the need for developing circuit-specific therapeutic tools in clinical research. Once circuit mechanisms are validated in a particular patient population, we must overcome the technical challenges associated with designing interventions that target specific brain circuits. For example, in preclinical studies, boosting MD excitability improves working memory in healthy mice, but we have no way yet to achieve this in humans with thalamic abnormalities. To specifically target the human MD without affecting other structures of the brain, we will need to capitalize on new technologies aimed at local drug delivery or local stimulation that are currently under development (e.g. [56,57]).
(4) Rodent findings provide testable hypotheses about the aetiology of specific pathophysiological changes observed in the disorder. In D2R-OE mice, an increase in striatal D2Rs caused a decrease in cortical dopamine. Thus in humans, subcortical dopaminergic hyper-function, that is associated with positive symptoms, could potentially also result in cortical dopaminergic hypofunction associated with cognitive deficits. So far, both observations have been made in different patient groups. The mouse data suggest that both findings could coexist in the same patients and that striatal abnormalities may precede and cause cortical abnormalities. PET imaging can determine whether this hypothesis is correct at least in a subpopulation of patients. We would further predict that patients that show both pathophysiological abnormalities should also display positive and cognitive symptoms. Understanding the aetiology of specific pathophysiological changes may help us to design earlier interventions that prevent the development of downstream changes and their associated behavioural symptomatology.
(5) Rodent findings give insight into whether individual risk factors that have been identified in humans interact to produce synergistic effects on neuropathology and behaviour. Epidemiological and genetic studies in humans frequently identify risk factors that in isolation show significant associations with schizophrenia, although their individual penetrance for the disorder is relatively low. It has been hypothesized that when two or more of these risk factors are present in the same individual, they may interact to synergistically increase the risk of developing the disorder. Animal studies can be used to test this hypothesis and to specifically narrow down risk factors that have a greater likelihood of interaction. An excellent example of this type of work by Giavanoli et al. showed that animals exposed to a low-level of maternal immune activation in utero, which on its own produced limited effects on adult behaviour, were particularly sensitive to subsequent exposure to peripubertal stress [58]. It is difficult, if not impossible, to screen for interactions like these in humans, but once an interaction is identified in rodents, focused studies to translate the findings to humans should be possible and extremely informative.
(6) Rodent findings should help identify pathophysiological changes occurring during development and guide early intervention. Recent high profile clinical trials (mGluR2/3 agonists for schizophrenia and mGluR4 agonists for autism) may have failed because the interventions occurred too late. For schizophrenia one of the most promising avenues is early intervention in subjects at high risk with the hope of preventing disease conversion. Although not directly discussed here, developmental models such as the PolyIC or D2R-OE mice should help to identify both pathophysiological changes occurring during development and optimal therapeutic windows that will prevent abnormalities in brain function from becoming persistent. Similar pathophysiological alterations may be identifiable in humans at risk for the disorder, and the animal models will then guide the timing and location of interventions.
Can we use mice to study schizophrenia? For the reasons described above, we are optimistic. We hope that this article will spur the scientific community to be mindful of not only the caveats, but also the strengths, of animal models, and that it will provoke continued dialogue between clinical and preclinical world as exemplified in the Royal Society meeting.
Acknowledgements
We thank Drs Joachim Scholz, Alexander Harris and Florencia Marcucci for their critical comments on the manuscript. We are grateful to the Royal Society for their support of the costs of attending this meeting ‘Of mice and mental health: facilitating dialogue between basic and clinical neuroscientists’ convened by Amy Milton and Emily A. Holmes.
Data accessibility
This article has no additional data.
Authors' contributions
S.C. and C.K. both conceptualized and wrote the manuscript.
Competing interests
We have no competing interests.
Funding
C.K.'s and S.C.'s salaries are supported by NIH RO1 MH093672 and MH107760, respectively.
References
- 1.Conn PJ, Roth BL. 2008. Opportunities and challenges of psychiatric drug discovery: roles for scientists in academic, industry, and government settings. Neuropsychopharmacology 33, 2048–2060. ( 10.1038/sj.npp.1301638) [DOI] [PubMed] [Google Scholar]
- 2.Nithianantharajah J, McKechanie AG, Stewart TJ, Johnstone M, Blackwood DH, St Clair D, Grant SG, Bussey TJ, Saksida LM. 2015. Bridging the translational divide: identical cognitive touchscreen testing in mice and humans carrying mutations in a disease-relevant homologous gene. Sci. Rep. 5, 14613 ( 10.1038/srep14613) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schiller D, Monfils MH, Raio CM, Johnson DC, Ledoux JE, Phelps EA. 2010. Preventing the return of fear in humans using reconsolidation update mechanisms. Nature 463, 49–53. ( 10.1038/nature08637) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tandon R, Nasrallah HA, Keshavan MS. 2009. Schizophrenia, ‘just the facts’ 4. Clinical features and conceptualization. Schizophr. Res. 110, 1–23. ( 10.1016/j.schres.2009.03.005) [DOI] [PubMed] [Google Scholar]
- 5.Green MF, Kern RS, Braff DL, Mintz J. 2000. Neurocognitive deficits and functional outcome in schizophrenia: are we measuring the ‘right stuff’? Schizophr. Bull. 26, 119–136. [DOI] [PubMed] [Google Scholar]
- 6.Keefe RS, et al. 2007. Neurocognitive effects of antipsychotic medications in patients with chronic schizophrenia in the CATIE Trial. Arch. Gen. Psychiatry 64, 633–647. [DOI] [PubMed] [Google Scholar]
- 7.Schizophrenia Working Group of the Psychiatric Genomics C. 2014. Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427. ( 10.1038/nature13595) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sekar A, et al. 2016. Schizophrenia risk from complex variation of complement component 4. Nature 530, 177–183. ( 10.1038/nature16549) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karayiorgou M, Simon TJ, Gogos JA. 2010. 22q11.2 microdeletions: linking DNA structural variation to brain dysfunction and schizophrenia. Nat. Rev. Neurosci. 11, 402–416. ( 10.1038/nrn2841) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Niklasson L, Rasmussen P, Oskarsdottir S, Gillberg C. 2001. Neuropsychiatric disorders in the 22q11 deletion syndrome. Genet. Med. 3, 79–84. [DOI] [PubMed] [Google Scholar]
- 11.Murphy KC, Jones LA, Owen MJ. 1999. High rates of schizophrenia in adults with velo-cardio-facial syndrome. Arch. Gen. Psychiatry 56, 940–945. [DOI] [PubMed] [Google Scholar]
- 12.Paylor R, Lindsay E. 2006. Mouse models of 22q11 deletion syndrome. Biol. Psychiatry 59, 1172–1179. ( 10.1016/j.biopsych.2006.01.018) [DOI] [PubMed] [Google Scholar]
- 13.Drew LJ, et al. 2011. The 22q11.2 microdeletion: fifteen years of insights into the genetic and neural complexity of psychiatric disorders. Int. J. Dev. Neurosci. 29, 259–281. ( 10.1016/j.ijdevneu.2010.09.007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Amelsvoort T, Henry J, Morris R, Owen M, Linszen D, Murphy K, Murphy D. 2004. Cognitive deficits associated with schizophrenia in velo-cardio-facial syndrome. Schizophr. Res. 70, 223–232. ( 10.1016/j.schres.2003.10.004) [DOI] [PubMed] [Google Scholar]
- 15.Stark KL, et al. 2008. Altered brain microRNA biogenesis contributes to phenotypic deficits in a 22q11-deletion mouse model. Nat. Genet. 40, 751–760. ( 10.1038/ng.138) [DOI] [PubMed] [Google Scholar]
- 16.Sigurdsson T, Stark KL, Karayiorgou M, Gogos JA, Gordon JA. 2010. Impaired hippocampal-prefrontal synchrony in a genetic mouse model of schizophrenia. Nature 464, 763–767. ( 10.1038/nature08855) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bahner F, Meyer-Lindenberg A. 2017. Hippocampal-prefrontal connectivity as a translational phenotype for schizophrenia. Eur. Neuropsychopharmacol. 27, 93–106. ( 10.1016/j.euroneuro.2016.12.007) [DOI] [PubMed] [Google Scholar]
- 18.Dierks T, Linden DE, Jandl M, Formisano E, Goebel R, Lanfermann H, Singer W. 1999. Activation of Heschl's gyrus during auditory hallucinations. Neuron 22, 615–621. [DOI] [PubMed] [Google Scholar]
- 19.Chun S, Westmoreland JJ, Bayazitov IT, Eddins D, Pani AK, Smeyne RJ, Yu J, Blundon JA, Zakharenko SS. 2014. Specific disruption of thalamic inputs to the auditory cortex in schizophrenia models. Science 344, 1178–1182. ( 10.1126/science.1253895) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brown AS, Derkits EJ. 2010. Prenatal infection and schizophrenia: a review of epidemiologic and translational studies. Am. J. Psychiatry 167, 261–280. ( 10.1176/appi.ajp.2009.09030361) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Canetta S, Sourander A, Surcel HM, Hinkka-Yli-Salomaki S, Leiviska J, Kellendonk C, McKeague IW, Brown AS. 2014. Elevated maternal C-reactive protein and increased risk of schizophrenia in a national birth cohort. Am. J. Psychiatry 171, 960–968. ( 10.1176/appi.ajp.2014.13121579) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Canetta SE, et al. 2014. Serological documentation of maternal influenza exposure and bipolar disorder in adult offspring. Am. J. Psychiatry 171, 557–563. ( 10.1176/appi.ajp.2013.13070943) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brown AS, Begg MD, Gravenstein S, Schaefer CA, Wyatt RJ, Bresnahan M, Babulas VP, Susser ES. 2004. Serologic evidence of prenatal influenza in the etiology of schizophrenia. Arch. Gen. Psychiatry 61, 774–780. ( 10.1001/archpsyc.61.8.774) [DOI] [PubMed] [Google Scholar]
- 24.Meyer U, Feldon J. 2012. To poly(I:C) or not to poly(I:C): advancing preclinical schizophrenia research through the use of prenatal immune activation models. Neuropharmacology 62, 1308–1321. ( 10.1016/j.neuropharm.2011.01.009) [DOI] [PubMed] [Google Scholar]
- 25.Shi L, Fatemi SH, Sidwell RW, Patterson PH. 2003. Maternal influenza infection causes marked behavioral and pharmacological changes in the offspring. J. Neurosci. 23, 297–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shi L, Patterson PH. 2005. Maternal influenza infection is likely to alter fetal brain development indirectly: the virus is not detected in the fetus. Int. J. Dev. Neurosci. 23, 299–305. ( 10.1016/j.ijdevneu.2004.05.005) [DOI] [PubMed] [Google Scholar]
- 27.Irving WL, et al. 2000. Influenza virus infection in the second and third trimesters of pregnancy: a clinical and seroepidemiological study. BJOG 107, 1282–1289. [DOI] [PubMed] [Google Scholar]
- 28.Lewis DA, Hashimoto T, Volk DW. 2005. Cortical inhibitory neurons and schizophrenia. Nat. Rev. Neurosci. 6, 312–324. ( 10.1038/nrn1648) [DOI] [PubMed] [Google Scholar]
- 29.Hoftman GD, Datta D, Lewis DA. 2017. Layer 3 excitatory and inhibitory circuitry in the prefrontal cortex: developmental trajectories and alterations in schizophrenia. Biol. Psychiatry 81, 862–873. ( 10.1016/j.biopsych.2016.05.022) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Frankle WG, Cho RY, Prasad KM, Mason NS, Paris J, Himes ML, Walker C, Lewis DA, Narendran R. 2015. In vivo measurement of GABA transmission in healthy subjects and schizophrenia patients. Am. J. Psychiatry 172, 1148–1159. ( 10.1176/appi.ajp.2015.14081031) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Uhlhaas PJ, Singer W. 2015. Oscillations and neuronal dynamics in schizophrenia: the search for basic symptoms and translational opportunities. Biol. Psychiatry 77, 1001–1009. ( 10.1016/j.biopsych.2014.11.019) [DOI] [PubMed] [Google Scholar]
- 32.Meyer U, Nyffeler M, Yee BK, Knuesel I, Feldon J. 2008. Adult brain and behavioral pathological markers of prenatal immune challenge during early/middle and late fetal development in mice. Brain Behav. Immun. 22, 469–486. ( 10.1016/j.bbi.2007.09.012) [DOI] [PubMed] [Google Scholar]
- 33.Canetta S, Bolkan S, Padilla-Coreano N, Song LJ, Sahn R, Harrison NL, Gordon JA, Brown A, Kellendonk C. 2016. Maternal immune activation leads to selective functional deficits in offspring parvalbumin interneurons. Mol. Psychiatry. 21, 956–968. ( 10.1038/mp.2015.222) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brown AS, et al. 2009. Prenatal exposure to maternal infection and executive dysfunction in adult schizophrenia. Am. J. Psychiatry 166, 683–690. ( 10.1176/appi.ajp.2008.08010089) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weinberger DR, Berman KF. 1996. Prefrontal function in schizophrenia: confounds and controversies. Phil. Trans. R. Soc. Lond. B 351, 1495–1503. [DOI] [PubMed] [Google Scholar]
- 36.Barch DM, Ceaser A. 2012. Cognition in schizophrenia: core psychological and neural mechanisms. Trends Cogn. Sci. 16, 27–34. ( 10.1016/j.tics.2011.11.015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kolb B, Whishaw IQ. 1983. Performance of schizophrenic patients on tests sensitive to left or right frontal, temporal, or parietal function in neurological patients. J. Nerv. Ment. Dis. 171, 435–443. [DOI] [PubMed] [Google Scholar]
- 38.Minzenberg MJ, Laird AR, Thelen S, Carter CS, Glahn DC. 2009. Meta-analysis of 41 functional neuroimaging studies of executive function in schizophrenia. Arch. Gen. Psychiatry 66, 811–822. ( 10.1001/archgenpsychiatry.2009.91) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Woodward ND, Karbasforoushan H, Heckers S. 2012. Thalamocortical dysconnectivity in schizophrenia. Am. J. Psychiatry 169, 1092–1099. ( 10.1176/appi.ajp.2012.12010056) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Anticevic A, et al. 2015. Association of thalamic dysconnectivity and conversion to psychosis in youth and young adults at elevated clinical risk. JAMA Psychiatry. 72, 882–891. ( 10.1001/jamapsychiatry.2015.0566) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Parnaudeau S, O'Neill PK, Bolkan SS, Ward RD, Abbas AI, Roth BL, Balsam PD, Gordon JA, Kellendonk C. 2013. Inhibition of mediodorsal thalamus disrupts thalamofrontal connectivity and cognition. Neuron 77, 1151–1162. ( 10.1016/j.neuron.2013.01.038) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bolkan SS, Stujenske JM, Parnaudeau S, Spellman TJ, Rauffenbart C, Abbas AI, Harris AZ, Gordon JA, Kellendonk C. 2017. Thalamic projections sustain prefrontal activity during working memory maintenance. Nat. Neurosci. 20, 987–996. ( 10.1038/nn.4568) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Spellman T, Rigotti M, Ahmari SE, Fusi S, Gogos JA, Gordon JA. 2015. Hippocampal-prefrontal input supports spatial encoding in working memory. Nature 522, 309–314. ( 10.1038/nature14445) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schmitt LI, Wimmer RD, Nakajima M, Happ M, Mofakham S, Halassa MM. 2017. Thalamic amplification of cortical connectivity sustains attentional control. Nature 545, 219–223. ( 10.1038/nature22073) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guo ZV, Inagaki HK, Daie K, Druckmann S, Gerfen CR, Svoboda K. 2017. Maintenance of persistent activity in a frontal thalamocortical loop. Nature 545, 181–186. ( 10.1038/nature22324) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Baumeister AA, Francis JL. 2002. Historical development of the dopamine hypothesis of schizophrenia. J. Hist. Neurosci. 11, 265–277. [DOI] [PubMed] [Google Scholar]
- 47.Howes OD, Kambeitz J, Kim E, Stahl D, Slifstein M, Abi-Dargham A, Kapur S. 2012. The nature of dopamine dysfunction in schizophrenia and what this means for treatment: meta-analysis of imaging studies. Arch. Gen. Psychiatry 66, 13–20. ( 10.1001/archgenpsychiatry.2012.169) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weinstein JJ, Chohan MO, Slifstein M, Kegeles LS, Moore H, Abi-Dargham A. 2017. Pathway-specific dopamine abnormalities in schizophrenia. Biol. Psychiatry 81, 31–42. ( 10.1016/j.biopsych.2016.03.2104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Demjaha A, et al. 2017. Antipsychotic treatment resistance in first-episode psychosis: prevalence, subtypes and predictors. Psychol. Med. 47, 1981–1989. ( 10.1017/S0033291717000435) [DOI] [PubMed] [Google Scholar]
- 50.Kellendonk C, Simpson EH, Polan HJ, Malleret G, Vronskaya S, Winiger V, Moore H, Kandel ER. 2006. Transient and selective overexpression of dopamine D2 receptors in the striatum causes persistent abnormalities in prefrontal cortex functioning. Neuron 49, 603–615. [DOI] [PubMed] [Google Scholar]
- 51.Simpson EH, Kellendonk C. 2017. Insights about striatal circuit function and schizophrenia from a mouse model of dopamine D2 receptor upregulation. Biol. Psychiatry 81, 21–30. ( 10.1016/j.biopsych.2016.07.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Goldman-Rakic PS, Castner SA, Svensson TH, Siever LJ, Williams GV. 2004. Targeting the dopamine D1 receptor in schizophrenia: insights for cognitive dysfunction. Psychopharmacology 174, 3–16. ( 10.1007/s00213-004-1793-y) [DOI] [PubMed] [Google Scholar]
- 53.Slifstein M, et al. 2015. Deficits in prefrontal cortical and extrastriatal dopamine release in schizophrenia: a positron emission tomographic functional magnetic resonance imaging study. JAMA Psychiatry. 72, 316–324. ( 10.1001/jamapsychiatry.2014.2414) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gold JM, Waltz JA, Frank MJ. 2015. Effort cost computation in schizophrenia: a commentary on the recent literature. Biol. Psychiatry. 78, 747–753. ( 10.1016/j.biopsych.2015.05.005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cazorla M, de Carvalho FD, Chohan MO, Shegda M, Chuhma N, Rayport S, Ahmari SE, Moore H, Kellendonk C. 2014. Dopamine D2 receptors regulate the anatomical and functional balance of basal ganglia circuitry. Neuron 81, 153–164. ( 10.1016/j.neuron.2013.10.041) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Grossman N, et al. 2017. Noninvasive deep brain stimulation via temporally interfering electric fields. Cell 169, 1029–1041. ( 10.1016/j.cell.2017.05.024) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Downs ME, Buch A, Sierra C, Karakatsani ME, Teichert T, Chen S, Konofagou EE, Ferrera VP. 2015. Long-term safety of repeated blood-brain barrier opening via focused ultrasound with microbubbles in non-human primates performing a cognitive task. PLoS ONE 10, e0125911 ( 10.1371/journal.pone.0125911) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Giovanoli S, et al. 2013. Stress in puberty unmasks latent neuropathological consequences of prenatal immune activation in mice. Science 339, 1095–1099. ( 10.1126/science.1228261) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This article has no additional data.