

ABSTRACT
In this study, an in vitro infection model for the hepatitis delta virus (HDV) was used to evaluate the antiviral effects of phosphorothioate nucleic acid polymers (NAPs) and investigate their mechanism of action. The results show that NAPs inhibit HDV infection at concentrations less than 4 μM in cultures of differentiated human hepatoma cells. NAPs were shown to be active at viral entry but inactive postentry on HDV RNA replication. Inhibition was independent of the NAP nucleotide sequence but dependent on both size and amphipathicity of the polymer. NAP antiviral activity was effective against HDV virions bearing the main hepatitis B virus (HBV) immune escape substitutions (D144A and G145R) and was pangenomic with regard to HBV envelope proteins. Furthermore, similar to immobilized heparin, immobilized NAPs could bind HDV particles, suggesting that entry inhibition was due, at least in part, to preventing attachment of the virus to cell surface glycosaminoglycans. The results document NAPs as a novel class of antiviral compounds that can prevent HDV propagation.
IMPORTANCE HDV infection causes the most severe form of viral hepatitis in humans and one of the most difficult to cure. Currently, treatments are limited to long-term administration of interferon at high doses, which provide only partial efficacy. There is thus an urgent need for innovative approaches to identify new antiviral against HDV. The significance of our study is in demonstrating that nucleic acid polymers (NAPs) are active against HDV by targeting the envelope of HDV virions. In an in vitro infection assay, NAP activity was recorded at concentrations less than 4 μM in the absence of cell toxicity. Furthermore, the fact that NAPs could block HDV at viral entry suggests their potential to control the spread of HDV in a chronically HBV-infected liver. In addition, NAP anti-HDV activity was pangenomic with regard to HBV envelope proteins and not circumvented by HBsAg substitutions associated with HBV immune escape.
KEYWORDS: HDV, hepatitis D virus, NAPs, nucleic acid polymers
INTRODUCTION
Worldwide, it is estimated that more than 240 million people are chronically infected with the hepatitis B virus (HBV) (1), approximately 15 to 20 million of whom are coinfected with the hepatitis D virus (HDV), an obligate satellite of HBV which uses the envelope proteins of the latter to assemble infectious HDV virions. As a consequence, only in the presence of the helper HBV can HDV propagate. HDV infection can occur either simultaneously with HBV (coinfection) or upon superinfection of a chronic HBV carrier (2). The latter scenario can lead to the most severe form of chronic viral liver disorder, characterized by an accelerated evolution toward cirrhosis and hepatocellular carcinoma in comparison to HBV monoinfection. In industrialized countries where HBV vaccination programs have been implemented, a decrease of HDV prevalence has been observed, but recent studies have revealed a return of the virus to some parts of Europe (3).
HDV is marked with the smallest of human viral genomes, a circular, single-stranded RNA of approximately 1,700 nucleotides (nt) in length. To ensure replication, the HDV RNA must hijack cellular enzymes, including DNA-dependent RNA polymerase II, for synthesis of cRNA intermediates using a rolling-circle mechanism of transcription similar to that utilized by plant viroids. Because the HDV genome encodes a single viral protein, expressed as two isoforms that are devoid of known enzymatic activity, the HDV replication cycle is hardly amenable to direct-acting antiviral targeting. As a consequence, the current treatment of chronic hepatitis D is limited to long-term administration of pegylated interferon, which is successful in less than 20% of cases given that a significant number of individuals who become HDV RNA negative at the end of treatment relapse at later time points (4, 5). Hence, new strategies are needed to overcome HDV infection, and nucleic acid polymers (NAPs) constitute an innovative concept.
NAPs are phosphorothioated oligonucleotides, which have been shown to exhibit a broad-spectrum antiviral activity. They are oligonucleotides in which phosphorothioation of a nonbridging oxygen atom in the phosphodiester linkage is applied to confer stability and amphipathic properties (6). The antiviral effect of NAPs observed in different viral species is independent of the nucleotide sequence but strictly dependent upon the presence of phosphorothioation/amphipathicity. As antiviral agents, NAPs can bind uncomplexed amphipathic α-helices in class I viral glycoproteins (7, 8). Such structures are present in surface proteins of many enveloped viruses, which might explain the broad-spectrum activity of NAPs against viruses such as HIV-1, herpes simplex viruses 1 and 2, cytomegalovirus, and lymphochoriomeningitis virus (7–11). NAPs were also reported to block entry of hepatitis C virus (HCV) at a postbinding step that may involve interactions with amphipathic domains of apolipoproteins associated with HCV particles (12). More recently, NAPs were proven active against hepadnaviruses in both in vitro and in vivo models: (i) antiviral effects were documented at viral entry and postentry steps of the duck hepatitis B virus (DHBV) replication cycle (13, 14), and in cultures of differentiated human hepatocytes, NAPs could block HBV infection (15); (ii) NAP treatment leads to a long-lasting control of DHBV infection in ducks, which was interpreted as a consequence of their ability to inhibit subviral particle assembly/secretion (16). In two recent clinical studies, NAP monotherapy of HBV chronic carriers led to 2- to 7-log reductions of serum HBsAg, 3- to 9-log reductions in serum HBV DNA, and the appearance of serum anti-HBsAg antibodies (10 to 1,712 mIU/ml) (17).
In the present study, an in vitro HDV infection model was used to assess the HDV-inhibitory activity of NAPs in human hepatocyte cultures. Three closely related NAPs were found to be active in blocking HDV infection in vitro at half-maximal inhibitory concentrations (IC50) of ≤4 μM.
RESULTS
For the present study, nine NAPs were selected, based on previously published demonstration of their potential as new antiviral compounds against Hepadnavirus infection (13, 14, 16, 17). The sequences and chemical properties of these NAPs are shown in Table 1. NAPs were 40 nt in length, except for the shorter REP 2152 (10 nt), REP 2151 (20 nt), and REP 2150 (30 nt) and the longer REP 2149 (60 nt), REP 2180 (80 nt), and REP 2181 (100 nt) versions of REP 2055 (40 nt). REP 2006 and REP 2107 have degenerate nucleic acid sequences, while all other NAPs have defined sequences devoid of CpG motifs. REP 2006, REP 2031, and REP 2055 are phosphorothioated oligodeoxyribonucleotides with amphipathic properties. REP 2107, 2139 and 2165 are also phosphorothioated oligoribonucleotides in which each nucleotide is further modified at the 2′ ribose position with an O-linked methyl moiety (2′ O methylation [2′OMe]). The 2′OMe modification stabilizes NAPs in the absence of phosphorothioation (8) and prevents recognition of the RNA by the innate immune response (18, 19). REP 2139, REP 2165, and REP 2147 have each cytosine base further methylated at the 5′ position (5′-MeC), which also blocks immune recognition of the oligonucleotides (20). REP 2138, REP 2147, and REP 2172 are nonphosphorothioated and contain only 2′OMe and/or 5′-MeC modifications.
TABLE 1.
Sequences and chemical properties of the NAPs used in this studya
| NAP | Sequence | Length (nt) | PS | 2′OMe | 5-MeC |
|---|---|---|---|---|---|
| REP 2006 | dN40 | 40 | + | ||
| REP 2031 | dC40 | 40 | + | ||
| REP 2181 | (dAdC)50 | 100 | + | ||
| REP 2180 | (dAdC)40 | 80 | + | ||
| REP 2149 | (dAdC)30 | 60 | + | ||
| REP 2055 | (dAdC)20 | 40 | + | ||
| REP 2150 | (dAdC)15 | 30 | + | ||
| REP 2151 | (dAdC)10 | 20 | + | ||
| REP 2152 | (dAdC)5 | 10 | + | ||
| REP 2107 | N40 | 40 | + | + | |
| REP 2138 | C40 | 40 | + | ||
| REP 2139 | (AC)20 | 40 | + | + | + |
| REP 2165 | (AC)20 | 40 | + | +b | + |
| REP 2147 | (AC)20 | 40 | + | + | |
| REP 2172 | (AC)20 | 40 | + |
Abbreviations: d, DNA; N, degenerate sequence; PS, phosphorothioation of phosphodiester linkage (increases amphipathicity); 2′OMe, O-linked methylation at the 2′ position in ribose (increased stability and reduced Toll-like receptor [TLR] reactivity); 5-MeC, methylation at the 5′ position in cytidine base (reduced TLR reactivity).
Positions 11, 21, and 31 have 2′OH ribose.
The anti-HDV evaluation was conducted using an HDV in vitro infection assay based on the susceptible Huh-106 cell line, which constitutively expresses the sodium taurocholate cotransporting polypeptide (NTCP), recently identified as a receptor for HBV and HDV at the surface of differentiated hepatocytes (21). In the HDV in vitro model, infection is nonproductive in the absence of HBV coinfection. Because the readout of infection is the amount of intracellular HDV RNA accumulated at 9 days postinoculation (dpi), any of the following steps can be affected by an antiviral compound: (i) attachment of viral particles to cell surface glycosaminoglycans (GAGs), (ii) binding to NTCP, (iii) membrane fusion and internalization, (iv) release of the inner HDV RNP cargo into the cytoplasm, (v) trafficking to the nuclear membrane, (vi) entry into the nucleus, and (vii) replication of the HDV RNA within the nucleus over a period of 9 dpi. Prior to antiviral analysis, NAPs were assayed for cytotoxicity in Huh-106 cell cultures, using the lactate dehydrogenase in culture supernatants as a reporter. Measurements were performed in triplicate cultures after cell incubation with 10 μM NAPs for 24 h for NAP-alone treatment or for 9 days in culture when coinoculated with HDV. No cytotoxic effect could be detected for any of the regular 40-mer NAPs used in this study (Table 2). Subsequent assays were thus performed at 1:2 serial dilutions of a 10 μM NAP solution.
TABLE 2.
Cytotoxicities of NAPs used in this study
| NAP (10 μM) | % cytotoxicity |
|||
|---|---|---|---|---|
| 24 h without HDV | 24 h with HDV | 9 days without HDV | 9 days with HDV | |
| REP 2006 | 1.75 ± 1.27 | −0.74 ± 0.87 | −0.39 ± 1.36 | −0.67 ± 0.99 |
| REP 2031 | 1.95 ± 1.11 | 0.12 ± 1.28 | 0.11 ± 0.92 | −0.17 ± 0.95 |
| REP 2055 | 1.7 ± 1.04 | 0.07 ± 1.26 | 0.18 ± 1.45 | −0.88 ± 0.8 |
| REP 2107 | 2.39 ± 1.34 | −0.33 ± 1.39 | −0.07 ± 1.17 | −0.68 ± 1.4 |
| REP 2138 | 1.99 ± 0.6 | −0.17 ± 1 | −0.33 ± 1.65 | −0.51 ± 0.5 |
| REP 2139 | 2.65 ± 0.96 | 0.17 ± 1.36 | −0.03 ± 0.58 | −0.42 ± 0.4 |
| REP 2147 | 2.17 ± 0.88 | −1.1 ± 0.28 | 0.35 ± 0.29 | −0.56 ± 1.28 |
| REP 2165 | 2.05 ± 1.26 | −1.04 ± 1.13 | 0.59 ± 1.21 | 0.43 ± 0.37 |
| REP 2172 | 1.77 ± 1.18 | 0.33 ± 1.32 | 0.61 ± 0.49 | 0.04 ± 0.71 |
NAPs can inhibit HDV infection of human hepatocytes in culture.
To directly assess NAP activity against HDV infection, Huh-106 cells were exposed to HDV virions at a multiplicity of infection (MOI) of 500 genome equivalents (GE) in the absence or presence of NAPs at 1:2 dilutions of 10 μM (Fig. 1). At 9 dpi, total cellular RNA was isolated, and the amounts of intracellular HDV RNA were measured by real-time reverse transcription-quantitative PCR (RT-qPCR) for infection readout. Phosphorothioated NAPs REP 2006, REP 2031, and REP 2055 all dose dependently reduced infection down to more than 2 logs for REP 2031 at 10 μM, whereas nonphosphorothioated controls REP 2138 REP 2147 and REP 2172 or phosphorothioated the NAPs REP 2107, REP 2139, and REP 2165 bearing a 2′OMe ribose had no activity against infection.
FIG 1.

NAPs are active against HDV infection in vitro. Huh-106 cells were exposed for 24 h to HDV (500 GE/cell) in the absence (0) or the presence of indicated 1:2 dilutions of 10 μM each NAP. Intracellular HDV RNA extracted at 9 days postinoculation (dpi) was quantified by real-time RT-qPCR for measurement of infection. The values in the histograms are shown as means ± SD, expressed as genome equivalents (ge) per 100 ng of total cellular RNA, obtained in three independent experiments. Asterisk, P < 0.05.
NAPs are internalized by human hepatocytes in culture.
To investigate whether NAP activity was related to cell internalization, uptake by Huh-106 cells was then assessed using Cy3-labeled REP 2031, REP 2055, REP 2139, REP 2147 and REP 2172. Cells were exposed for 24 h to 2.5 μM concentrations of each labeled NAP and then washed and fixed before examination by confocal microscopy. Internalization was documented for all five labeled NAPs, but two distinct patterns of intracellular localization were observed: (i) a uniform and diffuse distribution in the cytoplasm for the nonphosphorothioated NAPs REP 2147 and REP 2172 (Fig. 2) and (ii) a punctate, scattered pattern for the phosphorothioated NAPs REP 2031, REP 2055, and REP 2139, including a faint background of cytoplasmic diffuse staining with REP 2139 similar to that with REP 2147 and REP 2172. When labeled NAPs were coinoculated with HDV before confocal microscopy examination at 9 dpi, a similar cytoplasmic, punctate, scattered pattern was observed with REP 2031, REP 2055, and REP 2139 (Fig. 3A). In contrast, the inactive REP 2147 and REP 2172 could no longer be detected, whereas HDAg clearly localized to the nuclei, with no evidence of colocalization with NAPs. The most active NAPs, REP 2031 and REP 2055, were shown to reduce the percentage of HDAg-positive cells more than 3-fold at 9 dpi, consistent with an anti-HDV effect at viral entry (Fig. 3C). Whether the recorded patterns of internalization have any relevance to a biological activity remains to be elucidated.
FIG 2.
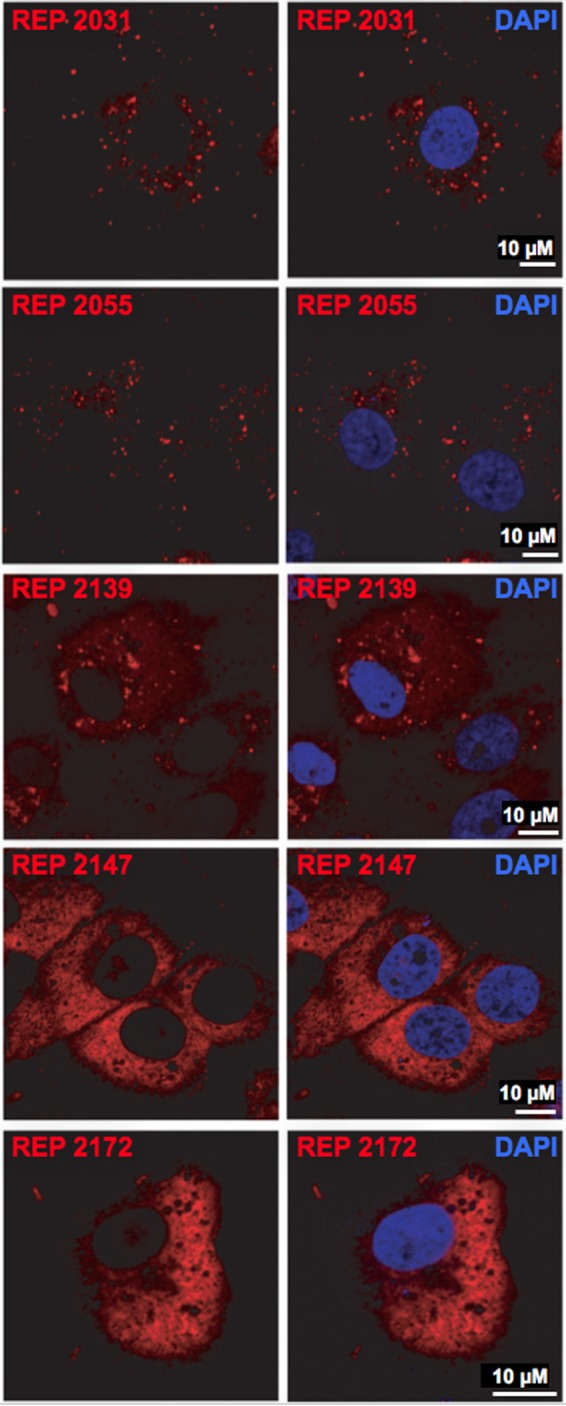
NAP internalization by Huh-106 cells. Cells were exposed for 24 h to Cy3-tagged versions of REP 2031, REP 2055, REP 2139, REP 2147, and REP 2172. Internalized NAPs were detected by virtue of their fluorescent tag (red) in cells in which nuclei were labeled by DAPI (blue) by confocal microscopy. Scale bars, 10 μm.
FIG 3.

NAP subcellular localization and anti-HDV activity in Huh-106 cells. Cells were inoculated with HDV in the presence of Cy3-tagged versions of REP 2031, REP 2055, REP 2139, REP 2147, and REP 2172 at 0.25 μM. Infected cells were counted at 9 dpi using a human anti-HDAg antibody-positive serum and Alexa Fluor 488-conjugated goat anti-human IgGs (green) (B). NAPs were detected at 9 dpi by virtue of their fluorescent tag (red), and nuclei were labeled with DAPI (blue) (A). Note that because spectral overlaps between Cy3 fluorophore (red) and Alex Fluor 488 (green), the yellow spots present in the merge panel demonstrate not colocalization between NAPs and HDAg but Cy3 emission detected in the Alexa Fluor 488 channel. Scale bars, 10 μm. The ratio of infected cells was plotted as percent HDV-infected cells (C). Asterisk, P < 0.05.
NAPs block HDV entry but do not interfere with HDV RNA replication postentry.
To sort between entry and postentry inhibitory activity, infection assays were carried out with the most active NAPs, REP 2006, REP 2031, and REP 2055, under two distinct conditions: (i) a coinoculation treatment, in which cells were exposed to virus in the presence of NAPs for 24 h (as in Fig. 1), and (ii) a postinoculation treatment, in which NAPs were added to the culture medium after the 24-h virus-cell exposure and left for a 24-h exposure to cells. A dose-dependent inhibition of infection was observed for all three NAPs in the coinoculation setting, demonstrating that NAPs are indeed active at the viral entry step. However, the same NAPs were inactive when administered postentry (Fig. 4A). IC50s for REP 2006, REP 2031, and REP 2055 in the coinoculation procedures were measured at 0.23, 3.68, and 3.66 μM, respectively (Fig. 4B). The antiviral effect of REP 2055 on HDV entry could also be confirmed in differentiated HepaRG cells (Fig. 4C).
FIG 4.
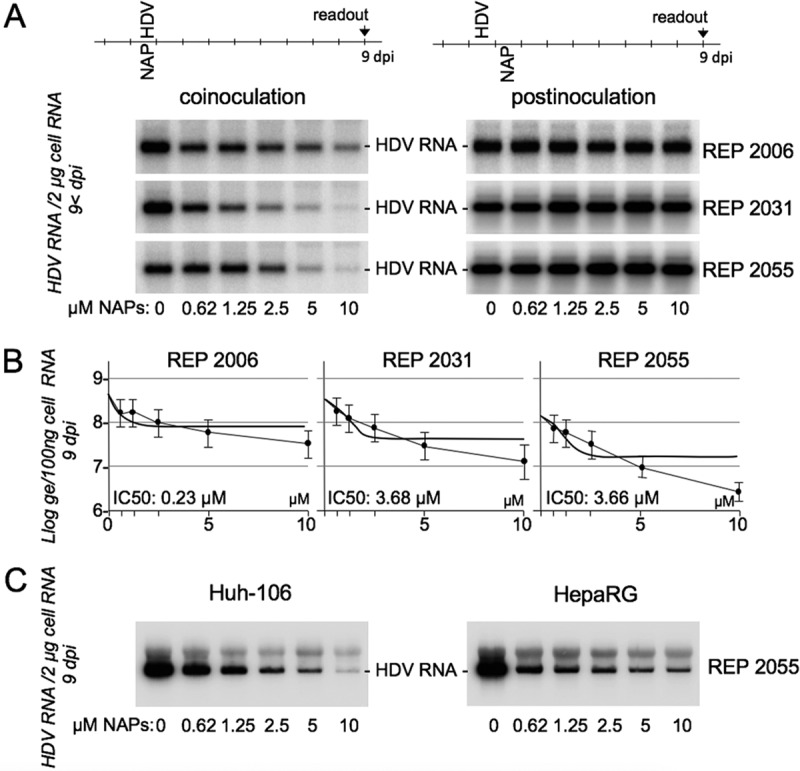
NAPs inhibit HDV entry into Huh-106 and HepaRG cells. (A) Huh-106 cells were inoculated with HDV at 500 GE per cell, in the absence (0) or the presence of the indicated concentrations of NAPs under two conditions: (i) a coinoculation treatment, in which cells were exposed to HDV in the presence of NAPs for 24 h, and (ii) postinoculation treatment, in which NAPs were added to cells for 24 h, after removal of the inoculum. At 9 dpi, total cellular RNA was isolated and analyzed for the presence of HDV RNA by Northern blotting (A). HDV RNA signals were quantified using a phosphorimager. The IC50 was calculated using GraphPad Prism (B). Results of the coinoculation treatment using REP 2055 were also assessed in cultures of differentiated HepaRG cells (C).
Treatment of HDV particles or susceptible cells with NAPs prior to inoculation does not prevent infection.
To further investigate the activity of NAPs at viral entry, a preinoculation treatment of cells at a NAP concentration of 10 μM for 24 h prior to inoculation was conducted in parallel with both postinoculation and coinoculation treatments. As shown in Fig. 5A, none of the selected NAPs was able to reduce susceptibility of Huh-106 cells to infection with HDV when provided prior to virus inoculation, demonstrating that NAPs would not act in binding to or modifying a cell surface receptor. To determine whether NAPs could alter infectivity of HDV particles, preparations of HDV virions were mock treated or treated with 1:2 dilutions of 100 μM REP 2055. After treatment, particles were sedimented by ultracentrifugation as described previously (22), and the pelleted particles were resuspended in culture medium before inoculation to Huh-106 cells. As shown in Fig. 5B, infectivity of NAP-treated virions was not affected. To exclude the possibility that polyethylene glycol (PEG)-mediated precipitation would be affected by NAPs, preparations of HDV virions were mock treated or treated with 1:2 dilutions of 25 μM REP 2055 before adding PEG 8000 at a 10% (wt/vol) final concentration. The analysis of HDV RNA in the PEG precipitates or supernatants shows no effect of NAPs on the precipitation efficiency of PEG (Fig. 5B).
FIG 5.
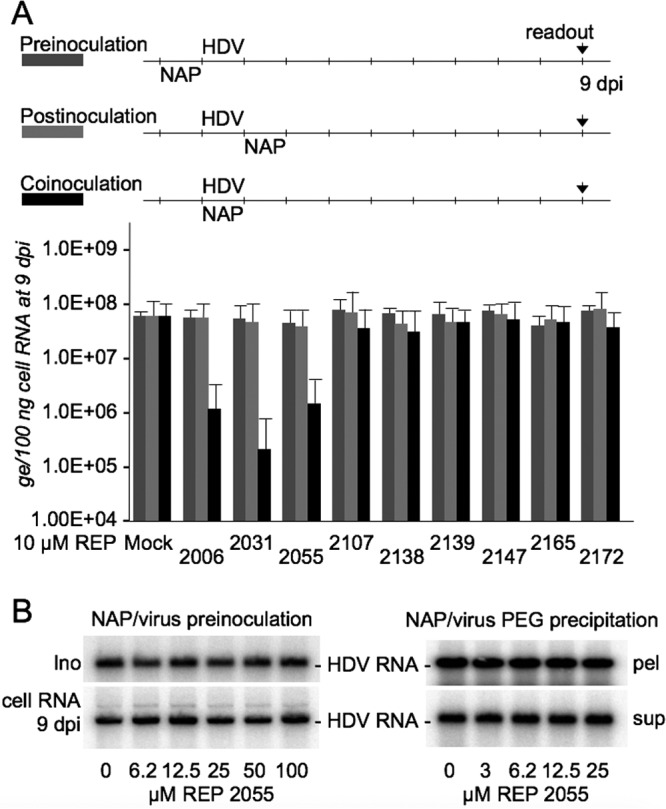
Exposure of Huh-106 cells to NAPs prior to HDV inoculation does not prevent infection. (A) Huh-106 cells were inoculated with HDV at 500 GE per cell, in the absence or the presence of NAPs at 10 μM, under three conditions: (i) treatment of cells with NAPs for 24 h prior to inoculation with HDV, (ii) treatment of cells with NAPs for 24 h, after removal of the inoculum, and (iii) treatment in which cells were exposed to HDV and NAPs for 24 h. Intracellular HDV RNA extracted at 9 dpi was quantified by real-time RT-qPCR for measurement of infection. The values in the histograms are shown as means ± SD, expressed as genome equivalents per 100 ng of total cellular RNA, obtained in three independent experiments. (B) HDV virions were mock treated or treated with 1:2 dilutions of 100 μM REP 2055 and then sedimented by ultracentrifugation. Pelleted particles were resuspended in culture medium before inoculation of Huh-106 cells. As shown on the left, integrity (Ino) and infectivity (cell RNA 9 dpi) of NAP-treated HDV virions were not affected. HDV virions were mock treated or treated with 1:2 dilutions of 25 μM REP 2055 before PEG precipitation. As shown on the right, the amount of HDV RNA in the PEG precipitates (pel) or PEG supernatants (sup) is not affected by the presence of NAPs.
Antiviral activity of NAPs at viral entry is size dependent.
The size-dependent nature of NAP antiviral activity has been demonstrated previously with a broad range of enveloped viruses, including herpesviruses and DHBV. To directly assess NAP size effect on antiviral activity, a series of phosphorothioated poly(dAdC) NAPs differing from the active REP 2055 only in size were tested in parallel (Table 1). Huh-106 cells were inoculated with HDV in the presence of each NAP at 10 μM, and infection was assessed by measuring intracellular HDV RNA at 9 dpi (Fig. 6). A biphasic inhibition/NAP size curve was observed with a high ratio for NAPs between 20- and 40-mer in length and a lower ratio for NAPs longer than 40-mer. In brief, shortening of REP 2055 length translates into a significant loss of antiviral activity, whereas increasing its size beyond 40-mer only marginally improved antiviral activity. This is in agreement with previously reported observations in different viral models (23).
FIG 6.
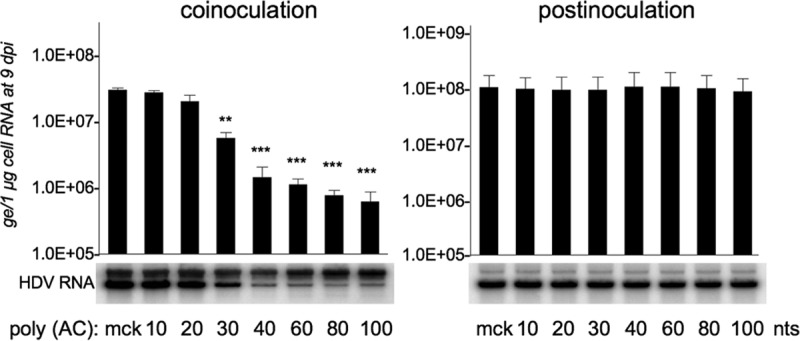
NAP inhibitory activity on HDV entry is size dependent. Cells were inoculated with HDV at 500 GE per cell, in the absence or the presence of 10 μM 100- to 10-mer versions of REP 2055 [poly (AC)] under two conditions: (i) a coinoculation treatment, in which cells were exposed to HDV in the presence of NAPs for 24 h, and (ii) postinoculation treatment, in which NAPs were added to cells for 24 h, after removal of the inoculum. At 9 dpi, total cellular RNA was isolated and analyzed for the presence of HDV RNA by Northern blotting. HDV RNA signals were quantified using a phosphorimager. The values in the histograms are shown as means ± SD, expressed as genome equivalents per microgram of cellular RNA, obtained in three independent experiments. Results were analyzed by Student's t test for unpaired data. Single asterisk, P < 0.05; double asterisks, P < 0.01; triple asterisks, P < 0.001.
To investigate whether a postentry activity could be conferred by NAPs longer that 40-mer, the same set of 10- to 100-mer poly(dAdC) NAPs was utilized in a postinoculation setting. Results show no evidence of anti-HDV activity gain at a postentry step (Fig. 6).
NAP antiviral activity is conserved against HDV coated with envelope proteins of HBV genotypes A to F and HDV bearing two of the main immune escape variants.
At present, 10 HBV genotypes (A to J) with 40 subtypes and four major serotypes (adr, adw, ayr, and ayw) have been documented. To investigate whether NAPs possess a pangenomic activity against HBV, preparations of HDV particles bearing the HBV envelope proteins of the genotypes A ayw1, B ayw1, B adw2, C adrq+, D ayw2, D ayw3, E ayw4, and F were produced, along with HDV virions bearing envelope proteins of HBV genotype D with immune escape-associated D144A and G145R substitutions in the S domain (24). Huh-106 cells were exposed to virions in the presence of 10 μM REP 2055 or REP 2172 as negative control. As shown in Fig. 7, infection and susceptibility to REP 2055 antiviral activity were observed irrespective of the HBV envelope proteins genotype or immune escape-associated substitution.
FIG 7.
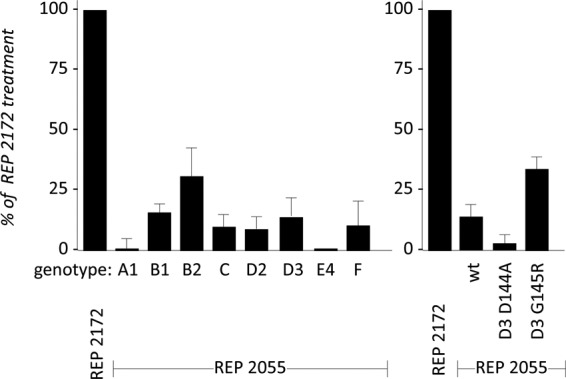
HDV, containing envelope proteins of different HBV genotypes, is susceptible to the inhibitory effect of NAPs. Huh-106 cells were inoculated with HDV virions (500 GE/cell) bearing the envelope proteins of the indicated HBV genotypes, or genotype D envelope proteins bearing the substitutions D144A or G145R, in the presence of 10 μM REP 2055 or REP 2172. Intracellular HDV RNA extracted at 9 dpi was quantified by Northern blot analysis for measurement of infection. Values in the histograms are shown as percent infection (mean ± SD) in the presence of REP 2172 control, obtained in three independent experiments.
HDV and HBV virions bind to immobilized NAPs.
To directly assay for an interaction between NAPs and HDV or HBV virions, NAP affinity chromatography columns were generated in which streptavidin-Sepharose was used to immobilize biotinylated NAPs. HDV and HBV preparations in serum-free medium were then applied to the columns, and their elution behavior in the presence of 1 M NaCl was determined. The flowthrough under physiological salt conditions was recovered, and bound particles were eluted at 1 M NaCl. HDV RNA was isolated and measured by Northern blotting in both flowthrough and eluate fractions. As shown in Fig. 8, HDV, and HBV to a lesser extent, bound to immobilized entry inhibitors REP 2055 and REP 2031 and to immobilized heparin. However, neither HDV nor HBV particles bound to immobilized REP 2172, REP 2147, or REP 2139. The results suggest that NAPs could inhibit infection by interfering with the early step of HDV/HBV attachment to heparan sulfate proteoglycans (HSPGs) at the surface of human hepatocytes.
FIG 8.
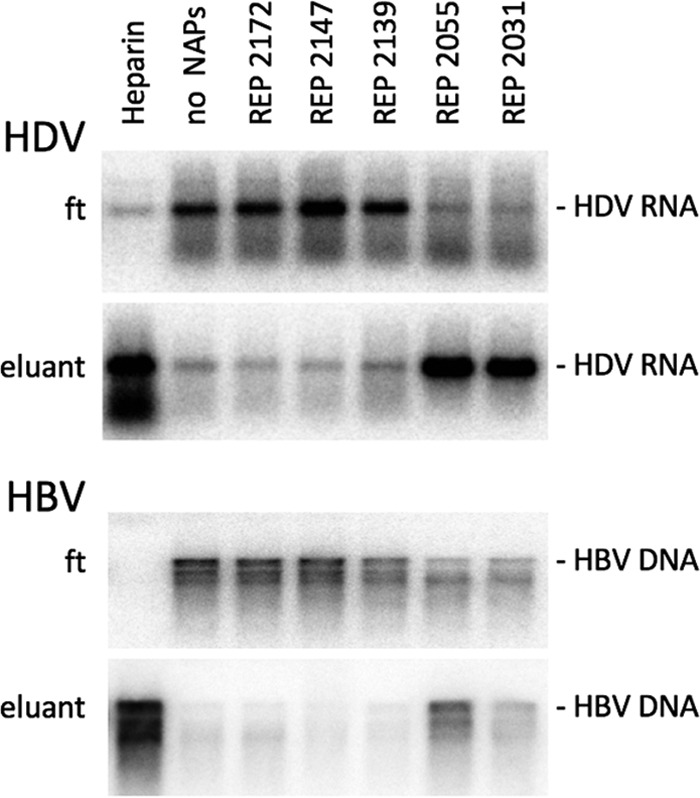
Immobilized NAPs bind HDV and HBV virions. Biotinylated NAPs (REP 2172, REP 2147, REP 2139, REP 2055, and REP 2031) were bound to streptavidin-Sepharose in a 400-μl volume column. After extensive washes of the column, HDV- or HBV-containing supernatants were loaded. The flowthrough was recovered and columns were washed 5 times with wash buffer. Bound particles were then eluted at 1 M NaCl. HDV RNA or HBV DNA was isolated from flowthrough (ft) and eluate (eluant) fractions and measured by Northern or Southern blot analysis, respectively. Four hundred-microliter heparin Sepharose columns (Heparin) were use as a control.
DISCUSSION
The present study demonstrates that NAPs can prevent infection of susceptible cells with HDV. The block occurs at viral entry and is independent of the NAP nucleotide sequence but strictly dependent upon phosphorothioation. NAP size was shown to be important, with the longest NAPs (40-mer and longer) displaying the strongest activity. NAPs blocked HDV entry only upon coadministration with the virus. NAPs were efficiently internalized upon a single exposure to cells in culture and directed to specific subcellular compartments with no apparent cytotoxicity. However, since our in vitro HDV infection assay was conducted in the absence of HBV coinfection, NAPs could not be evaluated for their activity against HDV particle assembly and secretion.
The coinoculation dependency for activity of NAPs is compatible with a blockade at the initial attachment of HDV virions to cell surface HSPGs (24–26). GAG side chains of HSPG are polysaccharides composed of multiple disaccharide units that are negatively charged upon sulfation. They are used by HBV and HDV as attachment receptor at the surface of human hepatocytes prior to virus binding to NTCP. NAPs and GAGs can bind HDV virions. Yet, exposure of the latter to soluble NAPs prior to inoculation did not lead to loss of infectivity. Our interpretation is that NAPs in solution would individually bind HDV with low affinity, but this binding could only be observed with NAPs immobilized on streptavidin beads (Fig. 8) so as to reach sufficient avidity for a stable interaction with viral particles.
It is known that an efficient in vitro infection of susceptible cells with HDV, or HBV, is dependent upon a lengthy virus-to-cell exposure, usually 16 to 24 h. The reason for this is not clear, but it is likely that the initial attachment to HSPGs is the limiting factor. In this setting, NAPs could be internalized more rapidly than the virus and thus be preferentially directed to endosomes devoid of viral particles. In fact, cells exposed to NAPs prior to inoculation do not lose susceptibility to infection. The requirement for NAP coinoculation with the virus indicates the need for either high concentration to prevent HSPG binding or cointernalization with the virus in the endocytic vesicles.
In contrast to other entry inhibitors, such as membrane-impermeant alkylating or reducing agents (22), which inhibit infectivity by inducing a conformational change in the HSPG binding domain of the HBV envelope proteins, NAPs do not affect HDV infectivity when applied to the virus prior to inoculation. As indicated above, this can be due to a suboptimal affinity/avidity in the culture medium conditions. Eventually, NAPs, such as REP 2031 and REP 2055, could act at viral entry after cointernalization with virions, within vesicles of the endocytic pathway; whether the binding to NTCP is affected is uncertain.
Although the backbone of phosphorothioated oligonucleotides is polyanionic, phosphorothioation confers an increased hydrophobic character on oligonucleotides (27), which confers on NAPs the ability to interact with the hydrophobic surfaces of amphipathic α-helices such as that of HIV-1 gp41 protein (8) or structural proteins of other viruses (7) or prion proteins (28). In the case of HDV, NAPs could block viral entry through polyanionic interactions preventing virus attachment to HSPGs. However, since the entry inhibition is phosphorothioation specific, NAPs could also interfere with HDV entry through hydrophobic/amphipathic interactions. REP 2139 was not active in blocking HDV entry in the current study or in a previous study with HBV (15), but it was demonstrated to be active against HBV and HDV in clinical studies (17, 29). The latter observation suggests that REP 2139 may be active against assembly and/or secretion of subviral particles (SVPs). This interpretation is consistent with the data obtained in the DHBV model, in which NAP treatment lead to inhibition of SVP release (16).
Fluorescence microscopy examinations show that NAPs are efficiently internalized in cultured hepatocytes, but it is unclear whether they accumulate in subcellular compartments that are involved in particle assembly and release when HBV envelope proteins are expressed. Intracellular trafficking of phosphorothioated oligonucleotides, which normally occurs in hepatocytes in vivo, is known to be deficient in vitro (30), leading to sequestration in endosomal compartments. Primary cultures of hepatocytes similarly take up phosphorothioated oligonucleotides but rapidly lose the characteristics for productive uptake. Contrary to inactive NAPs REP 2147 and REP 2172, the in vitro-active REP 2031 and REP 2055 (15; this study) and in vivo-active REP 2139 (17) are still detected in the cytoplasm of Huh-106 cells (Fig. 3) at day 9 posttreatment, with no apparent cytotoxicity, indicating their long-term intracellular stability, a property that would be crucial provided that they reach cellular compartments involved in SVP or HDV morphogenesis. This is being investigated in an HDV-permissive model to fully understand the scope of NAP activity against HDV. If indeed NAPs were to interfere with assembly/secretion of SVPs, they would likely affect assembly and secretion of HDV. What is clear, however, is that NAPs do not interfere with HDV RNA replication within the nucleus over a period of 9 dpi, either for lack of affinity to components directly involved in viral RNA transcription or inability to reach the nucleus.
Although our results clearly show that NAPs can interfere with HDV entry, it is difficult to estimate the impact of such antientry activity in HDV chronic carriers. HDV RNA has been shown to persist in cells for more than 6 weeks (31), but it is unlikely that it is as stable as HBV covalently closed circular DNA (cccDNA). Hence, chronic HDV infection would require a rate of propagation within the liver which would be greater than that necessary for chronic HBV infection. As a consequence, long-term treatment with antientry inhibitor, such as REP 2055, might be more efficient in eliminating HDV than HBV. Whatever precise mechanism is engaged by NAPs, it is important to note that their antiviral activity is pangenomic with regard to the helper HBV and not circumvented by substitutions in the HBV envelope proteins associated with immune escape (Fig. 7).
MATERIALS AND METHODS
Synthesis of NAPs.
All NAPs used in this study were prepared under standard solid-phase reaction conditions for synthesis of phosphorothioate oligonucleotides. NAPs were generated as sodium salts either by salt exchange in 3 M NaCl overnight at room temperature, followed by desalting by ultrahigh-pressure filtration with water, or by simultaneous salt exchange and desalting on Sephadex columns. The identity and purity of each NAP were confirmed by liquid chromatography-mass spectrometry (LC-MS) analysis. The chemical properties of the different NAPs used in this study are summarized in Table 1.
Cell culture and in vitro HDV infection assay.
The HDV-susceptible cell line Huh-106 (21) is a Huh-7-derived cell line that stably expresses the HBV receptor sodium taurocholate cotransporting polypeptide (NTCP) (32). Cells were cultured in William's medium E (WME) supplemented with 10% fetal bovine serum, HEPES (25 mM), gentamicin (10 μg/ml), Fungin (10 g/ml), and neomycin (250 μg/ml). NAP treatments of Huh-106 cells were carried out in serum-free growth medium supplemented with epidermal growth factor (EGF; 10 ng/ml), hydrocortisone (18 μg/ml), dexamethasone (40 ng/ml) and insulin-transferin-selenium (ITS) (5 ng/ml). HepaRG cells were grown and differentiated as described previously (33). Infection assays were performed as described previously (34). In brief, Huh-106 cells were seeded in 24-well plates at 1.00E + 05 cells per well and inoculated with HDV at a multiplicity of infection (MOI) of 500 HDV GE per cell on day 1 postseeding. Cells were harvested at 9 days postinoculation (dpi) for measurement of intracellular HDV RNA, which served as marker of infection.
RNA isolation and Northern blot hybridization.
Total RNA was isolated using a NucleoSpin RNA isolation kit (Macherey-Nagel GmbH & Co.) or a QIAamp viral RNA isolation kit (Qiagen) according to the manufacturer's protocol, then precipitated in the presence of isopropanol, and resuspended in RNase-free water. The purified RNA was subjected to electrophoresis through a 2.2 M formaldehyde, 1.2% agarose gel and transferred to a nylon membrane. The membrane-bound RNA was hybridized to a 32P-labeled RNA probe specific for detection of genomic HDV RNA. Quantification of radioactive signals was achieved using a phosphorimager (BAS-1800 II; Fuji).
HDV RNA quantification by reverse transcription and real-time PCR.
Reverse transcription of HDV RNA and qPCR amplification were performed using 10 to 100 ng of total cellular RNA and the One-step-qRT-PCR MasterMix (Eurogentec RT-QPRT-032X), according to the manufacturer's instructions. Reactions were performed in quadruplicate using an Eppendorf Mastercycler RealPlex. PCR conditions were one step of denaturation (10 min at 95°C) followed by 50 cycles (each cycle consisted of 15 s at 95°C and 1 min at 60°C). The following oligonucleotide primer and probe were used: HDV-835-851 (5′-TGGACGTGCGTCCTCCT-3′), HDV-905-889 (5′-TCTTCGGGTCGGCATGG-3′), and HDV-PROBE-856-870 (6-carboxyfluorescein [FAM]-ATGCCCAGGTCGGAC-black hole quencher [BHQ-1]).
HDV binding to immobilized NAPs and heparin-affinity chromatography.
A 50% slurry of streptavidin-agarose (Molecular Probes) was loaded on 0.22-μm Corning Costar Spin-X centrifuge tube filters. After five washes with phosphate-buffered saline (PBS), 10 μl of 100 μM biotinylated NAPs in 400 μl of PBS were added and incubated overnight at 4°C under shaking. Beads were washed 5 times with PBS before 100 μl of a suspension of HDV or HBV virions at 1.1E + 09 GE/ml was added and incubated at room temperature on a rotary agitator for 2 h. Postbinding, bead-containing tubes were drained and the flowthrough was harvested. Beads were washed 3 times with PBS before elution of the bound virus in 1 M NaCl Tris HCl (pH 7.4). Heparin Sepharose 6 Fast Flow (GE Healthcare) was used as the immobilized heparin substrate for a control.
Fluorescence microscopy.
NAP internalization in Huh-106 cells was visualized by confocal fluorescence microscopy using 5′ Cy3-labeled REP 2031, REP 2055, REP 2139, REP 2147, and REP 2172. Cells were seeded on glass coverslips and incubated with Cy3-labeled NAPs for 24 h or 9 days. Cells were then fixed with 4% paraformaldehyde for 10 min at room temperature and examined under confocal microscopy. For costaining with anti-HDAg antibodies, cells harvested at 9 dpi were fixed as described above, then washed with PBS, and permeabilized with 0.2% Triton X-100 in PBS for 30 min. Detection of intracellular HDAg was achieved after incubation with an anti-HDAg antibody-positive human serum at 1:1,000 dilution for 1 h at room temperature. Coverslips were then washed twice with PBS and incubated with secondary antibody for 1 h and mounted directly in Prolong gold antifade reagent (Molecular Probes) containing 1 μg/ml of 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI). Coverslips were examined using a Zeiss LSM 710 Meta confocal microscope.
Cytotoxicity assay.
NAP cytotoxicity was measured using a CytoTox96 nonradioactive cytotoxicity assay (Promega) according to the manufacturer's instructions. The activity of lactate dehydrogenase released into culture supernatants when toxicity is present was measured on triplicate cultures of NAP-treated cells.
Statistical analysis.
Results are expressed as the mean ± standard deviations (SD) from at least three independent experiments and were analyzed by Student's t test for unpaired data. P values of <0.05, <0.01, and <0.001 were considered statistically significant.
ACKNOWLEDGMENTS
The work was funded by Replicor Inc. M.B. is an employee of Replicor Inc. A.V. is an employee of Replicor Inc. with financial interests. C.S. is a CNRS investigator at INSERM U1134 at INTS.
REFERENCES
- 1.Schweitzer A, Horn J, Mikolajczyk RT, Krause G, Ott JJ. 2015. Estimations of worldwide prevalence of chronic hepatitis B virus infection: a systematic review of data published between 1965 and 2013. Lancet 386:1546–1555. doi: 10.1016/s0140-6736(15)61412-x. [DOI] [PubMed] [Google Scholar]
- 2.Sureau C, Negro F. 2016. The hepatitis delta virus: replication and pathogenesis. J Hepatol 64:S102–S116. doi: 10.1016/j.jhep.2016.02.013. [DOI] [PubMed] [Google Scholar]
- 3.Rizzetto M. 2015. Hepatitis D virus: introduction and epidemiology. Cold Spring Harb Perspect Med 5:a021576. doi: 10.1101/cshperspect.a021576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heidrich B, Yurdaydin C, Kabacam G, Ratsch BA, Zachou K, Bremer B, Dalekos GN, Erhardt A, Tabak F, Yalcin K, Gurel S, Zeuzem S, Cornberg M, Bock CT, Manns MP, Wedemeyer H, HIDIT-1 Study Group. 2014. Late HDV RNA relapse after peginterferon alpha-based therapy of chronic hepatitis delta. Hepatology 60:87–97. doi: 10.1002/hep.27102. [DOI] [PubMed] [Google Scholar]
- 5.Wedemeyer H, Yurdaydin C, Dalekos GN, Erhardt A, Cakaloglu Y, Degertekin H, Gurel S, Zeuzem S, Zachou K, Bozkaya H, Koch A, Bock T, Dienes HP, Manns MP, HIDIT Study Group. 2011. Peginterferon plus adefovir versus either drug alone for hepatitis delta. N Engl J Med 364:322–331. doi: 10.1056/NEJMoa0912696. [DOI] [PubMed] [Google Scholar]
- 6.Agrawal S, Tang JY, Brown DM. 1990. Analytical study of phosphorothioate analogues of oligodeoxynucleotides using high-performance liquid chromatography. J Chromatogr 509:396–399. doi: 10.1016/S0021-9673(01)93098-5. [DOI] [PubMed] [Google Scholar]
- 7.Lee AM, Rojek JM, Gundersen A, Stroher U, Juteau JM, Vaillant A, Kunz S. 2008. Inhibition of cellular entry of lymphocytic choriomeningitis virus by amphipathic DNA polymers. Virology 372:107–117. doi: 10.1016/j.virol.2007.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vaillant A, Juteau JM, Lu H, Liu S, Lackman-Smith C, Ptak R, Jiang S. 2006. Phosphorothioate oligonucleotides inhibit human immunodeficiency virus type 1 fusion by blocking gp41 core formation. Antimicrob Agents Chemother 50:1393–1401. doi: 10.1128/AAC.50.4.1393-1401.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bernstein DI, Goyette N, Cardin R, Kern ER, Boivin G, Ireland J, Juteau JM, Vaillant A. 2008. Amphipathic DNA polymers exhibit antiherpetic activity in vitro and in vivo. Antimicrob Agents Chemother 52:2727–2733. doi: 10.1128/AAC.00279-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cardin RD, Bravo FJ, Sewell AP, Cummins J, Flamand L, Juteau JM, Bernstein DI, Vaillant A. 2009. Amphipathic DNA polymers exhibit antiviral activity against systemic murine cytomegalovirus infection. Virol J 6:214. doi: 10.1186/1743-422X-6-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guzman EM, Cheshenko N, Shende V, Keller MJ, Goyette N, Juteau JM, Boivin G, Vaillant A, Herold BC. 2007. Amphipathic DNA polymers are candidate vaginal microbicides and block herpes simplex virus binding, entry and viral gene expression. Antivir Ther 12:1147–1156. [PubMed] [Google Scholar]
- 12.Matsumura T, Hu Z, Kato T, Dreux M, Zhang YY, Imamura M, Hiraga N, Juteau JM, Cosset FL, Chayama K, Vaillant A, Liang TJ. 2009. Amphipathic DNA polymers inhibit hepatitis C virus infection by blocking viral entry. Gastroenterology 137:673–681. doi: 10.1053/j.gastro.2009.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Noordeen F, Vaillant A, Jilbert AR. 2013. Nucleic acid polymers prevent the establishment of duck hepatitis B virus infection in vivo. Antimicrob Agents Chemother 57:5299–5306. doi: 10.1128/AAC.01005-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Noordeen F, Vaillant A, Jilbert AR. 2013. Nucleic acid polymers inhibit duck hepatitis B virus infection in vitro. Antimicrob Agents Chemother 57:5291–5298. doi: 10.1128/AAC.01003-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guillot C, Martel N, Berby F, Bordes I, Hantz O, Blanchet M, Sureau C, Vaillant A, Chemin I. 2017. Inhibition of hepatitis B viral entry by Nucleic acid polymers in HepaRG cells and primary human hepatocytes. PLoS One 12:e0179697. doi: 10.1371/journal.pone.0179697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Noordeen F, Scougall CA, Grosse A, Qiao Q, Ajilian BB, Reaiche-Miller G, Finnie J, Werner M, Broering R, Schlaak JF, Vaillant A, Jilbert AR. 2015. Therapeutic antiviral effect of the nucleic acid polymer REP 2055 against persistent duck hepatitis B virus infection. PLoS One 10:e0140909. doi: 10.1371/journal.pone.0140909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Al-Mahtab M, Bazinet M, Vaillant A. 2016. Safety and efficacy of nucleic acid polymers in monotherapy and combined with immunotherapy in treatment-naive Bangladeshi patients with HBeAg+ chronic hepatitis B infection. PLoS One 11:e0156667. doi: 10.1371/journal.pone.0156667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Judge AD, Bola G, Lee AC, MacLachlan I. 2006. Design of noninflammatory synthetic siRNA mediating potent gene silencing in vivo. Mol Ther 13:494–505. doi: 10.1016/j.ymthe.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 19.Robbins M, Judge A, Liang L, McClintock K, Yaworski E, MacLachlan I. 2007. 2′-O-methyl-modified RNAs act as TLR7 antagonists. Mol Ther 15:1663–1669. doi: 10.1038/sj.mt.6300240. [DOI] [PubMed] [Google Scholar]
- 20.Karikó K, Buckstein M, Ni H, Weissman D. 2005. Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity 23:165–175. doi: 10.1016/j.immuni.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 21.Verrier ER, Colpitts CC, Bach C, Heydmann L, Weiss A, Renaud M, Durand SC, Habersetzer F, Durantel D, Abou-Jaoude G, Lopez Ledesma MM, Felmlee DJ, Soumillon M, Croonenborghs T, Pochet N, Nassal M, Schuster C, Brino L, Sureau C, Zeisel MB, Baumert TF. 2015. A targeted functional RNAi screen uncovers Glypican 5 as an entry factor for hepatitis B and D viruses. Hepatology doi: 10.1002/hep.28013. [DOI] [PubMed] [Google Scholar]
- 22.Abou-Jaoudé G, Sureau C. 2007. Entry of hepatitis delta virus requires the conserved cysteine residues of the hepatitis B virus envelope protein antigenic loop and is blocked by inhibitors of thiol-disulfide exchange. J Virol 81:13057–13066. doi: 10.1128/JVI.01495-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vaillant A. 2016. Nucleic acid polymers: broad spectrum antiviral activity, antiviral mechanisms and optimization for the treatment of hepatitis B and hepatitis D infection. Antiviral Res 133:32–40. doi: 10.1016/j.antiviral.2016.07.004. [DOI] [PubMed] [Google Scholar]
- 24.Sureau C, Salisse J. 2013. A conformational heparan sulfate binding site essential to infectivity overlaps with the conserved hepatitis B virus A-determinant. Hepatology 57:985–994. doi: 10.1002/hep.26125. [DOI] [PubMed] [Google Scholar]
- 25.Lamas Longarela O, Schmidt TT, Schoneweis K, Romeo R, Wedemeyer H, Urban S, Schulze A. 2013. Proteoglycans act as cellular hepatitis delta virus attachment receptors. PLoS One 8:e58340. doi: 10.1371/journal.pone.0058340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leistner CM, Gruen-Bernhard S, Glebe D. 2008. Role of glycosaminoglycans for binding and infection of hepatitis B virus. Cell Microbiol 10:122–133. [DOI] [PubMed] [Google Scholar]
- 27.Agrawal S, Temsamani J, Tang JY. 1991. Pharmacokinetics, biodistribution, and stability of oligodeoxynucleotide phosphorothioates in mice. Proc Natl Acad Sci U S A 88:7595–7599. doi: 10.1073/pnas.88.17.7595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kocisko DA, Vaillant A, Lee KS, Arnold KM, Bertholet N, Race RE, Olsen EA, Juteau JM, Caughey B. 2006. Potent antiscrapie activities of degenerate phosphorothioate oligonucleotides. Antimicrob Agents Chemother 50:1034–1044. doi: 10.1128/AAC.50.3.1034-1044.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bazinet M, Pântea V, Cebotarescu V, Cojuhari L, Jimbei P, Albrecht J, Schmid P, Le Gal F, Gordien E, Krawczyk A, Mijočević H, Karimzadeh H, Roggendorf M, Vaillant A. 2017. Safety and efficacy of REP 2139 and pegylated interferon alfa-2a for treatment-naive patients with chronic hepatitis B virus and hepatitis D virus coinfection (REP 301 and REP 301-LTF): a non-randomised, open-label, phase 2 trial. Lancet Gastroenterol Hepatol doi: 10.1016/S2468-1253(17)30288-1. [DOI] [PubMed] [Google Scholar]
- 30.Koller E, Vincent TM, Chappell A, De S, Manoharan M, Bennett CF. 2011. Mechanisms of single-stranded phosphorothioate modified antisense oligonucleotide accumulation in hepatocytes. Nucleic Acids Res 39:4795–4807. doi: 10.1093/nar/gkr089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Giersch K, Helbig M, Volz T, Allweiss L, Mancke LV, Lohse AW, Polywka S, Pollok JM, Petersen J, Taylor J, Dandri M, Lutgehetmann M. 2014. Persistent hepatitis D virus mono-infection in humanized mice is efficiently converted by hepatitis B virus to a productive coinfection. J Hepatol 60:538–544. doi: 10.1016/j.jhep.2013.11.010. [DOI] [PubMed] [Google Scholar]
- 32.Yan H, Zhong G, Xu G, He W, Jing Z, Gao Z, Huang Y, Qi Y, Peng B, Wang H, Fu L, Song M, Chen P, Gao W, Ren B, Sun Y, Cai T, Feng X, Sui J, Li W. 2012. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. eLife 1:e00049. doi: 10.7554/eLife.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gripon P, Rumin S, Urban S, Le Seyec J, Glaise D, Cannie I, Guyomard C, Lucas J, Trepo C, Guguen-Guillouzo C. 2002. Infection of a human hepatoma cell line by hepatitis B virus. Proc Natl Acad Sci U S A 99:15655–15660. doi: 10.1073/pnas.232137699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sureau C. 2010. The use of hepatocytes to investigate HDV infection: the HDV/HepaRG model. Methods Mol Biol 640:463–473. doi: 10.1007/978-1-60761-688-7_25. [DOI] [PubMed] [Google Scholar]