

ABSTRACT
Increasing incidences of multidrug resistance in pathogenic bacteria threaten our ability to treat and manage bacterial infection. The development and FDA approval of novel antibiotics have slowed over the past decade; therefore, the adoption and improvement of alternative therapeutic strategies are critical for addressing the threat posed by multidrug-resistant bacteria. Host-directed therapies utilize small-molecule drugs and proteins to alter the host response to pathogen infection. Here, we highlight strategies for modulating the host inflammatory response to enhance bacterial clearance, small-molecule potentiation of innate immunity, and targeting of host factors that are exploited by pathogen virulence factors. Application of state-of-the-art “omic” technologies, including proteomics, transcriptomics, and image-omics (image-based high-throughput phenotypic screening), combined with powerful bioinformatics tools will enable the modeling of key signaling pathways in the host-pathogen interplay and aid in the identification of host proteins for therapeutic targeting and the discovery of host-directed small molecules that will regulate bacterial infection. We conclude with an outlook on research needed to overcome the challenges associated with transitioning host-directed therapies into a clinical setting.
KEYWORDS: antibiotic resistance, fluorescent image analysis, inflammation, innate immunity, multidrug resistance, proteomics
INTRODUCTION
Therapeutic strategies for the treatment of bacterial infections have historically relied on the antibiotics that target bacterial protein, DNA, RNA, or cell wall synthesis. Although antibiotics have been successfully used for decades, the discovery rate of novel antibiotics is unable to keep pace with the emergence of antibiotic-resistant bacteria. For example, the appearance of carbapenem-resistant Enterobacteriaceae has thwarted the therapeutic benefit of the carbapenem class of antibiotics, which are reserved as a last-line defense (1, 2). Widespread antibiotic use in medical treatment facilities has generated selection pressure to give rise to multidrug-resistant (MDR) bacteria, further driving the need for antibacterial treatments that employ novel molecular mechanisms (3). Each year in the United States, issues related to antibacterial resistance are projected to result in over 23,000 deaths, 2 million illnesses, and costs amounting to 20 billion dollars (4). To address the MDR problem, combination therapy is an emerging option. Combinations of two antibiotics, of an antibiotic with a drug targeting the antibiotic resistance mechanism, or of an antibiotic with an adjuvant are promising new therapeutic approaches (5, 6). However, the clinical manifestation of infections caused by the bacterial pathogens reflects a complex interaction between the pathogen, host, and antibiotics. As the innate immune system plays a critical role in battling the bacterial infection, strategic targeting of the host together with an appropriate antimicrobial treatment of the pathogen(s) by rational combination therapy may suppress antibacterial resistance, lead to successful treatment and resolution of antimicrobial-resistant infections, and thereby overcome some of the impediments to antibiotic therapies. The focus of this review is on host-directed therapies, including the use of immunomodulatory agents and tool compounds that target critical host signaling enzymes exploited by bacteria for their intracellular invasion, replication, and/or dissemination. We also describe the use of high-throughput systems biology and phenotypic compound screening approaches, which complement current hypothesis-driven efforts and shed light on the discovery of novel host targets required for bacterial replication.
Intracellular survival of bacteria by evading host defense. Wide varieties of pathogenic bacteria have evolved sophisticated mechanisms to hijack the host factors for their invasion, replication, or spread and evade the host immune surveillance. There are several benefits for bacteria in adapting an intracellular lifestyle. For example, the intracellular niche protects bacteria from the complement or adaptive immune system. Intracellular bacteria also have less competition from other resident bacteria for nutrients. In fact, few bacterial species have evolved to live within professional phagocytic cells such as macrophages (7).
Once a bacterium is taken up into a cell by either phagocytosis or receptor-mediated endocytosis, it traffics along the endocytic pathway toward lysosomal fusion and destruction. The pH decreases upon maturation of the phagosome into a phagolysosome. Phagolysosome acidification, which itself contributes to target degradation, is also required for the activation of lysosomal hydrolases, such as cathepsins, that function optimally at low pH. In addition, lysosomes harbor antimicrobial peptides and natural resistance-associated macrophage protein 1, which excludes divalent cations that are essential for microbial function (8). Intracellular bacteria that have adapted to life within a host vacuole can halt trafficking (e.g., Mycobacterium tuberculosis) or tailor their environment to avoid destruction (e.g., Coxiella burnetii) (9, 10). How Salmonella evades lysosomal degradation remains controversial. Multiple studies demonstrated that Salmonella prevents the fusion of a Salmonella-containing vacuole with lysosomes. An alternative mechanism proposed is the delay rather than avoidance of lysosome fusion, which is the key step in the establishment of a replicative niche by Salmonella enterica serovar Typhimurium (11, 12). Pathogens that escape from the phagosome and gain access to the cytosol can evade autophagy (e.g., Shigella flexneri, Burkholderia pseudomallei, Listeria monocytogenes, and Francisella tularensis) (13–16). Certain bacteria (e.g., S. flexneri, B. pseudomallei, L. monocytogenes, and Rickettsia spp.) polymerize host actin and form an actin-tail structure (17). Actin tails enable bacteria to propel through the cytoplasm and protrude from the host plasma membrane. These protrusions are internalized by surrounding host cells, resulting in bacteria enclosed in double-membrane vacuoles. Bacteria secrete proteins that disrupt both membranes, allowing the bacteria to escape into the cytosol and neighboring cells (Fig. 1). One characteristic feature of the B. pseudomallei and Burkholderia mallei intracellular life cycle is the fusion of infected mononuclear cells, forming multinucleated giant cells (MNGCs). Although the role of B. pseudomallei-induced MNGCs is unclear, it is believed that cell fusion facilitates localized dissemination of the bacteria (18).
FIG 1 .
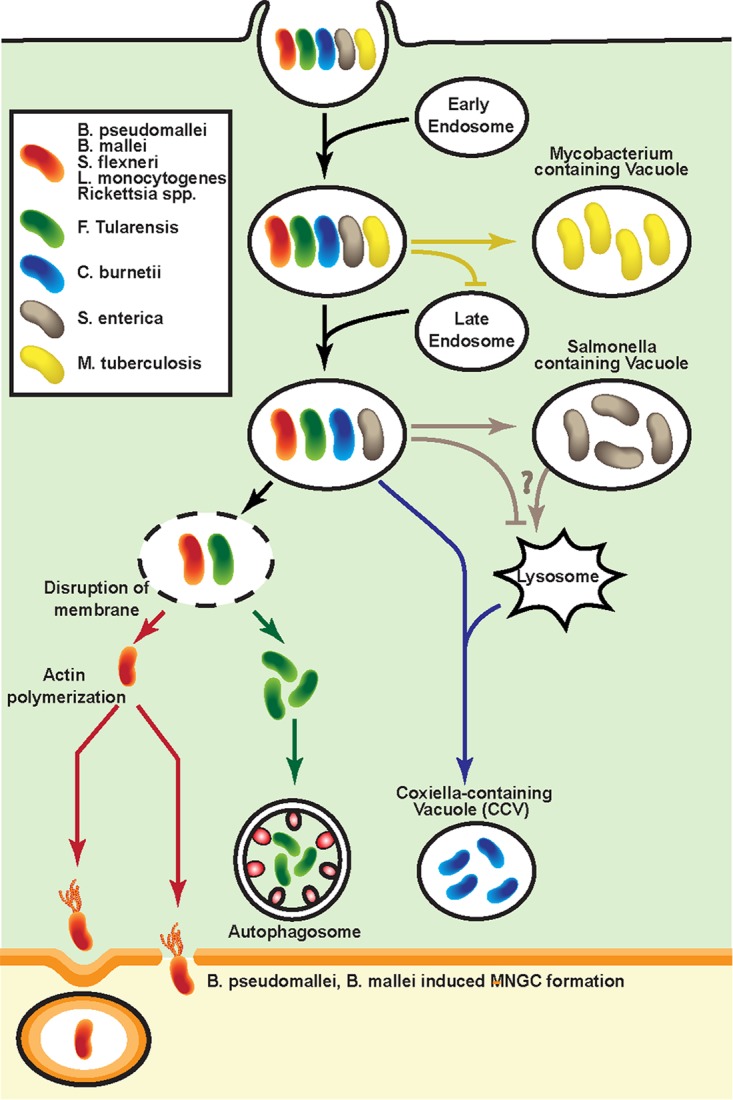
Life cycle of intracellular bacteria. M. tuberculosis inhibits the fusion of late endosomes with mycobacterium-containing vacuoles. S. enterica survives and replicates within a Salmonella-containing vacuole by avoiding or delaying fusion with lysosomes. C. burnetii develops various strategies to resist hostile host defense within the lysosomes and allows phagosomal trafficking to proceed all the way to lysosomal fusion. F. tularensis escapes the vacuole and replicates within the cytosol. F. tularensis can reenter the endosomal compartment by entering an autophagosome. Bacteria that escape into the cytosol can gain intracellular motility by forming actin tails, which also helps bacteria to spread into adjacent cells through membrane protrusion. B. pseudomallei and B. mallei can induce MNGC formation and promote cell-to-cell spread.
The interactions between bacteria and the mammalian host cells that they infect are multifaceted. Protection of host cells from intracellular bacterial infection relies on the appropriate timing, expression, location, and function of host defense mechanisms. While host cells strive to restrict bacterial infection, bacteria have evolved complex protein secretion systems that have diverse roles and perform numerous physiological functions that are required for bacterial survival and replication. Some of the secretion systems also deliver virulence factors into the mammalian host cells that allow the bacteria to replicate and spread in the host population (19, 20). Bacteria express a range of virulence factors that thwart host responses at various stages of their life cycle and usurp cellular functions to their advantage. Details with regard to the virulence factors’ mechanisms in facilitating the progression of the bacterial intracellular life cycle and counteracting host immune responses can be found elsewhere (http://www.mgc.ac.cn/VFs/). Failure to mount a robust immune response may result in the development of acute disease and, in some instances, reactivation of the latent infection. Harnessing the innate host defenses thus provides a rational basis for the development of host-directed antimicrobial therapeutics.
PROMOTING BACTERIAL CLEARANCE THROUGH MODULATING HOST INFLAMMATORY RESPONSES
Regulating PRR signaling pathways.
Pattern recognition receptors (PRRs), such as Toll-like receptors (TLRs), NOD-like receptors (NLRs), and RIG-I-like receptors, have evolved to detect conserved features of microbes known as pathogen-associated molecular patterns (PAMPs). This evolutionary strategy of the host enables a small number of germ line-encoded PRRs to recognize a vast variety of molecular structures associated with the pathogens. Thirteen TLRs have been reported. TLR1 to -9 are conserved in both mice and humans. Humans do not express TLR11 to -13. Studies using TLR-knockout mice revealed that each TLR has a distinct function in terms of PAMP recognition and the immune responses (21). For example, TLR2 heterodimerizes with either TLR1 or TLR6 in recognizing bacterial lipopeptides. TLR4 detects the presence of lipopolysaccharide (LPS), whereas TLR9 recognizes CpG islands that are enriched in the bacterial genome (22). With the exception of TLR3, stimulation of TLRs triggers a MyD88-mediated signaling cascade, which leads to NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells) activation and upregulation of proinflammatory gene expression (Fig. 2A) (22). Stimulating with TLR ligands promptly potentiates the production of proinflammatory cytokines and chemokines which facilitate the clearance of bacterial infections (22). In the mouse model, intranasal inoculation of monophosphoryl lipid A, a chemically modified derivative of the lipid A moiety of LPS, showed a significant reduction in the number of Haemophilus influenzae and Moraxella catarrhalis bacteria recovered from the nasopharynx (23). PRR ligands are also pursued as vaccine adjuvants. Five conserved antigens derived from Staphylococcus aureus, each having different roles in pathogenesis, were formulated with a TLR7 agonist and adsorbed onto alum adjuvant (4CT7-Staph). In the mouse peritonitis model, 80% to 90% protection against four different Staphylococcal strains was observed in mice vaccinated with 4CT7-Staph (24).
FIG 2 .

Strategies to promote antibacterial responses by modulating host immune responses. (A) Regulating pattern recognition receptor signaling pathways. CASP1, caspase 1; IKK, IκB kinase. (B) Targeting negative regulators of the autophagy machinery. CTSB, cathepsin B. (C) Stabilizing hypoxia-inducible factor 1α. (D) Modulating the production of ROS and RNS.
In addition to TLRs, PAMPs can be detected by NLRs residing in the cytosol, such as NLRP3 and NLRC4 (25). NLRP3 is an inflammasome-forming NLR, which involves the oligomerization of procaspase 1 through an adapter protein, the apoptosis-associated speck-like protein containing a CARD (ASC) (26). Autoproteolytic cleavage of procaspase 1 results in its activation and can subsequently convert pro-interleukin-1β (pro-IL-1β) and pro-IL-18 to their active forms (27). While NLRC4 can oligomerize with procaspase 1 directly, ASC is required for maximal NLRC4 inflammasome activation (28). NLRC4 inflammasome is important for clearing a variety of bacterial infections, including those by Salmonella Typhimurium, S. flexneri, Pseudomonas aeruginosa, and B. pseudomallei (29). B. pseudomallei induces NLRC4-dependent pyroptosis, a programmed cell death mechanism to contain intracellular pathogen infections, which restricts intracellular bacterial growth. Activation of NLRP3, on the other hand, upregulates IL-1β, promotes the replication of B. pseudomallei, recruits excessive neutrophils to the lung, and leads to tissue damage (30). In the murine model of B. pseudomallei infection, intraperitoneal (i.p.) delivery of glyburide, a small molecule that inhibits IL-1β production, followed by intranasal infection with B. pseudomallei significantly reduces bacterial dissemination into both liver and spleen (31). However, the therapeutic benefit of glyburide in patients infected with B. pseudomallei is not clear, as the correlation between glyburide-mediated anti-inflammatory responses and the disease outcomes remains controversial (32–34). MCC950 was also identified as a small molecule that potently inhibits NLRP3-induced ASC oligomerization but not NLRC4 signaling activation (35). The role of MCC950 in regulating bacterial infection requires further characterization. Identifying small molecules that selectively prevent cytokine secretion upon NLRP3 inflammasome activation thus appears as a promising new therapeutic strategy.
Boosting autophagy activity enhances killing of intracellular bacteria.
Autophagy is a cellular process that enables the digestion of cytoplasmic contents (i.e., toxic protein aggregates and dysfunctional organelles) in lysosomes. Autophagy is amplified in response to cellular stresses, including hypoxia, energy loss, and nutrient deprivation. By recycling cellular components, this process provides a mechanism for the adaptation to starvation and regulates cellular metabolism and homeostasis (36). In addition to its role in homeostatic maintenance, it is evident that cells also utilize autophagy as an innate immune mechanism for the clearance of intracellular pathogens (37). Autophagy starts with the assembly of a membrane sac, known as the phagophore, which elongates to enclose cytoplasmic components. The phagophore expands and grows into a double-membrane compartment, known as an autophagosome, which sequesters cytoplasmic targets (38). Central to the autophagy pathway is LC3B protein. Through a series of proteolytic modifications, pro-LC3 is converted to LC3B-II, which contributes to the closure of autophagosomes and mediates cargo docking (39). Fusion of autophagosome with lysosome results in autophagolysosomes, which contain a variety of proteases and acid hydrolases that enable the degradation of the cargo (40). Xenophagy, a selective degradation of intracellular microorganisms through autophagy, can be achieved in sequestosome 1-like receptors or NOD2-ATG16L1-mediated interactions (41). An alternative mechanism for bacterial clearance is through LC3-associated phagocytosis (LAP), which is mediated through single-membrane phagocytic vesicles that contain engulfed bacteria and are transiently coated with LC3-II. LAP is induced by multiple bacterial pathogens, including Escherichia coli, S. Typhimurium, Mycobacterium marinum, and B. pseudomallei (42). Induction of LAP with sirolimus, an mTOR inhibitor, increases the colocalization of B. pseudomallei with LC3 in phagosomes, thereby augmenting phagosomal maturation and further phagocytosis. Concomitantly, intracellular survival of B. pseudomallei decreases upon sirolimus treatment (14). In contrast, survival of intracellular B. pseudomallei was significantly increased after blocking autophagy induction (43). Intracellular pathogens have evolved strategies to counteract various stages of the autophagy pathway through inhibiting the autophagy induction signal, preventing recognition by the autophagy machinery, interfering with autophagy components, or blocking autophagosome fusion with the lysosome (38).
Boosting autophagy activity by inhibiting negative regulators of the pathway (i.e., p38 and cathepsin B) serves as a therapeutic route for bacterial clearance. Mitogen-activated protein kinase (MAPK) p38 negatively regulates autophagy (44). Treatment of macrophages with AMG548, a p38 inhibitor, promotes clearance of M. tuberculosis by inducing autophagy (45). Consistent with these observations, gefitinib (inhibitor of epidermal growth factor receptor) reduced M. tuberculosis-mediated p38 phosphorylation, thereby enhancing autophagy by increasing LC3 expression and restricting the growth of M. tuberculosis (Fig. 2B) (45).
Lysosomes mediate the degradation of endocytosed extracellular materials or sequestered intracellular components. Fundamental cellular processes orchestrated by lysosomes require cathepsin proteases. Cathepsin B is one of the 11 human lysosomal cathepsins (46). Cathepsin B plays an important role in immune responses such as TLR activation and antigen processing and presentation (46). The leakage of lysosomal cathepsin B into the cytoplasm can also trigger NLRP3 inflammasome in a cell-based assay model (47). In addition, lysosomal cathepsin B has also been demonstrated to be a negative regulator of autophagy and lysosomal biogenesis (48). Cathepsin-B-deficient macrophages upregulate genes associated with lysosome biogenesis and autophagy. Cathepsin-B-deficient mice subcutaneously infected with Francisella novicida showed a 40% increase in survival rate over the wild-type mice. Bacterial loads in the liver and spleen of wild-type mice were approximately 2 logs higher than those in cathepsin-deficient mice (48). Optimizing currently available cathepsin B inhibitors may provide an alternative venue in host-directed antimicrobial therapy.
Potentiating innate immune signaling pathways by stabilizing HIF.
Bacterial infections are often associated with hypoxic conditions which can stimulate the inflammatory response and improve infection clearance (49). The host response to hypoxic conditions is regulated at the transcriptional level by hypoxia-inducible factor (HIF). HIF-1β heterodimerizes with one of two HIF-α isoforms (HIF-1α and HIF-2α) and drives the expression of proinflammatory cytokines that mediate macrophage aggregation, invasion, and motility. The stability of HIF-α subunits is regulated by oxygen availability. Under normoxia conditions, prolyl-hydroxylases (PHDs) hydroxylate HIF-α and mark it for proteasomal degradation in a process mediated by von Hippel-Lindau tumor suppressor protein (VHL)-dependent ubiquitination. Under hypoxic conditions, PHDs are inactive, which allows for the accumulation of HIF-α (Fig. 2C). In contrast to VHL-null macrophages, which showed an enhanced intracellular killing of P. aeruginosa, HIF-1α-null macrophages failed to clear P. aeruginosa replication (50, 51).
Mice infected with uropathogenic Escherichia coli (UPEC) followed by the administration of an HIF-1α-stabilizing agent (AKB-4924) had a 10-fold reduction in UPEC colonization of the bladder (52). Consistent with this discovery, HIF-1α-deficient mice are more susceptible to UPEC urinary tract colonization (52). Given HIF-1α’s role in modulating host responses to promote bacterial clearance, it follows that preventing HIF-1α degradation may provide a therapeutic target against bacterial infection. However, the therapeutic benefit of HIF induction in unclear in the situation where the disease pathology is driven by an overactive immune response to bacterial components (53).
Redox mechanisms in the host defense/immune response.
Host immune cells such as macrophages and neutrophils produce reactive oxygen species (ROS) and reactive nitrogen species (RNS) molecules that act as a defense mechanism to trigger the clearance of the phagocytosed microorganisms. At physiological concentrations, ROS molecules act as secondary signaling messengers that regulate the expression of inflammatory mediators. However, an imbalance in the production and elimination of ROS is associated with human diseases (54). ROS molecules are produced by the host enzyme NADPH oxidase (NOX). Seven NOX homologues are encoded in the human genome. NOX2 (otherwise known as gp91Phox) is the best-characterized family member in terms of its regulation. gp91Phox is the catalytic unit of this multicomponent oxidase, whereas p22Phox, p40Phox, p47Phox, p67Phox, and the small GTPase RAC are regulatory subunits. Exposure of cells to pathogens triggers the assembly of cytosolic regulatory subunits (p40Phox, p47Phox, p67Phox, and RAC) with the transmembrane protein complex formed by gp91Phox and p22Phox (55). NOX2 mediates the transfer of an electron from NADPH to O2, forming the superoxide radical (O2˙−) (55). The superoxide radical is converted to hydrogen peroxide by superoxide dismutase. In the phagolysosome of neutrophils, myeloperoxidase (MPO) catalyzes the generation of hypochlorous acid (HOCI) from hydrogen peroxide and chloride ion. HOCl dramatically enhances the microbicidal activity of hydrogen peroxide (56). Proinflammatory cytokines such as IL-1β and tumor necrosis factor alpha (TNF-α) upregulate the expression of inducible nitric oxide synthase (iNOS), which produces nitric oxide from the amino acid l-arginine. Nitric oxide reacts with superoxide to generate peroxynitrite (OONO−), a potent nitrating agent and oxidant (Fig. 2D) (57). Both RNS and ROS cause damage to bacterial proteins and DNA by stimulating a variety of modifications, which can lead to cleavage of polypeptide chains, proteolytic degradation, enzyme inactivation, and protein cross-linking/aggregation, which inactivates invasive bacteria (58). Bacterial pathogens develop countermeasures against ROS and RNS production (59, 60). S. Typhimurium prevents phagocyte NADPH oxidase from trafficking toward Salmonella-containing vacuoles (61). F. tularensis produces antioxidant enzymes (SodB and SodC) that have ROS scavenging capacity, which suppresses innate immune activation and proinflammatory cytokine production (59). Host cells with impaired ROS production are susceptible to bacterial infections. It is estimated that 1 in 200,000 people are afflicted with chronic granulomatous disease (CGD), an immunodeficiency syndrome characterized by a profound susceptibility to bacterial infection due to the lack of NOX2 activity (62). The p47Phox-deficient mouse model of CGD succumbed to Burkholderia cepacia infection when given via the i.p. route, whereas the wild-type mice survived. Higher B. cepacia loads were observed in the peritoneum, spleen, kidney, and lungs of p47Phox mutant mice (63). Interestingly, HIF-1α stabilization enhances production of nitric oxide during UPEC infection. An increase in nitrite production was observed in AKB-4929-treated cells, which correlated with the upregulation of iNOS transcription (52). Although they have not had their antimicrobial activities demonstrated, multiple anticancer drugs are also shown to increase ROS levels within the cells (64). Further characterization of these drugs may provide novel platforms in treating bacterial infections.
Dampening host proinflammatory responses and thereby reducing bacterial load.
While mounting of an inflammatory response is critical in eradicating bacterial infection, a systemic bacterial infection can lead to an acute proinflammatory response, which may result in sepsis. Currently, no effective therapy is available to inhibit the activation phase of the acute inflammatory response to infection. A new group of host-protective lipids termed 13-series resolvins were demonstrated to promote bacterial phagocytosis, reduce recruitment of neutrophils to the site of inflammation, reduce inflammasome activation, and augment host recovery from systemic infection by accelerating the resolution of the acute inflammatory response (65). i.p. injection of resolvins into mice infected with E. coli showed a 40% increase in survival rate. Importantly, the resolvins did not exert direct bactericidal or bacteriostatic activity. The systemic administration of atorvastatin, which increased resolvin biosynthesis by means of S-nitrosylation-mediated activation of endothelial COX-2, significantly accelerated the resolution of infection and promoted survival in mice inoculated with E. coli (66). The discovery of bioactive lipids that target the resolution phase of the acute inflammatory response warrants further investigation as a new host-directed therapeutic strategy.
Alternatively, clavanin-MO, a synthetic peptide derived from clavanin A, demonstrated both host immune modulatory functions and direct antimicrobial activity against MDR strains of E. coli and methicillin-resistant S. aureus (67). In the cell-based study, clavanin-MO modulated immune responses by inducing anti-inflammatory cytokines (e.g., IL-10) and reduced LPS-mediated upregulation of proinflammatory cytokines (e.g., TNF-α and IL-12). In a murine animal model, i.p. infection by E. coli followed by i.p. treatment with clavanin-MO showed an 80% survival rate. This correlates with a significant decrease (~8 logs) in the number of viable bacterial counts found in the peritoneal fluid of clavanin-MO-treated groups (67). This illustrates the ability of the antibacterial/immunomodulatory peptide to control inflammation and promote survival of the host following bacterial infection.
MULTIOMICS APPROACHES TO IDENTIFY HOST TARGETS THAT MEDIATE BACTERIAL INFECTION
Advanced high-throughput technological developments in the fields of transcriptomics, proteomics, metabolomics, and imageomics (high-throughput, high-content imaging [HCI]) provide an unsurpassed opportunity for identifying host-pathogen interactors and characterizing gene functions in the context of bacterial infection. Transcriptomics studies enable quantitative measurements of the dynamic expression of the mRNA molecules and their variation in different states at the genome scale (68, 69). Proteomics studies (e.g., mass spectrometry [MS] and reverse-phase protein microarray [RPMA]) facilitate the characterization and quantitation of proteome changes from complex samples (70, 71). Metabolomics studies reveal metabolites (including lipids and small molecules) that are generated in response to infection (72). Importantly, technological advances related to assay miniaturization, high-throughput and automated image acquisition, and quantitative analysis have made it possible to extract hundreds of functional and morphological features that are associated with bacterial infections (73) (Fig. 3). Specific HCI parameters have been applied in host-directed therapeutic discovery to study effects of perturbations in the bacterial infection cycle (74–78). Analysis of the data derived from the omics studies will require bioinformatics tools that range from simple statistical analysis to sophisticated algorithms for large-scale analysis such as machine learning approaches that will help assign functional and biological information to the data set. Integration of the data from the different omics platforms will provide valuable correlation and correction of inferred models; help generate robust mechanistic models of molecular processes of the pathogen, the host, and their interactions; and enable a multidimensional view of functional host-pathogen interplay (79).
FIG 3 .

Identifying host targets that mediate bacterial infection through multiomics approaches. Integrative multiomics approaches encompass the following. (Top left) Transcriptomic studies enable quantitative measurements of the dynamic changes in mRNA expression at the genome scale. (Top right) The cell-based HCI assay enables the extraction of hundreds of cellular and subcellular morphological features that are associated with bacterial infections at the single-cell level. (Bottom left) Metabolomic studies reveal metabolites that are generated in response to infection. (Bottom right) Proteomic studies facilitate the characterization and quantitation of proteome changes from samples.
Host kinases exploited during bacterial infection.
The AKT1 signaling pathway is critical in modulating host responses to multiple bacterial infections and, hence, it is a suitable target for host-directed antibacterial therapeutics (80). A small interfering RNA (siRNA) kinome screen revealed that S. Typhimurium produces an effector protein, SopB, that causes misregulation of phagosome-lysosome fusion through its inhibition of AKT1 activation in the host (81). Screening a phosphatase-specific siRNA library in the context of S. Typhimurium- and M. tuberculosis-infected host cells revealed that approximately half the phosphatases identified impinged on AKT-related pathways (82). Phosphatidylinositol 3-kinase (PI3K)-mediated generation of phosphatidylinositol 2,4,5-triphosphate upon P. aeruginosa infection leads to the recruitment of AKT1, which facilitates bacterial transcytosis (83). Klebsiella pneumoniae induces an anti-inflammatory response by triggering a signaling axis that involves AKT1, which negatively regulates the NF-κB pathway (84). Given that the AKT1 kinase network is critical in modulating intracellular bacterial growth, an inhibitor that targets the PI3K/AKT pathway can be effective in treating bacterial infections. AR-12 is a celecoxib derivative that is devoid of cyclooxygenase 2 (COX-2) inhibitory activities (85). AR-12 inhibits the phosphorylation of PDK-1; subsequently, the phosphorylation and activation of AKT1 are inhibited, which may result in inhibition of the PI3K/AKT signaling pathway (85). It was also reported that AR-12 serves as an autophagy-inducing agent (86, 87). Oral administration of AR-12 in mice infected with S. Typhimurium reduced bacterial burden in spleens and livers by >90%, delayed the onset of the disease, and prolonged survival (88). Notably, AR-12 sensitized intracellular S. Typhimurium to aminoglycosides both in vitro and in vivo and prolonged the survival of infected mice (87). AR12 also inhibits F. novicida and F. tularensis intracellular survival in the cell-based assay (86). Although antibiotic resistance has not been identified in F. tularensis, continued exposure of this bacterial species to antibiotics, including aminoglycosides (gentamicin) and fluoroquinolones (ciprofloxacin), has the potential to generate drug-resistant strains (89).
Nonreceptor tyrosine kinases and inositol-requiring enzyme 1 (IRE1α) are also exploited during bacterial invasion, the life cycle, and cellular survival (90, 91). The recruitment of nonreceptor tyrosine kinases and their signaling axes is an integral part of bacterial internalization. Utilizing a panel of double-stranded RNAs (dsRNAs) targeting conserved genes involved in the regulation of the actin cytoskeleton, Pielage et al. described that the uptake of P. aeruginosa is a series of orchestrated events that requires Abl tyrosine kinase, the adapter protein Crk, the small GTPases Rac1 and Cdc42, and p21-activated kinase (90). Similarly, IRE1α, a conserved transmembrane kinase that regulates the host cell unfolded-protein response, was identified in an RNA interference (RNAi) screen and shown to be required for Brucella melitensis infection (91).
During the course of pathogen infections, protein phosphorylation is a critical cellular regulatory mechanism as receptors, signaling intermediates, and transcription factors are activated/deactivated by phosphorylation/dephosphorylation events. It is conceivable that modulating phosphorylation states of signaling components could be valuable for therapeutic intervention. RPMA studies have enabled the discovery of host targets through high-throughput detection of changes in expression and phosphorylation of host proteins during the course of bacterial infection. Modulation of host pathways was interrogated in extracts of RAW 264.7 macrophages infected with multiple B. pseudomallei strains at numerous time points postinfection. Detection of phosphorylated species of AMPK-α-1, Src, and glycogen synthase kinase 3β (GSK-3β) by the RPMA method suggested the involvement of metabolic pathways and regulation of innate immune pathways upon Burkholderia infection (71).
Interrogating the functional host-pathogen interplay through systems biology approaches.
An alternative approach to gain an in depth understanding of host-pathogen interactions during infection is to construct a protein-protein interaction (PPI) network between host protein and bacterial virulence factors. Using a yeast two-hybrid (Y2H) library that encompasses both human and murine factors, Memišević et al. successfully identified either human or murine proteins that interact with multiple B. mallei virulence factors (92). The resulting host-pathogen PPI network included three previously uncharacterized B. mallei virulence factors which were highly connected to host protein, highlighting their functional importance to the virulence of B. mallei and supporting the validity of the interactions described by the network (92). Similarly, a Y2H study conducted by Yang et al. showed the involvement of focal adhesion, regulation of cytoskeleton, leukocyte transendoepithelial migration, and the TLR and MAPK signaling pathways during Yersinia pestis infection (93). By using chemical cross-linking of proteins in combination with large-scale mass spectrometry, Schweppe et al. identified interactions between host structural proteins that mediate host cell-to-cell adhesion and the virulence factors of Acinetobacter baumannii during infection (94). Furthermore, the use of targeted quantitative metabolomics and high-throughput, unbiased proteomics revealed metabolites and proteins whose expressions were altered in mucosae isolated from chinchillas with nontypeable Haemophilus influenzae (NTHi)-induced acute otitis media. Treatment of human epithelial cells with an inhibitor targeting the identified actin-related protein Arp2/3 prevented NTHi infection (95). Detailed mechanistic studies of these host proteins in the context of infection can reveal crucial host-pathogen interactions, providing a platform for discovery of potential therapeutic interventions through disrupting host-pathogen complexes.
Identifying host-directed small molecules that regulate bacterial infection through drug screening.
A library of 640 FDA-approved drugs was screened to identify compounds that inhibited intracellular replication of C. burnetii, Legionella pneumophila, Brucella abortus, and Rickettsia conorii within a human monocytic cell line. Compounds that targeted G-protein-coupled receptors (GPCR) and intracellular calcium signals inhibited C. burnetii, L. pneumophila, B. abortus, and R. conorii replication, whereas those that target membrane cholesterol distribution reduced C. burnetii and L. pneumophila replication (96). Other FDA-approved drug repurposing efforts have uncovered nonantibiotic drugs that could either inhibit cell wall synthesis or enhance cell membrane permeability of Gram-negative bacteria (97). One should be aware that the artifactual effects of drugs or “promiscuous inhibitors” can inhibit multiple enzymes that impinge on the function of lysosomes and ultimately impact the survival of intracellular bacteria (34, 35). The concentration of a drug used under the screening condition can also greatly alter its specificity and selectivity with the expected target. Finally, using host-directed therapeutics in conjunction with antibiotics may provide a more effective way to eradicate the microbes, reduce the duration of antibiotic treatment, and alleviate tissue damage due to infection/inflammation.
PERSPECTIVE
Host-directed therapeutics offer promising adjunct therapeutic strategies to antibiotics for treating infections caused by drug-resistant Gram-negative bacteria. Promising host targets include various biological processes that modify host cell function, modulate the inflammatory response, or affect bacterial replication and virulence, which may be specific to a particular bacterial pathogen. Several large in vitro cell-based data sets have been generated which identify critical host-pathogen interactions (90–93). Performing meta-analyses to integrate orthogonal data sets combined with rational hypothesis-driven studies may identify candidate host targets for therapeutic intervention. While many studies have provided critical host pathways and proteins for targeting, there is a significant gap between the hypotheses and preclinical data and a lack of tool compounds that are capable of bridging the two in animal models of infection.
Although promising, challenges to the development of host-directed agents should be considered. For example, in order to take advantage of the low-hanging fruit of a repurposed FDA-approved nonantibiotic as adjunct therapy, the unbound plasma trough levels at the approved dosing regimen of the nonantibiotic should be greater than the in vitro 50% effective concentration (EC50) (or MIC) value when tested in combination. This concept is also fundamental for the development of tool compounds. Once a host-directed hypothesis is validated in vivo, the results need to be translated to humans. As opposed to direct antibiotics that have more predictable pharmacokinetic/pharmacodynamics relationships, host-directed therapeutics rely on the drug’s interaction with the host and subsequent downstream events within the animal’s immune system. This not only complicates the interpretation of efficacy experiments but introduces many variables in the translation of the results to humans. For example, the genomic responses in mouse models have been argued to poorly mimic human inflammation (98). For serious infections, where it becomes unethical to not provide the standard of care to the patient, the clinical studies of host-directed therapeutic combinations (with an antibiotic) may be required to show an advantage over the standard of care (antibiotic alone). In these cases, the achievement of a statistically significant outcome may be challenging. Potential host-directed therapeutics may encompass a great diversity of drug classes and may be accompanied by risks and concerns that need to be considered. Many potential targets are involved in numerous pathways, which highlight the possibility of toxicity. Therapeutics that stimulate the immune system are associated with risks of excessive inflammation leading to cytokine storm or systemic inflammatory response syndrome, which is deleterious to the patient’s ability to clear the infection. Additionally, the time of administration of host-directed therapeutics is critical as they typically target a particular phase of infection. Therefore, the treatment window will need to be clearly defined to maximize the therapeutic benefits of the treatment. Despite these challenges, successful identification and development of host-directed therapeutics will lead to a promising adjunctive therapy to treat infections caused by multidrug-resistant bacteria.
ACKNOWLEDGMENTS
This work was supported by the Department of Defense Chemical Biological Defense Program through the Defense Threat Reduction Agency (DTRA) under United States Army Medical Research Institute of Infectious Diseases (USAMRIID) project number 13267645.
Opinions, interpretations, conclusions, and recommendations are those of the authors and are not necessarily endorsed by the U.S. Army.
Footnotes
Citation Chiang C-Y, Uzoma I, Moore RT, Gilbert M, Duplantier AJ, Panchal RG. 2018. Mitigating the impact of antibacterial drug resistance through host-directed therapies: current progress, outlook, and challenges. mBio 9:e01932-17. https://doi.org/10.1128/mBio.01932-17.
Contributor Information
Rino Rappuoli, GSK Vaccines.
Danielle A. Garsin, University of Texas Health Science Center at Houston.
REFERENCES
- 1.McGann P, Snesrud E, Maybank R, Corey B, Ong AC, Clifford R, Hinkle M, Whitman T, Lesho E, Schaecher KE. 2016. Escherichia coli harboring mcr-1 and blaCTX-M on a novel IncF plasmid: first report of mcr-1 in the United States. Antimicrob Agents Chemother 60:4420–4421. doi: 10.1128/AAC.01103-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen L, Todd R, Kiehlbauch J, Walters M, Kallen A. 2017. Notes from the field: pan-resistant New Delhi metallo-beta-lactamase-producing Klebsiella pneumoniae—Washoe County, Nevada, 2016. MMWR Morb Mortal Wkly Rep 66:33. doi: 10.15585/mmwr.mm6601a7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Magiorakos AP, Srinivasan A, Carey RB, Carmeli Y, Falagas ME, Giske CG, Harbarth S, Hindler JF, Kahlmeter G, Olsson-Liljequist B, Paterson DL, Rice LB, Stelling J, Struelens MJ, Vatopoulos A, Weber JT, Monnet DL. 2012. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: an international expert proposal for interim standard definitions for acquired resistance. Clin Microbiol Infect 18:268–281. doi: 10.1111/j.1469-0691.2011.03570.x. [DOI] [PubMed] [Google Scholar]
- 4.World Health Organization 2014. Antimicrobial resistance: global report on surveillance 2014. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 5.Hancock RE, Nijnik A, Philpott DJ. 2012. Modulating immunity as a therapy for bacterial infections. Nat Rev Microbiol 10:243–254. doi: 10.1038/nrmicro2745. [DOI] [PubMed] [Google Scholar]
- 6.Tamma PD, Cosgrove SE, Maragakis LL. 2012. Combination therapy for treatment of infections with gram-negative bacteria. Clin Microbiol Rev 25:450–470. doi: 10.1128/CMR.05041-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Di Russo Case E, Samuel JE. 2016. Contrasting lifestyles within the host cell. Microbiol Spectr 4(1). doi: 10.1128/microbiolspec.VMBF-0014-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blackwell JM, Searle S, Goswami T, Miller EN. 2000. Understanding the multiple functions of Nramp1. Microbes Infect 2:317–321. doi: 10.1016/S1286-4579(00)00295-1. [DOI] [PubMed] [Google Scholar]
- 9.Mehra A, Zahra A, Thompson V, Sirisaengtaksin N, Wells A, Porto M, Köster S, Penberthy K, Kubota Y, Dricot A, Rogan D, Vidal M, Hill DE, Bean AJ, Philips JA. 2013. Mycobacterium tuberculosis type VII secreted effector EsxH targets host ESCRT to impair trafficking. PLoS Pathog 9:e1003734. doi: 10.1371/journal.ppat.1003734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van Schaik EJ, Chen C, Mertens K, Weber MM, Samuel JE. 2013. Molecular pathogenesis of the obligate intracellular bacterium Coxiella burnetii. Nat Rev Microbiol 11:561–573. doi: 10.1038/nrmicro3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bakowski MA, Braun V, Brumell JH. 2008. Salmonella-containing vacuoles: directing traffic and nesting to grow. Traffic 9:2022–2031. doi: 10.1111/j.1600-0854.2008.00827.x. [DOI] [PubMed] [Google Scholar]
- 12.Eswarappa SM, Negi VD, Chakraborty S, Chandrasekhar Sagar BK, Chakravortty D. 2010. Division of the Salmonella-containing vacuole and depletion of acidic lysosomes in Salmonella-infected host cells are novel strategies of Salmonella enterica to avoid lysosomes. Infect Immun 78:68–79. doi: 10.1128/IAI.00668-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ogawa M, Yoshimori T, Suzuki T, Sagara H, Mizushima N, Sasakawa C. 2005. Escape of intracellular Shigella from autophagy. Science 307:727–731. doi: 10.1126/science.1106036. [DOI] [PubMed] [Google Scholar]
- 14.Cullinane M, Gong L, Li X, Lazar-Adler N, Tra T, Wolvetang E, Prescott M, Boyce JD, Devenish RJ, Adler B. 2008. Stimulation of autophagy suppresses the intracellular survival of Burkholderia pseudomallei in mammalian cell lines. Autophagy 4:744–753. doi: 10.4161/auto.6246. [DOI] [PubMed] [Google Scholar]
- 15.Butchar JP, Cremer TJ, Clay CD, Gavrilin MA, Wewers MD, Marsh CB, Schlesinger LS, Tridandapani S. 2008. Microarray analysis of human monocytes infected with Francisella tularensis identifies new targets of host response subversion. PLoS One 3:e2924. doi: 10.1371/journal.pone.0002924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Birmingham CL, Canadien V, Gouin E, Troy EB, Yoshimori T, Cossart P, Higgins DE, Brumell JH. 2007. Listeria monocytogenes evades killing by autophagy during colonization of host cells. Autophagy 3:442–451. doi: 10.4161/auto.4450. [DOI] [PubMed] [Google Scholar]
- 17.Ray K, Marteyn B, Sansonetti PJ, Tang CM. 2009. Life on the inside: the intracellular lifestyle of cytosolic bacteria. Nat Rev Microbiol 7:333–340. doi: 10.1038/nrmicro2112. [DOI] [PubMed] [Google Scholar]
- 18.Whiteley L, Meffert T, Haug M, Weidenmaier C, Hopf V, Bitschar K, Schittek B, Kohler C, Steinmetz I, West TE, Schwarz S. 2017. Entry, intracellular survival, and multinucleated-giant-cell-forming activity of Burkholderia pseudomallei in human primary phagocytic and nonphagocytic cells. Infect Immun 85:e00468-17. doi: 10.1128/IAI.00468-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baxt LA, Garza-Mayers AC, Goldberg MB. 2013. Bacterial subversion of host innate immune pathways. Science 340:697–701. doi: 10.1126/science.1235771. [DOI] [PubMed] [Google Scholar]
- 20.Diacovich L, Gorvel JP. 2010. Bacterial manipulation of innate immunity to promote infection. Nat Rev Microbiol 8:117–128. doi: 10.1038/nrmicro2295. [DOI] [PubMed] [Google Scholar]
- 21.Satoh T, Akira S. 2016. Toll-like receptor signaling and its inducible proteins. Microbiol Spectr 4(6). doi: 10.1128/microbiolspec.MCHD-0040-2016. [DOI] [PubMed] [Google Scholar]
- 22.Takeuchi O, Akira S. 2010. Pattern recognition receptors and inflammation. Cell 140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 23.Hirano T, Kodama S, Kawano T, Maeda K, Suzuki M. 2011. Monophosphoryl lipid A induced innate immune responses via TLR4 to enhance clearance of nontypeable Haemophilus influenzae and Moraxella catarrhalis from the nasopharynx in mice. FEMS Immunol Med Microbiol 63:407–417. doi: 10.1111/j.1574-695X.2011.00866.x. [DOI] [PubMed] [Google Scholar]
- 24.Bagnoli F, Fontana MR, Soldaini E, Mishra RP, Fiaschi L, Cartocci E, Nardi-Dei V, Ruggiero P, Nosari S, De Falco MG, Lofano G, Marchi S, Galletti B, Mariotti P, Bacconi M, Torre A, Maccari S, Scarselli M, Rinaudo CD, Inoshima N, Savino S, Mori E, Rossi-Paccani S, Baudner B, Pallaoro M, Swennen E, Petracca R, Brettoni C, Liberatori S, Norais N, Monaci E, Bubeck Wardenburg J, Schneewind O, O’Hagan DT, Valiante NM, Bensi G, Bertholet S, De Gregorio E, Rappuoli R, Grandi G. 2015. Vaccine composition formulated with a novel TLR7-dependent adjuvant induces high and broad protection against Staphylococcus aureus. Proc Natl Acad Sci U S A 112:3680–3685. doi: 10.1073/pnas.1424924112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saxena M, Yeretssian G. 2014. Nod-like receptors: master regulators of inflammation and cancer. Front Immunol 5:327. doi: 10.3389/fimmu.2014.00327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rathinam VA, Vanaja SK, Fitzgerald KA. 2012. Regulation of inflammasome signaling. Nat Immunol 13:333–342. doi: 10.1038/ni.2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Caruso R, Warner N, Inohara N, Núñez G. 2014. NOD1 and NOD2: signaling, host defense, and inflammatory disease. Immunity 41:898–908. doi: 10.1016/j.immuni.2014.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guo H, Callaway JB, Ting JP. 2015. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med 21:677–687. doi: 10.1038/nm.3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matusiak M, Van Opdenbosch N, Vande Walle L, Sirard JC, Kanneganti TD, Lamkanfi M. 2015. Flagellin-induced NLRC4 phosphorylation primes the inflammasome for activation by NAIP5. Proc Natl Acad Sci U S A 112:1541–1546. doi: 10.1073/pnas.1417945112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ceballos-Olvera I, Sahoo M, Miller MA, Del Barrio L, Re F. 2011. Inflammasome-dependent pyroptosis and IL-18 protect against Burkholderia pseudomallei lung infection while IL-1beta is deleterious. PLoS Pathog 7:e1002452. doi: 10.1371/journal.ppat.1002452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koh GC, Weehuizen TA, Breitbach K, Krause K, de Jong HK, Kager LM, Hoogendijk AJ, Bast A, Peacock SJ, van der Poll T, Steinmetz I, Wiersinga WJ. 2013. Glyburide reduces bacterial dissemination in a mouse model of melioidosis. PLoS Neglect Trop Dis 7:e2500. doi: 10.1371/journal.pntd.0002500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu X, Foo G, Lim WP, Ravikumar S, Sim SH, Win MS, Goh JG, Lim JH, Ng YH, Fisher D, Khoo CM, Tan G, Chai LY. 2014. Sulphonylurea usage in melioidosis is associated with severe disease and suppressed immune response. PLoS Negl Trop Dis 8:e2795. doi: 10.1371/journal.pntd.0002795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kewcharoenwong C, Rinchai D, Utispan K, Suwannasaen D, Bancroft GJ, Ato M, Lertmemongkolchai G. 2013. Glibenclamide reduces pro-inflammatory cytokine production by neutrophils of diabetes patients in response to bacterial infection. Sci Rep 3:3363. doi: 10.1038/srep03363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Koh GC, Maude RR, Schreiber MF, Limmathurotsakul D, Wiersinga WJ, Wuthiekanun V, Lee SJ, Mahavanakul W, Chaowagul W, Chierakul W, White NJ, van der Poll T, Day NP, Dougan G, Peacock SJ. 2011. Glyburide is anti-inflammatory and associated with reduced mortality in melioidosis. Clin Infect Dis 52:717–725. doi: 10.1093/cid/ciq192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Coll RC, Robertson AA, Chae JJ, Higgins SC, Muñoz-Planillo R, Inserra MC, Vetter I, Dungan LS, Monks BG, Stutz A, Croker DE, Butler MS, Haneklaus M, Sutton CE, Núñez G, Latz E, Kastner DL, Mills KH, Masters SL, Schroder K, Cooper MA, O’Neill LA. 2015. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med 21:248–255. doi: 10.1038/nm.3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ryter SW, Cloonan SM, Choi AM. 2013. Autophagy: a critical regulator of cellular metabolism and homeostasis. Mol Cells 36:7–16. doi: 10.1007/s10059-013-0140-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Deretic V, Saitoh T, Akira S. 2013. Autophagy in infection, inflammation and immunity. Nat Rev Immunol 13:722–737. doi: 10.1038/nri3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang J, Brumell JH. 2014. Bacteria-autophagy interplay: a battle for survival. Nat Rev Microbiol 12:101–114. doi: 10.1038/nrmicro3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mizushima N. 2007. Autophagy: process and function. Genes Dev 21:2861–2873. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- 40.Tjelle TE, Brech A, Juvet LK, Griffiths G, Berg T. 1996. Isolation and characterization of early endosomes, late endosomes and terminal lysosomes: their role in protein degradation. J Cell Sci 109:2905–2914. [DOI] [PubMed] [Google Scholar]
- 41.Deretic V. 2012. Autophagy as an innate immunity paradigm: expanding the scope and repertoire of pattern recognition receptors. Curr Opin Immunol 24:21–31. doi: 10.1016/j.coi.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lai SC, Devenish RJ. 2012. LC3-associated phagocytosis (LAP): connections with host autophagy. Cells 1:396–408. doi: 10.3390/cells1030396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rinchai D, Riyapa D, Buddhisa S, Utispan K, Titball RW, Stevens MP, Stevens JM, Ogawa M, Tanida I, Koike M, Uchiyama Y, Ato M, Lertmemongkolchai G. 2015. Macroautophagy is essential for killing of intracellular Burkholderia pseudomallei in human neutrophils. Autophagy 11:748–755. doi: 10.1080/15548627.2015.1040969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Webber JL, Tooze SA. 2010. Coordinated regulation of autophagy by p38alpha MAPK through mAtg9 and p38IP. EMBO J 29:27–40. doi: 10.1038/emboj.2009.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stanley SA, Barczak AK, Silvis MR, Luo SS, Sogi K, Vokes M, Bray MA, Carpenter AE, Moore CB, Siddiqi N, Rubin EJ, Hung DT. 2014. Identification of host-targeted small molecules that restrict intracellular Mycobacterium tuberculosis growth. PLoS Pathog 10:e1003946. doi: 10.1371/journal.ppat.1003946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kramer L, Turk D, Turk B. 2017. The future of cysteine cathepsins in disease management. Trends Pharmacol Sci 38:873–898. doi: 10.1016/j.tips.2017.06.003. [DOI] [PubMed] [Google Scholar]
- 47.Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, Fitzgerald KA, Latz E. 2008. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol 9:847–856. doi: 10.1038/ni.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Qi X, Man SM, Malireddi RK, Karki R, Lupfer C, Gurung P, Neale G, Guy CS, Lamkanfi M, Kanneganti TD. 2016. Cathepsin B modulates lysosomal biogenesis and host defense against Francisella novicida infection. J Exp Med 213:2081–2097. doi: 10.1084/jem.20151938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schaffer K, Taylor CT. 2015. The impact of hypoxia on bacterial infection. FEBS J 282:2260–2266. doi: 10.1111/febs.13270. [DOI] [PubMed] [Google Scholar]
- 50.Rius J, Guma M, Schachtrup C, Akassoglou K, Zinkernagel AS, Nizet V, Johnson RS, Haddad GG, Karin M. 2008. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature 453:807–811. doi: 10.1038/nature06905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Peyssonnaux C, Datta V, Cramer T, Doedens A, Theodorakis EA, Gallo RL, Hurtado-Ziola N, Nizet V, Johnson RS. 2005. HIF-1alpha expression regulates the bactericidal capacity of phagocytes. J Clin Invest 115:1806–1815. doi: 10.1172/JCI23865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lin AE, Beasley FC, Olson J, Keller N, Shalwitz RA, Hannan TJ, Hultgren SJ, Nizet V. 2015. Role of hypoxia inducible factor-1alpha (HIF-1alpha) in innate defense against uropathogenic Escherichia coli infection. PLoS Pathog 11:e1004818. doi: 10.1371/journal.ppat.1004818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bhandari T, Nizet V. 2014. Hypoxia-inducible factor (HIF) as a pharmacological target for prevention and treatment of infectious diseases. Infect Dis Ther 3:159–174. doi: 10.1007/s40121-014-0030-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rastogi R, Geng X, Li F, Ding Y. 2016. NOX activation by subunit interaction and underlying mechanisms in disease. Front Cell Neurosci 10:301. doi: 10.3389/fncel.2016.00301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lambeth JD. 2004. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol 4:181–189. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- 56.Winterbourn CC, Hampton MB, Livesey JH, Kettle AJ. 2006. Modeling the reactions of superoxide and myeloperoxidase in the neutrophil phagosome: implications for microbial killing. J Biol Chem 281:39860–39869. doi: 10.1074/jbc.M605898200. [DOI] [PubMed] [Google Scholar]
- 57.Fang FC. 2004. Antimicrobial reactive oxygen and nitrogen species: concepts and controversies. Nat Rev Microbiol 2:820–832. doi: 10.1038/nrmicro1004. [DOI] [PubMed] [Google Scholar]
- 58.Vatansever F, de Melo WC, Avci P, Vecchio D, Sadasivam M, Gupta A, Chandran R, Karimi M, Parizotto NA, Yin R, Tegos GP, Hamblin MR. 2013. Antimicrobial strategies centered around reactive oxygen species—bactericidal antibiotics, photodynamic therapy, and beyond. FEMS Microbiol Rev 37:955–989. doi: 10.1111/1574-6976.12026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rabadi SM, Sanchez BC, Varanat M, Ma Z, Catlett SV, Melendez JA, Malik M, Bakshi CS. 2016. Antioxidant defenses of Francisella tularensis modulate macrophage function and production of proinflammatory cytokines. J Biol Chem 291:5009–5021. doi: 10.1074/jbc.M115.681478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nathan C, Shiloh MU. 2000. Reactive oxygen and nitrogen intermediates in the relationship between mammalian hosts and microbial pathogens. Proc Natl Acad Sci U S A 97:8841–8848. doi: 10.1073/pnas.97.16.8841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vazquez-Torres A, Xu Y, Jones-Carson J, Holden DW, Lucia SM, Dinauer MC, Mastroeni P, Fang FC. 2000. Salmonella pathogenicity island 2-dependent evasion of the phagocyte NADPH oxidase. Science 287:1655–1658. doi: 10.1126/science.287.5458.1655. [DOI] [PubMed] [Google Scholar]
- 62.Kuhns DB, Alvord WG, Heller T, Feld JJ, Pike KM, Marciano BE, Uzel G, DeRavin SS, Priel DA, Soule BP, Zarember KA, Malech HL, Holland SM, Gallin JI. 2010. Residual NADPH oxidase and survival in chronic granulomatous disease. N Engl J Med 363:2600–2610. doi: 10.1056/NEJMoa1007097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pizzolla A, Hultqvist M, Nilson B, Grimm MJ, Eneljung T, Jonsson IM, Verdrengh M, Kelkka T, Gjertsson I, Segal BH, Holmdahl R. 2012. Reactive oxygen species produced by the NADPH oxidase 2 complex in monocytes protect mice from bacterial infections. J Immunol 188:5003–5011. doi: 10.4049/jimmunol.1103430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang L, Li J, Zong L, Chen X, Chen K, Jiang Z, Nan L, Li X, Li W, Shan T, Ma Q, Ma Z. 2016. Reactive oxygen species and targeted therapy for pancreatic cancer. Oxid Med Cell Longev 2016:1616781. doi: 10.1155/2016/1616781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lee CR, Zeldin DC. 2015. Resolvin infectious inflammation by targeting the host response. N Engl J Med 373:2183–2185. doi: 10.1056/NEJMcibr1511280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dalli J, Chiang N, Serhan CN. 2015. Elucidation of novel 13-series resolvins that increase with atorvastatin and clear infections. Nat Med 21:1071–1075. doi: 10.1038/nm.3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Silva ON, de la Fuente-Núñez C, Haney EF, Fensterseifer IC, Ribeiro SM, Porto WF, Brown P, Faria-Junior C, Rezende TM, Moreno SE, Lu TK, Hancock RE, Franco OL. 2016. An anti-infective synthetic peptide with dual antimicrobial and immunomodulatory activities. Sci Rep 6:35465. doi: 10.1038/srep35465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Leroy Q, Raoult D. 2010. Review of microarray studies for host-intracellular pathogen interactions. J Microbiol Methods 81:81–95. doi: 10.1016/j.mimet.2010.02.008. [DOI] [PubMed] [Google Scholar]
- 69.Westermann AJ, Gorski SA, Vogel J. 2012. Dual RNA-seq of pathogen and host. Nat Rev Microbiol 10:618–630. doi: 10.1038/nrmicro2852. [DOI] [PubMed] [Google Scholar]
- 70.Jean Beltran PM, Federspiel JD, Sheng X, Cristea IM. 2017. Proteomics and integrative omic approaches for understanding host-pathogen interactions and infectious diseases. Mol Syst Biol 13:922. doi: 10.15252/msb.20167062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chiang CY, Uzoma I, Lane DJ, Memišević V, Alem F, Yao K, Kota KP, Bavari S, Wallqvist A, Hakami RM, Panchal RG. 2015. A reverse-phase protein microarray-based screen identifies host signaling dynamics upon Burkholderia spp. infection. Front Microbiol 6:683. doi: 10.3389/fmicb.2015.00683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Eisenreich W, Heesemann J, Rudel T, Goebel W. 2013. Metabolic host responses to infection by intracellular bacterial pathogens. Front Cell Infect Microbiol 3:24. doi: 10.3389/fcimb.2013.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bray MA, Singh S, Han H, Davis CT, Borgeson B, Hartland C, Kost-Alimova M, Gustafsdottir SM, Gibson CC, Carpenter AE. 2016. Cell painting, a high-content image-based assay for morphological profiling using multiplexed fluorescent dyes. Nat Protoc 11:1757–1774. doi: 10.1038/nprot.2016.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kota KP, Eaton B, Lane D, Ulrich M, Ulrich R, Peyser BD, Robinson CG, Jaissle JG, Pegoraro G, Bavari S, Panchal RG. 2013. Integrating high-content imaging and chemical genetics to probe host cellular pathways critical for Yersinia pestis infection. PLoS One 8:e55167. doi: 10.1371/journal.pone.0055167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chiang CY, Ulrich RL, Ulrich MP, Eaton B, Ojeda JF, Lane DJ, Kota KP, Kenny TA, Ladner JT, Dickson SP, Kuehl K, Raychaudhuri R, Sun M, Bavari S, Wolcott MJ, Covell D, Panchal RG. 2015. Characterization of the murine macrophage response to infection with virulent and avirulent Burkholderia species. BMC Microbiol 15:259. doi: 10.1186/s12866-015-0593-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Serio AW, Jeng RL, Haglund CM, Reed SC, Welch MD. 2010. Defining a core set of actin cytoskeletal proteins critical for actin-based motility of Rickettsia. Cell Host Microbe 7:388–398. doi: 10.1016/j.chom.2010.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pegoraro G, Bavari S, Panchal RG. 2012. Shedding light on filovirus infection with high-content imaging. Viruses 4:1354–1371. doi: 10.3390/v4081354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Casanova A, Low SH, Emmenlauer M, Conde-Alvarez R, Salcedo SP, Gorvel JP, Dehio C. 2016. Microscopy-based assays for high-throughput screening of host factors involved in Brucella infection of Hela cells. J Vis Exp doi: 10.3791/54263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Poultney CS, Greenfield A, Bonneau R. 2012. Integrated inference and analysis of regulatory networks from multi-level measurements. Methods Cell Biol 110:19–56. doi: 10.1016/B978-0-12-388403-9.00002-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Krachler AM, Woolery AR, Orth K. 2011. Manipulation of kinase signaling by bacterial pathogens. J Cell Biol 195:1083–1092. doi: 10.1083/jcb.201107132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kuijl C, Savage ND, Marsman M, Tuin AW, Janssen L, Egan DA, Ketema M, van den Nieuwendijk R, van den Eeden SJ, Geluk A, Poot A, van der Marel G, Beijersbergen RL, Overkleeft H, Ottenhoff TH, Neefjes J. 2007. Intracellular bacterial growth is controlled by a kinase network around PKB/AKT1. Nature 450:725–730. doi: 10.1038/nature06345. [DOI] [PubMed] [Google Scholar]
- 82.Albers HM, Kuijl C, Bakker J, Hendrickx L, Wekker S, Farhou N, Liu N, Blasco-Moreno B, Scanu T, den Hertog J, Celie P, Ovaa H, Neefjes J. 2014. Integrating chemical and genetic silencing strategies to identify host kinase-phosphatase inhibitor networks that control bacterial infection. ACS Chem Biol 9:414–422. doi: 10.1021/cb400421a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kierbel A, Gassama-Diagne A, Rocha C, Radoshevich L, Olson J, Mostov K, Engel J. 2007. Pseudomonas aeruginosa exploits a PIP3-dependent pathway to transform apical into basolateral membrane. J Cell Biol 177:21–27. doi: 10.1083/jcb.200605142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Frank CG, Reguerio V, Rother M, Moranta D, Maeurer AP, Garmendia J, Meyer TF, Bengoechea JA. 2013. Klebsiella pneumoniae targets an EGF receptor-dependent pathway to subvert inflammation. Cell Microbiol 15:1212–1233. doi: 10.1111/cmi.12110. [DOI] [PubMed] [Google Scholar]
- 85.Zhu J, Huang JW, Tseng PH, Yang YT, Fowble J, Shiau CW, Shaw YJ, Kulp SK, Chen CS. 2004. From the cyclooxygenase-2 inhibitor celecoxib to a novel class of 3-phosphoinositide-dependent protein kinase-1 inhibitors. Cancer Res 64:4309–4318. doi: 10.1158/0008-5472.CAN-03-4063. [DOI] [PubMed] [Google Scholar]
- 86.Chiu HC, Soni S, Kulp SK, Curry H, Wang D, Gunn JS, Schlesinger LS, Chen CS. 2009. Eradication of intracellular Francisella tularensis in THP-1 human macrophages with a novel autophagy inducing agent. J Biomed Sci 16:110. doi: 10.1186/1423-0127-16-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lo JH, Kulp SK, Chen CS, Chiu HC. 2014. Sensitization of intracellular Salmonella enterica serovar Typhimurium to aminoglycosides in vitro and in vivo by a host-targeted antimicrobial agent. Antimicrob Agents Chemother 58:7375–7382. doi: 10.1128/AAC.03778-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chiu HC, Kulp SK, Soni S, Wang D, Gunn JS, Schlesinger LS, Chen CS. 2009. Eradication of intracellular Salmonella enterica serovar Typhimurium with a small-molecule, host cell-directed agent. Antimicrob Agents Chemother 53:5236–5244. doi: 10.1128/AAC.00555-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sutera V, Levert M, Burmeister WP, Schneider D, Maurin M. 2014. Evolution toward high-level fluoroquinolone resistance in Francisella species. J Antimicrob Chemother 69:101–110. doi: 10.1093/jac/dkt321. [DOI] [PubMed] [Google Scholar]
- 90.Pielage JF, Powell KR, Kalman D, Engel JN. 2008. RNAi screen reveals an Abl kinase-dependent host cell pathway involved in Pseudomonas aeruginosa internalization. PLoS Pathog 4:e1000031. doi: 10.1371/journal.ppat.1000031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Qin QM, Pei J, Ancona V, Shaw BD, Ficht TA, de Figueiredo P. 2008. RNAi screen of endoplasmic reticulum-associated host factors reveals a role for IRE1alpha in supporting Brucella replication. PLoS Pathog 4:e1000110. doi: 10.1371/journal.ppat.1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Memišević V, Zavaljevski N, Pieper R, Rajagopala SV, Kwon K, Townsend K, Yu C, Yu X, DeShazer D, Reifman J, Wallqvist A. 2013. Novel Burkholderia mallei virulence factors linked to specific host-pathogen protein interactions. Mol Cell Proteomics 12:3036–3051. doi: 10.1074/mcp.M113.029041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yang H, Ke Y, Wang J, Tan Y, Myeni SK, Li D, Shi Q, Yan Y, Chen H, Guo Z, Yuan Y, Yang X, Yang R, Du Z. 2011. Insight into bacterial virulence mechanisms against host immune response via the Yersinia pestis-human protein-protein interaction network. Infect Immun 79:4413–4424. doi: 10.1128/IAI.05622-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Schweppe DK, Harding C, Chavez JD, Wu X, Ramage E, Singh PK, Manoil C, Bruce JE. 2015. Host-microbe protein interactions during bacterial infection. Chem Biol 22:1521–1530. doi: 10.1016/j.chembiol.2015.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Harrison A, Dubois LG, St John-Williams L, Moseley MA, Hardison RL, Heimlich DR, Stoddard A, Kerschner JE, Justice SS, Thompson JW, Mason KM. 2016. Comprehensive proteomic and metabolomic signatures of nontypeable Haemophilus influenzae-induced acute otitis media reveal bacterial aerobic respiration in an immunosuppressed environment. Mol Cell Proteomics 15:1117–1138. doi: 10.1074/mcp.M115.052498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Czyż DM, Potluri LP, Jain-Gupta N, Riley SP, Martinez JJ, Steck TL, Crosson S, Shuman HA, Gabay JE. 2014. Host-directed antimicrobial drugs with broad-spectrum efficacy against intracellular bacterial pathogens. mBio 5:e01534-14. doi: 10.1128/mBio.01534-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Brown D. 2015. Antibiotic resistance breakers: can repurposed drugs fill the antibiotic discovery void? Nat Rev Drug Discov 14:821–832. doi: 10.1038/nrd4675. [DOI] [PubMed] [Google Scholar]
- 98.Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, Richards DR, McDonald-Smith GP, Gao H, Hennessy L, Finnerty CC, López CM, Honari S, Moore EE, Minei JP, Cuschieri J, Bankey PE, Johnson JL, Sperry J, Nathens AB, Billiar TR, West MA, Jeschke MG, Klein MB, Gamelli RL, Gibran NS, Brownstein BH, Miller-Graziano C, Calvano SE, Mason PH, Cobb JP, Rahme LG, Lowry SF, Maier RV, Moldawer LL, Herndon DN, Davis RW, Xiao W, Tompkins RG, Inflammation and Host Response to Injury, Large Scale Collaborative Research Program . 2013. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci U S A 110:3507–3512. doi: 10.1073/pnas.1222878110. [DOI] [PMC free article] [PubMed] [Google Scholar]