

ABSTRACT
Coronavirus replication is associated with intracellular membrane rearrangements in infected cells, resulting in the formation of double-membrane vesicles (DMVs) and other membranous structures that are referred to as replicative organelles (ROs). The latter provide a structural scaffold for viral replication/transcription complexes (RTCs) and help to sequester RTC components from recognition by cellular factors involved in antiviral host responses. There is increasing evidence that plus-strand RNA (+RNA) virus replication, including RO formation and virion morphogenesis, affects cellular lipid metabolism and critically depends on enzymes involved in lipid synthesis and processing. Here, we investigated the role of cytosolic phospholipase A2α (cPLA2α) in coronavirus replication using a low-molecular-weight nonpeptidic inhibitor, pyrrolidine-2 (Py-2). The inhibition of cPLA2α activity, which produces lysophospholipids (LPLs) by cleaving at the sn-2 position of phospholipids, had profound effects on viral RNA and protein accumulation in human coronavirus 229E-infected Huh-7 cells. Transmission electron microscopy revealed that DMV formation in infected cells was significantly reduced in the presence of the inhibitor. Furthermore, we found that (i) viral RTCs colocalized with LPL-containing membranes, (ii) cellular LPL concentrations were increased in coronavirus-infected cells, and (iii) this increase was diminished in the presence of the cPLA2α inhibitor Py-2. Py-2 also displayed antiviral activities against other viruses representing the Coronaviridae and Togaviridae families, while members of the Picornaviridae were not affected. Taken together, the study provides evidence that cPLA2α activity is critically involved in the replication of various +RNA virus families and may thus represent a candidate target for broad-spectrum antiviral drug development.
IMPORTANCE Examples of highly conserved RNA virus proteins that qualify as drug targets for broad-spectrum antivirals remain scarce, resulting in increased efforts to identify and specifically inhibit cellular functions that are essential for the replication of RNA viruses belonging to different genera and families. The present study supports and extends previous conclusions that enzymes involved in cellular lipid metabolism may be tractable targets for broad-spectrum antivirals. We obtained evidence to show that a cellular phospholipase, cPLA2α, which releases fatty acid from the sn-2 position of membrane-associated glycerophospholipids, is critically involved in coronavirus replication, most likely by producing lysophospholipids that are required to form the specialized membrane compartments in which viral RNA synthesis takes place. The importance of this enzyme in coronavirus replication and DMV formation is supported by several lines of evidence, including confocal and electron microscopy, viral replication, and lipidomics studies of coronavirus-infected cells treated with a highly specific cPLA2α inhibitor.
KEYWORDS: coronavirus
INTRODUCTION
Coronavirinae is a subfamily of enveloped, positive-sense RNA (+RNA) viruses in the family Coronaviridae that, together with the Arteri-, Roni-, and Mesoniviridae, belongs to the order Nidovirales (1, 2). Coronavirus infections in humans are mainly associated with (common cold-like) upper respiratory tract infections and are caused by 4 coronavirus species that have been classified as members of the genera Alphacoronavirus (Human coronavirus 229E [HCoV-229E] and Human coronavirus NL63 [HCoV-NL63]) and Betacoronavirus (Human coronavirus OC43 [HCoV-OC43] and Human coronavirus HKU1 [HCoV-HKU1]) (1, 3). In contrast to these common human coronaviruses, infections with zoonotic coronaviruses, such as severe acute respiratory syndrome (SARS) coronavirus (SARS-CoV) (4, 5) and Middle East respiratory syndrome (MERS) coronavirus (MERS-CoV) (6), may lead to much more severe or even fatal respiratory disease in humans, as illustrated by the SARS outbreak in 2002-2003 (5, 7) and, more recently, a significant number of cases with acute respiratory distress syndrome caused by MERS-CoV, which have been recorded over the past 5 years (8).
Similar to other +RNA viruses, coronavirus replication involves extensive membrane rearrangements in infected cells, resulting in the formation of large, organelle-like “virus factories” to which the multisubunit viral replication/transcription complexes (RTCs) are anchored (reviewed in references 9 and 10). These replicative organelles (ROs) are thought to provide a structural scaffold for the viral RNA synthesis machinery and contribute to sequestering components of this machinery from host defense mechanisms, suggesting important roles for ROs in viral replication (9, 11–13). RO formation in coronavirus-infected cells requires three replicase gene-encoded nonstructural proteins (nsp's), called nsp3, nsp4, and nsp6, that all contain conserved transmembrane domains (14–17). Electron microscopy/tomography studies revealed that coronavirus-induced membrane rearrangements result in multiple paired-membrane structures, including double-membrane vesicles (DMVs) and convoluted membranes that appear to be connected to the rough endoplasmic reticulum (ER) (18–24). These ROs show remarkable parallels among different +RNA viruses that belong, for example, to the Flaviviridae, Picornaviridae, and Togaviridae families and perhaps also DNA viruses that replicate in the cytoplasm, such as members of the Poxviridae (11, 25). The molecular mechanisms and factors involved in the formation of these membranous structures are poorly understood, and the roles of specific membrane structures, lipid and protein components, and enzymes involved in their production remain to be studied in more detail. Consistent with the ER being the most likely membrane donor for coronaviral DMVs, cellular factors associated with ER-to-Golgi complex trafficking and early secretory pathways (e.g., PDI, Sec61a, EDEM1, and OS-9) have been reported to be involved in SARS-CoV and mouse hepatitis virus (MHV)-induced RTC formation (22, 26, 27). Also, a potential role of autophagy in coronaviral DMV formation has been discussed, even though a number of conflicting data make it difficult to draw definitive conclusions at this stage (28). At least in part, the observed differences may be related to the different cell lines and viruses used in these studies (29).
Given the major membrane rearrangements occurring in virus-infected cells, enzymes involved in cellular lipid metabolism have been suggested to play a major role in this process. In line with this, fatty acid synthase (FASN), which is a key enzyme in the fatty acid biosynthetic pathway, was shown to be recruited to dengue virus (DENV) replication complexes (30). Moreover, pharmacological inhibition of FASN by C75 (trans-4-carboxy-5-octyl-3-methylenebutyrolactone) or cerulenin resulted in impaired DENV, hepatitis C virus (HCV), West Nile virus (WNV), yellow fever virus (YFV), and vaccinia virus (VV) replication (30–35).
In this work, we extend these studies by reporting an essential role for cytosolic phospholipase A2α (cPLA2α) in the production of DMV-associated coronaviral RTCs. This enzyme belongs to the phospholipase A2 (PLA2) superfamily of lipolytic enzymes, which (among several other families) includes the secretory PLA2s (sPLA2s), Ca2+-independent PLA2s (iPLA2s), and Ca2+-dependent cPLA2s, with the latter including cPLA2α (36, 37). PLA2s catalyze the hydrolysis of glycerophospholipids at the sn-2 ester bond, generating a free fatty acid and a lysophospholipid (LPL) (37, 38). cPLA2α has a molecular mass of 85 kDa and preferentially hydrolyzes phospholipids that carry arachidonic acid (AA) at the sn-2 position. The activity of cPLA2α and its translocation to intracellular membranes is regulated by Ca2+ binding and phosphorylation at Ser-505 by mitogen-activated protein (MAP) kinase (39). In a previous study, cPLA2α activity was shown to be critically involved in the production of infectious progeny of HCV and DENV, while the activity was dispensable for vesicular stomatitis virus (VSV), a member of the Rhabdoviridae, suggesting distinct requirements for this lipolytic enzyme in the replication and assembly of different families of RNA viruses (40).
In this study, we report that the specific inhibition of cPLA2α activity has detrimental effects on coronavirus replication. In the presence of pyrrolidine-2 (Py-2) (41), a highly specific inhibitor of cPLA2α, the formation of DMVs and DMV-associated RTCs was significantly reduced in HCoV-229E-infected Huh-7 cells. Also, viral protein and RNA accumulation and production of infectious virus progeny were drastically diminished in the presence of noncytotoxic concentrations of Py-2. Moreover, using confocal microscopy, viral RTCs were shown to colocalize with LPL-containing membrane structures. Lipidomics studies revealed that LPL concentrations were increased in coronavirus-infected cells and that this phenotype was suppressed by Py-2. Taken together, our data suggest that LPLs produced by cPLA2α may be involved in DMV formation. The study also shows that cPLA2α activity is required for efficient replication of MERS-CoV and Semliki forest virus (SFV), while poliovirus (PV), human rhinovirus 1A (HRV1A), VV, and influenza A virus (IAV) replication was not affected in the presence of the cPLA2α inhibitor. In summary, our data lead us to conclude that cPLA2α is an important cellular factor acting at specific steps of the replication cycle of viruses from different +RNA virus families.
RESULTS
cPLA2α activity is required for HCoV-229E replication.
The inhibition of cPLA2α by Py-2 (20 μM) was previously reported to reduce the production of infectious virus progeny of different members of the Flaviviridae, as shown for HCV and DENV (40). We here investigated whether Py-2 also affects the replication of other +RNA viruses, such as coronaviruses, using HCoV-229E and MERS-CoV. First, we sought to confirm that Py-2 has no cytotoxic effects in Huh-7, MRC-5, and BEAS-B2 cells at concentrations shown previously to have strong antiviral affects (40). We found that cell viability was not affected by Py-2 concentrations of up to 40 μM (Fig. 1A). Next, we determined the effect of Py-2 on HCoV-229E reproduction. As shown by the results in Fig. 1B, treatment with Py-2 resulted in reduced viral titers in a dose-dependent manner, with nearly 100-fold reduction of HCoV-229E titers produced from Huh-7 cells treated with 20 μM Py-2. HCoV-229E replication in the presence of 20 μM Py-2 was also found to be reduced using MRC-5 (Fig. 1C) and BEAS-B2 (Fig. 1D) cells, suggesting that the observed antiviral effect of Py-2 is not cell type specific. As shown by the results in Fig. 1E, the reduced production of infectious virus progeny at 12 h postinfection (p.i.) could also be confirmed for later time points (15, 21, and 24 h p.i.), suggesting a profound inhibition (rather than a delay) of viral replication by this cPLA2α inhibitor.
FIG 1.

Production of infectious HCoV-229E progeny in cell culture is inhibited by the cPLA2α inhibitor Py-2. (A) MTT assay of Huh-7, BEAS-B2, and MRC-5 cells that were treated with the indicated concentrations of Py-2 for 12 h. Cell viability was determined using a tetrazolium-based reagent. (B to D) Virus titers (PFU/ml) in cell culture supernatants collected from Py-2-treated and HCoV-229E-infected Huh-7 (B), MRC-5 (C), and BEAS-B2 (D) cells at 12 h p.i. Cells were infected at an MOI of 3. At 2 h p.i., the virus inocula were replaced with cell culture medium containing the indicated concentrations of Py-2. (E) Growth curves of HCoV-229E in the presence or absence of Py-2 using Huh-7 cells infected at an MOI of 3. At 2 h p.i., the virus inocula were replaced with cell culture medium containing the indicated concentrations of Py-2. Supernatants were collected at 10, 12, 15, 21, and 24 h p.i., and virus titers were determined by plaque assay. Error bars show standard deviations.
In line with the Py-2 inhibition data, a second PLA2 inhibitor, arachidonyltrifluoromethane (AACOCF3; C21H31F3O), was confirmed to reduce HCoV-229E replication at nontoxic concentrations (Fig. 2A and B). Most likely, the slightly lower efficacy of AACOCF3 resulted from its lower specificity, as discussed previously (40). Similar antiviral effects on HCoV-229E replication were also observed for inhibitors of p38 MAP kinase and MEK, two important activators of cPLA2α (Fig. 2C), but not for inhibitors of enzymes acting downstream from cPLA2α in arachidonic acid (AA)-dependent pathways, such as cyclooxygenases 1 and 2 (COX1/2) and lipoxygenase (LOX) (Fig. 2D) (42), which metabolize AA to produce important inflammation mediators. Taken together, the data support a critical role of (activated) cPLA2α in HCoV-229E replication, suggesting that this lipolytic enzyme has a more general role in +RNA virus replication than previously thought.
FIG 2.
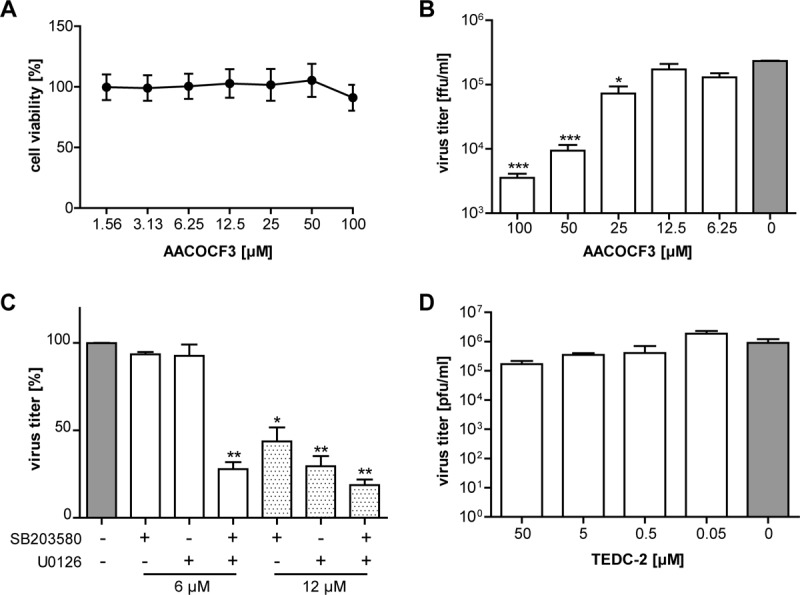
Antiviral activities of the PLA2 inhibitor AACOCF3 and of p38 and MEK inhibitors in coronavirus replication. (A) MTT assay of Huh-7 cells treated for 12 h with the indicated concentrations of AACOCF3. Cell viability (compared to that of untreated cells) was determined using a tetrazolium-based reagent. (B) Huh-7 cells were infected with HCoV-229E (MOI of 3). After 2 h, the virus inoculum was replaced with cell culture medium containing the indicated concentrations of AACOCF3. At 12 h p.i., cell culture supernatants were collected and used to determine virus titers by focus-forming assay. (C, D) Huh-7 cells were infected with HCoV-229E (MOI of 1) and treated with the indicated concentrations of the p38 inhibitor SB203580 and/or the MEK inhibitor U0126 as indicated (C) or the lipoxygenase inhibitor TEDC-2 (D). Virus titers in cell culture supernatants collected at 24 h p.i. were determined by plaque assay. Significance levels compared to the results for untreated cells were determined by two-tailed unpaired Student's t test and are indicated in panels B and C as follows: *, P < 0.05; **, P < 0.005; ***, P < 0.0005. Error bars show standard deviations.
To identify critical steps of the coronavirus life cycle that are affected by cPLA2α inhibitors, we characterized viral protein accumulation in infected cells by Western blotting and immunofluorescence analysis. As shown by the results in Fig. 3A and B, the accumulation of HCoV-229E structural (N) and nonstructural (nsp8) proteins was reduced in the presence of the drug, with nearly no viral proteins being detectable at 20 μM Py-2. In line with this, we found a significantly reduced accumulation of viral RNAs in infected cells treated with the cPLA2α inhibitor (Fig. 3C). These data suggest that, unlike the situation in HCV and DENV (40), cPLA2α activity may be required for an early step in coronavirus replication. To investigate whether viral entry or other early steps in viral replication are affected by the drug, we performed a time-of-addition experiment (Fig. 3D). For this purpose, Py-2 (20 μM) was included in the cell culture medium during virus adsorption (until 2 h p.i.) or at later time points (from 2 to 6, 4 to 8, 6 to 10, and 8 to 12 h p.i.). To determine the total infectious virus progeny produced until 12 h p.i. (with Py-2 being added at different time points), cell culture supernatants collected over time for a given experiment were pooled and virus titers were determined by focus-forming assay. The presence of Py-2 in the culture medium between 0 and 2 h p.i. had no effect on virus titers, indicating that cPLA2α activity is not required for viral entry. In contrast, the presence of Py-2 from 2 to 6 h p.i. caused a massive reduction of virus titers, while less profound effects were observed if the drug was present in the culture medium later in infection (Fig. 3D). The observed time-dependent effects of Py-2 on the production of infectious HCoV-229E progeny lead us to suggest that cPLA2α activity is important for an early step of coronavirus replication but not for entry itself.
FIG 3.

Time- and concentration-dependent inhibition of HCoV-229E replication in Huh-7 cells by the cPLA2α inhibitor Py-2. (A) Immunofluorescence analysis of N protein expression in Huh-7 cells infected with HCoV-229E at an MOI of 3 in the presence or absence of 20 μM Py-2. (B) Western blot analysis of HCoV-229E nonstructural (nsp8) and structural (N) protein accumulation at 12 h p.i. in Py-2-treated Huh-7 cells (MOI of 3). Actin was used as loading control. (C) Northern blot analysis of viral RNA in HCoV-229E-infected Huh-7 cells (MOI of 3) at 12 h p.i. Cells were kept in medium containing the indicated concentrations of Py-2. The positions of viral genomic and major subgenomic RNAs (2, 4, 5, 6, and 7) are indicated. (D) Time-dependent antiviral effects of Py-2. HCoV-229E-infected cells were treated with cPLA2α inhibitor (20 μM Py-2) for different time periods p.i. as indicated below. Production of infectious virus progeny was determined using (pooled) cell culture supernatants collected until 12 h p.i. Virus titers were determined and compared to the titer determined for infected but untreated cells. Experiments were done in triplicate. Significance levels determined by two-tailed unpaired Student's t test are indicated as follows: n.s., not significant; *, P < 0.05. Error bars show standard deviations.
cPLA2α inhibition prevents the formation of viral RTCs.
As described above, a profound antiviral effect was observed for Py-2 when given between 2 and 6 h p.i., suggesting that the formation of RTCs and their integration into rearranged cellular membranes may be affected by the drug. We therefore investigated potential effects of Py-2 on the formation of ROs in HCoV-229E-infected cells. Coronavirus RTCs are known to produce a typical perinuclear staining pattern when analyzed by immunofluorescence microscopy using reagents that detect double-stranded RNA (dsRNA) and viral replicative proteins. In the presence of 20 μM Py-2, the typical punctate perinuclear staining described previously for HCoV-229E RTC components in virus-infected cells (43, 44) was greatly diminished (Fig. 4). The profound reductions in both size and numbers of viral ROs were even more evident in three-dimensional reconstructions of z-stacks obtained from infected, untreated and infected, treated cells (Fig. 5A).
FIG 4.

Inhibition of coronavirus RTC formation in Py-2-treated cells. Huh-7 cells were mock infected or infected with HCoV-229E (MOI of 3) and incubated in the presence or absence of 20 μM Py-2 as indicated. At 12 h p.i., the cells were fixed with 3.7% paraformaldehyde and analyzed by immunofluorescence microscopy using antibodies specific for dsRNA (red signal) and nonstructural protein 8 (nsp8; green signal) to monitor the formation of viral RTCs in infected cells. Insets indicate regions of interest displayed at higher magnification in the next row.
FIG 5.
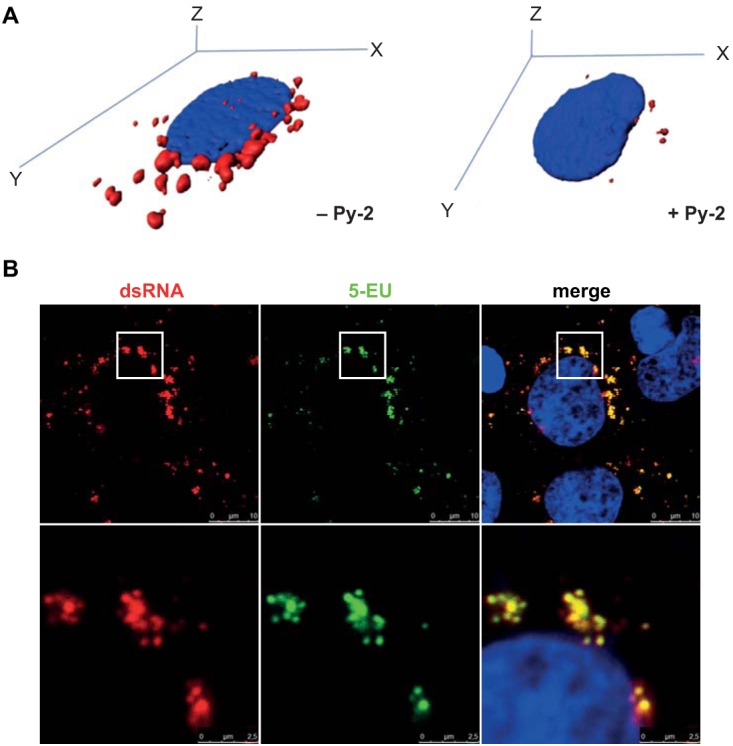
Immunofluorescence analysis of dsRNA and nascent RNA in HCoV-229E-infected cells. (A) Three-dimensional immunofluorescence analysis of z-stacks (Imaris) of representative HCoV-229E-infected Huh-7 cells (MOI of 3) incubated in the absence or presence of 20 μM Py-2 using a dsRNA-specific antibody (red signal). (B) Huh-7 cells were infected with HCoV-229E (MOI of 3). At 1 h p.i., the virus inoculum was replaced with medium containing 20 μM actinomycin D to inhibit cellular DNA-dependent RNA synthesis. At 11 h p.i., the cell culture medium was supplemented with 1 mM 5-EU. At 12 h p.i., the cells were fixed and incorporation of alkyne-modified 5-EU was detected using click chemistry (see Materials and Methods). Nuclei were stained with DAPI (4′,6-diamidino-2-phenylindole) (blue signal), and dsRNA was stained using a dsRNA-specific MAb (red signal). Insets indicate regions of interest displayed at higher magnification in the next row.
Although dsRNA and replicase gene-encoded nsp's are accepted as key components of viral RTCs and, therefore, have been widely used as markers to localize intracellular sites of viral RNA synthesis, a number of betacoronavirus studies showed that (i) dsRNA, (ii) newly synthesized RNA, (iii) RTC components, and (iv) (some) virus-induced membrane compartments may not always colocalize perfectly, particularly, at later time points p.i. (20, 24). We therefore decided to perform an additional experiment to answer the question of whether dsRNA and nascent RNA are equally suitable for detecting intracellular sites of RNA synthesis at 12 h p.i., that is, the time point used in the present study for HCoV-229E-infected Huh-7 cells. To this end, we conducted an immunofluorescence study of nascent RNA synthesis by click chemistry using the uridine analog 5-ethynyl uridine (5-EU) (45) and (co-)stained the sites of dsRNA accumulation using a dsRNA-specific antibody. As shown by the results in Fig. 5B, we were able to show that dsRNA and nascent RNA colocalize very well, suggesting that (most) dsRNA-containing structures represent ROs actively engaged in viral RNA synthesis, at least at this particular time point. Taken together, our data lead us to suggest that the formation of DMV-associated RTCs in HCoV-229E-infected cells is reduced in Py-2-treated cells.
To further corroborate this hypothesis, we studied DMV formation by transmission electron microscopy (Fig. 6). Intracellular virions (ICVs) were detected in approximately 50% of the cells analyzed per section and DMVs in approximately 40% of the cells analyzed per section in HCoV-229E-infected Huh-7 cells at 12 h p.i. (Fig. 6A, B, and E). In contrast, significantly fewer DMVs were detected if the virus-infected cells were treated with Py-2 (Fig. 6C and D), with only 20% of the cells containing DMVs and 10 to 15% containing ICVs (Fig. 6E). In infected, untreated cells, we regularly observed clusters of around 15 DMVs. As a result of Py-2 treatment, the number of DMVs per cell section dropped significantly, to about 5 per cell (Fig. 6F). This phenotype was most evident in cells treated with Py-2 early in infection, starting at 2 h p.i. There was no major difference observable between cells treated with Py-2 for 5 and 10 h (Fig. 6E and F, compare 2 to 7 and 2 to 12 h p.i.). In both cases, the numbers of DMVs and ICVs per cell were significantly reduced. In contrast, if cPLA2α activity was inhibited at later time points (between 7 and 12 h p.i.), DMV formation was not significantly affected compared to that in untreated HCoV-229E-infected cells. The data suggest an important role for cPLA2α activity in the process of DMV formation occurring early in infection, while this activity appears to be less critical if sufficient numbers of DMVs have already been formed (Fig. 6E and F).
FIG 6.

Py-2 reduces the formation of DMVs. Huh-7 cells were infected with HCoV-229E (MOI of 3) and incubated in the absence (A, B) or presence (C, D) of 20 μM Py-2 in the cell culture medium. At 12 h p.i., the cells were fixed with glutaraldehyde, embedded, and analyzed by transmission electron microscopy (EM912a/b; Zeiss) at 120 kV. The representative images included in this figure were selected from >100 images captured in two independent experiments (see below). (A and C) Insets indicate regions of interest displayed at higher magnification in panels B and D. (E, F) Time-dependent effects of Py-2 on DMV and intracellular virus (ICV) production in HCoV-229E-infected cells. Py-2 (20 μM) was included in the cell culture medium for the indicated times p.i. (E) Percentages of cells in which DMVs and ICVs could be identified (for details, see Materials and Methods). (F) Numbers of DMVs detected per cell (for details, see Materials and Methods). Significance levels compared to the results for untreated cells were determined by two-tailed unpaired Student's t test and are indicated in panels E and F as follows: n.s., not significant; *, P < 0.05; ***, P < 0.0005. The number of images analyzed per condition was as follows: inf (HCoV-infected cells without inhibitor), n = 154; 2 to 12 h, n = 190; 2 to 7 h, n = 135; 7 to 12 h, n = 107. Error bars show standard deviations.
Colocalization of coronavirus RTCs with LPLs.
cPLA2α cleaves glycerophospholipids at the sn-2 position, generating an LPL and releasing AA. The latter is a key inflammatory intermediate and important precursor that is metabolized by multiple enzymes, including cyclooxygenases 1/2 (COX1/2) and 5-lipoxygenase (LOX), leading to the production of prostaglandins, thromboxanes, leukotrienes, and many other inflammation mediators. As shown by the results in Fig. 2D and a previous study (42), we failed to obtain evidence for anticoronaviral activities of LOX and COX1/2 inhibitors, arguing against a major role of AA (and its downstream metabolites) in coronavirus replication. We therefore considered it more likely that the other product of PLA2α activity (i.e., the LPL) has a role in supporting viral replication, for example, by providing specific lipid components required to form viral ROs. To address the latter possibility, we made use of a fluorogenic phosphatidylcholine [1-O-(6-BODIPY 558/568-aminohexyl)-2 BODIPY FL C5-sn-glycero-3-phosphocholine; PC-A2] with (quenched) fluorophores attached to each of the two fatty acids. Following liposome-mediated uptake of PC-A2 and PLA2 (including cPLA2α)-mediated cleavage of the fatty acid attached to the sn-2 position of this fluorogenic substrate, the subcellular localization of the resulting LPL was monitored by confocal laser-scanning microscopy. To investigate a possible colocalization of LPLs and viral RTCs, PC-A2-treated and infected cells were fixed and a dsRNA-specific antibody was used as a marker for RTCs. As shown by the results in Fig. 7, viral RTCs were detected with their typical perinuclear staining pattern (see also Fig. 4). A careful inspection revealed that, although LPL and dsRNA clusters vary with respect to size and spatial distribution, there was a high degree of colocalization of dsRNA with LPL signals (but not vice versa), which is also supported by our calculations of Pearson's colocalization coefficients (PCCs) and colocalization rates. This colocalization of RTCs with LPLs is also illustrated by the signals displayed in the extra colocalization channel shown to the right (Fig. 7, rightmost column), with the colocalization signals strongly resembling the signals obtained for the dsRNA clusters alone. The high colocalization rates and PCC values revealed by our analysis strongly support the hypothesis that LPLs produced by cPLA2α are integral parts of DMVs. It should also be noted that the colocalization rates indicated in Fig. 7 are based on an automated and rather cautious method of calculation (see Materials and Methods). LPL signals that do not colocalize with dsRNA presumably originate from other intracellular membrane structures, such as endocytic vesicles.
FIG 7.

Coronavirus RTCs colocalize with LPLs produced by cPLA2α activity. Huh-7 cells were infected with HCoV-229E (MOI of 3) and incubated with PC-A2, a fluorogenic PLA2α substrate suitable to detect LPLs produced by cPLA2α cleavage at the sn-2 position of phospholipids. At 12 h p.i., the cells were fixed and immunostained for dsRNA. Colocalization signals were calculated for the total images shown in each of the 3rd to 6th rows and are displayed separately (right column). Colocalization rates and PCCs are indicated (for details, see Materials and Methods). Insets indicate regions of interest displayed at higher magnification in the next row.
Lipidome analysis of infected cells.
To gain more insight into the roles of lipids in coronavirus replication and DMV formation, we performed comparative lipidome analyses of (i) Huh-7 cells, (ii) Huh-7 cells incubated with UV-inactivated HCoV-229E, (iii) Huh-7 cells infected with HCoV-229E, (iv) Huh-7 cells infected with HCoV-229E and treated with Py-2, and (v) Huh-7 cells treated with Py-2 (Fig. 8). Lipids were isolated at 12 h p.i., a time when, under optimal conditions, DMVs have been formed (Fig. 4, 5, and 6) and large amounts of viral genomic and subgenomic RNAs have been produced (Fig. 3C). We monitored the abundances of 359 lipids of 14 classes covering membrane lipid classes and neutral lipids (see Tables S1 and S2 in the supplemental material).
FIG 8.

Coronavirus replication is associated with an increase of the cellular LPL content. The quantities of selected lipid classes and categories were determined using shotgun lipidomics. Shown are the results for Huh-7 cells alone, incubated with UV-inactivated HCoV-229E, infected with HCoV-229E, and infected with HCoV-229E and treated with 20 μM Py-2 as indicated to the left and below. (A) Abundance of all membrane lipids detected in the samples. (B) Abundance of neutral lipids, representing the sum of triacylglycerols, diacylglycerols, and cholesterylesters. Also shown are the abundances of phosphatidic acids (C), ceramides (D), lysophospholipids (E), and phosphatidylglycerols (PG) (F). Significance levels were determined by two-tailed unpaired Student's t test and are indicated as follows: n.s., not significant; *, P < 0.05; **, P < 0.005; ***, P < 0.0005. For a complete data set of individual lipid species, see Tables S1 and S2 in the supplemental material. Abundances are given in arbitrary units (a.u.). Error bars show standard deviations.
The study provided evidence that a number of changes in the cellular lipidome occur in HCoV-229E-infected cells compared to the cellular lipidomes in both the mock control and cells incubated with UV-inactivated HCoV-229E. Our data revealed no significant change in the total abundance of membrane lipids (Fig. 8A) and neutral lipids (triacylglycerols, diacylglycerols, and cholesterylesters) (Fig. 8B). However, we observed that Py-2 treatment reduced the abundance of membrane lipids by approximately 25% compared to their abundance in untreated HCoV-229E-infected cells or mock-infected cells (Fig. 8A). Further analyses of the lipid profiles revealed that the phosphatidic acid (PA) abundance was decreased in Py-2-treated and in HCoV-229E-infected Huh-7 cells at 12 h p.i. (Fig. 8C). For ceramides (Cer), an increased abundance was detected in Py-2-treated and in HCoV-229E-infected cells (Fig. 8D). However, Py-2 treatment of infected cells did not alter the ceramide content any further, suggesting that Cer- and PA-associated metabolic pathways in HCoV-229E-infected cells are not (or only marginally) affected by the cPLA2α activity.
For many lysophospholipid species (LPL), most prominently lysophosphatidylethanolamine (LPE) and lysophosphatidylinositols (LPI), a correlation between Py-2 treatment and inhibition of viral replication and lipid quantities was observed (Fig. 8E; Table S1). The total cellular LPL content was found to be reduced upon treatment with Py-2, supporting a specific role of cPLA2α in generating these LPLs. For HCoV-229E-infected cells, the LPL content was increased in comparison to that in cells incubated with UV-treated HCoV-229E, indicating that increased LPL production occurs after viral entry and requires a replication-competent virus. Additionally, inhibition of cPLA2α activity in HCoV-229E-infected cells suppressed the replication-associated increase of LPLs, resulting in an LPL content that was similar to those of (i) cells inoculated with UV-treated HCoV-229E and (ii) the mock control. Finally, we found a correlation between the cellular phosphatidylglycerol (PG) content and viral replication (Fig. 8F). The level of PG was 2-fold increased in HCoV-229E-infected cells, while Py-2 treatment during viral infection resulted in a PG level similar to that of the control (using UV-inactivated virus). Py-2 treatment alone had a minor effect on (total) PG abundances. However, PG species with shorter acyl chain lengths, such as PG 30:1, PG 32:2, PG 32:1, and PG 32:0, were clearly affected by the inhibitor and also showed significantly increased levels in cells incubated with UV-treated HCoV-229E (Table S1), indicating that cellular responses in lipid metabolism to viral receptor binding and/or entry might, at least in part, overlap with redirections of specific metabolic networks caused by the inhibition of PLA2α activity.
Taken together, these results show that coronavirus replication stimulates cellular LPL production, which together with the PLA2α inhibition data presented above, supports the idea that LPLs play an important role in DMV formation and viral replication.
Effect of cPLA2α inhibition on the replication of other viruses.
The observed critical role of cPLA2α activity in modulating cellular membrane structures and total LPL content in HCoV-229E-infected cells prompted us to investigate potential antiviral effects of cPLA2α inhibitors on other viruses, including viruses that are known to rearrange intracellular membrane structures and compartments. First, we analyzed the effect of Py-2 treatment on another coronavirus, MERS-CoV, which due to its pathogenicity and the large number of MERS-related deaths, has attracted significant attention (8). For MERS-CoV, a drastic inhibition of viral replication in Huh-7 and Vero cells was observed in the presence of Py-2, demonstrating that cPLA2α has an equally important role for alpha- and betacoronavirus replication (Fig. 9A). An antiviral effect of Py-2 could also be confirmed for SFV (Fig. 9B), suggesting that cPLA2α activity may also be involved in the replication of members of the family Togaviridae. In contrast, the viral reproduction of other viruses included in this study was not affected by Py-2. Thus, for example, antiviral effects of the cPLA2α inhibitor were not confirmed for HRV and PV (family Picornaviridae) (Fig. 9C and D). Also, vaccinia virus, a DNA virus from the family Poxviridae that is known to induce major rearrangements of host cell membranes, was not found to be affected by Py-2 (Fig. 9E). As another control, we included IAV (family Orthomyxoviridae) in this study because this virus replicates in the nucleus and does not produce membranous ROs in the host cell cytoplasm. Even with 40 μM Py-2, IAV replication was not inhibited (Fig. 9F). Taken together, these data suggest critical (but different) functions of cPLA2α activity in the replication of viruses representing different virus families.
FIG 9.

Effects of cPLA2α inhibition on the replication of other viruses. Cells were infected with MERS-CoV (Huh-7 and Vero cells) (A), SFV (BHK-21 cells) (B), human rhinovirus 1A (HeLa cells) (C), poliovirus (Vero cells) (D), vaccinia virus (Huh-7 cells) (E), and influenza A virus (A549 cells) (F) at an MOI of 3. At 2 h p.i., the virus inocula were replaced with medium supplemented with the indicated concentrations of Py-2. At 12 h p.i., supernatants were collected and virus titers were determined by focus-forming assay (FFU/ml) or plaque assay (PFU/ml). Experiments were done in triplicate. Significance levels compared to titers obtained with untreated cells were determined by two-tailed unpaired Student's t test and are indicated as follows: *, P < 0.05; **, P < 0.005. Error bars show standard deviations.
DISCUSSION
Despite their enormous genetic diversity, virtually all +RNA viruses employ specialized membrane compartments (replicative organelles [ROs]) as structural scaffolds for their multisubunit replication machinery (46, 47). Because of their essential role in viral RNA synthesis, the viral and cellular factors involved in the formation of ROs are thought to represent potential drug targets for antiviral intervention, and in line with this, a number of small-molecule inhibitors of enzymes or signaling molecules involved in cellular lipid metabolism and membrane rearrangements have been reported to be effective against specific +RNA viruses or even a group of related viruses from the same genus or family (reviewed in reference 48). However, there is also evidence that the cellular factors and structures involved in the formation and function(s) of viral ROs are more diverse than previously thought, with significant differences being reported even for closely related viruses (49). The available evidence suggests that a detailed understanding of the RO structures of specific +RNA viruses and virus families will be required to identify suitable targets for therapeutic intervention. Obviously, the identification of essential factors and metabolic and/or signaling pathways conserved across different genera and families would be highly desirable, possibly paving the way for the development of broad-spectrum antivirals.
In this study, we have been able to show that the pharmacological inhibition of a cellular phospholipase, cPLA2α, using a specific small-molecule inhibitor, drastically reduces coronavirus RNA synthesis and, as a consequence, protein accumulation and the production of infectious virus progeny. The data suggest that the inhibition of cPLA2α activity blocks an early step in the viral replication cycle, most likely the formation of virus-induced ROs. The cPLA2α activity was confirmed to be required for coronavirus replication, as shown for HCoV-229E (genus Alphacoronavirus) and MERS-CoV (genus Betacoronavirus), but the cPLA2α inhibitor was also effective against SFV, a member of the family Togaviridae, demonstrating that this phospholipase activity produces specific lipid compounds that are essential for the replication of phylogenetically diverse +RNA viruses. The precise role of cPLA2α in the production of fully functional ROs remains to be established. One of the products generated by cPLA2α is AA, an important signaling molecule and precursor of the eicosanoid family of potent inflammatory mediators, such as prostaglandins, leukotrienes, lipoxins, and thromboxanes (50). Among other functions, AA might indirectly affect membrane formation and trafficking events by modulating specific signaling pathways in coronavirus-infected cells. To address this possibility, we performed a small number of experiments using inhibitors of COX1/2 and LOX, two key enzymes requiring AA as a precursor. None of these inhibitors was found to have an effect on coronavirus replication (Fig. 2A) (42), contradicting the idea of a major role of AA-dependent pathways involving COX1/2 and LOX and their products in coronavirus replication, at least in vitro. Consistent with this hypothesis, the addition of AA to the cell culture medium failed to restore coronavirus replication in Py-2-treated cells (unpublished data). Based on these data and despite the fact that we cannot formally exclude other roles of AA in coronavirus replication, we consider it more likely that the LPLs (rather than AA) produced by cPLA2α have critical functions in coronavirus replication and, particularly, in the formation of virus-induced ROs. LPLs are found in relatively small amounts in biological membranes, and yet, they play important roles in a wide range of processes involving membrane remodeling, as well as membrane-protein and membrane-membrane interactions (51). Also, it is known that changes in the lipid composition of membranes may be associated with membrane fusion and fission processes (52). According to the “bilayer couple hypothesis” (53), the two leaflets of a lipid bilayer are tightly coupled, with asymmetric changes in one leaflet having the potential to induce major structural changes, such as membrane bending, fission, and fusion (54–56). Thus, for example, asymmetric cleavage of phospholipids in a lipid bilayer by cPLA2α (the latter converting cylindrical phospholipids into cone-shaped LPLs) can be expected to induce membrane curvature, which may in turn trigger the formation of vesicular membrane structures, as demonstrated previously for cPLA2α-mediated membrane-modulating activities involved in tubulation and vesiculation processes of the Golgi complex, the vesiculation of CD59-containing endosomes, and lipid droplet formation (57–60).
There is also evidence that, independent of its enzymatic activity, cPLA2α may change the membrane phospholipid packing through its hydrophobic C2 domain to induce the membrane bending required for phagosome formation in macrophages (61, 62). In addition to cPLA2α and related phospholipases, a large number of other factors have been shown to induce membrane curvature in diverse biological systems (for recent reviews, see references 63, 64, and 65).
To provide additional evidence for specific lipid classes, particularly LPLs, playing an important role in coronavirus replication, whole-cell lipidome analyses of coronavirus-infected cells were performed in this study. We were able to show that phosphatidic acid (PA) species are downregulated, whereas ceramide (Cer) and LPL species are upregulated in HCoV-229E-infected cells (Fig. 8; Table S1 in the supplemental material). PA is a key intermediate in the synthesis of glycerophospholipids and triacylglycerides and an important lipid mediator that is involved in diverse cellular functions, including vesicular trafficking, cytoskeletal changes, secretion, and membrane alterations (66, 67). The possible biological implications of the observed downregulation of PA remain to be studied.
The observed upregulation of bioactive Cer may indicate a cellular response to coronavirus replication or even a possible role of Cer in supporting coronaviral replication. Cer are known to induce apoptosis and autophagy (reviewed in reference 68). It remains to be studied whether (and to what extent) Cer contribute to autophagy and apoptosis in coronavirus-infected cells. Both processes have been suggested to be involved in coronavirus replication and represent emerging fields of coronavirus research, with partially controversial information being reported for different viral and cellular systems (22, 27, 28, 69–75).
Cer-rich domains are also known to increase the rigidity and stability of membranes. Cer are cone-shaped lipids that are able to induce negative curvature, thereby promoting inward budding of membranes (76) and, thus, possibly facilitating the formation of DMVs. In support of this, Cer was reported to be redistributed to West Nile virus (WNV)-induced ROs in infected Vero cells (77), while DENV, another member of the family of Flaviviridae, was found to induce increases in both Cer and LPL abundances in infected C6/36 mosquito cells (33). Interestingly, the study by Aktepe et al. (77) also showed that inhibition of Cer synthesis has detrimental effects on WNV (strain Kunjin) replication, while the replication of DENV serotype 2 strain New Guinea C was found to be enhanced, suggesting that the effects of Cer and Cer-derived lipids on +RNA virus replication are complex and, potentially, virus (strain) specific. Clearly, more studies are needed to obtain a better understanding of possible roles of Cer in viral replication.
In the context of this study, it was of particular interest to show that there is a significant increase of the LPL content in coronavirus-infected cells. This increase was not detected in infected cells treated with cPLA2α inhibitor, thus strongly supporting a critical involvement of cPLA2α activity in producing these increased LPL levels in infected cells. Furthermore, a colocalization of dsRNA with sn-2-cleaved fluorogenic LPLs was observed by fluorescence microscopy in virus-infected cells at 12 h p.i. Together, these observations support the idea that LPLs generated by cPLA2α are functionally relevant components of ROs produced in coronavirus-infected cells.
In a previous study, inhibition of cPLA2α activity by Py-2 was shown to affect HCV replication in vitro (40). However, in this case, the formation of HCV-induced ROs (“membranous web”) and viral RNA synthesis were not evidently affected. Instead, the production of infectious virus progeny was found to be reduced, most probably, by a reduction of lipid droplets required for HCV particle formation (40, 59). Furthermore, changes in the cellular lipidomes, including LPLs, were reported for cells infected with HCV and DENV (33, 78), further corroborating the hypothesis that LPLs have important but diverse functions in different +RNA virus systems.
To explore potential inhibitory effects of Py-2, several other viruses known to rearrange cellular membranes were included in this study. As mentioned above, the replication of MERS-CoV (genus Betacoronavirus) and SFV (family Togaviridae) was inhibited in the presence of Py-2, identifying cPLA2α as an important host factor for +RNA virus replication. In contrast, poliovirus 1 and human rhinovirus A1 (family Picornaviridae) were not affected by the cPLA2α inhibitor. Likewise, vaccinia virus (family Poxviridae), a DNA virus that is known to induce major rearrangements of cytoplasmic membranes (25), was not inhibited by the cPLA2α inhibitor. Finally, influenza A virus, a negative-strand RNA virus that replicates in the nucleus and does not induce specific ROs in the cytoplasm, was not affected by the cPLA2α inhibitor. Taken together, these inhibition data lead us to suggest that the formation of ROs of coronaviruses and, possibly, several other +RNA viruses depends on specific LPLs produced by cellular cPLA2α activities. The selective inhibitory effects observed for members of only a few +RNA virus families suggest very specific lipid requirements for these viruses and contradict potential nonspecific/toxic effects being responsible for the observed antiviral effects of Py-2 against corona- and alphaviruses. To our knowledge, the study provides the first in-depth analysis of cellular lipidome changes in coronavirus-infected cells and adds to the list of lipids and lipid-metabolizing enzymes confirmed to be involved in +RNA virus replication and, possibly, suitable as targets for antiviral small-molecule inhibitors. However, given the diverse structures, origins, and lipid/protein compositions of virus-induced ROs, the study supports previous conclusions that the inhibition of viral RO formation by targeting highly (or even universally) conserved cellular factors remains a challenging goal (46, 47).
MATERIALS AND METHODS
Cells and viruses.
Human hepatoma cells (Huh-7), human lung fibroblasts (MRC-5), African green monkey kidney cells (Vero and CV-1), baby hamster kidney cells (BHK-21), human lung epithelial cells (A549), human bronchial epithelial cells (BEAS-B2), human cervix epithelial cells (HeLa), and Madin-Darby canine kidney cells (MDCK-II) were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin, and 100 μg/ml streptomycin at 37°C and in an atmosphere containing 5% CO2. HCoV-229E, human rhinovirus 1A (HRV1A), influenza virus A/Giessen/06/09 (H1N1), SFV, PV type 1 (strain Mahoney), and VV (strain WR) were obtained from the virus collection of the Institute of Medical Virology, Giessen, Germany. MERS-CoV (EMC/2012) was kindly provided by Christian Drosten, Bonn, Germany.
Drugs and assays.
The cell-permeable pyrrolidine derivative pyrrolidine-2 (Py-2) (C49H44F2N4O5S, 840 g/mol), a highly specific cPLA2α inhibitor, was purchased from Merck Millipore (compound 4d, catalog number 525143) (41). Arachidonyltrifluoromethane (AACOCF3) (C21H31F3O, 356.5 g/mol), an analog of AA that inhibits cPLA2 by direct binding (79), was obtained from Santa Cruz Biotechnology. The MEK inhibitor U0126 and the p38 MAP kinase inhibitor SB202190 were purchased from Selleckchem. The lipoxygenase inhibitor TEDC-2 [2-(1-thienyl)ethyl 3,4-dihydroxybenzylidenecyanoacetate] was purchased from Tocris Bioscience. Compounds were stored at −20°C as 2 mM (Py-2), 10 mM (AACOCF3), 20 mM (SB202190), and 50 mM (U0126 and TEDC-2) stock solutions in dethyl sulfoxide (DMSO).
Cell viability in the presence of specific drugs was determined in a 96-well format by MTT [3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide] assay (80). Briefly, nearly confluent cell monolayers were incubated with cell culture medium containing the respective drug at the concentrations indicated on Fig. 1A and B, respectively. Following incubation for 12 h, the culture medium was replaced with 200 μl MTT mixture (DMEM containing 10% FBS and 175 μg/ml tetrazolium bromide; Sigma). Following incubation for 90 min at 37°C, the cells were fixed with 3.7% paraformaldehyde (PFA; Roth) in phosphate-buffered saline (PBS) for 30 min. Then, the fixing solution was removed and 200 μl isopropanol was added to each well. Formazan formation was measured by determining the absorbance at 490 nm using a spectrophotometer (BioTek).
To determine the antiviral effects of drugs, confluent cell monolayers of Huh-7 (for HCoV-229E, MERS-CoV, and VV), MRC-5 and BEAS-B2 (for HCoV-229E), HeLa (for HRV1A), BHK-21 (for SFV), A549 (for IAV H1N1), and Vero (for PV and MERS-CoV) cells were infected at a multiplicity of infection (MOI) of 3. After 2 h, the virus inoculum was removed, cells were rinsed with PBS, and fresh medium containing the concentrations of Py-2 and AACOCF3 indicated or DMSO (solvent control) was added. At 12 h p.i., the cell culture supernatant was collected. The antiviral activities of SB202190, TEDC-2, and U0126 were determined using identical conditions except that cell culture supernatants were collected at 24 h p.i. After short-term storage at −80°C, the cell culture supernatants were used for virus titration.
Virus titration.
Focus-forming assays were used to determine the titers of IAV and coronaviruses. Briefly, Huh-7 (for CoVs) or MDCK (for IAV H1N1) cells were seeded in 96-well plates. At 90% confluence, the medium was removed, and the cells were washed with PBS++ (PBS containing 1 mM MgCl2 and 0.9 mM CaCl2) and inoculated for 1 h at 33°C with 10-fold serial dilutions of virus-containing cell culture supernatants in PBS++-BSA-P-S (PBS++ containing 0.2% bovine serum albumin [BSA], 100 U/ml penicillin, and 100 mg/ml streptomycin). Next, the virus inoculum was replaced with minimum essential medium (MEM) containing 1.25% Avicel (FMC Biopolymer) for 24 to 48 h. For IAV, 1 μg/ml trypsin was included in this medium. Next, the Avicel-containing medium was removed, and cells were washed with PBS and then fixed and permeabilized for 30 min with PBS containing 3.7% PFA and 1% Triton X-100. The cells were washed again with PBS and incubated with 50 μl of the appropriate primary antibody solution, each diluted in PBS containing 0.1% Tween 20 (PBST), as follows: anti-HCoV-229E-N mouse monoclonal antibody (MAb) (M.30.HCo.I1E7, 1:5,000 dilution; Ingenasa), rabbit anti-MERS-CoV-N polyclonal antiserum (100211-RP02-50, 1:200; Sino Biological, Inc.), and mouse anti-IAV-NP MAb (1:6,000; kindly provided by S. Ludwig, Münster, Germany). Following incubation for 1 h at room temperature, the cells were washed 3 times and incubated with the appropriate secondary antibody (goat anti-mouse IgG-horseradish peroxidase [HRP] [sc-2005] or goat anti-rabbit IgG-HRP [sc-2004], 1:1,000 in PBST; Santa Cruz Biotechnology) for 1 h at room temperature. The cells were washed again with PBS, stained with an AEC (3-amino-9-ethylcarbazole) staining kit (Sigma), and air dried, and focus numbers were determined.
To determine virus titers by plaque assay, Huh-7 (for HCoV-229E and MERS-CoV), HeLa (for HRV1A), Vero (for PV), CV-1 (for VV) and BHK-21 (for SFV) cells were seeded in 6-well plates and inoculated with 10-fold serial virus dilutions in PBS++-BSA-P-S for 1 h. Next, the virus inoculum was replaced with Avicel-containing medium (see above). At 2 to 4 days p.i., the medium was removed and cells were washed with PBS and fixed with 3.7% PFA in PBS. The cell monolayer was stained with 0.15% crystal violet in PBS, and plaques were counted.
Western blot analysis.
Huh-7 cells were infected with HCoV-229E at an MOI of 3 and incubated in medium containing the concentrations of cPLA2α inhibitor or DMSO (solvent control) indicated. At 12 h p.i., the cells were lysed in Triton lysis buffer (TLB; 20 mM Tris-HCl, pH 7.4, 137 mM NaCl, 10% glycerol, 1% Triton X-100, 2 mM EDTA, 50 mM sodium glycerophosphate, 20 mM sodium pyrophosphate, 5 μg/ml aprotinin, 5 μg/ml leupeptin, 1 mM sodium vanadate, 5 mM benzamidine). Proteins were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) in a 10% gel and transferred onto a 0.45-μm-pore-size nitrocellulose membrane (Protran; Amersham). Membranes were incubated for 1 h at room temperature with appropriate primary antibodies diluted in PBS containing 3% BSA, as follows: mouse anti-HCoV-229E-N-specific MAb (1:1,000, M.30.HCo.I1E7; Ingenasa), HCoV-229E nsp8-specific rabbit antiserum (1:1,000) (43), mouse anti-β-actin MAb (1:10,000, ab8226; Abcam), and rabbit anti-β-actin polyclonal antibody (1:10,000, ab8227; Abcam). After extensive washing with PBS, the membrane was incubated with goat anti-rabbit IRDye 800CW (1:10,000, 926-32211; LI-COR) and goat anti-mouse IRDye 680CW (1:10,000, 926-68070; LI-COR) polyclonal antibodies for 1 h at room temperature. After another wash step, the membranes were dried and the immunostained proteins were analyzed and quantified using a LI-COR Odyssey imaging system and software.
Northern blot analysis of intracellular viral RNA.
Total cellular RNA from infected cells was isolated by using TRIzol reagent (ThermoFisher Scientific) according to the manufacturer's instructions, and Northern hybridization was done as described previously (81) using a 32P-labeled DNA probe specific for HCoV-229E nucleotides 26857 to 27235.
Immunofluorescence analysis of viral RTCs.
Huh-7 cells were infected with HCoV-229E at an MOI of 3 and incubated in medium containing 20 μM Py-2 or DMSO (solvent control). At 12 h p.i., the cells were fixed and stained with mouse anti-dsRNA MAb (1:100, J2; SCICONS English & Scientific Consulting Kft.), mouse anti-HCoV-229E-N MAb (1:100, M.30.HCo.I1E7; Ingenasa), and rabbit anti-HCoV-229E-nsp8 polyclonal antiserum (1:100) (43). As secondary antibodies, Alexa Fluor 594 goat anti-mouse IgG (H+L) and Alexa Fluor 488 F(ab′)2-goat anti-rabbit IgG (H+L) (1:500, catalog numbers A11005 and A11070; Invitrogen) were used. Antibodies were diluted in PBS containing 3% BSA. For colocalization studies of viral RTCs with lysophospholipids produced by cellular PLA2 activities, Huh-7 cells were treated with the fluorogenic PLA2 substrate red/green BODIPY phospholipid PC-A2 (catalog number A10072; Invitrogen) according to the manufacturer's instructions. Briefly, 30 μl of 10 mM DOPC (1,2-dioleoyl-sn-glycero-3-phosphocholine; Avanti Polar Lipids), 30 μl of 10 mM DOPG {1,2-dioleoyl-sn-glycero-3-[phospho-rac-(1-glycerol)]; Avanti Polar Lipids}, and 30 μl of 1 mM PC-A2 (each prepared using ethanol as the solvent) were mixed. Seventy-seven microliters of this ethanolic lipid mixture was injected under rapid stirring into 5 ml of buffer containing 50 mM Tris-HCl, 100 mM NaCl, 1 mM CaCl2 (pH adjusted to 8.9). Next, this liposomally incorporated substrate mixture was added to the cells for 15 min at 10°C. Thereafter, the cells were infected with HCoV-229E at an MOI of 3. At 12 h p.i., the cells were fixed and stained using a dsRNA-specific MAb (see above). Colocalization studies of dsRNA with newly synthesized RNA were done according to the method of Hagemeijer et al. (20). Briefly, Huh-7 cells grown on glass coverslips were infected with HCoV-229E at an MOI of 3. At 2 h p.i., the virus inoculum was replaced with medium containing 20 μM actinomycin D (Sigma) to block cellular DNA-dependent RNA synthesis. At 11 h p.i., 1 mM 5-ethynyl uridine (5-EU; Invitrogen) was included in the medium. At 12 h p.i., the cells were fixed with 3.7% PFA in PBS and permeabilized using 0.1% Triton X-100. Incorporation of the alkyne-modified uridine analog, 5-EU, was visualized using click chemistry according to the manufacturer's instructions (Click-iT RNA Alexa Fluor 488 imaging kit; Invitrogen). In addition, the cells were stained using a dsRNA-specific antibody as described above.
Images were acquired by confocal laser-scanning microscopy (Leica TCS SP5). For colocalization analysis, hardware prerequisites and settings were carefully observed. For imaging, a 63× Plan-Apochromat objective (λ corrected; numeric aperture [NA] = 1.4) was used, and the pinhole aperture was set to 1 Airy unit (AU = 1), resulting in an optical section thickness of 0.772 μm. The intensities of laser lines used for excitation were kept rather low to minimize possible bleaching effects. The gain and offset of the photomultiplier tubes (PMTs) were optimized for each channel using lookup table (LUT) functions in order to adapt thresholds and to prevent clipping of high-intensity signals. The frames of the different channels were recorded sequentially, and care was taken to ensure clear spectral separation of the signals analyzed and to exclude any cross talk and bleed-through between channels.
Data were processed using the Imaris 8.4 software package (Bitplane). Colocalization analysis of dsRNA signals with signals of PC-A2 and cPLA2α-mediated cleavage products was based on correlating intensities in the different channels according to Pearson's colocalization coefficient (PCC), taking advantage of the PCC being highly independent from background levels and signal brightness (82). Values of 1, −1, and 0 indicate perfect colocalization, strict exclusion, and random localization, respectively, as described previously (83–86). Colocalization volumes and rates were calculated for total images using automated threshold settings (87).
Electron microscopy.
Huh-7 cells were grown on Aclar film (Agar Scientific). Following infection with HCoV-229E at an MOI of 3 (or mock infection) for 2 h, the cell culture medium was replaced with fresh medium containing 20 μM Py-2 (or the appropriate amount of DMSO as a control). After 12 h, at 33°C, the cell culture medium was removed. The cells were washed with PBS, fixed with 3% formaldehyde and 1% glutaraldehyde in 0.1 M PBS, and postfixed in 1% osmium tetroxide. After incubation in 1% aqueous uranyl acetate (Polysciences), specimens were dehydrated in an ethanol series (30, 50, 70, 80, 90, 96, and 100% [vol/vol], 20 min each) and embedded in Epon (Serva). From the heat-cured blocks, silver-to-gold ultrathin sections were cut and subsequently treated for contrast in uranyl acetate and lead citrate. Ultrathin sections were inspected in the transmission electron microscope (EM 912a/b; Zeiss) at 120 kV under zero-loss conditions and images were recorded at a slight underfocus using a cooled 2,000- by 2,000-pixel slow-scan charge-coupled device (CCD) camera (SharpEye/TRS) and the iTEM package (Olympus Soft Imaging Solutions). All experiments were done in biological duplicates. For statistical analysis, a total of >100 ultrathin sections from different cells were analyzed in each experiment.
Lipidome analysis.
Huh-7 cells were mock infected or infected with HCoV-229E at an MOI of 3 and incubated in medium containing (or lacking) Py-2 (20 μM, 2 to 12 h p.i.). As additional controls, (i) untreated/mock-infected cells and (ii) cells inoculated with UV-inactivated HCoV-229E were used. UV-inactivated virus was obtained by exposing an aliquot of the same virus stock to UV light (TUV15W/G15 T8; Philips) for 3 h. Inactivation of virus infectivity was confirmed by plaque assay. At 12 h p.i., cells were collected and subjected to cellular lipidome analysis. Briefly, lipids were extracted using methyl-tert-butyl ether as described previously (88) and analyzed with a Q-Exactive plus mass spectrometer (Thermo Scientific) using the shotgun lipidomics approach and LipidXplorer (89–92). Lipids were quantified using an internal standard mixture (Table S2) following the lipidomics screen approach. Lipid abundances were calculated using the respective standards and normalized to cell number (Tables S1 and S2). Experiments were performed using 8 biologically independent replicates for each group except for the group of HCoV-229E-infected, untreated cells (7 replicates). Statistical analyses were done using the two-tailed unpaired Student's t test.
Supplementary Material
ACKNOWLEDGMENTS
We thank Christian Drosten (Berlin, Germany) for providing MERS-CoV EMC/2012 and Nadja Karl, Anna Möbus, Sabine Agel, and Barbara Hönig for excellent technical assistance.
The work was supported by the German Center for Infection Research (DZIF), partner site Giessen, Germany (TTU Emerging Infections, to S.P. and J.Z.), and the Deutsche Forschungsgemeinschaft (KFO 309, project P3, to J.Z.; SFB 1021 RNA viruses: RNA metabolism, host response and pathogenesis; projects A01 and C01 to J.Z. and S.P., respectively).
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The authors declare no conflict of interest relating to this study.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.01463-17.
REFERENCES
- 1.de Groot RJ, Baker SC, Baric R, Enjuanes L, Gorbalenya AE, Holmes KV, Perlman S, Poon L, Rottier PJM, Talbot PJ, Woo PCY, Ziebuhr J. 2012. Family Coronaviridae, p 806–828. In King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ (ed), Virus taxonomy. Elsevier, Amsterdam, the Netherlands. [Google Scholar]
- 2.de Groot RJ, Cowley JA, Enjuanes L, Faaberg KS, Perlman S, Rottier PJM, Snijder EJ, Ziebuhr J, Gorbalenya AE. 2012. Order Nidovirales, p 785–795. In King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ (ed), Virus taxonomy. Elsevier, Amsterdam, the Netherlands. [Google Scholar]
- 3.van der Hoek L. 2007. Human coronaviruses: what do they cause? Antivir Ther 12:651–658. [PubMed] [Google Scholar]
- 4.Drosten C, Gunther S, Preiser W, van der Werf S, Brodt HR, Becker S, Rabenau H, Panning M, Kolesnikova L, Fouchier RA, Berger A, Burguiere AM, Cinatl J, Eickmann M, Escriou N, Grywna K, Kramme S, Manuguerra JC, Muller S, Rickerts V, Sturmer M, Vieth S, Klenk HD, Osterhaus AD, Schmitz H, Doerr HW. 2003. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med 348:1967–1976. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- 5.Peiris JS, Yuen KY, Osterhaus AD, Stohr K. 2003. The severe acute respiratory syndrome. N Engl J Med 349:2431–2441. doi: 10.1056/NEJMra032498. [DOI] [PubMed] [Google Scholar]
- 6.Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus ADME, Fouchier RAM. 2012. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med 367:1814–1820. doi: 10.1056/NEJMoa1211721. [DOI] [PubMed] [Google Scholar]
- 7.Peiris JS, Lai ST, Poon LL, Guan Y, Yam LY, Lim W, Nicholls J, Yee WK, Yan WW, Cheung MT, Cheng VC, Chan KH, Tsang DN, Yung RW, Ng TK, Yuen KY. 2003. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet 361:1319–1325. doi: 10.1016/S0140-6736(03)13077-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zumla A, Hui DS, Perlman S. 2015. Middle East respiratory syndrome. Lancet 386:995–1007. doi: 10.1016/S0140-6736(15)60454-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Angelini MM, Neuman BW, Buchmeier MJ. 2014. Untangling membrane rearrangement in the Nidovirales. DNA Cell Biol 33:122–127. doi: 10.1089/dna.2013.2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.V'Kovski P, Al-Mulla H, Thiel V, Neuman BW. 2015. New insights on the role of paired membrane structures in coronavirus replication. Virus Res 202:33–40. doi: 10.1016/j.virusres.2014.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ahlquist P. 2006. Parallels among positive-strand RNA viruses, reverse-transcribing viruses and double-stranded RNA viruses. Nat Rev Microbiol 4:371–382. doi: 10.1038/nrmicro1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.den Boon JA, Ahlquist P. 2010. Organelle-like membrane compartmentalization of positive-strand RNA virus replication factories. Annu Rev Microbiol 64:241–256. doi: 10.1146/annurev.micro.112408.134012. [DOI] [PubMed] [Google Scholar]
- 13.den Boon JA, Diaz A, Ahlquist P. 2010. Cytoplasmic viral replication complexes. Cell Host Microbe 8:77–85. doi: 10.1016/j.chom.2010.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Angelini MM, Akhlaghpour M, Neuman BW, Buchmeier MJ. 2013. Severe acute respiratory syndrome coronavirus nonstructural proteins 3, 4, and 6 induce double-membrane vesicles. mBio 4:e00524-13. doi: 10.1128/mBio.00524-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baliji S, Cammer SA, Sobral B, Baker SC. 2009. Detection of nonstructural protein 6 in murine coronavirus-infected cells and analysis of the transmembrane topology by using bioinformatics and molecular approaches. J Virol 83:6957–6962. doi: 10.1128/JVI.00254-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oostra M, Hagemeijer MC, van Gent M, Bekker CP, te Lintelo EG, Rottier PJ, de Haan CA. 2008. Topology and membrane anchoring of the coronavirus replication complex: not all hydrophobic domains of nsp3 and nsp6 are membrane spanning. J Virol 82:12392–12405. doi: 10.1128/JVI.01219-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Perlman S, Netland J. 2009. Coronaviruses post-SARS: update on replication and pathogenesis. Nat Rev Microbiol 7:439–450. doi: 10.1038/nrmicro2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ulasli M, Verheije MH, de Haan CAM, Reggiori F. 2010. Qualitative and quantitative ultrastructural analysis of the membrane rearrangements induced by coronavirus. Cell Microbiol 12:844–861. doi: 10.1111/j.1462-5822.2010.01437.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hagemeijer MC, Verheije MH, Ulasli M, Shaltiël IA, de Vries LA, Reggiori F, Rottier PJM, de Haan CAM. 2010. Dynamics of coronavirus replication-transcription complexes. J Virol 84:2134–2149. doi: 10.1128/JVI.01716-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hagemeijer MC, Vonk AM, Monastyrska I, Rottier PJM, de Haan CAM. 2012. Visualizing coronavirus RNA synthesis in time by using click chemistry. J Virol 86:5808–5816. doi: 10.1128/JVI.07207-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Knoops K, Bárcena M, Limpens RWAL, Koster AJ, Mommaas AM, Snijder EJ. 2012. Ultrastructural characterization of arterivirus replication structures: reshaping the endoplasmic reticulum to accommodate viral RNA synthesis. J Virol 86:2474–2487. doi: 10.1128/JVI.06677-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Snijder EJ, van der Meer Y, Zevenhoven-Dobbe J, Onderwater JJM, van der Meulen J, Koerten HK, Mommaas AM. 2006. Ultrastructure and origin of membrane vesicles associated with the severe acute respiratory syndrome coronavirus replication complex. J Virol 80:5927–5940. doi: 10.1128/JVI.02501-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gosert R, Kanjanahaluethai A, Egger D, Bienz K, Baker SC. 2002. RNA replication of mouse hepatitis virus takes place at double-membrane vesicles. J Virol 76:3697–3708. doi: 10.1128/JVI.76.8.3697-3708.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Knoops K, Kikkert M, Worm SH, Zevenhoven-Dobbe JC, van der Meer Y, Koster AJ, Mommaas AM, Snijder EJ. 2008. SARS-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol 6:e226. doi: 10.1371/journal.pbio.0060226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sodeik B, Krijnse-Locker J. 2002. Assembly of vaccinia virus revisited: de novo membrane synthesis or acquisition from the host? Trends Microbiol 10:15–24. doi: 10.1016/S0966-842X(01)02256-9. [DOI] [PubMed] [Google Scholar]
- 26.Knoops K, Swett-Tapia C, van den Worm SH, Te Velthuis AJ, Koster AJ, Mommaas AM, Snijder EJ, Kikkert M. 2010. Integrity of the early secretory pathway promotes, but is not required for, severe acute respiratory syndrome coronavirus RNA synthesis and virus-induced remodeling of endoplasmic reticulum membranes. J Virol 84:833–846. doi: 10.1128/JVI.01826-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reggiori F, Monastyrska I, Verheije MH, Cali T, Ulasli M, Bianchi S, Bernasconi R, de Haan CA, Molinari M. 2010. Coronaviruses hijack the LC3-I-positive EDEMosomes, ER-derived vesicles exporting short-lived ERAD regulators, for replication. Cell Host Microbe 7:500–508. doi: 10.1016/j.chom.2010.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maier HJ, Britton P. 2012. Involvement of autophagy in coronavirus replication. Viruses 4:3440–3451. doi: 10.3390/v4123440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reid CR, Airo AM, Hobman TC. 2015. The virus-host interplay: biogenesis of +RNA replication complexes. Viruses 7:4385–4413. doi: 10.3390/v7082825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heaton NS, Perera R, Berger KL, Khadka S, Lacount DJ, Kuhn RJ, Randall G. 2010. Dengue virus nonstructural protein 3 redistributes fatty acid synthase to sites of viral replication and increases cellular fatty acid synthesis. Proc Natl Acad Sci U S A 107:17345–17350. doi: 10.1073/pnas.1010811107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Greseth MD, Traktman P. 2014. De novo fatty acid biosynthesis contributes significantly to establishment of a bioenergetically favorable environment for vaccinia virus infection. PLoS Pathog 10:e1004021. doi: 10.1371/journal.ppat.1004021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martin-Acebes MA, Blazquez AB, Jimenez de Oya N, Escribano-Romero E, Saiz JC. 2011. West Nile virus replication requires fatty acid synthesis but is independent on phosphatidylinositol-4-phosphate lipids. PLoS One 6:e24970. doi: 10.1371/journal.pone.0024970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Perera R, Riley C, Isaac G, Hopf-Jannasch AS, Moore RJ, Weitz KW, Pasa-Tolic L, Metz TO, Adamec J, Kuhn RJ. 2012. Dengue virus infection perturbs lipid homeostasis in infected mosquito cells. PLoS Pathog 8:e1002584. doi: 10.1371/journal.ppat.1002584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sagan SM, Rouleau Y, Leggiadro C, Supekova L, Schultz PG, Su AI, Pezacki JP. 2006. The influence of cholesterol and lipid metabolism on host cell structure and hepatitis C virus replication. Biochem Cell Biol 84:67–79. doi: 10.1139/o05-149. [DOI] [PubMed] [Google Scholar]
- 35.Yang W, Hood BL, Chadwick SL, Liu S, Watkins SC, Luo G, Conrads TP, Wang T. 2008. Fatty acid synthase is up-regulated during hepatitis C virus infection and regulates hepatitis C virus entry and production. Hepatology 48:1396–1403. doi: 10.1002/hep.22508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Balsinde J, Winstead MV, Dennis EA. 2002. Phospholipase A(2) regulation of arachidonic acid mobilization. FEBS Lett 531:2–6. doi: 10.1016/S0014-5793(02)03413-0. [DOI] [PubMed] [Google Scholar]
- 37.Murakami M, Taketomi Y, Miki Y, Sato H, Hirabayashi T, Yamamoto K. 2011. Recent progress in phospholipase A(2) research: from cells to animals to humans. Prog Lipid Res 50:152–192. doi: 10.1016/j.plipres.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 38.Ghosh M, Tucker DE, Burchett SA, Leslie CC. 2006. Properties of the group IV phospholipase A2 family. Prog Lipid Res 45:487–510. doi: 10.1016/j.plipres.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 39.Lin LL, Wartmann M, Lin AY, Knopf JL, Seth A, Davis RJ. 1993. cPLA2 is phosphorylated and activated by MAP kinase. Cell 72:269–278. doi: 10.1016/0092-8674(93)90666-E. [DOI] [PubMed] [Google Scholar]
- 40.Menzel N, Fischl W, Hueging K, Bankwitz D, Frentzen A, Haid S, Gentzsch J, Kaderali L, Bartenschlager R, Pietschmann T. 2012. MAP-kinase regulated cytosolic phospholipase A2 activity is essential for production of infectious hepatitis C virus particles. PLoS Pathog 8:e1002829. doi: 10.1371/journal.ppat.1002829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seno K, Okuno T, Nishi K, Murakami Y, Watanabe F, Matsuura T, Wada M, Fujii Y, Yamada M, Ogawa T, Okada T, Hashizume H, Kii M, Hara S, Hagishita S, Nakamoto S, Yamada K, Chikazawa Y, Ueno M, Teshirogi I, Ono T, Ohtani M. 2000. Pyrrolidine inhibitors of human cytosolic phospholipase A(2). J Med Chem 43:1041–1044. doi: 10.1021/jm9905155. [DOI] [PubMed] [Google Scholar]
- 42.Müller C, Karl N, Ziebuhr J, Pleschka S. 2016. D, L-Lysine acetylsalicylate + glycine impairs coronavirus replication. J Antivir Antiretrovir 8:142–150. [Google Scholar]
- 43.Ziebuhr J, Siddell SG. 1999. Processing of the human coronavirus 229E replicase polyproteins by the virus-encoded 3C-like proteinase: identification of proteolytic products and cleavage sites common to pp1a and pp1ab. J Virol 73:177–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Heusipp G, Grotzinger C, Herold J, Siddell SG, Ziebuhr J. 1997. Identification and subcellular localization of a 41 kDa, polyprotein 1ab processing product in human coronavirus 229E-infected cells. J Gen Virol 78(Pt 11):2789–2794. doi: 10.1099/0022-1317-78-11-2789. [DOI] [PubMed] [Google Scholar]
- 45.Jao CY, Salic A. 2008. Exploring RNA transcription and turnover in vivo by using click chemistry. Proc Natl Acad Sci U S A 105:15779–15784. doi: 10.1073/pnas.0808480105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Romero-Brey I, Bartenschlager R. 2014. Membranous replication factories induced by plus-strand RNA viruses. Viruses 6:2826–2857. doi: 10.3390/v6072826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Strating JR, van Kuppeveld FJ. 2017. Viral rewiring of cellular lipid metabolism to create membranous replication compartments. Curr Opin Cell Biol 47:24–33. doi: 10.1016/j.ceb.2017.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Konan KV, Sanchez-Felipe L. 2014. Lipids and RNA virus replication. Curr Opin Virol 9:45–52. doi: 10.1016/j.coviro.2014.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dorobantu CM, Albulescu L, Harak C, Feng Q, van Kampen M, Strating JR, Gorbalenya AE, Lohmann V, van der Schaar HM, van Kuppeveld FJ. 2015. Modulation of the host lipid landscape to promote RNA virus replication: the Picornavirus Encephalomyocarditis virus converges on the pathway used by hepatitis C virus. PLoS Pathog 11:e1005185. doi: 10.1371/journal.ppat.1005185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Harizi H, Corcuff JB, Gualde N. 2008. Arachidonic-acid-derived eicosanoids: roles in biology and immunopathology. Trends Mol Med 14:461–469. doi: 10.1016/j.molmed.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 51.Mishima K, Nakajima M, Ogihara T. 2004. Effects of lysophospholipids on membrane order of phosphatidylcholine. Colloids Surf B Biointerfaces 33:185–189. doi: 10.1016/j.colsurfb.2003.10.004. [DOI] [Google Scholar]
- 52.Roux A, Cuvelier D, Nassoy P, Prost J, Bassereau P, Goud B. 2005. Role of curvature and phase transition in lipid sorting and fission of membrane tubules. EMBO J 24:1537–1545. doi: 10.1038/sj.emboj.7600631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sheetz MP, Singer SJ. 1974. Biological membranes as bilayer couples. A molecular mechanism of drug-erythrocyte interactions. Proc Natl Acad Sci U S A 71:4457–4461. doi: 10.1073/pnas.71.11.4457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Burger KN. 2000. Greasing membrane fusion and fission machineries. Traffic 1:605–613. doi: 10.1034/j.1600-0854.2000.010804.x. [DOI] [PubMed] [Google Scholar]
- 55.Huttner WB, Schmidt AA. 2002. Membrane curvature: a case of endofeelin'…. Trends Cell Biol 12:155–158. doi: 10.1016/S0962-8924(02)02252-3. [DOI] [PubMed] [Google Scholar]
- 56.Zimmerberg J, Gawrisch K. 2006. The physical chemistry of biological membranes. Nat Chem Biol 2:564–567. doi: 10.1038/nchembio1106-564. [DOI] [PubMed] [Google Scholar]
- 57.Brown WJ, Chambers K, Doody A. 2003. Phospholipase A2 (PLA2) enzymes in membrane trafficking: mediators of membrane shape and function. Traffic 4:214–221. doi: 10.1034/j.1600-0854.2003.00078.x. [DOI] [PubMed] [Google Scholar]
- 58.de Figueiredo P, Drecktrah D, Katzenellenbogen JA, Strang M, Brown WJ. 1998. Evidence that phospholipase A2 activity is required for Golgi complex and trans Golgi network membrane tubulation. Proc Natl Acad Sci U S A 95:8642–8647. doi: 10.1073/pnas.95.15.8642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gubern A, Casas J, Barcelo-Torns M, Barneda D, de la Rosa X, Masgrau R, Picatoste F, Balsinde J, Balboa MA, Claro E. 2008. Group IVA phospholipase A2 is necessary for the biogenesis of lipid droplets. J Biol Chem 283:27369–27382. doi: 10.1074/jbc.M800696200. [DOI] [PubMed] [Google Scholar]
- 60.Cai B, Caplan S, Naslavsky N. 2012. cPLA2alpha and EHD1 interact and regulate the vesiculation of cholesterol-rich, GPI-anchored, protein-containing endosomes. Mol Biol Cell 23:1874–1888. doi: 10.1091/mbc.E11-10-0881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gallop JL, McMahon HT. 2005. BAR domains and membrane curvature: bringing your curves to the BAR. Biochem Soc Symp 2005:223–231. doi: 10.1042/bss0720223. [DOI] [PubMed] [Google Scholar]
- 62.Zizza P, Iurisci C, Bonazzi M, Cossart P, Leslie CC, Corda D, Mariggio S. 2012. Phospholipase A2IValpha regulates phagocytosis independent of its enzymatic activity. J Biol Chem 287:16849–16859. doi: 10.1074/jbc.M111.309419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brown MF. 2012. Curvature forces in membrane lipid-protein interactions. Biochemistry 51:9782–9795. doi: 10.1021/bi301332v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jarsch IK, Daste F, Gallop JL. 2016. Membrane curvature in cell biology: an integration of molecular mechanisms. J Cell Biol 214:375–387. doi: 10.1083/jcb.201604003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McMahon HT, Boucrot E. 2015. Membrane curvature at a glance. J Cell Sci 128:1065–1070. doi: 10.1242/jcs.114454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang X, Devaiah SP, Zhang W, Welti R. 2006. Signaling functions of phosphatidic acid. Prog Lipid Res 45:250–278. doi: 10.1016/j.plipres.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 67.Athenstaedt K, Daum G. 2006. The life cycle of neutral lipids: synthesis, storage and degradation. Cell Mol Life Sci 63:1355–1369. doi: 10.1007/s00018-006-6016-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gault CR, Obeid LM, Hannun YA. 2010. An overview of sphingolipid metabolism: from synthesis to breakdown. Adv Exp Med Biol 688:1–23. doi: 10.1007/978-1-4419-6741-1_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fung TS, Liu DX. 2014. Coronavirus infection, ER stress, apoptosis and innate immunity. Front Microbiol 5:296. doi: 10.3389/fmicb.2014.00296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cong Y, Verlhac P, Reggiori F. 2017. The interaction between Nidovirales and autophagy components. Viruses 9:182. doi: 10.3390/v9070182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.de Haan CA, Reggiori F. 2008. Are nidoviruses hijacking the autophagy machinery? Autophagy 4:276–279. doi: 10.4161/auto.5241. [DOI] [PubMed] [Google Scholar]
- 72.Prentice E, Jerome WG, Yoshimori T, Mizushima N, Denison MR. 2004. Coronavirus replication complex formation utilizes components of cellular autophagy. J Biol Chem 279:10136–10141. doi: 10.1074/jbc.M306124200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhao Z, Thackray LB, Miller BC, Lynn TM, Becker MM, Ward E, Mizushima NN, Denison MR, Virgin HW IV. 2007. Coronavirus replication does not require the autophagy gene ATG5. Autophagy 3:581–585. doi: 10.4161/auto.4782. [DOI] [PubMed] [Google Scholar]
- 74.Cottam EM, Maier HJ, Manifava M, Vaux LC, Chandra-Schoenfelder P, Gerner W, Britton P, Ktistakis NT, Wileman T. 2011. Coronavirus nsp6 proteins generate autophagosomes from the endoplasmic reticulum via an omegasome intermediate. Autophagy 7:1335–1347. doi: 10.4161/auto.7.11.16642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Maier HJ, Cottam EM, Stevenson-Leggett P, Wilkinson JA, Harte CJ, Wileman T, Britton P. 2013. Visualizing the autophagy pathway in avian cells and its application to studying infectious bronchitis virus. Autophagy 9:496–509. doi: 10.4161/auto.23465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Utermohlen O, Herz J, Schramm M, Kronke M. 2008. Fusogenicity of membranes: the impact of acid sphingomyelinase on innate immune responses. Immunobiology 213:307–314. doi: 10.1016/j.imbio.2007.10.016. [DOI] [PubMed] [Google Scholar]
- 77.Aktepe TE, Pham H, Mackenzie JM. 2015. Differential utilisation of ceramide during replication of the flaviviruses West Nile and dengue virus. Virology 484:241–250. doi: 10.1016/j.virol.2015.06.015. [DOI] [PubMed] [Google Scholar]
- 78.Roe B, Kensicki E, Mohney R, Hall WW. 2011. Metabolomic profile of hepatitis C virus-infected hepatocytes. PLoS One 6:e23641. doi: 10.1371/journal.pone.0023641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Street IP, Lin HK, Laliberte F, Ghomashchi F, Wang Z, Perrier H, Tremblay NM, Huang Z, Weech PK, Gelb MH. 1993. Slow- and tight-binding inhibitors of the 85-kDa human phospholipase A2. Biochemistry 32:5935–5940. doi: 10.1021/bi00074a003. [DOI] [PubMed] [Google Scholar]
- 80.Mosmann T. 1983. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 81.Thiel V, Ivanov KA, Putics A, Hertzig T, Schelle B, Bayer S, Weissbrich B, Snijder EJ, Rabenau H, Doerr HW, Gorbalenya AE, Ziebuhr J. 2003. Mechanisms and enzymes involved in SARS coronavirus genome expression. J Gen Virol 84:2305–2315. doi: 10.1099/vir.0.19424-0. [DOI] [PubMed] [Google Scholar]
- 82.Dunn KW, Kamocka MM, McDonald JH. 2011. A practical guide to evaluating colocalization in biological microscopy. Am J Physiol Cell Physiol 300:C723–C742. doi: 10.1152/ajpcell.00462.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Manders EM, Stap J, Brakenhoff GJ, van Driel R, Aten JA. 1992. Dynamics of three-dimensional replication patterns during the S-phase, analysed by double labelling of DNA and confocal microscopy. J Cell Sci 103(Pt 3):857–862. [DOI] [PubMed] [Google Scholar]
- 84.Manders EM, Verbeek FJ, Aten JA. 1993. Measurement of co-localization of objects in dual-colour confocal images. J Microsc 169:375–382. doi: 10.1111/j.1365-2818.1993.tb03313.x. [DOI] [PubMed] [Google Scholar]
- 85.Adler J, Parmryd I. 2010. Quantifying colocalization by correlation: the Pearson correlation coefficient is superior to the Mander's overlap coefficient. Cytometry 77A:733–742. doi: 10.1002/cyto.a.20896. [DOI] [PubMed] [Google Scholar]
- 86.Barlow AL, Macleod A, Noppen S, Sanderson J, Guerin CJ. 2010. Colocalization analysis in fluorescence micrographs: verification of a more accurate calculation of Pearson's correlation coefficient. Microsc Microanal 16:710–724. doi: 10.1017/S143192761009389X. [DOI] [PubMed] [Google Scholar]
- 87.Costes SV, Daelemans D, Cho EH, Dobbin Z, Pavlakis G, Lockett S. 2004. Automatic and quantitative measurement of protein-protein colocalization in live cells. Biophys J 86:3993–4003. doi: 10.1529/biophysj.103.038422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Matyash V, Liebisch G, Kurzchalia TV, Shevchenko A, Schwudke D. 2008. Lipid extraction by methyl-tert-butyl ether for high-throughput lipidomics. J Lipid Res 49:1137–1146. doi: 10.1194/jlr.D700041-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Herzog R, Schuhmann K, Schwudke D, Sampaio JL, Bornstein SR, Schroeder M, Shevchenko A. 2012. LipidXplorer: a software for consensual cross-platform lipidomics. PLoS One 7:e29851. doi: 10.1371/journal.pone.0029851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Herzog R, Schwudke D, Shevchenko A. 2013. LipidXplorer: Software for quantitative shotgun lipidomics compatible with multiple mass spectrometry platforms. Curr Protoc Bioinformatics 43:14.12.1–14.12.30. doi: 10.1002/0471250953.bi1412s43. [DOI] [PubMed] [Google Scholar]
- 91.Schwudke D, Liebisch G, Herzog R, Schmitz G, Shevchenko A. 2007. Shotgun lipidomics by tandem mass spectrometry under data-dependent acquisition control. Methods Enzymol 433:175–191. doi: 10.1016/S0076-6879(07)33010-3. [DOI] [PubMed] [Google Scholar]
- 92.Schwudke D, Schuhmann K, Herzog R, Bornstein SR, Shevchenko A. 2011. Shotgun lipidomics on high resolution mass spectrometers. Cold Spring Harb Perspect Biol 3:a004614. doi: 10.1101/cshperspect.a004614. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.