

ABSTRACT
Coronavirus spike proteins from different genera are divergent, although they all mediate coronavirus entry into cells by binding to host receptors and fusing viral and cell membranes. Here, we determined the cryo-electron microscopy structure of porcine deltacoronavirus (PdCoV) spike protein at 3.3-Å resolution. The trimeric protein contains three receptor-binding S1 subunits that tightly pack into a crown-like structure and three membrane fusion S2 subunits that form a stalk. Each S1 subunit contains two domains, an N-terminal domain (S1-NTD) and C-terminal domain (S1-CTD). PdCoV S1-NTD has the same structural fold as alpha- and betacoronavirus S1-NTDs as well as host galectins, and it recognizes sugar as its potential receptor. PdCoV S1-CTD has the same structural fold as alphacoronavirus S1-CTDs, but its structure differs from that of betacoronavirus S1-CTDs. PdCoV S1-CTD binds to an unidentified receptor on host cell surfaces. PdCoV S2 is locked in the prefusion conformation by structural restraint of S1 from a different monomeric subunit. PdCoV spike possesses several structural features that may facilitate immune evasion by the virus, such as its compact structure, concealed receptor-binding sites, and shielded critical epitopes. Overall, this study reveals that deltacoronavirus spikes are structurally and evolutionally more closely related to alphacoronavirus spikes than to betacoronavirus spikes; it also has implications for the receptor recognition, membrane fusion, and immune evasion by deltacoronaviruses as well as coronaviruses in general.
IMPORTANCE In this study, we determined the cryo-electron microscopy structure of porcine deltacoronavirus (PdCoV) spike protein at a 3.3-Å resolution. This is the first atomic structure of a spike protein from the deltacoronavirus genus, which is divergent in amino acid sequences from the well-studied alpha- and betacoronavirus spike proteins. Here, we described the overall structure of the PdCoV spike and the detailed structure of each of its structural elements. Moreover, we analyzed the functions of each of the structural elements. Based on the structures and functions of these structural elements, we discussed the evolution of PdCoV spike protein in relation to the spike proteins from other coronavirus genera. This study combines the structure, function, and evolution of PdCoV spike protein and provides many insights into its receptor recognition, membrane fusion, and immune evasion.
KEYWORDS: viral spike glycoprotein, prefusion, receptor binding, membrane fusion, immune evasion, cryo-electron microscopy, single-particle reconstruction
INTRODUCTION
Coronaviruses are large enveloped RNA viruses that can be classified into four genera: Alpha-, Beta-, Gamma-, and Deltacoronavirus (1). Both alphacoronaviruses (hereinafter, α-coronaviruses) and β-coronaviruses infect mammals, γ-coronaviruses infect birds, and δ-coronaviruses infect mammals and birds (1). Representative coronaviruses include human NL63 coronavirus (HCoV-NL63) and porcine transmissible gastroenteritis coronavirus (TGEV) from the Alphacoronavirus genus (α genus); mouse hepatitis coronavirus (MHV), bovine coronavirus (BCoV), severe acute respiratory syndrome coronavirus (SARS-CoV), and Middle East respiratory syndrome coronavirus (MERS-CoV) from the β genus; avian infectious bronchitis virus (IBV) from the γ genus; and porcine deltacoronavirus (PdCoV) from the δ genus (2). Coronaviruses from different genera demonstrate distinct serotypes, mainly due to the divergence of their envelope-anchored spike proteins (3). The spike proteins mediate viral entry into host cells by first binding to host receptors through their S1 subunit and then fusing host and viral membranes through their S2 subunit (4). Hence, they are critical determinants of viral host range and tissue tropism and also induce most of the host immune responses (5). Knowing the structure and function of the spike proteins from different genera is critical for understanding cell entry, pathogenesis, evolution, and immunogenicity of coronaviruses (6).
The receptor recognition pattern by coronaviruses is complicated (7). The S1 subunits from α- and β-coronavirus spikes contain two domains, the N-terminal domain (S1-NTD) and C-terminal domain (S1-CTD). Depending on the virus, either one or both of the S1 domains can function as the receptor-binding domain (RBD) by binding to host receptors. On the one hand, S1-CTDs from α- and β-coronaviruses have different tertiary structures, but they share a structural topology, indicating a common evolutionary origin and subsequent divergent evolution of S1-CTDs (7). α-Coronavirus S1-CTDs recognize either angiotensin-converting enzyme 2 (ACE2) or aminopeptidase-N (APN) as their protein receptor, whereas β-coronavirus S1-CTDs recognize either ACE2 or dipeptidyl peptidase 4 (DPP4) (8–16). Hence, S1-CTDs likely have undergone further divergent evolution to recognize different receptors. On the other hand, S1-NTDs from α- and β-coronaviruses both have the same structural fold as human galectins, and they recognize either sugar receptors or a protein receptor, CEACAM1 (17–23). Hence, it has been suggested that coronavirus S1-NTDs originated from host galectins and have undergone divergent evolution to recognize different receptors (7). These studies on receptor recognition by coronaviruses have revealed complex evolutionary relationships among the spikes from different genera.
The membrane fusion mechanism for coronavirus spikes is believed to be similar to those used by class 1 viral membrane fusion proteins (24, 25). The best-studied such protein is hemagglutinin (HA) from influenza virus (26, 27). Influenza HA exists in two structurally distinct conformations. Its prefusion conformation on mature virions is a trimer, already cleaved by host proteases into receptor-binding subunit HA1 and membrane fusion subunit HA2, which remain associated. During the membrane fusion process, HA1 dissociates and HA2 undergoes a dramatic conformational change to reach its postfusion conformation: two heptad repeat (HR) regions from each HA2 subunit, HR-N and HR-C, refold into a six-helix bundle, and a previously buried hydrophobic fusion peptide (FP) becomes exposed and inserts into host membrane. The cryo-electron microscopy (cryo-EM) structures of α- and β-coronavirus spikes in the prefusion conformation have recently been determined (28–31). The overall architecture of α- and β-coronavirus spikes is similar to, albeit more complex than, that of influenza HA. Biochemical studies have identified parts of S2 that form six-helix bundle structures and likely correspond to HR-N and HR-C (32–34) and another part of S2 that associates with membranes and likely corresponds to FP (35, 36). It was demonstrated that α-coronavirus spikes are heavily glycosylated, with S2 being more heavily glycosylated than S1, as a viral strategy for immune evasion (29). These studies on membrane fusion by α- and β-coronavirus spikes have suggested a common molecular mechanism for membrane fusion shared by coronavirus spikes and other class 1 viral membrane fusion proteins (37, 38).
PdCoV from the δ genus is a highly lethal viral pathogen in piglets (39–41). Compared to the extensive studies on α- and β-coronavirus spikes, much less is known about the structure and function of δ-coronavirus spikes. It is not clear which of their S1 domains functions as the RBD, where the structural elements of S2 are located, how δ-coronavirus spikes are structurally and evolutionarily related to the spikes from other genera, or what strategies δ-coronavirus spikes use to evade host immune surveillance. This study fills in these critical gaps by determining the cryo-EM structure of PdCoV spike and revealing its functions in receptor binding, viral entry, and immune evasion.
RESULTS AND DISCUSSION
Overall structure of PdCoV spike.
To capture PdCoV spike in the prefusion conformation, we constructed and prepared PdCoV spike ectodomain (S-e) without the transmembrane anchor or intracellular tail (Fig. 1A). We also excluded a short pretransmembrane region, because this region is hydrophobic and can adversely affect protein solubility (42). Instead, we replaced these regions with a GCN4 trimerization tag followed by a His6 tag. We expressed PdCoV S-e in insect cells and purified it to homogeneity. We collected cryo-EM data on PdCoV S-e and determined its structure at 3.3-Å resolution (Table 1 and Fig. 1B and 2).
FIG 1.

Overall structure of PdCoV S-e in the prefusion conformation. (A) Schematic drawing of PdCoV S-e (spike ectodomain). S1, receptor-binding subunit. S2, membrane fusion subunit. GCN4-His6, GCN4 trimerization tag followed by His6 tag. S1-NTD, N-terminal domain of S1. S1-CTD, C-terminal domain of S1. CH-N and CH-C, central helices N and C. FP, fusion peptide. HR-N and HR-C, heptad repeats N and C. Residues in shaded regions (N terminus, GCN4 tag, and His6 tag) were not traced in the structure. (B) Cryo-EM maps of PdCoV S-e with atomic model fitted in. The maps have a contour of 6.6 σ. (C) Cryo-EM structure of prefusion PdCoV S-e. Each of the monomeric subunits is colored differently. (D) Structure of a monomeric subunit in the prefusion conformation. The structural elements are colored in the same way as those in panel A.
TABLE 1.
Data and model statistics
| Parametera | Value |
|---|---|
| Data collection | |
| Microscope | Titan Krios |
| Voltage (kV) | 300 |
| Defocus range (μm) | 1.0 to 4.0 |
| No. of movies | 2,168 |
| Frames per movie | 55 |
| Dose rate (e−/Å2/s) | 4.7 |
| Total dose per movie (e−/Å2) | 51.7 |
| Data processing | |
| No. of particles | 87,002 |
| Symmetry | C3 |
| Provided B factor (Å2) | −150 |
| Map resolution (Å) | 3.3 |
| Model validation | |
| UCSF Chimera CC (57) | 0.865 |
| EMRinger score (54) | 2.77 |
| MolProbity score (53) | 1.91 |
| All-atom clash score (53) | 5.48 |
| Rotamers outliers (%) | 0.78 |
| Ramachandran allowed (%) | 99.59 |
| Ramachandran outliers (%) | 0.41 |
| RMSD | |
| Bond length (Å) | 0.009 |
| Bond angles (°) | 1.437 |
RMSD, root mean square deviations.
FIG 2.

Cryo-EM data analysis of PdCoV S-e. (A) Representative micrographs of frozen-hydrated PdCoV S-e particles and representative 2D class averages in different orientations. The arrow indicates a poorly ordered tail region in some of the particles. (B) Gold-standard Fourier shell correlation (FSC) curves. The resolution was determined to be 3.3 Å. The 0.143 and 0.5 cutoff values are indicated by horizontal gray bars. (C) Final cryo-EM map of PdCoV S-e colored according to the local resolution.
The atomic structure of prefusion PdCoV S-e contains residues 52 to 1017, covering all of the key structural elements except HR-C (Fig. 1A). The overall trimeric structure of PdCoV spike is similar to, but more compact than, those of α- and β-coronavirus spikes: PdCoV spike has a length of 130 Å from S1 to S2 and a width of 50 Å at S2 (Fig. 1C). S2 itself spans 100 Å in length (Fig. 1D). Three S1 subunits form a crown-like structure and sit on top of the trimeric S2 stalk (Fig. 1C and D). Three S1-CTDs are located at the top and center of the spike trimer, whereas three S1-NTDs are located on the lower and outer side of S1-CTDs (Fig. 3A, B, C, and D). The S1-CTD mainly stacks with the S1-NTD from the same monomeric subunit, although there also exist intersubunit interactions between S1-CTDs from different subunits and between S1-CTD and S1-NTD from different subunits. In contrast, the S1 trimer of β-genus MHV spike has an intertwined quaternary structure, with S1-CTD from one subunit mainly stacking with S1-NTD from another subunit (Fig. 4A) (30). Like PdCoV spike, the S1-CTD in α-genus HCoV-NL63 spike also mainly stacks with the S1-NTD from the same subunit (Fig. 4B) (29). Moreover, whereas each subunit of PdCoV S1 contains only one S1-NTD, each subunit of HCoV-NL63 S1 contains two, possibly resulting from gene duplication (Fig. 4B) (29). Connecting S1 and S2 are two subdomains, SD1 and SD2, and a long loop (Fig. 3A and B). The structure of PdCoV S2 is in the prefusion conformation and can be aligned well with those of α- and β-coronavirus S2 fragments (Fig. 4A and B). HR-C is missing from both the current PdCoV S2 structure and previously published α- and β-coronavirus S2 structures, suggesting that this region is poorly ordered. Our structural model also includes glycans N-linked to 39 residues on the trimer (13 on each monomeric subunit). In this article, we will illustrate the structures and functions of each of the structural elements in PdCoV spike.
FIG 3.

Structure of PdCoV S1. (A) Schematic drawing of PdCoV S1. SD1, subdomain 1. SD2, subdomain 2. SD1 consists of two discontinuous regions, SD1′ and SD1″. SD2 consists of two discontinuous regions, SD2′ and SD″. (B) Structure of monomeric S1. Domains and subdomains are colored in the same way as those in panel A. Residue ranges for each of the domains and subdomains are labeled. (C) Structure of trimeric S1, viewed from the side. Each of the monomeric subunits is colored differently. The empty space under S1 is occupied by S2, which is not shown here. (D) Structure of trimeric S1, viewed from the top. Each of the monomeric subunits is colored differently.
FIG 4.
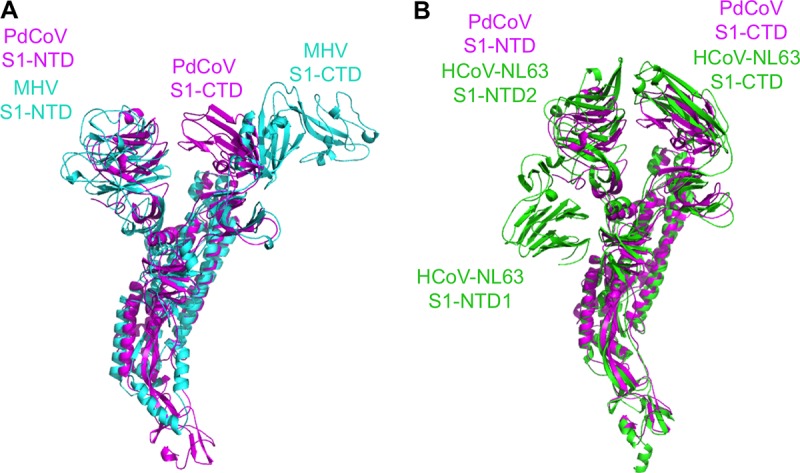
Structural alignments of PdCoV spike with the spikes from other coronavirus genera. (A) Alignment of PdCoV and β-genus MHV spikes. PdCoV spike is colored magenta. MHV spike (PDB entry 3JCL) is colored cyan. (B) Alignment of PdCoV and α-genus HCoV-NL63 spikes. PdCoV spike is colored magenta. HCoV-NL63 spike (PDB entry 5SZS) is colored green. Each subunit of PdCoV S1 contains only one S1-NTD, whereas each subunit of HCoV-NL63 S1 contains two.
Structure, function, and evolution of PdCoV S1-NTD.
PdCoV S1-NTD adopts a β-sandwich fold identical to that of human galectins (Fig. 5A). Its core structure consists of two anti-parallel β-sheet layers: one is seven stranded and the other is six stranded. On top of the core structure is a short α-helix. Underneath the core structure is another three-stranded β-sheet and another α-helix. The S1-NTDs from α- and β-coronaviruses have the same galectin fold (Fig. 5B and C). Like PdCoV S1-NTD, α-coronavirus S1-NTDs contain a short α-helix on top of the core structure, but β-coronavirus S1-NTDs contain a ceiling-like structure in the same location. The galectin fold of PdCoV S1-NTD suggests that like some of the α- and β-coronavirus S1-NTDs, PdCoV S1-NTD recognizes sugars as host receptors to facilitate initial viral attachment to cells; hence, it may function as a viral lectin.
FIG 5.

Structure and function of PdCoV S1-NTD. (A) Structure of PdCoV S1-NTD. The putative sugar-binding site is indicated by the question mark. (B) Structure of α-genus HCoV-NL63 S1-NTD (PDB entry 5SZS). (C) Structure of β-genus BCoV S1-NTD (PDB entry 4H14). (D) ELISA sugar-binding assay for PdCoV S1-NTD. Here, the ELISA plates were precoated with sugar-rich mucin, and then PdCoV S1-NTD was added and incubated with mucin. Mucin-bound S1-NTD was detected using antibodies recognizing its C-terminal His6 tag. Porcine epidemic diarrhea virus (PEDV) S1 was used as the positive control. PdCoV S1-CTD, SARS-CoV S1-CTD, and BSA were used as negative controls. A plate without mucin was used as an additional negative control. Statistical analyses were performed using two-tailed t test. Error bars indicate standard errors of the means (SEM) (n = 5). ***, P < 0.001.
We investigated the sugar-binding capability of PdCoV S1-NTD. To this end, we expressed and purified recombinant PdCoV S1-NTD containing a C-terminal His6 tag and carried out an enzyme-linked immunosorbent assay (ELISA) to examine whether it binds sugar (Fig. 5D). More specifically, PdCoV S1-NTD was incubated with mucin, which contains a variety of sugar chains on its surface; subsequently, the mucin-bound PdCoV S1-NTD was detected using antibodies recognizing its His6 tag. The result showed that PdCoV S1-NTD bound to mucin. Thus, PdCoV S1-NTD bound to the sugar moiety of mucin and potentially can recognize sugar as its receptor. The sugar-binding site in PdCoV S1-NTD is currently unknown. Because the sugar-binding site in β-genus BCoV S1-NTD and the galactose-binding site in human galectins both are located on top of the core structure (18, 43), the sugar-binding site in PdCoV S1-NTD also may be located in the same region (Fig. 5A and C).
The above-described structural and functional analyses of PdCoV S1-NTD provide insight into the evolution of coronavirus S1-NTDs from different genera. Previously, based on the structures and functions of β-coronavirus S1-NTDs, we hypothesized that ancestral coronaviruses acquired a galectin gene from the host and incorporated it into their spike gene, which began to encode S1-NTD; we further predicted that the S1-NTDs from other genera also contain the galectin fold. Both the structure of PdCoV S1-NTD presented here and the structures of α-coronavirus S1-NTDs determined by recent studies confirmed our earlier prediction and lent further support to our previous hypothesis. Hence, coronavirus S1-NTDs from different genera likely all have the same evolutionary origin, which might be the host galectin, and have conserved the galectin fold through evolution.
Structure, function, and evolution of PdCoV S1-CTD.
PdCoV S1-CTD adopts a β-sandwich fold also containing two β-sheet layers: one is a three-stranded anti-parallel β-sheet, and the other is a three-stranded mixed β-sheet (Fig. 6A). Its structure is similar to the β-sandwich core structure of α-coronavirus S1-CTDs but is different from the core structure of β-coronavirus S1-CTDs that contain a single β-sheet layer (Fig. 6B and C). We previously showed that despite their different structural folds, α- and β-coronavirus S1-CTDs share the same structural topology (i.e., connectivity of secondary structural elements) (7). Similarly, PdCoV S1-CTD also shares the same structural topology with β-coronavirus S1-CTDs. Because α- and β-coronaviruses widely use their S1-CTD as the main RBD by recognizing protein receptors, PdCoV S1-CTD also may recognize a protein receptor and function as the main RBD.
FIG 6.
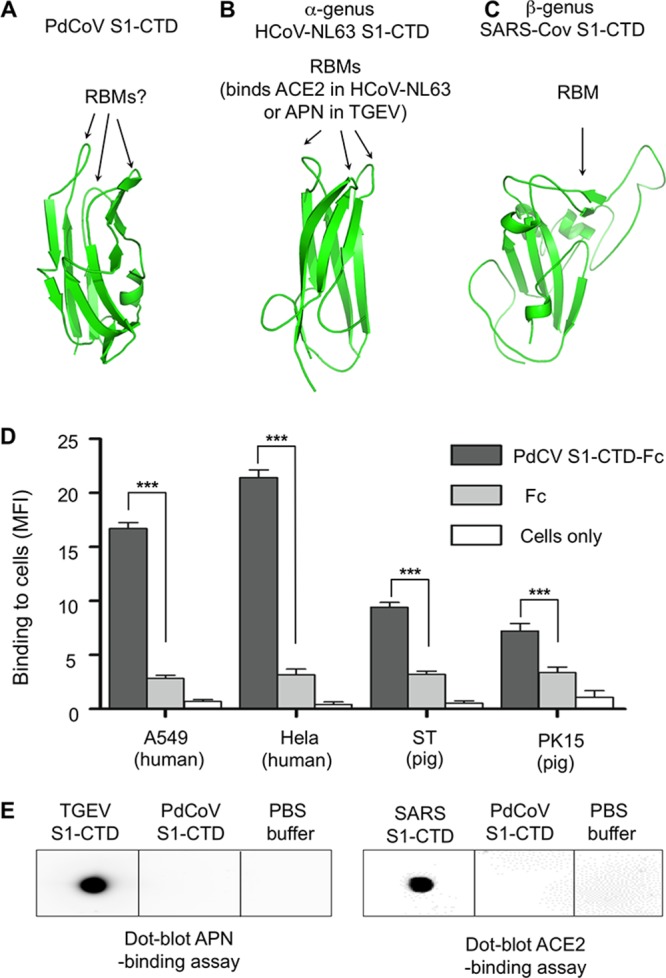
Structure and function of PdCoV S1-CTD. (A) Structure of PdCoV S1-CTD. The putative RBM loops are indicated by the question mark. (B) Structure of α-genus HCoV-NL63 S1-CTD (PDB entry 3KBH). (C) Structure of β-genus SARS-CoV S1-CTD (PDB entry 2AJF). (D) Flow cytometry assay for the binding of PdCoV S1-CTD to the surface of mammalian cells. Cell-bound PdCoV S1-CTD was detected using antibodies recognizing its C-terminal Fc tag. Fc or cells only were used as negative controls. Statistical analyses were performed using two-tailed t test. Error bars indicate SEM (n = 4). ***, P < 0.001. (E) Dot blot receptor-binding assay for PdCoV S1-CTD. Here, the receptor (either APN or ACE2) was first dotted onto a membrane. Subsequently, PdCoV S1-CTD was dotted and incubated with the receptor. Receptor-bound S1-CTD was detected using antibodies recognizing its C-terminal His6 tag. TGEV and SARS-CoV S1-CTDs were used as positive controls. PBS buffer was used as a negative control.
We examined the possibility of PdCoV S1-CTD recognizing a receptor on the surface of mammalian cells. To this end, we expressed and purified recombinant PdCoV S1-CTD containing a C-terminal Fc tag and performed a flow cytometry assay to detect the binding of PdCoV S1-CTD-Fc to mammalian cells (Fig. 6D). Here, the cell-bound PdCoV S1-CTD was detected using antibodies recognizing its Fc tag. The result showed that PdCoV S1-CTD-Fc bound to both human and pig cells with significantly higher affinity than Fc alone, suggesting that PdCoV S1-CTD binds to a receptor on the surface of both human and pig cells. Although PdCoV S1-CTD demonstrates higher affinity for human cells than for pig cells, it is unknown whether PdCoV infects human cells, since receptor recognition is only one of several factors that can impact coronavirus infections. We further investigated whether PdCoV S1-CTD recognizes ACE2 or APN, two known protein receptors for α-coronavirus S1-CTDs. To this end, we prepared and purified recombinant PdCoV S1-CTD containing a C-terminal His6 tag and carried out a dot blot assay to examine whether it binds ACE2 or APN (Fig. 6E). The results showed that PdCoV S1-CTD does not bind ACE2 or APN. As positive controls, TGEV S1-CTD binds APN, whereas SARS-CoV S1-CTD binds ACE2. Taken together, these results demonstrate that PdCoV S1-CTD likely functions as the main RBD and binds to a yet-to-be-identified receptor on the surface of human and pig cells.
The receptor-binding site in PdCoV S1-CTD is currently unknown. In α-coronavirus S1-CTDs, the three loops on the top of the β-sandwich core function as receptor-binding motifs (RBMs) by binding to their respective protein receptor, ACE2 for HCoV-NL63 and APN for TGEV. In PdCoV S1-CTD, the same three loops are structurally similar to their counterparts in α-coronavirus S1-CTDs. Hence, these three loops in PdCoV S1-CTD may bind to a protein receptor and function as RBMs. In the current structure, the S1-CTD is in a closed conformation, with its putative RBMs pointing toward the S1-NTD and unavailable for receptor binding. To bind its receptor, the S1-CTD would need to switch to an open conformation by “standing up” on the spike trimer and rendering the putative RBMs available for receptor binding.
Based on the above-described structural and functional analyses, we discuss the evolution of coronavirus S1-CTDs. Because S1-CTD is located on the tip of the prefusion spike trimer, it is the most exposed region on the surface of virions and thereby is under heavy immune pressure to evolve. Possibly as a consequence of immune pressure, S1-CTD is structurally divergent among different coronavirus genera: α- and δ-coronavirus S1-CTDs have a β-sandwich core, whereas β-coronavirus S1-CTDs have a β-sheet core. The RBMs are located on the very tip of S1-CTDs and are even more structurally divergent than the core structure of S1-CTDs. The RBMs in α- and δ-coronavirus S1-CTDs are three short discontinuous loops; depending on the virus, their RBM loops can bind APN (as in TGEV), ACE2 (as in HCoV-NL63), or a yet-to-be-identified receptor (as in PdCoV). The RBM in β-coronavirus S1-CTDs is a long continuous subdomain; depending on the virus, their RBM can bind ACE2 (as in SARS-CoV) or DPP4 (as in MERS-CoV). Despite their structural divergence, the S1-CTDs from different genera share the same structural topology in their cores (7). These results suggest that these S1-CTDs have a common evolutionary origin and have undergone divergent evolution. Moreover, our study demonstrates that PdCoV S1-CTD is structurally and evolutionarily more closely related to α-coronavirus S1-CTDs than to β-coronavirus S1-CTDs.
Structures, functions, and evolution of S1 subdomains.
The structures of SD1 and SD2 are similar to those of their counterparts in α- and β-coronavirus spikes (Fig. 3B). SD1 adopts a small β-sandwich fold containing two antiparallel β-sheets: one is two stranded and the other is five stranded. SD2 also adopts a small β-sandwich fold containing two three-stranded β-sheets: one is antiparallel and the other is mixed. Interestingly, both SD1 and SD2 consist of discontinuous regions: the majority of their sequences are to the C terminus of S1-CTD, but they also each contain a region to the N terminus of S1-CTD. Based on these structural data, SD1 and SD2 might have evolved later than S1-NTD and S1-CTD. The main function of the two S1 subdomains is to connect S1 and S2, but SD1 also plays a role in membrane fusion, as discussed below.
Structure, function, and evolution of S2.
The overall structure of the prefusion trimeric PdCoV S2 is similar to those of α- and β-coronaviruses. Two central helices, CH-N and CH-C, from each subunit form a six-helix intersubunit interface. Based on previous biochemical and structural studies using isolated regions in S2, HR-N corresponds to a region consisting of four helices and connecting loops and HR-C corresponds to a disordered region (Fig. 7A and B) (30). The exact location of FP is uncertain, but it may correspond to a region consisting of two helices and a connecting loop (30). Examination of the prefusion and postfusion structures of influenza HA2 suggests that during the conformational changes of PdCoV S2, HR-N from each subunit in the prefusion conformation would need to fold into one long central helix as part of the six-helix bundle of the postfusion structure (Fig. 7C). Hence, like influenza HA2, part of CH-C in PdCoV S2 also should be part of HR-N, such that the other parts of HR-N can anchor upon CH-C and extend toward the membrane-distal direction (Fig. 7A). Like the FP in influenza HA2, the FP in PdCoV S2 also would need to change its conformation, spring out toward the membrane-distal direction, and insert into the target membrane. The reason why HR-N and FP are locked in their prefusion conformation is likely because S1-CTD and SD1 from another subunit sit on top of them and prevent them from extending in the membrane-distal direction. The stacking between S1 and S2 from two different subunits contributes to the compact structure of PdCoV spike trimer. Two protease cleavages, one at the S1/S2 boundary and the other on the N terminus of FP, potentially can remove the structural restraint of S1 on S2, allowing the conformational changes of S2 to occur (30, 37, 44). Both the structural and mechanistic similarities between coronavirus S2 and influenza HA2 suggest that the two viral membrane fusion proteins are evolutionarily related (4). The above-described analysis will need to be confirmed by the atomic structure of postfusion PdCoV S2.
FIG 7.
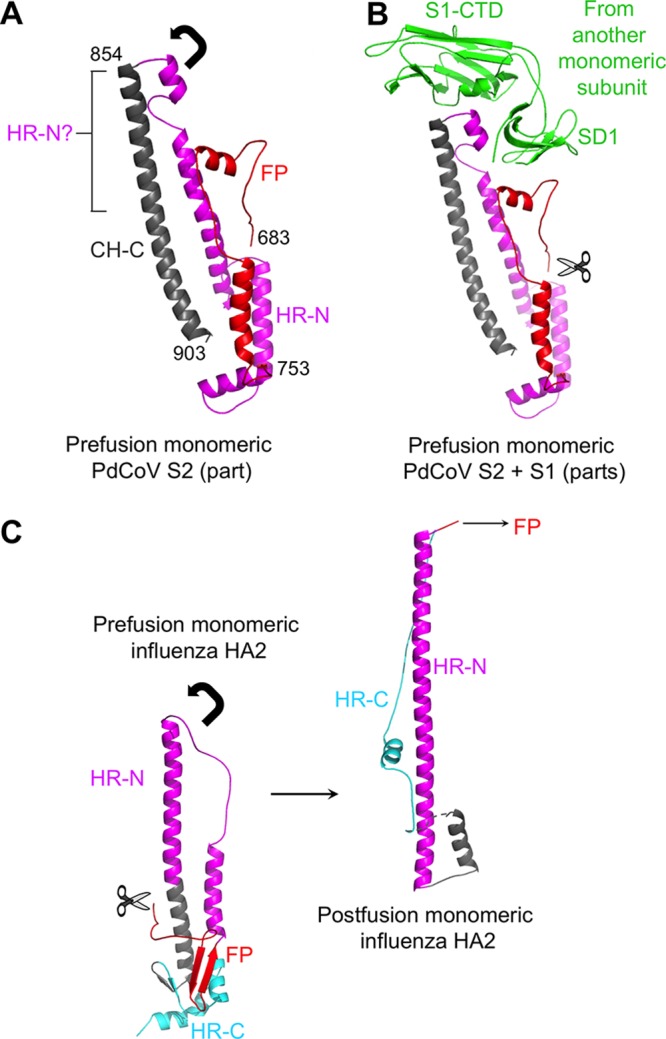
Structure and function of PdCoV S2. (A) Structure of the prefusion monomeric PdCoV S2 only including CH-C, HR-N, and FP. The arrow indicates the direction in which HR-N would need to extend to reach the postfusion conformation. The question mark indicates part of CH-C that likely is part of HR-N. Residue ranges for each of the structural elements are labeled. (B) S1-CTD and SD1 from a different subunit stack with HR-N and FP, respectively, preventing them from switching to their postfusion conformation. The scissor indicates the proteolysis site at the N terminus of FP. (C) Structures of influenza HA2 in the prefusion and postfusion conformations (PDB entries 2YPG and 1QU1). The arrow indicates the direction in which HR-N would need to extend to reach the postfusion conformation. The scissor indicates the proteolysis site at the N terminus of FP.
Immune evasion strategies by PdCoV spike.
The structure of PdCoV spike suggests immune evasion strategies by PdCoV spike. First, the PdCoV spike has a compact structure. The six domains and six subdomains of trimeric S1 are tightly packed (Fig. 3B and C), which reduces the surface area of the spike protein. Despite its compact structure, S1 maintains the two-RBD system, giving the virus more options in receptor selections than a single-RBD system would do. Second, in the current structure, PdCoV S1-CTD is in a closed conformation with its putative RBM loops facing S1-NTD and inaccessible to the host receptor (Fig. 3D). Upon infecting host cells, S1-CTD would need to switch to an open conformation to render the putative RBM loops accessible to the host receptor. The closed-to-open conformational change of S1-CTD has been observed for β-genus MERS-CoV and SARS-CoV spikes (28). This mechanism can minimize the exposure of the putative RBM loops to the immune system. Third, our structural model of PdCoV spike contains glycans N-linked to 39 residues (13 on each subunit); there are also another 24 predicted, but not observed, N-linked glycosylation sites (8 on each subunit) (Fig. 8A and B). Most of these sites are located on the surface of S1, which is in contrast to α-genus HCoV-NL63 spike, where S2 is more heavily glycosylated than S1. Thus, while it was previously suggested that HCoV-NL63 spike evades host immune surveillance mainly by glycan shielding its S2 epitopes (29), PdCoV spike appears to evade host immune surveillance mainly by glycan shielding its S1 epitopes. For example, the putative sugar-binding site in PdCoV S1-NTD is surrounded by glycans, which reduces the accessibility of this site to the immune system (Fig. 8C). As a comparison, the sugar-binding site in β-genus BCoV S1-NTD is also shielded, not by glycans but by the ceiling-like structure on top of the core structure (18). Taken together, PdCoV spike has several structural features that may facilitate viral immune evasion, such as reducing surface areas, concealing receptor-binding sites, and shielding critical S1 epitopes.
FIG 8.

Glycosylation sites on the surface of PdCoV spike. (A) Distribution of N-linked glycosylation sites on the one-dimensional structure of PdCoV spike. Ψ indicates N-linked glycosylate site. Those on the top indicate glycans observed in the structure. Those at the bottom indicate predicted, but not observed, glycosylation sites. Predicted glycosylation sites in the N-terminal region and HR-C were not included because these two regions were not traced in the structure. (B) Distribution of N-linked glycosylation sites on the three-dimensional structure of PdCoV spike. Observed glycans are in dark blue. Predicted, but not observed, glycosylation sites are in light blue. (C) Distribution of N-linked glycosylation sites in monomeric S1. Question marks indicate the putative sugar-binding site in S1-NTD and putative RBMs in S1-CTD.
Conclusions.
In this study, we determined the cryo-EM structure of PdCoV spike at a 3.3-Å resolution. To our knowledge, this is the first atomic structure of a spike protein from the δ genus, which is divergent in amino acid sequences from the well-studied α- and β-coronavirus spikes. Our study reveals a compact PdCoV spike trimer locked in the prefusion conformation. The trimeric S1 contains six domains (three copies of S1-NTD and S1-CTD each) and six subdomains (three copies of SD1 and SD2 each) that tightly pack into a crown-like structure. PdCoV S1-NTD has the same galectin fold as α- and β-coronavirus S1-NTDs; it binds sugar and potentially can recognize sugars as its receptors. These results expand our knowledge on the structures and functions of S1-NTDs from different coronavirus genera and provide further evidence on the common host origin of coronavirus S1-NTDs. PdCoV S1-CTD has the same β-sandwich fold as α-coronavirus S1-CTDs, and this structural fold differs from the β-sheet fold of β-coronavirus S1-CTDs. However, S1-CTDs from all coronavirus genera share the same structural topology, suggesting a common evolutionary origin of coronavirus S1-CTDs. PdCoV S1-CTD binds to an unidentified receptor on mammalian cell surfaces and may function as the main RBD. Moreover, PdCoV S1-CTD is in a closed conformation, with its putative receptor-binding sites buried; it would need to switch to an open conformation for receptor binding. The structures of both S1-NTD and S1-CTD of PdCoV are more similar to those of α-coronaviruses than to those of β-coronaviruses and, hence, PdCoV spike is evolutionarily more closely related to α-coronavirus spikes than to β-coronavirus spikes. The trimeric PdCoV S2 forms the stalk of the spike protein. Each of the S2 subunits is locked in the prefusion conformation by structural constraint of S1 from a different monomeric subunit. More specifically, HR-N and FP are prevented from refolding into their postfusion conformation by the steric restrictions from S1-CTD and SD1, respectively, of another subunit. PdCoV spike possesses several structural features that appear to facilitate its evasion from host immune surveillance, such as its compact structure, the closed conformation of its S1-CTD, and heavy glycosylation near critical epitopes in S1. Overall, our study combines the structure and function of PdCoV spike and provides many insights into the receptor recognition, membrane fusion, immune evasion, and evolution of PdCoV spike as well as coronavirus spikes in general.
MATERIALS AND METHODS
Expression, purification, and treatment of PdCoV spike ectodomain.
PdCoV spike ectodomain (S-e) (residues 18 to 1077) was cloned into pFastBac vector (Life Technologies Inc.) with an N-terminal honeybee melittin signal peptide and C-terminal GCN4 and His6 tags. It was expressed in SF9 insect cells using the Bac-to-Bac system (Life Technologies Inc.) and purified as previously described (15). Briefly, the protein was harvested from cell culture medium and purified sequentially on a nickel-nitrilotriacetic acid (Ni-NTA) column and Superdex 200 gel filtration column (GE Healthcare). Because we showed earlier that low pH could facilitate trimer formation (45), we incubated PdCoV S-e in buffer containing 0.1 M sodium citrate (pH 5.6) at room temperature for 1 h and then repurified it on a Superdex 200 gel filtration column in buffer containing 20 mM Tris, pH 7.2, and 200 mM NaCl.
Cryo-electron microscopy.
For sample preparation, aliquots of PdCoV S-e (3 μl; 0.35 mg/ml; in buffer containing 2 mM Tris, pH 7.2, and 20 mM NaCl) were applied to glow-discharged CF-2/1-4C C-flat grids (Protochips). The grids then were plunge-frozen in liquid ethane using an FEI MarkIII Vitrobot system (FEI Company).
For data collection, images were recorded using a Gatan K2 Summit direct electron detector in the direct electron counting mode (Gatan), attached to a Titan-Krios transmission electron microscope (FEI Company), at Purdue University. The automated software Leginon (46) was used to collect ∼2,100 movies at ×22,500 magnification and at a defocus range of between 0.5 and 3 μm. Each movie had a total accumulated exposure of 52 e/Å2 fractionated in 55 frames of 200 ms of exposure. Data collection statistics are summarized in Table 1.
For data processing, the recorded movies were corrected for beam-induced motion using MotionCor2 (47). The final image was bin averaged to give a pixel size of 1.3 Å. The parameters of the microscope contrast transfer function were estimated for each micrograph using GCTF (48). Particles were automatically picked and extracted using RELION 2.0 on a GPU workstation with a box size of 256 pixels. Initially, particles were subjected to two-dimensional (2D) alignment and clustering using RELION 2.0, and the best classes were selected for an additional 2D alignment. Some of the particles on 2D class averages appear to have a tail (Fig. 2A), which may correspond to HR-C. Nevertheless, the weak density of the tail region suggests that this region is poorly ordered and, hence, this region was not included in subsequent map calculation and model building. All of the particles, with or without the tail, were subjected to 3D autorefine with a mask covering the overall shape of the particles (excluding the tail region) to yield the map. The orientations of the particles used in the final reconstruction map sufficiently covered the whole sphere in the Fourier space to allow calculation of a 3D map with isotropic resolution. The map was sharpened with the modulation transfer function of K2 operated at 300 kV using RELION 2.0 postprocessing. Reported resolution was based on the gold-standard Fourier shell correlation (FSC) of 0.143 criterion, and Fourier shell correction curves were corrected for the effects of soft masking by high-resolution noise substitution (49). Data processing statistics are summarized in Table 1.
Model building and refinement.
For atomic model building, the cryo-EM structure of HCoV-NL63 spike (PDB entry 5SZS) was divided into 7 parts (S1-NTD, SD2′, SD1′, S1-CTD, SD1″, SD2″, and S2) and fitted into the cryo-EM map of PdCoV S-e individually using UCSF Chimera (50) and Coot (51). Model rebuilding was performed manually in Coot based on the well-defined continuous density of the main chain, and sequence register assignment was guided mainly by the density of N-linked glycans and of bulky amino acid residues. The structural model was refined using Phenix (52) with geometry restraints and 3-fold noncrystallographic symmetry constraints. Refinement and manual model correction in Coot were carried out iteratively until there was no more improvement in geometry parameters. The quality of the final model was analyzed using MolProbity (53) and EMRinger (54). The validation statistics of the structural model are summarized in Table 1.
ELISA sugar-binding assay.
PdCoV S1-NTD containing a C-terminal His6 tag was expressed and purified in the same way as PdCoV S-e and assayed for its sugar-binding capability using an ELISA as previously described (18). Briefly, ELISA plates were precoated with bovine mucin (1 mg/ml) at 37°C for 1 h. After blocking with 1% bovine serum albumin (BSA) at 37°C for 1 h, PdCoV S1-NTD (1 μg/ml) was added to the plates and incubated with mucin at 37°C for 1 h. After washes with PBS buffer, the plates were incubated with anti-His6 antibody (Santa Cruz) at 37°C for 1 h. The plates then were washed with PBS and incubated with horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG antibody (1:5,000) at 37°C for 1 h. After more washes with PBS, enzymatic reaction was carried out using ELISA substrate (Life Technologies Inc.) and stopped with 1 M H2SO4. Absorbance at 450 nm (A450) was measured using a Tecan Infinite M1000 PRO microplate reader (Tecan Group Ltd.). Five replicates were done for each sample. Porcine epidemic diarrhea virus (PEDV) S1 and SARS-CoV S1-CTD were prepared as previously described (15, 55), and PdCoV S1-CTD was prepared as described below; these three proteins were used in the assay as controls.
Dot blot receptor-binding assay.
PdCoV S1-CTD containing a C-terminal His6 tag was expressed and purified in the same way as PdCoV S-e and assayed for its receptor-binding capability using a dot blot receptor-binding assay as previously described (55). Briefly, 5 μM receptor (human ACE2 or porcine APN) was dotted onto nitrocellulose membranes. The membranes were dried and blocked with 1% BSA and then incubated with 1 μM PdCoV S1-CTD at 4°C for 2 h. After washes with PBS buffer, the membranes were incubated with anti-His6 antibody (Life Technologies Inc.) at 4°C for 2 h, washed with PBS, incubated with HRP-conjugated goat anti-mouse IgG antibody (1:5,000) at 4°C for 2 h, and washed with PBS. Finally, the receptor-bound proteins were detected using a chemiluminescence reagent (ECL plus; GE Healthcare). Recombinant human ACE2 and porcine APN were prepared as previously described (13, 15).
Flow cytometry cell-binding assay.
PdCoV S1-CTD containing a C-terminal Fc tag was expressed, purified, and assayed for its cell-binding capability by flow cytometry as previously described (56). Briefly, human (HeLa and A549) and pig (ST and PK15) cells were incubated with PdCoV S1-CTD-Fc (40 μg/ml), or human IgG-Fc control, at room temperature for 30 min, followed by incubation with fluorescein isothiocyanate (FITC)-labeled anti-human IgG-Fc antibody for 30 min. The cells then were analyzed for the binding using flow cytometry.
Accession number(s).
The cryo-EM map has been deposited in the Electron Microscopy Data Bank (EMDB) under accession code 7063 (https://pdbj.org/emnavi/quick.php?id=emdb-7063). The atomic model has been deposited in the Protein Data Bank (PDB) under accession code 6B7N.
ACKNOWLEDGMENTS
This work was supported by NIH grant R01AI089728 (to F.L.) and funding from the University of Minnesota (to W.Z.).
Initial cryo-EM images were collected using FEI Tecnai TEMs maintained at the Characterization Facility of the University of Minnesota. Final cryo-EM data were collected at the EM facility of Purdue University. We thank Valorie Bowman, Thomas Klose, Yue Liu, and Steve Wilson for help in data collection and image processing and Hinh Ly for comments on the manuscript. Initial image analysis and computation work was performed using the workstations at the Basic Sciences Computing Laboratory of the University of Minnesota Supercomputing Institute.
Footnotes
For a companion article on this topic, see https://doi.org/10.1128/JVI.01628-17.
REFERENCES
- 1.Gonzalez JM, Gomez-Puertas P, Cavanagh D, Gorbalenya AE, Enjuanes L. 2003. A comparative sequence analysis to revise the current taxonomy of the family Coronaviridae. Arch Virol 148:2207–2235. doi: 10.1007/s00705-003-0162-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Perlman S, Netland J. 2009. Coronaviruses post-SARS: update on replication and pathogenesis. Nat Rev Microbiol 7:439–450. doi: 10.1038/nrmicro2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Du LY, He YX, Zhou YS, Liu SW, Zheng BJ, Jiang SB. 2009. The spike protein of SARS-CoV–a target for vaccine and therapeutic development. Nat Rev Microbiol 7:226–236. doi: 10.1038/nrmicro2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li F. 2016. Structure, function, and evolution of coronavirus spike proteins. Annu Rev Virol 3:237–261. doi: 10.1146/annurev-virology-110615-042301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Graham RL, Baric RS. 2010. Recombination, reservoirs, and the modular spike: mechanisms of coronavirus cross-species transmission. J Virol 84:3134–3146. doi: 10.1128/JVI.01394-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Graham RL, Donaldson EF, Baric RS. 2013. A decade after SARS: strategies for controlling emerging coronaviruses. Nat Rev Microbiol 11:836–848. doi: 10.1038/nrmicro3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li F. 2015. Receptor recognition mechanisms of coronaviruses: a decade of structural studies. J Virol 89:1954–1964. doi: 10.1128/JVI.02615-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li WH, Moore MJ, Vasilieva N, Sui JH, Wong SK, Berne MA, Somasundaran M, Sullivan JL, Luzuriaga K, Greenough TC, Choe H, Farzan M. 2003. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 426:450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Raj VS, Mou HH, Smits SL, Dekkers DHW, Muller MA, Dijkman R, Muth D, Demmers JAA, Zaki A, Fouchier RAM, Thiel V, Drosten C, Rottier PJM, Osterhaus A, Bosch BJ, Haagmans BL. 2013. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature 495:251–254. doi: 10.1038/nature12005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Delmas B, Gelfi J, Lharidon R, Vogel LK, Sjostrom H, Noren O, Laude H. 1992. Aminopeptidase-N is a major receptor for the enteropathogenic coronavirus TGEV. Nature 357:417–420. doi: 10.1038/357417a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hofmann H, Pyrc K, van der Hoek L, Geier M, Berkhout B, Pohlmann S. 2005. Human coronavirus NL63 employs the severe acute respiratory syndrome coronavirus receptor for cellular entry. Proc Natl Acad Sci U S A 102:7988–7993. doi: 10.1073/pnas.0409465102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reguera J, Santiago C, Mudgal G, Ordono D, Enjuanes L, Casasnovas JM. 2012. Structural bases of coronavirus attachment to host aminopeptidase N and its inhibition by neutralizing antibodies. PLoS Pathog 8:e1002859. doi: 10.1371/journal.ppat.1002859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen L, Lin YL, Peng G, Li F. 2012. Structural basis for multifunctional roles of mammalian aminopeptidase N. Proc Natl Acad Sci U S A 109:17966–17971. doi: 10.1073/pnas.1210123109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu K, Li W, Peng G, Li F. 2009. Crystal structure of NL63 respiratory coronavirus receptor-binding domain complexed with its human receptor. Proc Natl Acad Sci U S A 106:19970–19974. doi: 10.1073/pnas.0908837106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li F, Li WH, Farzan M, Harrison SC. 2005. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science 309:1864–1868. doi: 10.1126/science.1116480. [DOI] [PubMed] [Google Scholar]
- 16.Lu G, Hu Y, Wang Q, Qi J, Gao F, Li Y, Zhang Y, Zhang W, Yuan Y, Bao J, Zhang B, Shi Y, Yan J, Gao GF. 2013. Molecular basis of binding between novel human coronavirus MERS-CoV and its receptor CD26. Nature 500:227–231. doi: 10.1038/nature12328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peng GQ, Sun DW, Rajashankar KR, Qian ZH, Holmes KV, Li F. 2011. Crystal structure of mouse coronavirus receptor-binding domain complexed with its murine receptor. Proc Natl Acad Sci U S A 108:10696–10701. doi: 10.1073/pnas.1104306108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peng GQ, Xu LQ, Lin YL, Chen L, Pasquarella JR, Holmes KV, Li F. 2012. Crystal structure of bovine coronavirus spike protein lectin domain. J Biol Chem 287:41931–41938. doi: 10.1074/jbc.M112.418210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tan KM, Zelus BD, Meijers R, Liu JH, Bergelson JM, Duke N, Zhang R, Joachimiak A, Holmes KV, Wang JH. 2002. Crystal structure of murine sCEACAM1a 1,4: a coronavirus receptor in the CEA family. EMBO J 21:2076–2086. doi: 10.1093/emboj/21.9.2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dveksler GS, Pensiero MN, Cardellichio CB, Williams RK, Jiang GS, Holmes KV, Dieffenbach CW. 1991. Cloning of the mouse hepatitis-virus (Mhv) receptor–expression in human and hamster-cell lines confers susceptibility to Mhv. J Virol 65:6881–6891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Williams RK, Jiang GS, Holmes KV. 1991. Receptor for mouse hepatitis-virus is a member of the carcinoembryonic antigen family of glycoproteins. Proc Natl Acad Sci U S A 88:5533–5536. doi: 10.1073/pnas.88.13.5533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schultze B, Gross HJ, Brossmer R, Herrler G. 1991. The S-protein of bovine coronavirus is a hemagglutinin recognizing 9-o-acetylated sialic-acid as a receptor determinant. J Virol 65:6232–6237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schultze B, Krempl C, Ballesteros ML, Shaw L, Schauer R, Enjuanes L, Herrler G. 1996. Transmissible gastroenteritis coronavirus, but not the related porcine respiratory coronavirus, has a sialic acid (N-glycolylneuraminic acid) binding activity. J Virol 70:5634–5637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harrison SC. 2015. Viral membrane fusion. Virology 479-480:498–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bosch BJ, van der Zee R, de Haan CAM, Rottier PJM. 2003. The coronavirus spike protein is a class I virus fusion protein: structural and functional characterization of the fusion core complex. J Virol 77:8801–8811. doi: 10.1128/JVI.77.16.8801-8811.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Skehel JJ, Wiley DC. 2000. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu Rev Biochemistry 69:531–569. doi: 10.1146/annurev.biochem.69.1.531. [DOI] [PubMed] [Google Scholar]
- 27.Colman PM, Lawrence MC. 2003. The structural biology of type I viral membrane fusion. Nat Rev Mol Cell Biol 4:309–319. doi: 10.1038/nrm1076. [DOI] [PubMed] [Google Scholar]
- 28.Yuan Y, Cao D, Zhang Y, Ma J, Qi J, Wang Q, Lu G, Wu Y, Yan J, Shi Y, Zhang X, Gao GF. 2017. Cryo-EM structures of MERS-CoV and SARS-CoV spike glycoproteins reveal the dynamic receptor binding domains. Nat Commun 8:15092. doi: 10.1038/ncomms15092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Walls AC, Tortorici MA, Frenz B, Snijder J, Li W, Rey FA, DiMaio F, Bosch BJ, Veesler D. 2016. Glycan shield and epitope masking of a coronavirus spike protein observed by cryo-electron microscopy. Nat Struct Mol Biol 23:899–905. doi: 10.1038/nsmb.3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Walls AC, Tortorici MA, Bosch BJ, Frenz B, Rottier PJ, DiMaio F, Rey FA, Veesler D. 2016. Cryo-electron microscopy structure of a coronavirus spike glycoprotein trimer. Nature 531:114–117. doi: 10.1038/nature16988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kirchdoerfer RN, Cottrell CA, Wang N, Pallesen J, Yassine HM, Turner HL, Corbett KS, Graham BS, McLellan JS, Ward AB. 2016. Pre-fusion structure of a human coronavirus spike protein. Nature 531:118–121. doi: 10.1038/nature17200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Duquerroy S, Vigouroux A, Rottier PJ, Rey FA, Bosch BJ. 2005. Central ions and lateral asparagine/glutamine zippers stabilize the post-fusion hairpin conformation of the SARS coronavirus spike glycoprotein. Virology 335:276–285. doi: 10.1016/j.virol.2005.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu YH, Lou ZY, Liu YW, Pang H, Tien P, Gao GF, Rao ZH. 2004. Crystal structure of severe acute respiratory syndrome coronavirus spike protein fusion core. J Biol Chem 279:49414–49419. doi: 10.1074/jbc.M408782200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Supekar VM, Bruckmann C, Ingallinella P, Bianchi E, Pessi A, Carfi A. 2004. Structure of a proteolytically resistant core from the severe acute respiratory syndrome coronavirus S2 fusion protein. Proc Natl Acad Sci U S A 101:17958–17963. doi: 10.1073/pnas.0406128102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ou X, Zheng W, Shan Y, Mu Z, Dominguez SR, Holmes KV, Qian Z. 2016. Identification of the fusion peptide-containing region in betacoronavirus spike glycoproteins. J Virol 90:5586–5600. doi: 10.1128/JVI.00015-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Basso LG, Vicente EF, Crusca E Jr, Cilli EM, Costa-Filho AJ. 2016. SARS-CoV fusion peptides induce membrane surface ordering and curvature. Sci Rep 6:37131. doi: 10.1038/srep37131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Belouzard S, Chu VC, Whittaker GR. 2009. Activation of the SARS coronavirus spike protein via sequential proteolytic cleavage at two distinct sites. Proc Natl Acad Sci U S A 106:5871–5876. doi: 10.1073/pnas.0809524106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heald-Sargent T, Gallagher T. 2012. Ready, set, fuse! The coronavirus spike protein and acquisition of fusion competence. Viruses 4:557–580. doi: 10.3390/v4040557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ma Y, Zhang Y, Liang X, Lou F, Oglesbee M, Krakowka S, Li J. 2015. Origin, evolution, and virulence of porcine deltacoronaviruses in the United States. mBio 6:e00064. doi: 10.1128/mBio.00064-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hu H, Jung K, Vlasova AN, Chepngeno J, Lu Z, Wang Q, Saif LJ. 2015. Isolation and characterization of porcine deltacoronavirus from pigs with diarrhea in the United States. J Clin Microbiol 53:1537–1548. doi: 10.1128/JCM.00031-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dong N, Fang L, Yang H, Liu H, Du T, Fang P, Wang D, Chen H, Xiao S. 2016. Isolation, genomic characterization, and pathogenicity of a Chinese porcine deltacoronavirus strain CHN-HN-2014. Vet Microbiol 196:98–106. doi: 10.1016/j.vetmic.2016.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mahajan M, Bhattacharjya S. 2015. NMR structures and localization of the potential fusion peptides and the pre-transmembrane region of SARS-CoV: Implications in membrane fusion. Biochim Biophys Acta 1848:721–730. doi: 10.1016/j.bbamem.2014.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Seetharaman J, Kanigsberg A, Slaaby R, Leffler H, Barondes SH, Rini JM. 1998. X-ray crystal structure of the human galectin-3 carbohydrate recognition domain at 2.1-angstrom resolution. J Biol Chem 273:13047–13052. doi: 10.1074/jbc.273.21.13047. [DOI] [PubMed] [Google Scholar]
- 44.Millet JK, Whittaker GR. 2014. Host cell entry of Middle East respiratory syndrome coronavirus after two-step, furin-mediated activation of the spike protein. Proc Natl Acad Sci U S A 111:15214–15219. doi: 10.1073/pnas.1407087111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li F, Berardi M, Li WH, Farzan M, Dormitzer PR, Harrison SC. 2006. Conformational states of the severe acute respiratory syndrome coronavirus spike protein ectodomain. J Virol 80:6794–6800. doi: 10.1128/JVI.02744-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Carragher B, Kisseberth N, Kriegman D, Milligan RA, Potter CS, Pulokas J, Reilein A. 2000. Leginon: an automated system for acquisition of images from vitreous ice specimens. J Struct Biol 132:33–45. doi: 10.1006/jsbi.2000.4314. [DOI] [PubMed] [Google Scholar]
- 47.Li X, Mooney P, Zheng S, Booth CR, Braunfeld MB, Gubbens S, Agard DA, Cheng Y. 2013. Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat Methods 10:584–590. doi: 10.1038/nmeth.2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang K. 2016. Gctf: real-time CTF determination and correction. J Struct Biol 193:1–12. doi: 10.1016/j.jsb.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen S, McMullan G, Faruqi AR, Murshudov GN, Short JM, Scheres SH, Henderson R. 2013. High-resolution noise substitution to measure overfitting and validate resolution in 3D structure determination by single particle electron cryomicroscopy. Ultramicroscopy 135:24–35. doi: 10.1016/j.ultramic.2013.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Goddard TD, Huang CC, Ferrin TE. 2007. Visualizing density maps with UCSF Chimera. J Struct Biol 157:281–287. doi: 10.1016/j.jsb.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 51.Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen VB, Arendall WB III, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. 2010. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Barad BA, Echols N, Wang RY, Cheng Y, DiMaio F, Adams PD, Fraser JS. 2015. EMRinger: side chain-directed model and map validation for 3D cryo-electron microscopy. Nat Methods 12:943–946. doi: 10.1038/nmeth.3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu C, Tang J, Ma Y, Liang X, Yang Y, Peng G, Qi Q, Jiang S, Li J, Du L, Li F. 2015. Receptor usage and cell entry of porcine epidemic diarrhea coronavirus. J Virol 89:6121–6125. doi: 10.1128/JVI.00430-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Du L, Tai W, Yang Y, Zhao G, Zhu Q, Sun S, Liu C, Tao X, Tseng CK, Perlman S, Jiang S, Zhou Y, Li F. 2016. Introduction of neutralizing immunogenicity index to the rational design of MERS coronavirus subunit vaccines. Nat Commun 7:13473. doi: 10.1038/ncomms13473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]