

ABSTRACT
Hepatitis C virus (HCV) RNA replication occurs in tight association with remodeled host cell membranes, presenting as cytoplasmic accumulations of single-, double-, and multimembrane vesicles in infected cells. Formation of these so-called replication organelles is mediated by a complex interplay of host cell factors and viral replicase proteins. Of these, nonstructural protein 4B (NS4B), an integral transmembrane protein, appears to play a key role, but little is known about the molecular mechanisms of how this protein contributes to organelle biogenesis. Using forward and reverse genetics, we identified glycine zipper motifs within transmembrane helices 2 and 3 of NS4B that are critically involved in viral RNA replication. Foerster resonance energy transfer analysis revealed the importance of the glycine zippers in NS4B homo- and heterotypic self-interactions. Additionally, ultrastructural analysis using electron microscopy unraveled a prominent role of glycine zipper residues for the subcellular distribution and the morphology of HCV-induced double-membrane vesicles. Notably, loss-of-function NS4B glycine zipper mutants prominently induced single-membrane vesicles with secondary invaginations that might represent an arrested intermediate state in double-membrane vesicle formation. These findings highlight a so-far-unknown role of glycine residues within the membrane integral core domain for NS4B self-interaction and functional as well as structural integrity of HCV replication organelles.
IMPORTANCE Remodeling of the cellular endomembrane system leading to the establishment of replication organelles is a hallmark of positive-strand RNA viruses. In the case of HCV, expression of the nonstructural proteins induces the accumulation of double-membrane vesicles that likely arise from a concerted action of viral and coopted cellular factors. However, the underlying molecular mechanisms are incompletely understood. Here, we identify glycine zipper motifs within HCV NS4B transmembrane segments 2 and 3 that are crucial for the protein's self-interaction. Moreover, glycine residues within NS4B transmembrane helices critically contribute to the biogenesis of functional replication organelles and, thus, efficient viral RNA replication. These results reveal how glycine zipper motifs in NS4B contribute to structural and functional integrity of the HCV replication organelles and, thus, viral RNA replication.
KEYWORDS: NS4B, glycine zipper, hepatitis C virus, membrane proteins, membranous web, positive-strand RNA virus, replication organelle
INTRODUCTION
Hepatitis C virus (HCV) is a major global health problem, accounting for about 71 million chronically infected individuals worldwide. While acute infections by HCV most often are asymptomatic, they are characterized by a high rate of chronicity, frequently resulting in hepatosteatosis, liver cirrhosis, and eventually hepatocellular carcinoma. The development of potent direct-acting antiviral drugs has revolutionized the treatment of chronic hepatitis C; however, owing to their high costs and limited availability in most countries with high HCV prevalence as well as the large number of undiagnosed infections, the global burden of chronic hepatitis C is expected to drop only slowly (1).
HCV is a positive-strand RNA virus and the prototype member of the Hepacivirus genus within the family Flaviviridae, which additionally comprises the genera Flavivirus, Pestivirus, and Pegivirus (2). Unique to HCV is the association of the virus particle with components of the lipoprotein system, giving rise to so-called lipoviroparticles (3, 4). HCV enters hepatocytes by clathrin-mediated endocytosis and utilizes a plethora of attachment factors and entry receptors (5). Upon glycoprotein-mediated fusion of the viral envelope with the endosomal membrane, the released RNA genome is used for protein synthesis at the rough endoplasmic reticulum (ER), giving rise to an approximately 3,000-amino-acids-long polyprotein precursor that is co- and posttranslationally processed by cellular and viral proteases into 10 proteins (6). The structural proteins core, envelope protein 1 (E1), and E2, as well as the accessory proteins p7 and nonstructural protein 2 (NS2), are required for the assembly of infectious virus progeny. The proteins NS3, NS4A, NS4B, NS5A, and NS5B form the viral replicase required for the amplification of the viral RNA genome (7).
All steps of the viral replication cycle occur on the surface of intracellular membranes derived from the ER. This requires extensive remodeling of the ER, which is mediated by nonstructural proteins in concert with coopted host cell factors, giving rise to so-called viral replication factories or replication organelles, an evolutionarily conserved feature of positive-strand RNA viruses (8–10). Interestingly, replication organelles can be morphologically subdivided into two major classes, the invaginated vesicles/spherule type and the double-membrane vesicle (DMV) type. Of note, this classification does not follow taxonomy but is believed to reflect common molecular principles underlying replication organelle formation among evolutionarily unrelated virus families (11, 12).
Replication of genomic or subgenomic HCV RNAs or expression of NS3-5B induce the formation of predominantly DMVs in the cytosol, the accumulation of which correlates with the kinetics of viral RNA replication (13, 14). Moreover, DMVs contain viral RNA, nonstructural proteins, and enzymatically active replicase, arguing that they are bona fide viral replication organelles (15). Regulated polyprotein cleavage as well as determinants in NS4B and NS5A were shown to be required for membrane rearrangements (16–19), but their biogenesis remains incompletely understood, also due to the lack of detectable intermediate structures.
NS4B is a highly hydrophobic integral membrane protein, containing two amphipathic α-helices each in its N- and C-terminal domains and flanking the central hydrophobic core of the protein that is composed most likely of four transmembrane helices (reviewed in reference 6) (Fig. 1A). Residues within the N- and C-terminal domains mediate NS4B self-interaction, which in turn facilitates DMV formation (16, 19). The central transmembrane helices contain an unusually high proportion of glycine and small-side-chain amino acid residues, some of which were shown to be required for HCV RNA replication and possibly for the formation of the viral replication complex (20); however, their exact role has remained uncharacterized.
FIG 1.

Glycine zipper constituting residues in NS4B transmembrane segments 2 and 3 are required for HCV RNA replication. (A) NS4B membrane topology. The N-terminal amphipathic α-helices AH1 and AH2 and the C-terminal α-helices H1 and H2 are depicted, while the four transmembrane segments (TMS1-4) are shown as cylinders. Red numbers give NS4B amino acid residues. The arrow indicates the reported dual topology of the N-terminal domain, resulting from the posttranslational flip of the N terminus into the ER lumen. (B) Amino acid sequence of NS4B TMS2 and TMS3 of the HCV isolate JFH-1. Fully conserved amino acid residues are marked with asterisks. Residues targeted in this study are underlined and highlighted in red. (C) Helical wheel representations of NS4B TMS2 (left) and TMS3 (right) are depicted. Bulky hydrophobic amino acids are shown in blue, glycine residues are depicted in yellow, and red circles highlight positions targeted in this study. (D) The design of the subgenomic luciferase reporter replicon used for transient replication assays is shown in the top panel. The firefly luciferase (Fluc) coding region is translated under the control of the HCV internal ribosome entry site (IRES) contained in the 5′ untranslated region (UTR). The second cistron (NS3 to NS5B) is translated via the IRES of the encephalomyocarditis virus (EMCV-IRES). Huh7-Lunet cells were transfected with in vitro-transcribed replicon RNAs specified on the bottom. Cells were lysed 4, 24, 48, and 72 h after transfection, and luciferase activity in cell lysates was determined. Data were normalized for transfection efficiency using the 4-h values. The active-site NS5B polymerase mutant (ΔGDD) indicates the assay background. Mean values of two independent experiments, each measured in duplicate are shown. Error bars indicate standard deviations. The significance of differences between wild-type (wt) and mutant A151L for each time point was calculated by using the unpaired, two-tailed Student's t test (*, P = 0.0056; ns, not significant). (E) The design of the subgenomic replicon used for selection assays is shown in the top panel. It is analogous to the one shown in panel D but encodes the neomycin phosphotransferase (neoR) instead of luciferase. Huh7-Lunet cells were transfected with replicon RNAs specified above each image and subjected to selection with G418. After 3 to 4 weeks, single-cell clones were stained and counted. CFU per μg RNA are given.
Transmembrane proteins frequently exhibit GxxxG motifs, proposed to mediate their self-interaction within the lipid bilayer (21). Of note, this motif is frequently extended into a (G,A,S)x3Gx2–3(G,S,T) sequence, which has been designated glycine zipper motif (22). By exhibiting a distinct right-handed packaging, glycine zipper motifs have been proposed to mediate transmembrane helix bundling, facilitated by backbone carbonyl and Cα hydrogen (23) interactions of the opposing helices.
Here, we identify and characterize conserved glycine zipper motifs within HCV NS4B transmembrane helices 2 and 3 that are involved in NS4B self-interaction and contribute critically to HCV-induced membrane rearrangements.
RESULTS
Conserved glycine zipper residues in NS4B transmembrane helices 2 and 3 are required for HCV RNA replication.
HCV NS4B is a polytopic transmembrane protein containing two amphipathic α-helices each in its N- and C-terminal domains (18, 24, 25), while the central hydrophobic core (amino acids 75 to 191) of NS4B is believed to be composed of four transmembrane segments (26) (Fig. 1A). Despite the lack of precise structural information of the integral membrane domain of NS4B, a striking feature of transmembrane segments (TMSs) 2 and 3 is the high content of glycine residues and small side chain amino acids, many of which are conserved among HCV genotypes (Fig. 1B). The occurrence of repeated glycine residues separated by three amino acids (GxxxG motifs) is observed in the transmembrane segments of many proteins (21). Its extension into a (G,A,S)x3Gx2–3(G,S,T) configuration, designated the glycine zipper motif, is thought to attribute certain features to transmembrane domains via specific interactions of TMSs (22). Indeed, NS4B TMS2 and TMS3 each contain one conserved glycine zipper motif, comprising amino acids S121 to G129 in TMS2 and G143 to A151L in TMS3 (Fig. 1B). Moreover, Heliquest-generated (27) α-helix projections of NS4B TMSs revealed the alignment of glycine and small-side-chain amino acid residues on one particular side of TMS2 and TMS3 (Fig. 1C). Interestingly, the projections suggested a segregation of glycine and bulky hydrophobic amino acids on opposing sides of the helices, which was most apparent for TMS3.
With the aim to test the functional relevance of glycine zipper motifs for HCV RNA replication, we replaced single residues with the bulky amino acid leucine and monitored transient RNA replication by using a subgenomic genotype 2a luciferase reporter replicon (Fig. 1D). All three single point mutations in TMS2 (S121L, G125L, and G129L) decreased HCV RNA replication by more than three orders of magnitude and, thus, to a similar degree as that observed for the nonreplicative ΔGDD polymerase mutant (Fig. 1D), consistent with an earlier report (20). In a comparable fashion, replacement of individual residues that constitute the glycine zipper in TMS3 (G143, G147, and G150) (Fig. 1C) by leucine residues decreased HCV RNA replication dramatically (Fig. 1D). Interestingly, mutant A151L exhibited only a minor delay at the onset of RNA replication visible at 24 h after transfection, but at later time points it was indistinguishable from the wild type (wt).
Although the very subtle phenotype of the A151L mutation suggested that it is not involved in the glycine zipper motif of TMS3, we assessed the impact of this mutation on long-term HCV RNA replication. To this end, we inserted the A151L mutation into a selectable genotype 2a replicon containing the neomycin phosphotransferase gene in the first cistron (Fig. 1E). Selection for G418-resistant colonies, i.e., cells containing a stably replicating HCV RNA, revealed an approximately 10-fold decrease for mutant A151L compared to the wild type, while cells transfected with the replication-defective RNA (ΔGDD) or mock-transfected cells did not yield cell colonies (Fig. 1E). This decreased replication fitness of mutant A151L in the long-term selection assay, but not in the transient replication experiment, likely reflected the impaired replication competence observed 24 h after transfection (Fig. 1D). In any case, this rather minor replication defect of the A151L mutant suggested that A151 is not part of the glycine zipper motif in NS4B TMS3. Consistent with this idea, this residue is positioned toward the hydrophobic side of this TMS (Fig. 1C, right).
To address further the possible role of conserved glycine zipper interfaces of NS4B TMS2 and TMS3, we reasoned that insertion of an alanine residue within those regions should disrupt glycine zipper motifs by altering the conserved glycine residue-exposing interface (Fig. 2A and B). Indeed, insertion of alanine residues at NS4B positions 122 and 129 in TMS2 as well as 144 and 150 in TMS3 reduced HCV RNA replication to background levels (Fig. 2C). This result underscored the important role of glycine residue motifs within NS4B TMS2 and TMS3 for HCV RNA replication.
FIG 2.

Single-amino-acid residue insertions into NS4B TMS2 and TMS3 abolish HCV RNA replication. Helical wheel representations of NS4B TMS2 (A) and TMS3 (B) are depicted. Bulky hydrophobic amino acids are indicated in blue, glycine residues are depicted in yellow, and red lines highlight the glycine zipper interface. Alanine insertions are highlighted in red and disrupt the glycine zipper interfaces, as indicated by the dashed black lines. (C) Huh7-Lunet cells were transfected with subgenomic luciferase reporter replicon RNAs specified on the bottom. Cells were lysed 4, 24, 48, and 72 h after transfection, and luciferase activity in cell lysates was determined. Data were normalized for transfection efficiency by using the 4-h values. The active-site NS5B polymerase mutant (ΔGDD) was used to determine the assay background. Mean values of two independent experiments are shown. Error bars indicate standard deviations.
Identification of pseudoreversions rescuing the replication defect caused by mutations in NS4B transmembrane segments.
We next selected for second-site compensatory mutations (pseudoreversions) that phenotypically rescue the replication defect of the primary mutations in the glycine zipper motifs. To this end, we inserted those single-amino-acid substitutions that profoundly suppressed HCV RNA replication into the selectable subgenomic replicon (Fig. 1E). wt and mutant replicon RNA-transfected cells were cultured in the presence of the neomycin analogue G418 to select for cells containing continuously replicating HCV RNA, and after about 4 weeks single cell clones emerged. However, cell colony numbers were above the background of the assay only in the cases of mutant S121L and G143L, as determined by the ΔGDD negative-control mutant (Fig. 3A). Nucleotide sequence analysis of viral cDNA spanning the NS4B-NS5A coding region confirmed the presence of the primary mutation S121L and identified the conserved pseudoreversion S93R in NS4B (Fig. 3B and C). In contrast, no conserved second-site mutation was identified in the replicon quasispecies RNA pool of cells transfected with mutant G143L. Thus, we further constructed and tested mutants G147A and G150A, reasoning that replacements of glycine residues with alanine residues are subtler alterations than insertion of bulky leucine residues. Indeed, mutant G150A yielded an uncountable number of G418-resistant cell colonies (Fig. 3A), arguing for high replication competence, which is in accordance with previously published data (20). In contrast, mutant G147A yielded only a few G418-resistant cell colonies (Fig. 3A). Nucleotide sequence analysis revealed the conserved pseudoreversion I140F, in addition to the primary G147A mutation (Fig. 3B). Interestingly, residue 140 is located 7 residues N terminal of G147, corresponding to two turns in an α-helix, providing genetic evidence that this part of NS4B indeed adopts an α-helical fold (Fig. 3E).
FIG 3.

Pseudoreversions located in close proximity of the primary mutation in NS4B TMS2 and TMS3 rescue replication of glycine zipper mutants. (A) Huh7-Lunet cells were transfected with G418-selectable subgenomic replicon RNAs encoding either the wild-type (wt) replicase (NS3-5B) or a replicase containing a mutation in NS4B as specified at the bottom. Transfected cells were cultured in medium containing either 0.25 or 0.5 mg/ml G418, as specified at the top, and after 3 to 4 weeks single-cell clones were counted. #, confluence of the cell layer; *, no G418-resistant cell clones obtained. (B) Single-cell clones were expanded, and compensatory mutations contained in replicon RNAs in these cells were identified by nucleotide sequence analysis of replicon RNAs after amplification by reverse transcription-PCR. (C) Membrane topology of the NS4B N-terminal fragment (amino acids 40 to 132) indicating the positions of the primary mutation (S121L) in TMS2 and the second-site mutation (S93R) in the ER-luminal loop between TMS1 and TMS2. (D) Huh7-Lunet cells were transfected with subgenomic luciferase reporter replicon RNAs specified at the bottom. Cells were lysed 4, 24, 48, and 72 h after transfection, and luciferase activity in cell lysates was determined. Data were normalized for transfection efficiency using the 4-h values. The active-site NS5B polymerase mutant (ΔGDD) was used to determine the assay background. Mean values from two independent experiments are shown. Error bars indicate standard deviations. (E) The helical wheel representation of TMS3 highlights bulky hydrophobic amino acid residues in blue, glycine residues in yellow, the primary mutation G147A in TMS2 in red, and the corresponding pseudoreversion I140F, localizing two turns up in the same helical TMS, in green. (F) HCV RNA replication kinetics of NS4B mutants as determined by transient assays specified for panel D. Note the replication rescue of the G147A mutant by the I140F pseudoreversion.
We validated the functional relevance of these pseudoreversions by inserting them either alone or in combination with the respective primary mutation into the luciferase reporter replicon (Fig. 1D). In the context of the wild-type replicon, the S93R mutation, residing in the putative ER-luminal loop between TMS1 and TMS2, supported HCV RNA replication. Importantly, this mutation rescued the replication defect of the primary S121L mutant (Fig. 3D). However, S93R failed to restore the replication defect caused by primary mutation G125L or G129L within the same glycine zipper interface, arguing against a generic effect of S93R-mediated enhancement of HCV replication fitness. Moreover, pseudorevertant I140F, located two α-helical turns N terminal of primary mutation G147A (Fig. 3E), restored the replication capacity of the primary mutant while not exhibiting an obvious phenotype when introduced into the wild-type replicon (Fig. 3F).
We next corroborated the impact of NS4B glycine zipper motifs on HCV replication and virus production by using a full-length HCV genome. To this end, we introduced a panel of NS4B mutants and corresponding pseudoreversions into the genotype 2a chimeric virus JcR2a, encoding a Renilla luciferase reporter (Fig. 4A) (28). Primary mutations in TMS2 and TMS3 (S121L, G125L, G147L, G147A, and G150L) decreased full-length HCV RNA replication by more than two orders of magnitude to a degree similar to that observed for the nonreplicative ΔGDD polymerase mutant (Fig. 4B). In contrast, NS4B mutant A151L, as well as the primary mutants S121L and G147A, each combined with the corresponding pseudoreversion (S93R and I140F, respectively), supported full-length HCV RNA replication, albeit to a lesser extent than that observed for the wild type or the envelope glycoprotein-deficient ΔE1E2 mutant.
FIG 4.
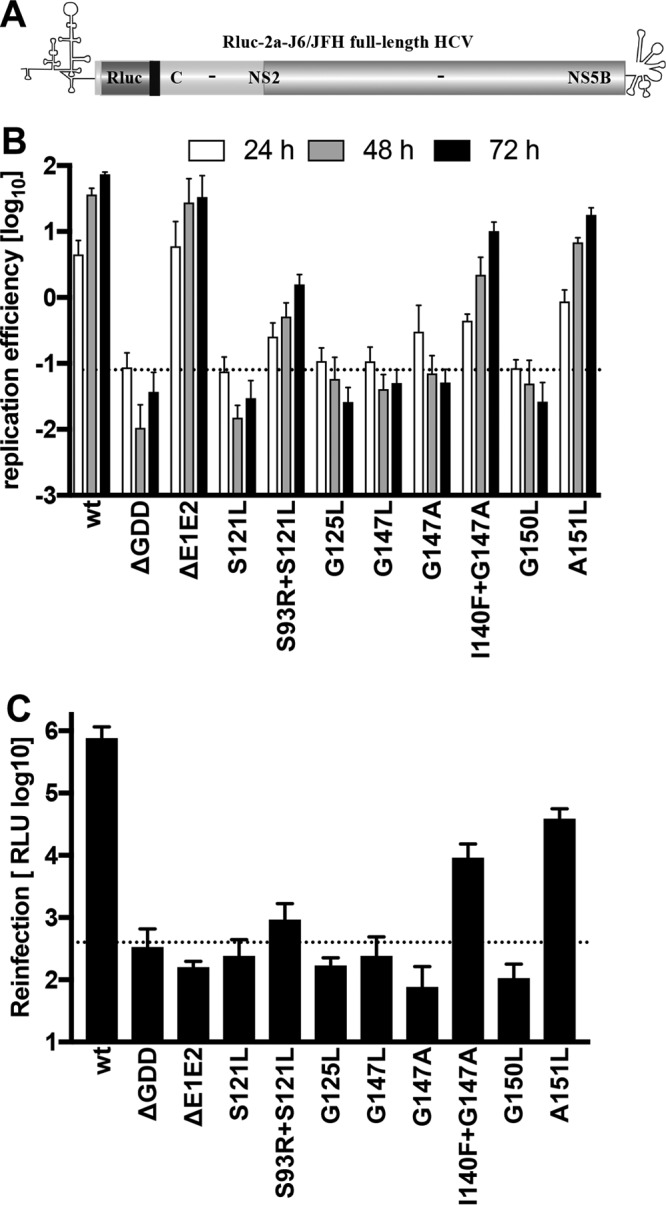
Impact of mutations and pseudoreversions in TMS2 and TMS3 on full-length HCV RNA replication and particle production. (A) Huh7-Lunet cells were transfected with in vitro-transcribed HCV full-length RNAs derived from the reporter virus genome specified at the bottom (JcR2a) (28). (B) Cells were lysed 4, 24, 48, and 72 h after transfection, and luciferase activity in cell lysates was determined. Data were normalized for transfection efficiency using the 4-h values. The active-site NS5B polymerase mutant (ΔGDD) indicates the assay background. Mean values from two independent experiments, each measured in duplicate, are shown. Error bars indicate standard deviations. (C) Culture supernatants harvested 72 h after transfection of cells described for panel B were used to infect naive Huh7.5 cells. After 72 h cells were lysed and luciferase activity in cell lysates was determined. The envelope-deficient mutant (ΔE1E2) indicates the assay background (dotted line). Mean values from two independent experiments, each measured in duplicate, are shown. Error bars indicate standard deviations.
To assess the impact of these mutations on virus production, supernatants of transfected cells harvested 72 h after transfection were used to infect naive cells, and reporter activity in infected cells was determined as a measure of HCV particle production. Consistent with their replication phenotype, primary glycine zipper mutants S121L, G125L, G147L, G147A, and G150L did not produce infectious HCV progeny, while low-level virus production was detected for the primary mutant A151L and the combinations of primary mutations with the corresponding pseudoreversions (S93R+S121L and I140F+G147A) (Fig. 4C). Importantly, suppression of virus production correlated well with suppression of RNA replication, indicating that the glycine zipper mutations impair replication but not HCV particle production. In summary, these results suggested an important role of the glycine zipper motifs within TMS2 and TMS3 of the NS4B transmembrane domain for HCV RNA replication.
Impact of glycine zipper mutations on NS4B protein steady-state levels.
To assess the underlying mechanism, we analyzed the impact of these mutations on NS4B stability. To this end, we determined NS4B steady-state levels in a replication-independent system that is based on the expression of the minimal set of viral replicase proteins (NS3-5B) via T7 RNA polymerase-based cytoplasmic transcription, thus excluding confounding effects arising from differences in replication efficiencies (Fig. 5A). Huh7-Lunet/T7 cells were transfected with the wild-type construct or the various mutants derived thereof, and steady-state levels of NS4B and NS5A were determined by Western blotting using monospecific antibodies (Fig. 5B to D). While several mutations in NS4B TMS2 and TMS3 slightly decreased NS4B abundance, as determined by the ratio of NS4B to NS5A steady-state levels, this reduction did not correlate with the extent of replication impairment caused by a given mutation (Fig. 5B). Indeed, mutant A151L, exhibiting the most striking decrease in NS4B steady-state protein level, was only slightly impaired in HCV RNA replication, whereas NS4B abundance of replication-defective mutants was unaffected (compare NS4B/NS5A ratios in Fig. 1D and 5B). Moreover, selected second-site compensatory mutations did not affect decreased abundance of NS4B (Fig. 5C and D). Therefore, we concluded that mutations in the NS4B glycine zipper motifs suppressed HCV RNA replication by mechanisms other than reduced protein stability.
FIG 5.

Impact of mutations in TMS2 and TMS3 on NS4B protein stability. (A to C) Huh7-Lunet/T7 cells were transfected with the pTM expression vector (empty) or the same vector encoding the NS3-5B polyprotein fragment, schematically depicted in panel A, corresponding to wild-type (wt) NS4B or an NS4B mutant specified on the top. Cells were harvested 24 h posttransfection, and proteins were detected by Western blotting using antibodies monospecific for NS4B, NS5A, or GAPDH. Numbers below each lane refer to the relative abundance of NS4B as determined by densitometry. After subtraction of the background and normalization for equal loading by using GAPDH, NS4B-specific signals were normalized to those obtained for NS5A. Representative results of two independent experiments are shown in each panel.
Effect of glycine zipper mutations in TMS2 and TMS3 on NS4B homo- and heterotypic self-interactions.
NS4B is thought to form oligomeric complexes. These are mediated by homo- and heterotypic self-interactions, requiring residues within the NS4B N- and C-terminal domains, and are critically involved in replication organelle biogenesis (16, 19). However, a contribution of the central transmembrane domain of NS4B has not yet been reported. Given the well-established function of glycine zipper motifs in transmembrane helix interactions, we sought to analyze the effect of TMS2 and TMS3 mutations on NS4B homo- and heterotypic self-interactions. To this end, we used a previously reported Foerster resonance energy transfer (FRET)-based assay allowing us to determine NS4B oligomerization within the cellular membrane environment (16, 19). Since NS4B interactions are mediated by multiple determinants, we carried out FRET experiments using N- and C-terminal subdomains of NS4B, which are more likely to reveal subtle alterations in self-interaction caused by single-amino-acid substitutions (16, 19). Of note, when we probed the homotypic interaction of the NS4B N-terminal fragment spanning amino acids 40 to 130, the G125L mutation significantly decreased FRET efficiency, while the mutations S121L and G129L did not (Fig. 6A). Additionally, for the homotypic self-interaction of the NS4B C-terminal fragment spanning amino acids 130 to 261, we observed a significant drop exerted by the TMS3 glycine zipper mutation G150L, while other mutations in TMS3 had no effect (Fig. 6B). Likewise, only mutations G125L and G150L significantly decreased the NS4B heterotypic interaction of the N- and C-terminal domain when mutant fragments were probed against the corresponding wild-type counterpart (Fig. 6C and D). Thus, we concluded that in addition to determinants in the NS4B N- and C-terminal cytoplasmic domains, TMS2 and TMS3 also contribute to NS4B self-interactions. Notably, interaction-decreasing mutations affected (central) residues of the glycine zipper motifs, likely mediating intra- and intermolecular packaging of NS4B transmembrane helices, thus contributing to the oligomeric state of the protein.
FIG 6.

Impact of mutations in glycine zipper motifs on NS4B homo- and heterotypic self-interactions. U2OS cells were transfected with plasmids encoding NS4B N- and C-terminal fragments, each C-terminally fused to cyan fluorescent protein (CFP) or yellow fluorescent protein (YFP), as indicated at the top of each panel. The NS4B coding sequence was either wild type (wt) or contained the mutation specified at the bottom of the diagrams. Coexpression of CFP and YFP only (CFP+YFP) and expression of a CFP-YFP fusion protein (CFP-YFP) served as negative and positive controls, respectively. Twenty-four hours after transfection, the interaction of protein fragments was assessed by FRET acceptor photobleaching. Box-and-whisker diagrams show the median FRET efficiency (middle line) from at least 30 measurements from 2 independent experiments. Bottom and top lines of the boxes represent the 25th and 75th percentile, respectively. Vertical lines indicate minimum and maximum values. (A) Impact of mutations in TMS2 on homotypic interaction of the NS4B N-terminal fragment. (B) Influence of mutations in TMS3 on homotypic interaction of the NS4B C-terminal half. The heterotypic interaction of the NS4B N-terminal with the C-terminal fragment, containing mutations in TMS2 and TMS3, are shown in panels C and D, respectively. The significance of differences compared to the wild type was calculated by using the unpaired, two-tailed Student's t test (***, P < 0.0001; **, P < 0.001).
Mutations affecting glycine zipper motifs in NS4B TMS2 and TMS3 disturb subcellular distribution and morphology of HCV-induced double membrane vesicles, which are reversed by second-site mutations.
HCV genome replication most likely takes place at membranous virus-induced organelle-like structures composed predominantly of DMVs (13–15, 29–31). These are derived from the ER and appear in fluorescence microscopy as cytoplasmic punctae of various sizes. To determine a possible impact of mutations in NS4B on its subcellular localization, we generated a series of NS3-5B polyprotein expression constructs encoding NS4B glycine zipper mutants. These polyproteins were expressed in Huh7-Lunet/T7 cells that were analyzed by NS4B-specific immunofluorescence 24 h after transfection. With one exception, all NS4B variants displayed an ER-like/punctate staining pattern that was indistinguishable from that of the wild type (Fig. 7), consistent with an earlier report describing the analogous finding for the G129L mutant but with an NS4B focus disruption in the case of the G125L mutant (20). While the reason for this discrepancy is unclear, in our study the exception was the G125L+G150L double mutant that displayed a doughnut-like NS4B subcellular distribution that is typical for an accumulation around lipid droplets (32).
FIG 7.

Influence of mutations in NS4B TMS2 and TMS3 on subcellular localization of NS4B. Huh7-Lunet/T7 cells were transfected with the pTM expression vector encoding NS3-5B of either wild-type (wt) sequence or containing mutations in NS4B specified at the top right of each panel. After 24 h, cells were fixed and NS4B was detected by immunofluorescence. Representative images for each condition are displayed. Magnified views of dashed-line boxed areas are shown in the lower left of each panel. The scale bars represent 25 μm.
Given the previously reported NS4B membrane remodeling capacity (33) and the requirement of NS4B self-interaction for DMV formation (16), we next investigated the influence of mutations in NS4B TMS2 and TMS3 on membrane rearrangements induced by expression of the NS3-5B viral replicase proteins. While DMVs induced by expression of wild-type NS3-5B were scattered throughout the cytoplasm of transfected cells (Fig. 8A, left) and exhibited an average diameter of about 200 nm (Fig. 8A, right, and I), NS4B mutations S121L, G125L, G129L, G143L, and G147L caused a clustering of DMVs in distinct regions of the cytoplasm and significantly decreased the average DMV diameter to about 50% (Fig. 8B to F and I). In contrast, the replication-competent NS4B mutation A151L induced DMVs with wild-type-like diameter of about 200 nm, and they were scattered throughout the cytoplasm (Fig. 8H). Of note, the significantly reduced DMV diameter phenotype exhibited by mutants S121L, G125L, G129L, G143L, and G147L (Fig. 8I) correlated with their loss of RNA replication, while the replication-competent mutant NS4B A151L induced DMVs with diameters indistinguishable from those of the wild type. In contrast, expression of the viral replicase containing the replication-inactivating G150L mutation did not induce DMVs but rather single-membrane vesicles (SMVs) that, in many instances, exhibited an apparently stalled invagination (Fig. 8G). Interestingly, similar SMVs with a horseshoe-like appearance were observed for the replication-deficient mutant G129L, albeit to a lesser extent (Fig. 8D), arguing for a concerted action of the glycine zipper motifs for DMV biogenesis.
FIG 8.

Effects of mutations in NS4B TMS2 and TMS3 on subcellular distribution and morphology of HCV-induced replication organelles. Huh7-Lunet/T7 cells were transfected with the pTM expression vector encoding NS3-5B of either the wild-type (wt) sequence or containing NS4B mutations specified at the top left of each panel. After 24 h cells were fixed, processed, and analyzed by TEM as described in Materials and Methods. (A to H) Subcellular distribution and morphology of DMVs induced by expression of wild-type NS3-5B or NS3-5B containing a mutation in NS4B as specified at the top left corner. White and black scale bars in each panel represent 500 nm and 100 nm, respectively. Localization of cellular organelles is indicated. ER, endoplasmic reticulum; LD, lipid droplet; M, mitochondrion; N, nucleus. White arrows in panels A and G refer to regular DMVs and SMVs, respectively, with stalled invaginations. (I) Quantitation of DMV diameter. The scatter plot depicts the size (diameter) of at least 500 individual DMVs from at least 10 different transfected cells per condition. Horizontal lines indicate mean values; na, not applicable. Each data set was compared to that of the wild type, and significance of differences was calculated by using the unpaired, two-tailed Student's t test (***, P < 0.0001; ns, nonsignificant).
Owing to their impact on NS4B self-interaction, we also analyzed DMV formation upon expression of the NS3-5B polyprotein containing the combination of NS4B mutations G125L in TMS2 and G150L in TMS3, since they both decreased NS4B self-interaction (Fig. 6). This allowed us to investigate DMV biogenesis for mutant NS4B protein with compromised glycine zipper motifs in both TMS2 and TMS3 simultaneously. Interestingly, this double mutant (G125L+G150L) induced vesicular structures prominently clustering around cytoplasmic lipid droplets (Fig. 9A), consistent with the distribution observed by immunofluorescence (Fig. 7). In addition to small SMVs with diameters ranging from 50 to 150 nm, larger SMVs with characteristic invaginations, comparable to those induced by mutants G129L and G150L, also were observed. In summary, we found that the glycine zipper motifs in NS4B are required for proper DMV biogenesis in three different ways: (i) for correct subcellular distribution, (ii) for correct morphology (diameter), and possibly (iii) for a transition from SMVs to DMVs as proposed for poliovirus (34).
FIG 9.

Pseudoreversions restore regular subcellular distribution and morphology of HCV replication factories of glycine zipper mutants. Huh7-Lunet/T7 cells were transfected with the NS3-5B expression vector containing either the wild type (wt) or given mutation(s) in NS4B. After 24 h cells were fixed, processed, and analyzed by TEM as described in Materials and Methods. (A) Cells transfected with the NS3-5B expression vector containing mutations G125L and G150L in NS4B exhibited prominent accumulations of small single-membrane vesicles around lipid droplets (middle), in addition to bigger single-membrane vesicles with secondary invaginations. Subcellular distribution and DMV morphology of primary mutant S121L, the psdeudorevertant S93R, and the double mutant is shown in panels B to D and for primary mutant G147A, the psdeudorevertant I140F, and the double mutant is shown in panels E to G. White and black scale bars in each panel represent 500 nm and 100 nm, respectively. (H) Quantitation of DMV diameters. The scatter plot depicts the size (diameter) of at least 500 individual DMVs from at least 10 different transfected cells per condition. Data sets for the wild-type and mutant S121L are the same as those described for Fig. 8I and are shown for comparison. Horizontal lines indicate mean values; na, not applicable. Each data set was compared to the wild type, and significance of differences was calculated by using the unpaired, two-tailed Student's t test (***, P < 0.0001; ns, nonsignificant).
Having identified pseudoreversions compensating for replication loss of NS4B mutants, we investigated these mutations for restoration of proper subcellular localization of DMVs and organelle morphology. When analyzed in the context of a wild-type replicase, pseudoreversion S93R did not affect DMV morphology (Fig. 9C) but, importantly, restored DMV morphology of the otherwise deficient S121L mutant (Fig. 9B and D). Moreover, the NS4B mutation G147A, causing alterations in subcellular distribution and size of DMVs similar to those of G147L (Fig. 9E and 8F and I), was rescued to the wild-type level by the I140F pseudoreversion (Fig. 9G), whereas it was neutral in the context of the wild-type replicase (Fig. 9F). Quantitation of DMV size revealed consistently that replication-abrogating mutations in NS4B TMS2 and TMS3 significantly decreased DMV diameters, which were restored by selected pseudoreversions (Fig. 8I and 9H), underscoring a central role of the glycine zipper motifs in NS4B transmembrane helices for the biogenesis of functional HCV replication organelles.
DISCUSSION
In this study, we provide evidence that glycine residues within NS4B TMS2 and TMS3 critically contribute to HCV RNA replication by facilitating the establishment of functional replication organelles. Notably, these glycine and small-side-chain amino acid residues are part of a (G,A,S)x3Gx2–3(G,S,T) sequence signature, which has been designated the glycine zipper motif (22). Importantly, we report that glycine zipper sequences within NS4B TMS2 and TMS3 appear to function in a fashion similar to that previously described for proteins containing these motifs: they mediate protein self-interactions via membrane-integral TMSs.
The importance of NS4B glycine zipper motifs for HCV RNA replication was demonstrated by single-amino acid substitutions and insertion mutations, which effectively blocked replication of subgenomic HCV replicons and full-length HCV. These results are consistent with an earlier report by Han and coworkers showing that leucine substitutions for glycine residues at positions 125 and 129 block HCV replication (20). Interestingly, the very low occurrence of pseudoreversions in the long-term replication/selection assay that we observed underscored the importance of glycine residues within NS4B TMS2 and TMS3 and, thus, their high conservation. Therefore, we conclude that both the properties of small-side-chain glycine residues and their 2 to 3 spacing amino acid residues appear to be required to build up glycine zipper interfaces in TMS2 and TMS3 that are indispensable for NS4B function(s) in HCV RNA replication. Furthermore, the selected second-site compensatory mutation I140F in NS4B TMS3 is consistent with its predicted helical structure, as this pseudoreversion localized 7 amino acid residues N terminal of the primary G147A mutation, corresponding to two turns of an α-helix. The occurrence of this pseudoreversion on the same side of the α-helical TMS, affecting a bulky amino acid, supported the notion of a zippering interaction mechanism involving small-side-chain amino acids and glycine residues flanked by bulky side chain amino acids, which might enable TMS interactions within the lipid bilayer. Moreover, pseudoreversion S93R, localized within the ER luminal loop between TMS1 and TNS2 (Fig. 3C), might rescue the deleterious effect of primary mutation S121L by stabilizing TMS2 positioning in the membrane via a presumably tighter interaction of the positively charged arginine residue and negatively charged phospholipid head groups. Of note, mutations conferring resistance to NS4B-specific small-molecule inhibitors frequently give rise to an arginine residue at amino acid position 94 (35, 36), suggesting an effect of NS4B inhibitors on glycine zipper-mediated functions of the protein. In support of this notion, a recent study reported decreased NS4B self-interaction upon NS4B inhibitor treatment (37). However, whether this inhibitor effect depends on glycine zipper-mediated self-interactions remains to be determined.
While all mutations targeting glycine zipper residues were required for HCV RNA replication, we identified a single mutation in each TMS2 and TMS3 that significantly decreased NS4B self-interactions. This was likely due to the fact that NS4B oligomerization is mediated by multiple determinants, previously reported to reside in the N-terminal AH2 (19), the cytoplasmic C-terminal domain (16), and in TMS2 and TMS3 (20). Hence, only mutations that severely compromised NS4B self-interactions caused significantly decreased FRET signal, while subtler alterations were not revealed by FRET but still were detrimental to HCV RNA replication. Interestingly, in the TMS2 glycine zipper motif, only the mutation targeting the central glycine residue affected NS4B interactions, while the mutation targeting the last glycine residue within the TMS3 motif decreased NS4B interactions. Similar observations have been made previously for glycine zipper motifs residing in the TMSs of glycophorin A and the atypical kinase COQ8A (21, 38). While glycine zipper-mediated TMS2-TMS2 and TMS3-TMS3 homotypic interactions occur in an intermolecular fashion, the heterotypic TMS2-TMS3 interaction occurs inter- or intramolecularly, arguing for an intricate glycine zipper-mediated architecture of NS4B TMSs. However, unraveling NS4B TMS arrangement(s) requires detailed structural information of the protein within its native membrane environment, which might be uncovered by using a combination of NS4B in vitro expression and solid-state nuclear magnetic resonance spectroscopy (39). Taken together, our data suggest novel determinants within NS4B TMSs contributing to its complex network of inter- and possibly intramolecular interactions that are indispensable for proper NS4B function(s).
In any case, the results presented here suggest a direct correlation between NS4B TMS2 and TMS3 glycine zipper-mediated induction of functional HCV replication organelles essential for viral RNA replication. Of note, DMVs induced by replication-abrogating glycine zipper mutations resembled the reduced diameter and clustered phenotype of DMVs observed upon knockdown of phosphatidylinositol 4-kinase III alpha (PI4KIIIα) and oxysterol-binding protein 1 (OSBP), which are two major HCV dependency factors (40, 41). However, the possibility that an NS4B glycine zipper-mediated activity merges into pathways that modulate (via PI4KIIIα and OSBP) the lipid composition of the HCV replication organelle remains to be determined.
Remarkably, the NS4B mutant G150L and the double mutant G125L+G150L failed to induce DMVs but instead led to the formation of SMVs, frequently exhibiting a prominent invagination. Hence, it is tempting to speculate that these structures represent arrested intermediates. In this case, HCV-induced DMVs would arise from SMVs that undergo secondary invagination, similar to what has been proposed for poliovirus (34). Such striking parallels of membrane remodeling underscore the fundamental functional conservation employed by positive-strand RNA viruses of distant phylogenetic origin to build up their membranous replication organelles.
MATERIALS AND METHODS
Antibodies.
Mouse monoclonal antibody 9E10, recognizing NS5A domain III of the HCV isolates Con1 and JFH1, was a kind gift of Charles M. Rice; the rabbit polyclonal antibody raised against NS4B has been described (42). The primary antibody recognizing human glyceraldehyde-3-phosphate dehydrogenase (GAPDH; sc-47724/0411) was purchased from Santa Cruz. Secondary horseradish peroxidase-conjugated antibodies were purchased from Sigma-Aldrich.
Cell lines.
Huh7-Lunet cells (43), Huh7-Lunet/T7 cells (44), Huh7.5 cells (45), and U2OS cells (46) were maintained in Dulbecco's modified Eagle (DMEM) medium (Invitrogen) supplemented with 2 mM l-glutamine, nonessential amino acids, 100 U/ml penicillin, 100 μg/ml streptomycin, and 10% fetal calf serum (DMEM cplt). Huh7-Lunet/T7 cells were cultured in the presence of 5 μg/ml zeocin. G418 (Geneticin; Invitrogen) was added at a final concentration of 250 or 500 μg/ml for replicon selection assays.
Plasmid constructs.
All nucleotide and amino acid numbers refer to the genotype 2a isolate JFH-1 (GenBank accession no. AB047639). Plasmids encoding the subgenomic replicons, pFK_i389LucNS3-3′_JFH_δg, and the nonreplicative mutant, pFK_i389LucNS3-3′_NS5BΔGDD_JFH_δg, have been described previously (47). Full-length Rluc-expressing HCV reporter virus carried by pFK_i389-RLuc2aCore-3′_Jc1_δg was described in reference 28. Single-amino-acid substitutions and insertions into the NS4B coding region were generated by PCR-based site-directed mutagenesis. Selectable replicons were engineered by replacing the first cistron encoding the firefly luciferase with the gene encoding neomycin phosphotransferase by using flanking AgeI and KpnI restriction sites. Plasmids used in FRET assays (pCMVJFH4B40–130-CFP, pCMVJFH4B40–130-YFP, pCMVJFH4B130–261-CFP, and pCMVJFH4B130–261-YFP) as well as control plasmids were described earlier (19). The T7-based expression plasmid encoding the HCV NS3-NS5B replicase proteins (pTM-NS3-3′_JFH) was reported previously (48). Sequence integrity of all constructs was confirmed by nucleotide sequence analysis.
In vitro transcription, RNA transfection, and virus assay. In vitro transcripts of HCV subgenomic replicons and JcR2a full-length constructs were generated, purified, and transfected into Huh7 cells as described previously (16, 49). Viral RNA replication was determined by measuring luciferase activity in cell lysates as described earlier (16, 49). Virus production was quantified by inoculating naive Huh7.5 cells with supernatants of cells harvested 72 h after transfection and measuring luciferase activity in these cells 72 h after infection (49).
FRET analysis.
NS4B interaction experiments were measured by using Foerster resonance energy transfer (FRET) acceptor bleaching as described previously (19). In brief, U2OS cells were seeded into a 12-well plate onto glass coverslips at a density of 1.5 × 105 cells per well. The next day, cells were transfected with 1 μg of each NS4B-CFP and -YFP expression constructs by using a calcium phosphate transfection kit (Clontech). After 24 h, cells were washed with phosphate-buffered saline (PBS) and fixed with 4% paraformaldehyde solution in PBS (1.8 mM KH2PO4, 5.9 mM Na2HPO4 × 2H2O, 2.7 mM KCl, 137 mM NaCl [pH 7.4]) for 10 min at room temperature. After rinsing with PBS, cells were mounted on glass slides with Fluoromount G (Southern Biotech, Birmingham, AL), and FRET measurements were carried out using a Leica SP2 confocal laser scanning microscope. FRET efficiency was determined by using the following formula: FRETeff = [(EDpost −EDpre)/EDpost] × 100, where ED represents the emitted donor fluorescence before (EDpre) and after (EDpost) photobleaching of the acceptor fluorophore. FRET efficiency was determined at two different view fields per cell by using the Leica software package RK03032000. Measurements of at least 15 cells from two independent transfections were used to calculate the box plots.
Immunoblot analysis.
Samples were lysed in radioimmunoprecipitation assay (RIPA) buffer for 30 min on ice and centrifuged for 10 min at 20,000 × g, followed by quantitation of protein concentration in the supernatant using the bicinchoninic acid (BCA) protein assay kit (Thermo Fischer) according to the recommendations of the manufacturer. Equal amounts of protein were denatured by addition of the same volume of 2× protein sample buffer (200 mM Tris [pH 8.8], 5 mM EDTA, 0.1% bromophenol blue, 10% sucrose, 3% SDS, 2% β-mercaptoethanol) and incubated for 5 min at 95°C. Proteins were separated by SDS-PAGE and electrotransferred onto nitrocellulose membranes. After blocking of the membranes with 5% nonfat milk, they were incubated with primary antibodies (NS4B, 1:1,000; NS5A 9E10, 1:10,000; GAPDH, 1:10,000) by overnight incubation at 4°C and after washing with PBS–0.1% Tween 20 with secondary horseradish peroxidase-conjugated antibodies. Membranes were developed with the Western Lightning plus ECL reagent (PerkinElmer) and imaged using an Intas ChemoCam Imager 3.2 (Intas, Göttingen, Germany).
Immunofluorescence analysis.
For indirect immunofluorescence, Huh7-Lunet/T7 cells were seeded onto glass coverslips. The next day, the cells were transfected with pTM-NS3-3′-based expression vectors by using the TransIT-LT1 transfection reagent (Mirus Bio) according to the manufacturer's instructions. After 24 h, the cells were fixed by 10 min of treatment with 4% paraformaldehyde, washed with PBS, and permeabilized by incubation with 0.3% Triton X-100 in PBS for 5 min. After washing with PBS, cells were incubated with 3% bovine serum albumin (BSA) in PBS for 30 min, followed by a 1-h treatment at room temperature in 1% BSA–PBS containing the NS4B-specific antibody (diluted 1:100). Cells were washed three times with PBS and incubated in 1% BSA–PBS containing the secondary antibody (donkey-anti-rabbit 488; diluted 1:1,000) for 1 h. After washing with PBS, nuclear DNA was stained with 4′,6-diamidino-2-phenylindole (DAPI), washed again, and mounted with Vectashield (Vector Laboratories Inc.). Images were acquired using a PerkinElmer ERS-6 spinning disc confocal microscope.
Sample preparation and ultrastructural analysis by TEM.
Huh7-Lunet/T7 cells were seeded onto glass coverslips. The next day, the cells were transfected with pTM-NS3-3′-based expression vectors by using the TransIT-LT1 transfection reagent (Mirus Bio) according to the manufacturers' instructions. After 24 h the cells were washed with PBS, fixed for 30 min with 2.5% glutaraldehyde in 50 mM sodium cacodylate buffer [pH 7.2] containing 100 mM KCl, 10 mM MgCl2, 10 mM CaCl2, and 2% sucrose, and processed for transmission electron microscopy (TEM) as described previously (16). Samples were sectioned and examined using a Zeiss EM 10 transmission electron microscope with a built-in Mega View camera (Olympus).
Selection for and identification of second-site compensatory mutations.
Huh7-Lunet cells transfected with a selectable replicon RNA were resuspended in 20 ml DMEM cplt and seeded onto two 10-cm-diameter culture dishes. After 24 h G418 was added to a final concentration of 250 or 500 μg/ml. Medium was exchanged at least twice a week until single-cell clones became visible. Two cell clones per 10-cm-diameter dish were isolated and expanded for further analyses. Total RNA of each cell clone was obtained by using the NucleoSpin RNAII kit (Macherey-Nagel) according to the instructions of the manufacturer. Five micrograms of total RNA served as the template for cDNA synthesis using oligonucleotide A/2A/9842 (5′-GGAACAGTTAGCTATGGAGTGTACC-3′) as the primer and SuperScript III (Invitrogen) as specified by the manufacturer. A region spanning NS3 to NS5A was amplified using oligonucleotide primers f_Ns3_NsiI (5′-TACCAATGAGGTCACCCTCACACAC-3′) and r_NS5A_RsrII (5′-AGGCGGAACCTGTCTCTGAGG-3′) and the Expand long template PCR system (Roche). After digestion with NsiI and RsrII, amplicons were inserted into pFK_i389LucNS3-3′_JFH_δg, and at least two DNA clones per cell clone were sequenced to confirm the presence of the primary mutation and to identify conserved pseudoreversions.
Statistical analysis.
Statistical analyses were performed as specified in the figure legends. Significance values were calculated by applying the two-tailed, unpaired Student's t test for normally distributed data sets, available in the GraphPad Prism 6 software package (GraphPad Software Inc.).
ACKNOWLEDGMENTS
We thank Marie Bartenschlager, Ulrike Herian, Stephanie Kallis, and Fredy Huschmand for excellent technical assistance. We are grateful to Anja Böckmann, University of Lyon, and Beat Meier, ETH Zurich, for their continuous support and to Hilmar Bading, Department of Neurobiology, Heidelberg University, in whose laboratory work by O.R. was carried out. We also acknowledge the permanent support provided by the University of Heidelberg Electron Microscopy Core Facility (EMCF Heidelberg). We also thank all members of the Molecular Virology unit for continuous stimulating discussions.
This work was supported by grants from the Deutsche Forschungsgemeinschaft (TRR83, TP13) to R.B. and a postdoctoral fellowship (CONICYT BECAS CHILE-POSTDOCTORADO 74150080) to O.R. D.P. acknowledges current support from EMBO LTF 2015-1380.
REFERENCES
- 1.Yamane D, McGivern DR, Masaki T, Lemon SM. 2013. Liver injury and disease pathogenesis in chronic hepatitis C. Curr Top Microbiol Immunol 369:263–288. [DOI] [PubMed] [Google Scholar]
- 2.International Committee on Taxonomy of Viruses (ICTV). 2016. The online (10th) report of the International Committee on Taxonomy of Viruses. https://talk.ictvonline.org/ictv-reports/ictv_online_report/positive-sense-rna-viruses/w/flaviviridae?Redirected=true.
- 3.Bartenschlager R, Penin F, Lohmann V, Andre P. 2011. Assembly of infectious hepatitis C virus particles. Trends Microbiol 19:95–103. doi: 10.1016/j.tim.2010.11.005. [DOI] [PubMed] [Google Scholar]
- 4.Lindenbach BD, Rice CM. 2013. The ins and outs of hepatitis C virus entry and assembly. Nat Rev Microbiol 11:688–700. doi: 10.1038/nrmicro3098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ding Q, von Schaewen M, Ploss A. 2014. The impact of hepatitis C virus entry on viral tropism. Cell Host Microbe 16:562–568. doi: 10.1016/j.chom.2014.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moradpour D, Penin F. 2013. Hepatitis C virus proteins: from structure to function. Curr Top Microbiol Immunol 369:113–142. [DOI] [PubMed] [Google Scholar]
- 7.Paul D, Madan V, Bartenschlager R. 2014. Hepatitis C virus RNA replication and assembly: living on the fat of the land. Cell Host Microbe 16:569–579. doi: 10.1016/j.chom.2014.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.den Boon JA, Ahlquist P. 2010. Organelle-like membrane compartmentalization of positive-strand RNA virus replication factories. Annu Rev Microbiol 64:241–256. doi: 10.1146/annurev.micro.112408.134012. [DOI] [PubMed] [Google Scholar]
- 9.Belov GA, van Kuppeveld FJ. 2012. (+)RNA viruses rewire cellular pathways to build replication organelles. Curr Opin Virol 2:740–747. doi: 10.1016/j.coviro.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nagy PD, Strating JR, van Kuppeveld FJ. 2016. Building viral replication organelles: close encounters of the membrane types. PLoS Pathog 12:e1005912. doi: 10.1371/journal.ppat.1005912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Paul D, Bartenschlager R. 2013. Architecture and biogenesis of plus-strand RNA virus replication factories. World J Virol 2:32–48. doi: 10.5501/wjv.v2.i2.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Paul D, Bartenschlager R. 2015. Flaviviridae replication organelles: oh, what a tangled web we weave. Annu Rev Virol 2:289–310. doi: 10.1146/annurev-virology-100114-055007. [DOI] [PubMed] [Google Scholar]
- 13.Romero-Brey I, Merz A, Chiramel A, Lee JY, Chlanda P, Haselman U, Santarella-Mellwig R, Habermann A, Hoppe S, Kallis S, Walther P, Antony C, Krijnse-Locker J, Bartenschlager R. 2012. Three-dimensional architecture and biogenesis of membrane structures associated with hepatitis C virus replication. PLoS Pathog 8:e1003056. doi: 10.1371/journal.ppat.1003056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ferraris P, Beaumont E, Uzbekov R, Brand D, Gaillard J, Blanchard E, Roingeard P. 2013. Sequential biogenesis of host cell membrane rearrangements induced by hepatitis C virus infection. Cell Mol Life Sci 70:1297–1306. doi: 10.1007/s00018-012-1213-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paul D, Hoppe S, Saher G, Krijnse-Locker J, Bartenschlager R. 2013. Morphological and biochemical characterization of the membranous hepatitis C virus replication compartment. J Virol 87:10612–10627. doi: 10.1128/JVI.01370-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Paul D, Romero-Brey I, Gouttenoire J, Stoitsova S, Krijnse-Locker J, Moradpour D, Bartenschlager R. 2011. NS4B self-interaction through conserved C-terminal elements is required for the establishment of functional hepatitis C virus replication complexes. J Virol 85:6963–6976. doi: 10.1128/JVI.00502-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Romero-Brey I, Berger C, Kallis S, Kolovou A, Paul D, Lohmann V, Bartenschlager R. 2015. NS5A domain 1 and polyprotein cleavage kinetics are critical for induction of double-membrane vesicles associated with hepatitis C virus replication. mBio 6:e00759. doi: 10.1128/mBio.00759-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gouttenoire J, Montserret R, Paul D, Castillo R, Meister S, Bartenschlager R, Penin F, Moradpour D. 2014. Aminoterminal amphipathic alpha-helix AH1 of hepatitis C virus nonstructural protein 4B possesses a dual role in RNA replication and virus production. PLoS Pathog 10:e1004501. doi: 10.1371/journal.ppat.1004501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gouttenoire J, Roingeard P, Penin F, Moradpour D. 2010. Amphipathic alpha-helix AH2 is a major determinant for the oligomerization of hepatitis C virus nonstructural protein 4B. J Virol 84:12529–12537. doi: 10.1128/JVI.01798-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Han Q, Aligo J, Manna D, Belton K, Chintapalli SV, Hong Y, Patterson RL, van Rossum DB, Konan KV. 2011. Conserved GXXXG- and S/T-like motifs in the transmembrane domains of NS4B protein are required for hepatitis C virus replication. J Virol 85:6464–6479. doi: 10.1128/JVI.02298-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Russ WP, Engelman DM. 2000. The GxxxG motif: a framework for transmembrane helix-helix association. J Mol Biol 296:911–919. doi: 10.1006/jmbi.1999.3489. [DOI] [PubMed] [Google Scholar]
- 22.Kim S, Jeon TJ, Oberai A, Yang D, Schmidt JJ, Bowie JU. 2005. Transmembrane glycine zippers: physiological and pathological roles in membrane proteins. Proc Natl Acad Sci U S A 102:14278–14283. doi: 10.1073/pnas.0501234102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mueller BK, Subramaniam S, Senes A. 2014. A frequent, GxxxG-mediated, transmembrane association motif is optimized for the formation of interhelical Calpha-H hydrogen bonds. Proc Natl Acad Sci U S A 111:E888–E895. doi: 10.1073/pnas.1319944111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gouttenoire J, Montserret R, Kennel A, Penin F, Moradpour D. 2009. An amphipathic alpha-helix at the C terminus of hepatitis C virus nonstructural protein 4B mediates membrane association. J Virol 83:11378–11384. doi: 10.1128/JVI.01122-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gouttenoire J, Castet V, Montserret R, Arora N, Raussens V, Ruysschaert JM, Diesis E, Blum HE, Penin F, Moradpour D. 2009. Identification of a novel determinant for membrane association in hepatitis C virus nonstructural protein 4B. J Virol 83:6257–6268. doi: 10.1128/JVI.02663-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lundin M, Monne M, Widell A, Von Heijne G, Persson MA. 2003. Topology of the membrane-associated hepatitis C virus protein NS4B. J Virol 77:5428–5438. doi: 10.1128/JVI.77.9.5428-5438.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gautier R, Douguet D, Antonny B, Drin G. 2008. HELIQUEST: a web server to screen sequences with specific alpha-helical properties. Bioinformatics 24:2101–2102. doi: 10.1093/bioinformatics/btn392. [DOI] [PubMed] [Google Scholar]
- 28.Poenisch M, Metz P, Blankenburg H, Ruggieri A, Lee JY, Rupp D, Rebhan I, Diederich K, Kaderali L, Domingues FS, Albrecht M, Lohmann V, Erfle H, Bartenschlager R. 2015. Identification of HNRNPK as regulator of hepatitis C virus particle production. PLoS Pathog 11:e1004573. doi: 10.1371/journal.ppat.1004573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miyanari Y, Hijikata M, Yamaji M, Hosaka M, Takahashi H, Shimotohno K. 2003. Hepatitis C virus non-structural proteins in the probable membranous compartment function in viral genome replication. J Biol Chem 278:50301–50308. doi: 10.1074/jbc.M305684200. [DOI] [PubMed] [Google Scholar]
- 30.Gosert R, Egger D, Lohmann V, Bartenschlager R, Blum HE, Bienz K, Moradpour D. 2003. Identification of the hepatitis C virus RNA replication complex in Huh-7 cells harboring subgenomic replicons. J Virol 77:5487–5492. doi: 10.1128/JVI.77.9.5487-5492.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Quinkert D, Bartenschlager R, Lohmann V. 2005. Quantitative analysis of the hepatitis C virus replication complex. J Virol 79:13594–13605. doi: 10.1128/JVI.79.21.13594-13605.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miyanari Y, Atsuzawa K, Usuda N, Watashi K, Hishiki T, Zayas M, Bartenschlager R, Wakita T, Hijikata M, Shimotohno K. 2007. The lipid droplet is an important organelle for hepatitis C virus production. Nat Cell Biol 9:1089–1097. doi: 10.1038/ncb1631. [DOI] [PubMed] [Google Scholar]
- 33.Egger D, Wolk B, Gosert R, Bianchi L, Blum HE, Moradpour D, Bienz K. 2002. Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J Virol 76:5974–5984. doi: 10.1128/JVI.76.12.5974-5984.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Belov GA, Nair V, Hansen BT, Hoyt FH, Fischer ER, Ehrenfeld E. 2012. Complex dynamic development of poliovirus membranous replication complexes. J Virol 86:302–312. doi: 10.1128/JVI.05937-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bryson PD, Cho NJ, Einav S, Lee C, Tai V, Bechtel J, Sivaraja M, Roberts C, Schmitz U, Glenn JS. 2010. A small molecule inhibits HCV replication and alters NS4B's subcellular distribution. Antiviral Res 87:1–8. doi: 10.1016/j.antiviral.2010.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shotwell JB, Baskaran S, Chong P, Creech KL, Crosby RM, Dickson H, Fang J, Garrido D, Mathis A, Maung J, Parks DJ, Pouliot JJ, Price DJ, Rai R, Seal JW III, Schmitz U, Tai VW, Thomson M, Xie M, Xiong ZZ, Peat AJ. 2012. Imidazo[1,2-a]pyridines that directly interact with hepatitis C NS4B: initial preclinical characterization. ACS Med Chem Lett 3:565–569. doi: 10.1021/ml300090x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Choi M, Lee S, Choi T, Lee C. 2013. A hepatitis C virus NS4B inhibitor suppresses viral genome replication by disrupting NS4B's dimerization/multimerization as well as its interaction with NS5A. Virus Genes 47:395–407. doi: 10.1007/s11262-013-0956-5. [DOI] [PubMed] [Google Scholar]
- 38.Khadria AS, Mueller BK, Stefely JA, Tan CH, Pagliarini DJ, Senes A. 2014. A Gly-zipper motif mediates homodimerization of the transmembrane domain of the mitochondrial kinase ADCK3. J Am Chem Soc 136:14068–14077. doi: 10.1021/ja505017f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fogeron ML, Jirasko V, Penzel S, Paul D, Montserret R, Danis C, Lacabanne D, Badillo A, Gouttenoire J, Moradpour D, Bartenschlager R, Penin F, Meier BH, Bockmann A. 2016. Cell-free expression, purification, and membrane reconstitution for NMR studies of the nonstructural protein 4B from hepatitis C virus. J Biomol NMR 65:87–98. doi: 10.1007/s10858-016-0040-2. [DOI] [PubMed] [Google Scholar]
- 40.Reiss S, Rebhan I, Backes P, Romero-Brey I, Erfle H, Matula P, Kaderali L, Poenisch M, Blankenburg H, Hiet MS, Longerich T, Diehl S, Ramirez F, Balla T, Rohr K, Kaul A, Buhler S, Pepperkok R, Lengauer T, Albrecht M, Eils R, Schirmacher P, Lohmann V, Bartenschlager R. 2011. Recruitment and activation of a lipid kinase by hepatitis C virus NS5A is essential for integrity of the membranous replication compartment. Cell Host Microbe 9:32–45. doi: 10.1016/j.chom.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang H, Perry JW, Lauring AS, Neddermann P, De Francesco R, Tai AW. 2014. Oxysterol-binding protein is a phosphatidylinositol 4-kinase effector required for HCV replication membrane integrity and cholesterol trafficking. Gastroenterology 146:1373–1385. doi: 10.1053/j.gastro.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lohmann V, Korner F, Herian U, Bartenschlager R. 1997. Biochemical properties of hepatitis C virus NS5B RNA-dependent RNA polymerase and identification of amino acid sequence motifs essential for enzymatic activity. J Virol 71:8416–8428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Friebe P, Boudet J, Simorre JP, Bartenschlager R. 2005. Kissing-loop interaction in the 3′ end of the hepatitis C virus genome essential for RNA replication. J Virol 79:380–392. doi: 10.1128/JVI.79.1.380-392.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Appel N, Pietschmann T, Bartenschlager R. 2005. Mutational analysis of hepatitis C virus nonstructural protein 5A: potential role of differential phosphorylation in RNA replication and identification of a genetically flexible domain. J Virol 79:3187–3194. doi: 10.1128/JVI.79.5.3187-3194.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Blight KJ, McKeating JA, Rice CM. 2002. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J Virol 76:13001–13014. doi: 10.1128/JVI.76.24.13001-13014.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ponten J, Saksela E. 1967. Two established in vitro cell lines from human mesenchymal tumours. Int J Cancer 2:434–447. doi: 10.1002/ijc.2910020505. [DOI] [PubMed] [Google Scholar]
- 47.Schaller T, Appel N, Koutsoudakis G, Kallis S, Lohmann V, Pietschmann T, Bartenschlager R. 2007. Analysis of hepatitis C virus superinfection exclusion by using novel fluorochrome gene-tagged viral genomes. J Virol 81:4591–4603. doi: 10.1128/JVI.02144-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Backes P, Quinkert D, Reiss S, Binder M, Zayas M, Rescher U, Gerke V, Bartenschlager R, Lohmann V. 2010. Role of annexin A2 in the production of infectious hepatitis C virus particles. J Virol 84:5775–5789. doi: 10.1128/JVI.02343-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stoeck IK, Lee JY, Tabata K, Romero-Brey I, Paul D, Schult P, Lohmann V, Kaderali L, Bartenschlager R. 18 October 2017. Hepatitis C virus replication depends on endosomal cholesterol homeostasis. J Virol doi: 10.1128/JVI.01196-17. [DOI] [PMC free article] [PubMed] [Google Scholar]