

ABSTRACT
Merkel cell polyomavirus (MCPyV) is the first polyomavirus to be associated with human cancer. Mechanistic studies attempting to fully elucidate MCPyV's oncogenic mechanisms have been hampered by the lack of animal models for MCPyV infection. In this study, we examined the ability of MCPyV-GFP pseudovirus (containing a green fluorescent protein [GFP] reporter construct), MCPyV recombinant virions, and several MCPyV chimeric viruses to infect dermal fibroblasts isolated from various model animals, including mouse (Mus musculus), rabbit (Oryctolagus cuniculus), rat (Rattus norvegicus), chimpanzee (Pan troglodytes), rhesus macaque (Macaca mulatta), patas monkey (Erythrocebus patas), common woolly monkey (Lagothrix lagotricha), red-chested mustached tamarin (Saguinus labiatus), and tree shrew (Tupaia belangeri). We found that MCPyV-GFP pseudovirus was able to enter the dermal fibroblasts of all species tested. Chimpanzee dermal fibroblasts were the only type that supported vigorous MCPyV gene expression and viral replication, and they did so to a level beyond that of human dermal fibroblasts. We further demonstrated that both human and chimpanzee dermal fibroblasts produce infectious MCPyV virions that can successfully infect new cells. In addition, rat dermal fibroblasts supported robust MCPyV large T antigen expression after infection with an MCPyV chimeric virus in which the entire enhancer region of the MCPyV early promoter has been replaced with the simian virus 40 (SV40) analog. Our results suggest that viral transcription and/or replication events represent the major hurdle for MCPyV cross-species transmission. The capacity of rat dermal fibroblasts to support MCPyV early gene expression suggests that the rat is a candidate model organism for studying viral oncogene function during Merkel cell carcinoma (MCC) oncogenic progression.
IMPORTANCE MCPyV plays an important role in the development of a highly aggressive form of skin cancer, Merkel cell carcinoma (MCC). With the increasing number of MCC diagnoses, there is a need to better understand the virus and its oncogenic potential. However, studies attempting to fully elucidate MCPyV's oncogenic mechanisms have been hampered by the lack of animal models for MCPyV infection. To pinpoint the best candidate for developing an MCPyV infection animal model, we examined MCPyV's ability to infect dermal fibroblasts isolated from various established model animals. Of the animal cell types we tested, chimpanzee dermal fibroblasts were the only isolates that supported the full MCPyV infectious cycle. To overcome the infection blockade in the other model animals, we constructed chimeric viruses that achieved robust MCPyV entry and oncogene expression in rat fibroblasts. Our results suggest that the rat may serve as an in vivo model to study MCV oncogenesis.
KEYWORDS: Merkel cell polyomavirus, animal dermal fibroblasts, viral life cycle
INTRODUCTION
Merkel cell carcinoma (MCC) was first described by Cyril Toker in 1972 as a poorly differentiated carcinoma of the dermis and subcutaneous tissues (1). MCC has a mortality of approximately 33% at 3 years, higher than that of melanoma (2). The incidence of MCC has tripled over the past 20 years as the aging population with prolonged sunlight exposure increases (3, 4).
Merkel cell polyomavirus (MCPyV), a member of the Polyomaviridae family, is a nonenveloped virus with a circular, double-stranded DNA genome of ∼5.4 kb (5). The viral genome contains the viral origin of replication and transcription regulatory elements, as well as the early and late coding regions. The early region encodes large T (LT) antigen, small T (sT) antigen, the 57kT antigen, and a protein called alternative LT open reading frame (ORF) (ALTO) (5, 6). The late region encodes the capsid proteins VP1 and VP2 (7–9). In the present study, we used the expression of early and late genes encoding LT and VP1 as a marker of established viral transcription and LT-associated replication foci as a marker of robust viral DNA replication.
About 80% of recorded MCCs have detectable MCPyV genomic DNA integrated into the tumor cell genome (10). It is well established that clonal integration of MCPyV genomic DNA into the host genome precedes the development of the majority of MCC cases (11–13). The continued expression of the MCPyV viral oncogenes, sT and LT with a C-terminal truncation, is required for MCC tumor cells to survive (14, 15). These findings provide strong evidence that MCPyV is a major causative agent of this skin cancer.
Most aspects of MCPyV virology and oncogenic mechanisms have been studied using transfected cells, preventing the full investigation of these topics. Animal models are needed to overcome this limitation. We recently discovered that MCPyV successfully infects human dermal fibroblasts (16). In the present study, we explored the ability of MCPyV and MCPyV-derived viruses to infect dermal fibroblasts isolated from various model animals. MCPyV-GFP pseudovirus (containing a green fluorescent protein [GFP] reporter construct) and recombinant MCPyV virus produced using previously reported methods (7) were used to examine MCPyV infection in dermal fibroblasts isolated from mouse (Mus musculus), rabbit (Oryctolagus cuniculus), rat (Rattus norvegicus), chimpanzee (Pan troglodytes), rhesus macaque (Macaca mulatta), patas monkey (Erythrocebus patas), common woolly monkey (Lagothrix lagotricha), red-chested mustached tamarin (Saguinus labiatus), and tree shrew (Tupaia Belangeri) skin specimens. We found that chimpanzee dermal fibroblasts were the only isolates tested that supported the full MCPyV cycle, including viral entry, DNA replication, gene transcription, and virion production. This novel cross-species analysis of MCPyV infection suggests that the virus has a narrow range of tropism and that relatively minor shifts in the host cell environment may be protective against productive MCPyV infection. Additionally, we identified robust virus entry and MCPyV oncogene expression in rat dermal fibroblasts infected with a chimeric virus that carries the enhancer region of simian virus 40 (SV40) in place of the analogous region of the MCPyV genome. In the future, the rat may serve as an in vivo model to study MCPyV oncogene expression and the transition from active MCPyV infection to MCC tumorigenesis.
RESULTS
MCPyV infection of mouse dermal fibroblasts.
The mouse (Mus musculus) has become the foremost animal model system for biomedical research due to its relatively low cost, short life span, and genetic tractability and the wealth of existing research using this model. Therefore, we tested mouse cells in vitro, using previously established methods, to gauge the potential of an in vivo mouse model of MCPyV infection. First, we used MCPyV-GFP pseudovirions, which contain a GFP reporter construct, to transduce mouse dermal fibroblasts. In this experiment, a cytoplasmic GFP signal under fluorescence microscopy serves as a marker for effective viral entry. Human dermal fibroblasts, infected in parallel with mouse cells, were used as a positive control. Consistent with past reports showing that MCPyV-GFP pseudovirions can deliver encapsidated reporter DNA to a wide variety of different cell types (7, 8, 17–21), GFP expression was observed in both mouse and human dermal fibroblasts exposed to the pseudovirions (Fig. 1A and Table 1). However, far fewer GFP-positive (GFP+) cells were observed in the mouse dermal fibroblast samples than in human dermal fibroblasts (Fig. 1A and B and Table 1). Second, we treated mouse and human dermal fibroblasts with recombinant MCPyV virions and performed immunofluorescence microscopy to assess early and late gene expression. LT-positive (LT+) and VP1-positive cells were detected among human dermal fibroblasts at the expected levels (∼40%), while none of the MCPyV virion-treated mouse dermal fibroblasts exhibited LT or VP1 expression (Fig. 1C and D and Table 1). These results corroborate earlier findings that MCPyV infectious entry is relatively pervasive but suggest that mouse dermal fibroblasts cannot support MCPyV gene expression.
FIG 1.
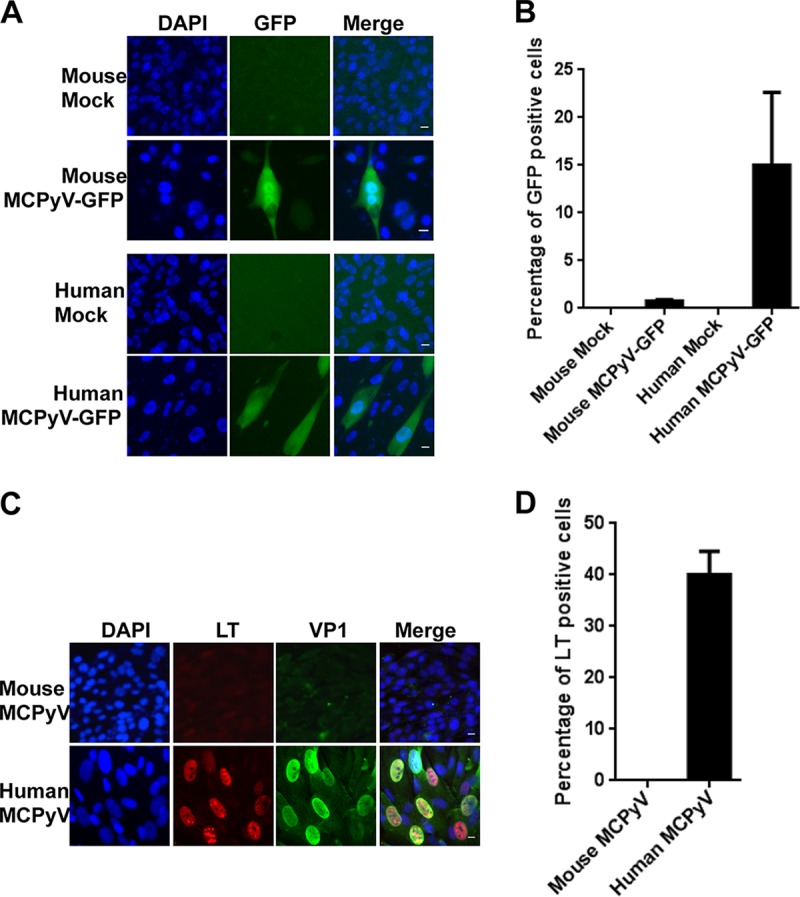
MCPyV-GFP and MCPyV infection of dermal fibroblasts isolated from mouse and human skin samples. (A) Dermal fibroblasts isolated from mouse and human skin samples were treated with MCPyV-GFP pseudovirions in DMEM–F-12 medium containing EGF, bFGF, CHIR99021, and 1 mg/ml collagenase IV for 2 days. After switching to fresh DMEM–F-12 medium containing 20% FBS for three more days, cells were counterstained with Hoechst 33458. Bar, 10 μm. (B) Percentages of GFP+ cells in the experiment whose results are shown in panel A. (C) Dermal fibroblasts isolated from mouse and human skin samples were treated with MCPyV virions in DMEM–F-12 medium containing EGF, bFGF, CHIR99021, and 1 mg/ml collagenase IV for 2 days. After switching to fresh DMEM–F-12 medium containing 20% FBS for three more days, cells were immunostained using the indicated antibodies and counterstained with DAPI. Bar, 10 μm. (D) Percentages of LT+ cells in the experiment whose results are shown in panel C. Error bars represent standard errors of the means (SEM) of the results of at least three independent experiments.
TABLE 1.
Percentages of GFP- or LT-positive cells in dermal fibroblasts treated with MCPyV pseudovirus, MCPyV, or MCPyV chimeric viruses
| Source of fibroblasts | % of GFP+ fibroblasts (mean ± SEMa) following exposure to MCPyV-GFP | % of LT+ fibroblasts (mean ± SEMa) following exposure to: |
|||
|---|---|---|---|---|---|
| MCPyV | MCPyV-MuPyV | MCPyV-SV40 A | MCPyV-SV40 B | ||
| Human | 15.13 ± 7.49 | 40.17 ± 4.80 | 4.00 ± 1.61 | 21.80 ± 3.39 | 33.33 ± 5.64 |
| Mouse | 0.87 ± 0.23 | 0 | <0.01 | 0.07 ± 0.02 | 0.13 ± 0.04 |
| Rabbit | 46.30 ± 8.32 | 0 | 0.12 ± 0.04 | <0.01 | 0.57 ± 0.23 |
| Rat | 33.33 ± 5.76 | 5.80 ± 2.48 | 3.17 ± 1.13 | 3.83 ± 1.10 | 52.77 ± 6.68 |
| Chimpanzee | 47.17 ± 9.59 | 43.67 ± 4.60 | NA | NA | NA |
| Rhesus macaque | 31.50 ± 6.37 | 0.81 ± 0.17 | 0.67 ± 0.32 | 3.16 ± 1.01 | 16.43 ± 5.57 |
| Patas monkey | 15.63 ± 3.23 | 0.40 ± 0.15 | NA | NA | NA |
| Common woolly monkey | 10.40 ± 2.58 | 0 | NA | NA | NA |
| Red-chested mustached tamarin | 9.47 ± 2.12 | 0 | NA | NA | NA |
| Tree shrew | 3.83 ± 1.34 | 0 | NA | NA | NA |
Data are from the results of at least three independent experiments. NA, not applicable.
MCPyV infection of rabbit and rat dermal fibroblasts.
The rabbit (Oryctolagus cuniculus) and rat (Rattus norvegicus) models are also commonly used in biomedical research, for many of the same reasons as the mouse model. We were able to isolate dermal fibroblasts from members of both species and assess their capacity to support MCPyV infection using the same strategy as described for mouse cells above. Robust GFP expression was observed in both rabbit and rat dermal fibroblasts exposed to MCPyV-GFP pseudovirions (Fig. 2A). Strong GFP signals could be detected in ∼46% of the rabbit dermal fibroblasts treated with MCPyV-GFP pseudovirions (Fig. 2B and Table 1). About 33% of the rat dermal fibroblasts also exhibited bright GFP signals (Fig. 2B and Table 1). These data show that both rabbit and rat dermal fibroblasts support efficient MCPyV-GFP pseudovirus entry to a higher degree than human dermal fibroblasts.
FIG 2.
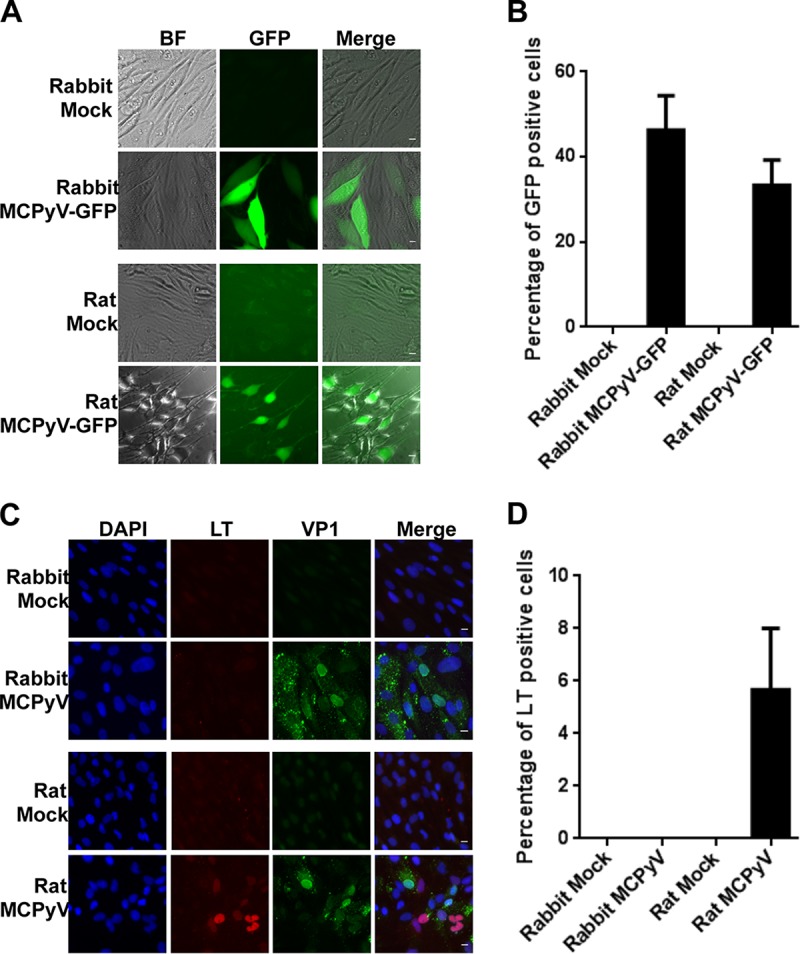
MCPyV-GFP and MCPyV infection of dermal fibroblasts isolated from rabbit and rat dermal fibroblasts. (A) Dermal fibroblasts isolated from rabbit and rat skin samples were treated with MCPyV-GFP pseudovirions as described in the legend to Fig. 1A and examined under a fluorescence microscope. Bar, 10 μm. (B) Percentages of GFP+ cells in the experiment whose results are shown in panel A. (C) Dermal fibroblasts isolated from rabbit and rat skin samples were treated with MCPyV virions and stained as described for Fig. 1C. Bar, 10 μm. (D) Percentages of LT+ cells in the experiment whose results are shown in panel C. Error bars represent SEM of the results of at least three independent experiments.
After infecting rabbit dermal fibroblasts with recombinant MCPyV, we observed few VP1-positive cells and none that were LT positive (Fig. 2C and D). Because no cells expressed detectable levels of the early gene LT, the low VP1 signal can be attributed to the capsids of input virions, rather than productive infection (Fig. 2C and D). Rat dermal fibroblasts, on the other hand, exhibited a low level of LT positivity when infected with MCPyV, about 5%, compared to 40% of human dermal fibroblasts (Fig. 2C and D and Table 1). Like mouse dermal fibroblasts, both rabbit and rat dermal fibroblasts allow efficient MCPyV entry, but among these cell types, only rat dermal fibroblasts support a low level of early gene expression and none exhibit late gene expression, produce replication foci, or yield infectious particles.
MCPyV infection of nonhuman primate dermal fibroblasts.
Because dermal fibroblasts from the small mammals tested thus far were unable to support MCPyV infection, we assessed the potential of MCPyV infection in mammals with more recent common ancestors to humans. We isolated dermal fibroblasts from nonhuman primates, including the chimpanzee (Pan troglodytes), Old World monkeys rhesus macaque (Macaca mulatta) and patas monkey (Erythrocebus patas), the New World monkeys common woolly monkey (Lagothrix lagotricha) and red-chested mustached tamarin (Saguinus labiatus), and the tree shrew (Tupaia Belangeri), and employed the same in vitro infection conditions as described above. As expected, we found that the GFP signals from MCPyV-GFP pseudovirions were readily detected in all nonhuman primate fibroblasts tested (Fig. 3A and B and Table 1), suggesting that MCPyV-GFP pseudovirions were able to enter the dermal fibroblasts isolated from these species. Significantly, after treatment with MCPyV virions, more than 43% of the chimpanzee fibroblasts exhibited strong MCPyV LT expression, as well as the formation of viral replication foci (Fig. 3C and D and Table 1). To better understand this robust infection phenotype in primary chimpanzee fibroblasts, we tested three primary chimpanzee cell lines from animals of different ages. We found that cell lines generated from newborn chimpanzee dermal fibroblasts supported a high degree of viral infection (>40%), whereas less than 10% of cells isolated from 10- or 16-year-old chimpanzees became infected (Fig. 3E). This finding suggests that the degree of stemness in primary dermal fibroblasts may play a role in MCPyV infection. Comparatively, less than 1% of rhesus macaque and patas monkey dermal fibroblasts supported MCPyV LT expression, and very few cells contained small viral replication foci (Fig. 3C and D and Table 1). In addition, telomerase-immortalized rhesus macaque dermal fibroblasts (Telo-RF) were infected by MCPyV to a similarly low level as the primary rhesus macaque dermal fibroblasts (data not shown). We did not detect any MCPyV LT expression in cells isolated from the common woolly monkey, red-chested mustached tamarin, or tree shrew (Fig. 3C and D and Table 1). In short, chimpanzee dermal fibroblasts support MCPyV entry, LT expression, and the formation of viral replication foci to the same degree as human dermal fibroblasts, while each of the other nonhuman primate dermal fibroblasts tested showed minimal evidence of successful viral infection.
FIG 3.

MCPyV-GFP and MCPyV infection of dermal fibroblasts isolated from nonhuman primate skin samples. (A) Dermal fibroblasts isolated from nonhuman primate skin samples were treated with MCPyV-GFP pseudovirions as described in the legend to Fig. 1A, and the cells' images were captured on day 5. (B) Percentages of GFP+ cells in the experiment whose results are shown in panel A. Bar, 10 μm. (C) Dermal fibroblasts isolated from nonhuman primates were treated with MCPyV virions and stained as described in the legend to Fig. 1C. Bar, 10 μm. (D) Percentages of LT+ cells in the experiment whose results are shown in panel C. (E) Chimpanzee fibroblast cell lines S033665 (female, sampled at 1 day old), S008861 (male, sampled at 16 years old), and S008933 (female, sampled at 10 years old) were treated with MCPyV virions and stained as described in the legend to Fig. 1C, and percentages of LT+ cells were determined. (F) Chimpanzee and human dermal fibroblasts can produce infectious MCPyV virions. The flow chart shows MCPyV's initial infection and reinfection of dermal fibroblasts. (G) Chimpanzee dermal fibroblasts were treated with MCPyV virions in DMEM–F-12 medium containing EGF, bFGF, CHIR99021, and 1 mg/ml collagenase IV or DMEM–F-12 medium containing 20% FBS to block viral entry (Control) for 2 days. After switching to fresh DMEM–F-12 medium containing 20% FBS for 9 more days, the medium was harvested and centrifuged at 12,000 rpm at 4°C for 10 min. The supernatants were used to treat fresh human dermal fibroblasts in DMEM–F-12 medium containing EGF, bFGF, CHIR99021, and 4 mg/ml collagenase IV for 2 days. After switching to fresh DMEM–F-12 medium containing 20% FBS for 3 more days, cells were analyzed by qPCR. (H) Human dermal fibroblasts treated with MCPyV virions as described in the legend to panel G were analyzed by qPCR. Error bars represent SEM of the results of at least three independent experiments.
Because chimpanzee and human dermal fibroblasts support MCPyV LT and VP1 expression, as well as the formation of viral replication foci, we next tested whether these cells could produce infectious MCPyV virions. We first infected chimpanzee and human dermal fibroblasts with MCPyV virions. After 9 days, the supernatants of the infected cells were harvested and used to infect newly isolated human dermal fibroblasts (Fig. 3F). To control for potential infectious capacity conferred by virions remaining from the initial infection, we blocked virus entry during the first virus infection under control conditions by adding fetal bovine serum (FBS) to the medium as we established previously (16). This approach establishes an input level of infection in order to subtract the impact of the inoculum virions. We found that supernatants from both chimpanzee and human dermal fibroblasts previously infected with MCPyV in the absence of FBS can infect fresh human dermal fibroblasts, as indicated by the LT and VP1 expression in the stained cells. In contrast, secondary inoculation with supernatants of cells originally treated with virus in the presence of FBS during the first infection did not produce LT- or VP1-positive cells. Furthermore, quantitative real-time PCR (qPCR) analysis showed that the MCPyV genome was significantly amplified in the reinfected human dermal fibroblasts, but not in the cells treated with supernatants of the control group in which inoculum virions were prevented from entering the cells by FBS (Fig. 3F to H). Our finding suggests that both chimpanzee and human dermal fibroblasts can produce infectious MCPyV virions in vitro.
Construction of chimeric MCPyV viruses.
The finding that MCPyV readily infects chimpanzee and human dermal fibroblasts in vitro, but not cells from other primates or lower mammals, implies that host cell environments across evolutionary clades are uniquely restrictive or permissive to MCPyV replication. Understanding these governing factors may assist in the prevention or treatment of MCPyV-positive MCC. In order to pursue this line of inquiry in vivo, we adopted an indirect approach, as chimpanzee model studies have been suspended globally. We generated MCPyV chimeric viruses, aiming to induce viral transcription in animal cells other than chimpanzee. Enhancers and upstream regulatory elements of polyomavirus promoters have some specific functions in conferring viral gene inducibility, tissue specificity, and/or a general enhancement of transcription (22, 23). We predicted that the enhancer regions of MCPyV also mediate its promoter activity and, consequently, its ability to transcribe viral genes essential for replication in specific host environments. We constructed a chimeric MCPyV virus in which the enhancer regions of the MCPyV early promoter were replaced by the analogous region of the murine polyomavirus (MuPyV) genome described previously (23). We hypothesized that this chimeric virus, MCPyV-MuPyV (GenBank accession number MG234276) (Fig. 4A), would be better able to initiate transcription in small mammal cells. In addition, others have found that SV40 can transcribe in human, mouse, rat, and rabbit cells (24). We also confirmed that the SV40 genome could express the SV40 LT protein in both human and rhesus macaque dermal fibroblasts (Fig. 4B). After considering this broad range of transcriptional activity, we constructed two additional MCPyV-SV40 chimeric viruses, in which either a portion of (MCpyV-SV40 A; GenBank accession number MG234277) or the entire (MCPyV-SV40 B; GenBank accession number MG234278) enhancer region of the MCPyV early promoter was replaced by the homologous regions of the SV40 genome described in a previous study (23) (Fig. 4C and D). The early promoter and origin regions of MCPyV remain intact in all three chimeric viruses in order to mimic MCPyV transcription and replication. After packaging these chimeric MCPyV viruses, we tested their titers. We found that the titer of MCPyV-SV40 A was quite similar to that of wild-type MCPyV, whereas MCPyV-SV40 B and MCPyV-MuPyV had titers that were about 10 times and 100 times less, respectively, than that of wild-type MCPyV (Fig. 4E).
FIG 4.

Construction of MCPyV-MuPyV, MCPyV-SV40 A, and MCPyV-SV40 B chimeric viruses. (A) The enhancer region of the MCPyV early promoter was replaced with the analogous region from the MuPyV genome. (B) Dermal fibroblasts isolated from human and rhesus macaque skin samples were transfected with the religated SV40 genome. After 5 days, all cells were immunostained using the indicated antibodies and counterstained with DAPI. Bar, 10 μm. (C) The enhancer region of the MCPyV early promoter was replaced with the analogous region of the SV40 genome. (D) The MCPyV late promoter region was deleted from the MCPyV-SV40 A genome. (E) The virus titers of MCPyV and the chimeric virions as represented by the genome copy numbers were determined by qPCR.
Infection of dermal fibroblasts using chimeric MCPyV viruses.
We then tested the activity of our newly generated chimeric virions in the set of dermal fibroblasts isolated from the model organisms described above. MCPyV-MuPyV, MCPyV-SV40 A, and MCPyV-SV40 B chimeric virions transcribed LT in human dermal fibroblasts, indicating that all of the chimeric viruses were infectious (Fig. 5 and Table 1). Although MCPyV-SV40 B's titer was about 10 times less than that of MCPyV-SV40 A, it exhibited a better transduction efficiency (Fig. 5). We also used the chimeric viruses to infect mouse dermal fibroblasts but did not observe an infection efficiency higher than 0.15% with any of the chimeric viruses, even MCPyV-MuPyV (Table 1). The viruses were also tested for their infectivity in rhesus macaque fibroblasts. LT expression was detected in about 16% of these cells treated with MCPyV-SV40 B virions, while MCPyV-MuPyV and MCPyV-SV40 A showed much lower infection efficiencies (Fig. 6 and Table 1). Notably, VP1 expression was not detected in any of the rhesus macaque fibroblasts treated with the chimeric virions (Fig. 6A). The infection efficiency and viral gene expression pattern exhibited by each of the chimeric viruses in immortalized rhesus macaque Telo-RF dermal fibroblasts were similar to those seen in primary rhesus macaque dermal fibroblasts (data not shown). Finally, all of the chimeric viruses resulted in very little LT expression in rabbit and mouse cells (Table 1).
FIG 5.
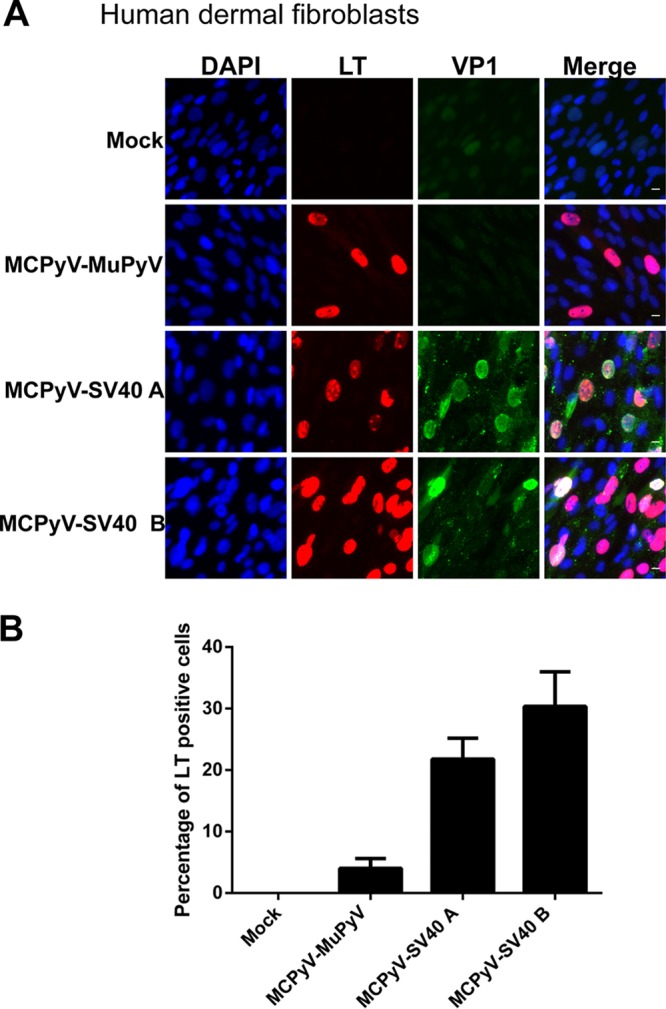
MCPyV-MuPyV, MCPyV-SV40 A, and MCPyV-SV40 B infection of dermal fibroblasts isolated from human skin samples. (A) Dermal fibroblasts isolated from human skin samples were infected with MCPyV-MuPyV, MCPyV-SV40 A, and MCPyV-SV40 B chimeric virions and stained as described in the legend to Fig. 1C. Bar, 10 μm. (B) Percentages of LT+ cells in the experiment whose results are shown in panel A. Error bars represent SEM of the results of at least three independent experiments.
FIG 6.
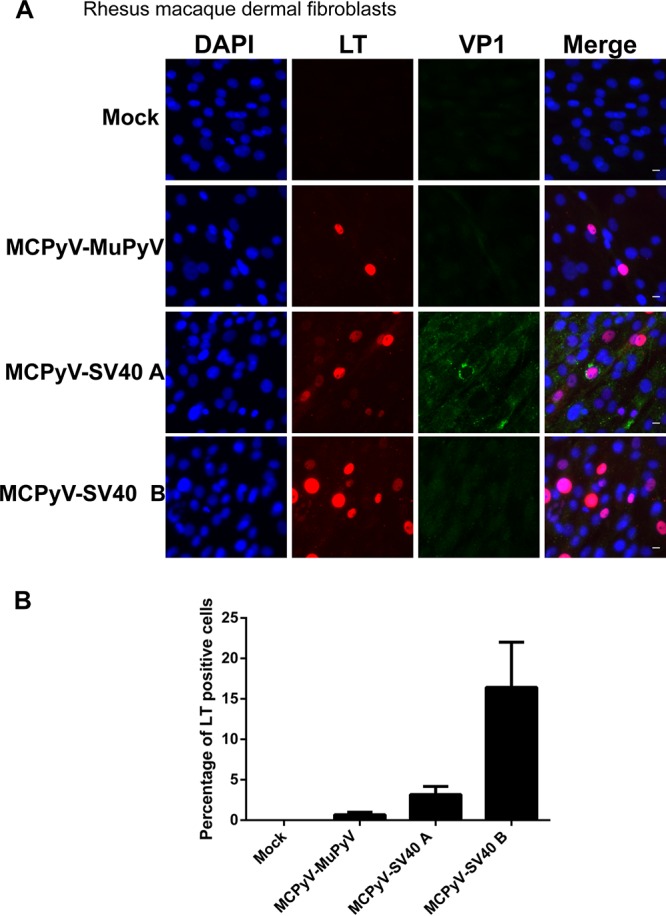
MCPyV chimeric virus infection of primary rhesus macaque dermal fibroblasts. (A) Primary rhesus macaque dermal fibroblasts were treated with MCPyV-MuPyV, MCPyV-SV40 A, and MCPyV-SV40 B chimeric virions and stained as described in the legend to Fig. 1C. Bar, 10 μm. (B) Percentages of LT+ cells in the experiment whose results are shown in panel A. Error bars represent SEM of the results of at least three independent experiments.
Since 5% of rat dermal fibroblasts treated with recombinant MCPyV virions display LT transcription (Fig. 2C and D), we tested whether the chimeric viruses we constructed could improve upon this promising phenotype. Although about 3% of the rat cells treated with MCPyV-MuPyV and MCPyV-SV40 A showed LT expression (Fig. 7 and Table 1), over 50% of rat dermal fibroblasts treated with MCPyV-SV40 B virions supported robust LT antigen expression (Fig. 7 and Table 1). Such levels of early transcription are even higher than those observed in human dermal fibroblasts infected with recombinant MCPyV in vitro. However, qPCR analysis showed that, for both MCPyV-SV40 B and MCPyV, the amounts of viral DNA in the treated cell samples decreased significantly between 54 and 120 h (Fig. 7C). The result suggests that MCPyV DNA replication does not take place in the rat cells.
FIG 7.
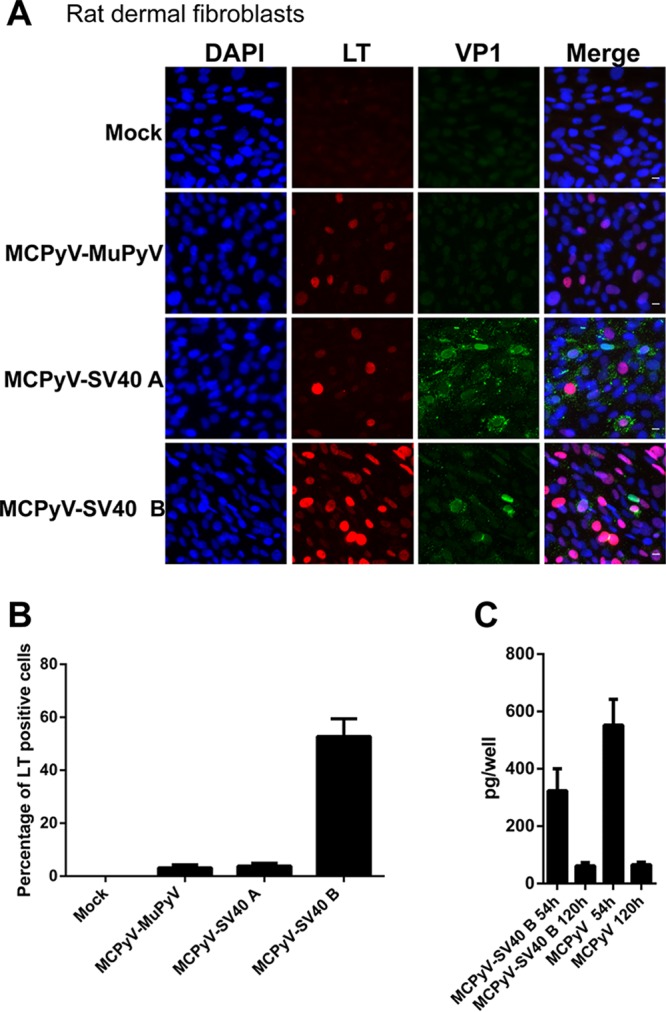
MCPyV-MuPyV, MCPyV-SV40 A, and MCPyV-SV40 B infection of dermal fibroblasts isolated from rat skin samples. (A) Dermal fibroblasts isolated from rat skin samples were treated with MCPyV-MuPyV, MCPyV-SV40 A, and MCPyV-SV40 B chimeric virions and stained as described in the legend to Fig. 1C. Bar, 10 μm. (B) Percentages of LT+ cells in the experiment whose results are shown in panel A. (C) Rat dermal fibroblasts were treated with MCPyV or MCPyV-SV40 B virions as described in the legend to Fig. 1C. After switching to fresh DMEM–F-12 medium containing FBS, the cells were harvest at 54 h and 120 h postinfection. The cells were analyzed by qPCR. Error bars represent SEM of the results of at least three independent experiments.
DISCUSSION
Currently, there is no animal model available for studying MCPyV virology. Consequently, it has not been possible to observe the MCPyV infection cycle and related viral events in vivo. The lack of an MCPyV animal infection model also hampers investigation of its mechanism of oncogenesis, leading to Merkel cell carcinoma (MCC). Because of the complex interaction between MCC and the host immune system (25), it is important to have an in vivo model in which the host immune response can be observed within the context of natural MCPyV infection. Our recent identification of human dermal fibroblasts as a natural MCPyV host cell type in humans and the concurrent establishment of a successful in vitro MCPyV infection model provide important clues for what may constitute a successful in vivo model (16). The finding also points to the likely cell type and skin layer in which the virus infects humans and potential model organisms. Building on this study, we investigated MCPyV's ability to infect dermal fibroblasts isolated from established model animals in order to pinpoint the best candidate for MCPyV research.
In this study, mouse dermal fibroblasts were shown to support MCPyV-GFP pseudovirus entry but not MCPyV transcription and replication. This result suggests that mice may lack key cellular factors necessary for supporting these important viral activities. Therefore, despite the many advantages inherent in mouse model systems, the mouse is not a promising model organism for studying MCPyV infection. Although both rabbit and rat dermal fibroblasts were shown to support robust MCPyV-GFP pseudovirus entry, almost none of the rabbit dermal fibroblasts and less than 6% of rat dermal fibroblasts supported viral transcription/replication. We therefore investigated nonhuman primates as potential candidates.
Chimpanzee dermal fibroblasts not only support efficient MCPyV viral entry, transcription, and replication but also produce infectious virions (Fig. 3F to H). All chimpanzee studies have been suspended, however, eliminating the possibility of their use as an in vivo model for MCPyV infection. The dermal fibroblasts of Old World monkeys, specifically the rhesus macaque and patas monkey, support low levels of MCPyV infection, but the dermal fibroblasts of the New World monkeys common woolly monkey and red-chested mustached tamarin do not support virus transcription and replication. Despite the fact that the genetic makeup of these nonhuman primates is more similar to that of humans than is the genetic makeup of lower mammals, none of them showed promise as a potential MCPyV animal model.
Together, the isolation and infection of animal dermal fibroblasts suggest that, among the small mammals and nonhuman primates tested, only chimpanzees support the full MCPyV replicative cycle. This narrow range of infection hosts suggests that MCPyV may have coevolved with its human hosts. In silico comparisons between the expression profiles of permissive and restrictive hosts tested in our in vitro infection system may identify the unique host cellular factors that support the MCPyV infectious life cycle.
Our results suggest that MCPyV infectious entry is relatively prevalent, while the viral transcription and/or replication events may represent the major barriers for MCPyV cross-species transmission. We therefore sought alternative means of advancing the MCPyV in vivo effort by constructing chimeric viruses to overcome species-specific restrictions. Although all three chimeric viruses we constructed, MCPyV-MuPyV, MCPyV-SV40 A, and MCPyV-SV40 B, were able to infect human dermal fibroblasts, none of them could accomplish robust viral transcription and replication in mouse, rhesus macaque, or rabbit dermal fibroblasts. In contrast, rat dermal fibroblasts, which were slightly more permissive to native MCPyV transcription, support robust viral infection and early gene transcription of chimeric MCPyV-SV40 B virus (Fig. 7). However, VP1 expression was quite low and very few viral replication foci were observed in rat dermal fibroblasts transduced with MCPyV-SV40 B (Fig. 7A). qPCR analysis confirmed that neither MCPyV nor MCPyV-SV40 B could replicate in rat dermal fibroblasts (Fig. 7C). These results suggest that additional modification(s) of the viral enhancers/promoters/origin of replication is needed in order to achieve all viral events of the MCPyV infection cycle in rat dermal fibroblasts. Along this line, novel rat polyomaviruses (RPyVs) with genome structures similar to that of MCPyV have been discovered recently (26, 27). In future studies, MCPyV-RPyV chimeric viruses will be developed to stimulate the viral replication and late gene expression potential in rat dermal fibroblasts. The ability of rat dermal fibroblasts to support MCPyV-SV40 B early gene transcription suggests that the rat has the potential to be used as an animal model for recapitulating MCPyV oncogene expression during MCC development. In addition, the metabolic physiology of rats is closer to that of humans than is the mouse metabolic physiology (28), making it a better species to study the pharmacokinetic characteristics of MCPyV immune responses and potential MCC drugs.
MATERIALS AND METHODS
Cell culture.
Tree shrew skin samples were kindly provided by David Fitzpatrick (Max Planck Florida Institute). Human neonatal foreskin samples were provided by Aimee S. Payne (University of Pennsylvania Skin Disease Research Center). Rabbit dermal fibroblast RAB-9 cells were purchased from ATCC. Rhesus macaque skin samples were obtained from Peter J. Didier (Tulane National Primate Research Center). Rat ear skin samples were provided by Margie Maronski (Neuron Culture Service Center, University of Pennsylvania). Mouse ear skin samples were provided by Sunny Shin (University of Pennsylvania). Telomerase-immortalized rhesus macaque dermal fibroblasts (Telo-RF) were provided by Maxine Linial (Fred Hutchinson Cancer Research Center). Chimpanzee dermal fibroblast cell lines S033665, S008861 and S008933, patas monkey dermal fibroblast cell line AG06116, red-chested mustached tamarin dermal fibroblast cell line AG05308, and common woolly monkey dermal fibroblast cell line AG05356 were obtained from the Coriell Institute for Medical Research. Human, mouse, rabbit, and rat dermal fibroblasts were maintained in Dulbecco's modified Eagle's medium (DMEM) (catalog number 11965084; Life Technologies) supplemented with 10% FBS (catalog number SH30071.03; HyClone). All nonhuman primate dermal fibroblasts were maintained in minimal essential medium (MEM) alpha (catalog number 12571048; Life Technologies) supplemented with 20% FBS (catalog number SH30071.03; HyClone). All protocols were approved by the University of Pennsylvania Institutional Review Board.
MCPyV virion, MCPyV-GFP, and MCPyV chimeric virus preparation.
Chimeric virus construction and infection were approved by the University of Pennsylvania Institutional Review Board. Recombinant MCPyV virions and MCPyV-GFP pseudovirus were prepared as previously described, with minor modifications (7). Briefly, 293TT cells were maintained in DMEM with 10% fetal calf serum, 1% nonessential amino acids, 1% GlutaMax-I (Invitrogen), and 250 μg/ml hygromycin (Roche). For recombinant MCPyV and MCPyV chimeric virion production, 293TT cells were transfected with plasmids pMtB, pADL*, and the religated recombinant genome of MCPyV isolate R17b or the chimeric viral genomes. Five days posttransfection, recombinant virions were harvested and purified over an OptiPrep gradient. For MCPyV-GFP pseudovirion production, 293TT cells were transfected with plasmids pXpressGFP, pwM2m, and ph2mΔGFP. Two days posttransfection, MCPyV-GFP virions were harvested and purified over an OptiPrep gradient.
Infection of dermal fibroblasts with MCPyV, MCPyV-GFP, and MCPyV chimeric viruses.
Dermal fibroblasts cultured in 10-cm dishes were washed once with 1× Dulbecco's phosphate-buffered saline (DPBS) (catalog number 14190136; Life Technologies) and incubated with 0.05% trypsin-EDTA (catalog number 25300-054; Life Technologies) at 37°C for 5 min. The cells were examined under the microscope to ensure that they were coming off the dish. After adding 10 ml DMEM–F-12 medium (catalog number 11330-032; Life Technologies) to the dish, the cell mixture was transferred to a 15-ml tube and centrifuged at 180 × g for 2 min. The cells were resuspended in DMEM–F-12 medium containing 20 ng/ml epidermal growth factor (EGF) (catalog number 236-EG-200; R&D Systems), 20 ng/ml basic fibroblast growth factor (bFGF) (product number 354060; Corning Life Sciences), 3 μM CHIR99021 (item number 13122; Cayman Chemical), and 1 mg/ml collagenase IV. A total of 4 × 103 cells resuspended in 100 μl medium were seeded into each well of a 96-well plate, and the virions were added to the cells. After the cells were incubated at 37°C in 5% CO2 for 48 h, FBS was added to each well to reach 20% final concentration. The cells were incubated for 72 h before immunofluorescence staining.
qPCR.
For analyzing the MCPyV genome copies by quantitative real-time PCR (qPCR), 1 μl of the viral capsid was digested in 100 μl of a solution containing 20 mM Tris, pH 8, 20 mM dithiothreitol (DTT), 20 mM EDTA, 0.5% SDS, and 0.2% proteinase K and incubated for 20 min at 50°C. After adding 600 μl of Qiagen buffer PB to the digestion reaction mixture, the viral DNA was purified over a QIAprep purple spin column and eluted in 50 μl Tris-EDTA (TE). Two microliters of the sample was analyzed by qPCR. The genome copy number was calculated using a standard curve generated using serial dilution of a sample containing a known copy number of plasmid pR17b. qPCR was performed using a CFX96 real-time PCR detection system (Bio-Rad) and IQ SYBR green supermix (Bio-Rad). The primers used to amplify the MCPyV genome have been described previously (7).
Immunofluorescent staining.
Cells were fixed with 3% paraformaldehyde in PBS for 20 min. Immunofluorescence staining was performed as previously described (29). The following primary antibodies were used: anti-MCPyV LT antibody (CM2B4) (sc-136172, 1:1,000; Santa Cruz), anti-MCPyV VP1 rabbit antibody (1:2,000), and anti-SV40 LT antibody (sc-147, 1:1,000; Santa Cruz). The secondary antibodies used were Alexa Fluor 594 goat anti-mouse IgG (A-11032, 1:1,000; Invitrogen) and Alexa Fluor 488 goat anti-rabbit IgG (A-11034, 1:500; Invitrogen). All immunofluorescence images were collected using an inverted fluorescence microscope (Olympus IX81) connected to a high-resolution charge-coupled-device camera (FAST1394; QImaging). Images were analyzed and presented using SlideBook software (version 5.0; Intelligent Imaging Innovations, Inc.). The scale bars were added using ImageJ software.
Statistical analyses.
Statistical analysis was performed using GraphPad Prism software to compare the data from the control and experimental groups. A two-tailed P value of less than 0.05 was considered significant.
Accession number(s).
The noncoding regulatory region (NCRR) sequences of MCPyV-MuPyV, MCPyV-SV40 A, and MCPyV-SV40 B have been deposited in GenBank under accession numbers MG234276, MG234277, and MG234278.
ACKNOWLEDGMENTS
We thank Peter J. Didier (Tulane National Primate Research Center) for rhesus macaque skin samples, David Fitzpatrick (Max Planck Florida Institute) for tree shrew skin samples, Maxine Linial (Fred Hutchinson Cancer Research Center) for rhesus macaque cell line Telo-RF, Aimee S. Payne (University of Pennsylvania Skin Disease Research Center) for human neonatal foreskin samples, Margie Maronski (Neuron Culture Service Center, University of Pennsylvania) for rat skin samples, Sunny Shin (University of Pennsylvania) for mouse skin samples, Coriell Institute for Medical Research for chimpanzee dermal fibroblast cell lines S033665, S008861, and S008933, patas monkey dermal fibroblast cell line AG06116, red-chested mustached tamarin dermal fibroblast cell line AG05308, and common woolly monkey dermal fibroblast cell line AG05356, and Christopher B. Buck (National Cancer Institute) for the murine polyomavirus genome. We also thank the members of our laboratories for helpful discussions and critical reviews of the manuscript.
This work was supported by National Institutes of Health (NIH) grants (grant numbers R01CA187718 and R01CA148768) and an NCI Cancer Center support grant (grant number NCI P30 CA016520).
REFERENCES
- 1.Toker C. 1972. Trabecular carcinoma of the skin. Arch Dermatol 105:107–110. doi: 10.1001/archderm.1972.01620040075020. [DOI] [PubMed] [Google Scholar]
- 2.Allen PJ, Bowne WB, Jaques DP, Brennan MF, Busam K, Coit DG. 2005. Merkel cell carcinoma: prognosis and treatment of patients from a single institution. J Clin Oncol 23:2300–2309. doi: 10.1200/JCO.2005.02.329. [DOI] [PubMed] [Google Scholar]
- 3.Hodgson NC. 2005. Merkel cell carcinoma: changing incidence trends. J Surg Oncol 89:1–4. doi: 10.1002/jso.20167. [DOI] [PubMed] [Google Scholar]
- 4.Albores-Saavedra J, Batich K, Chable-Montero F, Sagy N, Schwartz AM, Henson DE. 2010. Merkel cell carcinoma demographics, morphology, and survival based on 3870 cases: a population based study. J Cutan Pathol 37:20–27. doi: 10.1111/j.1600-0560.2009.01370.x. [DOI] [PubMed] [Google Scholar]
- 5.Gjoerup O, Chang Y. 2010. Update on human polyomaviruses and cancer. Adv Cancer Res 106:1–51. doi: 10.1016/S0065-230X(10)06001-X. [DOI] [PubMed] [Google Scholar]
- 6.Carter JJ, Daugherty MD, Qi X, Bheda-Malge A, Wipf GC, Robinson K, Roman A, Malik HS, Galloway DA. 2013. Identification of an overprinting gene in Merkel cell polyomavirus provides evolutionary insight into the birth of viral genes. Proc Natl Acad Sci U S A 110:12744–12749. doi: 10.1073/pnas.1303526110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schowalter RM, Pastrana DV, Buck CB. 2011. Glycosaminoglycans and sialylated glycans sequentially facilitate Merkel cell polyomavirus infectious entry. PLoS Pathog 7:e1002161. doi: 10.1371/journal.ppat.1002161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schowalter RM, Reinhold WC, Buck CB. 2012. Entry tropism of BK and Merkel cell polyomaviruses in cell culture. PLoS One 7:e42181. doi: 10.1371/journal.pone.0042181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schowalter RM, Buck CB. 2013. The Merkel cell polyomavirus minor capsid protein. PLoS Pathog 9:e1003558. doi: 10.1371/journal.ppat.1003558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feng H, Shuda M, Chang Y, Moore PS. 2008. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science 319:1096–1100. doi: 10.1126/science.1152586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Houben R, Schrama D, Becker JC. 2009. Molecular pathogenesis of Merkel cell carcinoma. Exp Dermatol 18:193–198. doi: 10.1111/j.1600-0625.2009.00853.x. [DOI] [PubMed] [Google Scholar]
- 12.Chang Y, Moore PS. 2012. Merkel cell carcinoma: a virus-induced human cancer. Annu Rev Pathol 7:123–144. doi: 10.1146/annurev-pathol-011110-130227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shuda M, Feng H, Kwun HJ, Rosen ST, Gjoerup O, Moore PS, Chang Y. 2008. T antigen mutations are a human tumor-specific signature for Merkel cell polyomavirus. Proc Natl Acad Sci U S A 105:16272–16277. doi: 10.1073/pnas.0806526105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Houben R, Shuda M, Weinkam R, Schrama D, Feng H, Chang Y, Moore PS, Becker JC. 2010. Merkel cell polyomavirus-infected Merkel cell carcinoma cells require expression of viral T antigens. J Virol 84:7064–7072. doi: 10.1128/JVI.02400-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu W, MacDonald M, You J. 2016. Merkel cell polyomavirus infection and Merkel cell carcinoma. Curr Opin Virol 20:20–27. doi: 10.1016/j.coviro.2016.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu W, Yang R, Payne AS, Schowalter RM, Spurgeon ME, Lambert PF, Xu X, Buck CB, You J. 2016. Identifying the target cells and mechanisms of Merkel cell polyomavirus infection. Cell Host Microbe 19:775–787. doi: 10.1016/j.chom.2016.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feng H, Kwun HJ, Liu X, Gjoerup O, Stolz DB, Chang Y, Moore PS. 2011. Cellular and viral factors regulating Merkel cell polyomavirus replication. PLoS One 6:e22468. doi: 10.1371/journal.pone.0022468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Neumann F, Borchert S, Schmidt C, Reimer R, Hohenberg H, Fischer N, Grundhoff A. 2011. Replication, gene expression and particle production by a consensus Merkel cell polyomavirus (MCPyV) genome. PLoS One 6:e29112. doi: 10.1371/journal.pone.0029112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang X, Li J, Schowalter RM, Jiao J, Buck CB, You J. 2012. Bromodomain protein Brd4 plays a key role in Merkel cell polyomavirus DNA replication. PLoS Pathog 8:e1003021. doi: 10.1371/journal.ppat.1003021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li J, Wang X, Diaz J, Tsang SH, Buck CB, You J. 2013. Merkel cell polyomavirus large T antigen disrupts host genomic integrity and inhibits cellular proliferation. J Virol 87:9173–9188. doi: 10.1128/JVI.01216-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tsang SH, Wang X, Li J, Buck CB, You J. 2014. Host DNA damage response factors localize to Merkel cell polyomavirus DNA replication sites to support efficient viral DNA replication. J Virol 88:3285–3297. doi: 10.1128/JVI.03656-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Serfling E, Jasin M, Schaffner W. 1985. Enhancers and eukaryotic gene transcription. Trends Genet 1:224–230. doi: 10.1016/0168-9525(85)90088-5. [DOI] [Google Scholar]
- 23.de Villiers J, Olson L, Tyndall C, Schaffner W. 1982. Transcriptional ‘enhancers’ from SV40 and polyoma virus show a cell type preference. Nucleic Acids Res 10:7965–7976. doi: 10.1093/nar/10.24.7965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pipas JM. 2009. SV40: cell transformation and tumorigenesis. Virology 384:294–303. doi: 10.1016/j.virol.2008.11.024. [DOI] [PubMed] [Google Scholar]
- 25.Bhatia S, Afanasiev O, Nghiem P. 2011. Immunobiology of Merkel cell carcinoma: implications for immunotherapy of a polyomavirus-associated cancer. Curr Oncol Rep 13:488–497. doi: 10.1007/s11912-011-0197-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ehlers B, Richter D, Matuschka FR, Ulrich RG. 2015. Genome sequences of a rat polyomavirus related to murine polyomavirus, Rattus norvegicus polyomavirus 1. Genome Announc 3:e00997-15. doi: 10.1128/genomeA.00997-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rigatti LH, Toptan T, Newsome JT, Moore PS, Chang Y. 2016. Identification and characterization of novel rat polyomavirus 2 in a colony of X-SCID rats by P-PIT assay. mSphere 1:e00334-16. doi: 10.1128/mSphere.00334-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Iannaccone PM, Jacob HJ. 2009. Rats! Dis Model Mech 2:206–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu W, Stein P, Cheng X, Yang W, Shao NY, Morrisey EE, Schultz RM, You J. 2014. BRD4 regulates Nanog expression in mouse embryonic stem cells and preimplantation embryos. Cell Death Differ 21:1950–1960. doi: 10.1038/cdd.2014.124. [DOI] [PMC free article] [PubMed] [Google Scholar]