

ABSTRACT
Restriction factors are intrinsic cellular defense proteins that have evolved to block microbial infections. Retroviruses such as HIV-1 are restricted by TRIM5 proteins, which recognize the viral capsid shell that surrounds, organizes, and protects the viral genome. TRIM5α uses a SPRY domain to bind capsids with low intrinsic affinity (KD of >1 mM) and therefore requires higher-order assembly into a hexagonal lattice to generate sufficient avidity for productive capsid recognition. TRIMCyp, on the other hand, binds HIV-1 capsids through a cyclophilin A domain, which has a well-defined binding site and higher affinity (KD of ∼10 μM) for isolated capsid subunits. Therefore, it has been argued that TRIMCyp proteins have dispensed with the need for higher-order assembly to function as antiviral factors. Here, we show that, consistent with its high degree of sequence similarity with TRIM5α, the TRIMCyp B-box 2 domain shares the same ability to self-associate and facilitate assembly of a TRIMCyp hexagonal lattice that can wrap about the HIV-1 capsid. We also show that under stringent experimental conditions, TRIMCyp-mediated restriction of HIV-1 is indeed dependent on higher-order assembly. Both forms of TRIM5 therefore use the same mechanism of avidity-driven capsid pattern recognition.
IMPORTANCE Rhesus macaques and owl monkeys are highly resistant to HIV-1 infection due to the activity of TRIM5 restriction factors. The rhesus macaque TRIM5α protein blocks HIV-1 through a mechanism that requires self-assembly of a hexagonal TRIM5α lattice around the invading viral core. Lattice assembly amplifies very weak interactions between the TRIM5α SPRY domain and the HIV-1 capsid. Assembly also promotes dimerization of the TRIM5α RING E3 ligase domain, resulting in synthesis of polyubiquitin chains that mediate downstream steps of restriction. In contrast to rhesus TRIM5α, the owl monkey TRIM5 homolog, TRIMCyp, binds isolated HIV-1 CA subunits much more tightly through its cyclophilin A domain and therefore was thought to act independently of higher-order assembly. Here, we show that TRIMCyp shares the assembly properties of TRIM5α and that both forms of TRIM5 use the same mechanism of hexagonal lattice formation to promote viral recognition and restriction.
KEYWORDS: pattern recognition, restriction factor, retrovirus
INTRODUCTION
TRIM5α and TRIMCyp proteins, here collectively called TRIM5 proteins, are restriction factors that intercept the incoming core particles of retroviruses such as HIV-1 and prevent infection by accelerating core dissociation and inhibiting reverse transcription (reviewed in references 1 and 2). TRIM5 proteins initiate restriction by recognizing the hexagonal capsid lattice that forms the outer shell of the retroviral core particle. Capsid binding is mediated by the TRIM5 C-terminal domains, which come in two forms: TRIM5α contains a SPRY domain (also known as PRYSPRY or B30.2), whereas TRIMCyp contains a cyclophilin A domain (Fig. 1A) (3–12). The two capsid-binding domains differ substantially in their intrinsic affinities for the individual CA subunits that comprise retroviral capsids. TRIM5α SPRY domains exhibit extremely weak pairwise affinities for isolated CA monomers (estimated dissociation constant [KD] of >1 mM) (7, 11, 13, 14). The SPRY domain binding site(s) on the capsid surface is also poorly defined: susceptibility and resistance determinants map to large swaths of the capsid surface (15–23), and single SPRY domains appear to contact multiple, adjacent CA subunits (14, 24, 25). Measurable interactions have been observed between the isolated SPRY domain of rhesus TRIM5α and a disulfide-stabilized HIV-1 CA hexamer, but even with this hexavalent CA assembly the SPRY binding affinity remains in the millimolar range (24).
FIG 1.

Primary sequences of TRIM5 proteins. (A) Schematics of the domain organization of TRIM5α and TRIMCyp. (B) Alignment of owl monkey TRIMCyp and rhesus monkey TRIM5α. Identical residues in the RBCC regions of the two proteins are shaded gray. The N-terminal boundaries of the C-terminal capsid-binding domains are indicated. For reference, zinc-coordinating residues in the RING and B-box 2 domains are colored in red and blue, respectively. (C) Alignment of the miniTRIMCyp and miniTRIM5α constructs. The exogenous coiled-coil hairpin linker is colored gray. The N-terminal boundary of the RING-less Bcc constructs is indicated by the black triangle. Black dots mark residues found within the trimerization interface. Asterisks indicate residues in the hairpin linker that were mutated to cysteines to facilitate crystallization of miniTRIMCyp.
Owing to the extremely weak intrinsic affinity of the SPRY domain for CA, TRIM5α requires an affinity amplification mechanism, provided by avidity, to recognize retroviral capsids and restrict viral replication efficiently. Avidity gains come from dimerization and higher-order assembly, which are mediated by the RBCC domains (RING, B-box, and coiled-coil; also called the tripartite motif) that precede the capsid-binding domains (11, 26–31). The TRIM5α coiled-coil domain makes an elongated, antiparallel dimer that displays two SPRY domains for simultaneous binding, likely in a close-packed configuration (29–33). The avidity generated by dimerization is sufficient to allow detection of capsid binding in vitro but not restriction in cells (11, 28, 34, 35). Full-affinity capsid binding and efficient restriction require additional higher-order interactions that are mediated by the B-box 2 domain, which forms trimers that connect the coiled-coil-mediated dimers into an extended hexagonal net (26, 27, 35, 36). The hexagonal TRIM net wraps about the capsid and arranges the associated SPRY domains to match the arrangement of the CA subunits on the capsid surface, thereby generating powerful avidity effects (28, 37). The N-terminal RING domain of TRIM5α does not appear to be required for capsid recognition, although it may contribute lattice-stabilizing interactions (38) and is required to synthesize the polyubiquitin chains that mediate subsequent steps of restriction (39–43).
In contrast to TRIM5α, TRIMCyp binds the HIV-1 capsid using a cyclophilin A domain, which binds relatively tightly to an exposed loop on the N-terminal lobe of the CA protein (KD of ∼10 μM) (44–48). Consistent with its higher intrinsic affinity for isolated CA subunits, TRIMCyp's restriction activity is reported to be independent of B-box 2 domain-mediated interactions and instead to simply require dimerization via the coiled coil (49–51). Indeed, artificial capsid-binding restriction factors have been created by appending exogenous dimerization motifs to the monomeric cyclophilin A protein (52, 53).
Despite the differences in their capsid-binding domains, the RBCC motifs of TRIM5α and TRIMCyp proteins share significant sequence similarity, with 74% amino acid identity between the prototypical isoforms, TRIM5α from rhesus monkey and TRIMCyp from owl monkey (Fig. 1B). This degree of sequence conservation implies conservation of function, and indeed, purified owl monkey TRIMCyp was recently shown to assemble into hexagonal lattices in vitro, similar to the rhesus TRIM5α protein (37). Here, we have analyzed in detail how higher-order assembly affects TRIMCyp recognition and restriction of HIV-1 capsids. We show that the TRIMCyp B-box 2 domain shares the propensity to self-associate and crystallizes as a trimer that is similar in structure to the TRIM5α B-box trimer. We further show that TRIMCyp-mediated restriction is enhanced by these B-box-mediated interactions under stringent experimental conditions.
RESULTS
Design and biophysical characterization of miniTRIMCyp proteins.
We previously described the design and characterization of “miniTRIM” proteins, which revealed the molecular details of how the B-box 2 domain of TRIM5α associates into trimers that form the vertices of the TRIM5α hexagonal lattice (35). The miniTRIM construction allowed us to examine B-box-mediated interactions while avoiding biochemical complications arising from higher-order assembly (seen in longer recombinant constructs) or aggregation (shorter constructs). We have used the same strategy to characterize the self-association behavior of the TRIMCyp B-box 2 domain. Briefly, an RBcc construct, encoding the RING and B-box regions of owl monkey TRIMCyp and the first 28 residues of the coiled coil (residues 1 to 158), was fused to the terminal residues of the coiled coil (residues 224 to 264), with an intervening hairpin sequence from a bacterial seryl-tRNA synthetase (35). To facilitate crystallization, a second miniTRIM, denoted Bcc, was created by deleting the RING domain sequence (residues 1 to 87) (Fig. 1C). As with their TRIM5α counterparts, the miniTRIMCyp constructs purified readily and behaved well in solution.
We and others have previously shown that the TRIM5α B-box 2 domain has a plastic oligomerization interface that can mediate both dimerization and trimerization (35, 36, 54). In the context of the mini-coiled coils, the TRIM5α B-box is predominantly dimeric at micromolar concentrations (35), even though it is the trimer form that facilitates hexagonal lattice assembly together with the native coiled-coil domain. Similarly, our analyses by using SEC-MALS (size exclusion chromatography coupled with multiangle light scattering) indicated that both the RBcc and Bcc miniTRIMCyp constructs could dimerize in solution. Importantly, however, the dimerization affinities of the miniTRIMCyp B-box constructs were significantly weaker than their miniTRIM5α counterparts, as evidenced by the relative proportions of monomer and dimer peaks (compare blue curves in Fig. 2A, B, C, and D).
FIG 2.
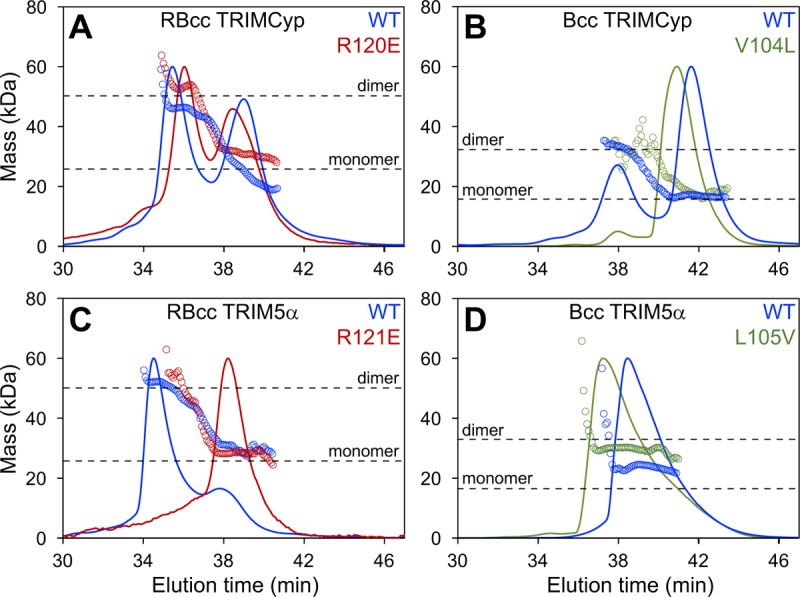
SEC-MALS analyses of miniTRIM proteins. Shown are profiles of the indicated constructs of miniTRIMCyp (A and B) and miniTRIM5α (C and D). All proteins were injected at the same concentration (100 μM), so that the population distributions can be directly compared. Curves show the UV trace, and open circles indicate the population-averaged molecular masses of the eluting species. Horizontal dashed lines indicate the expected monomer and dimer molecular masses for each construct.
We also analyzed oligomerization of the miniTRIM constructs by sedimentation velocity analytical centrifugation, which measures the rates at which the proteins move in solution in response to centrifugal force. Diffusion-corrected, model-independent distribution plots of the miniTRIM sedimentation distributions, obtained from van Holde-Weishet analyses (see Materials and Methods for details), had rising half-parabolic appearances, which are characteristic of proteins undergoing a reversible two-state equilibrium (Fig. 3A and B). However, plots of the RBcc constructs deviated from the parabolic shape at higher sedimentation coefficient values, indicating the presence of higher-molecular-weight species (Fig. 3A). Accordingly, parametrically constrained spectrum analysis (PCSA) revealed that the two major components of each of the miniTRIM samples were likely to be monomeric and dimeric species (Fig. 3C and D). Minor higher-order species were also present in the PCSA plots for the RBcc constructs (Fig. 3C, asterisks); these two samples were prone to aggregation, however, and the biological significance of the additional higher-order peaks therefore is unclear. In contrast, the Bcc distributions appeared more cleanly two state (Fig. 3D), and we therefore performed finite element analysis fitting of the raw data (see Materials and Methods). Both the miniTRIMCyp and miniTRIM5α Bcc data gave excellent fits to simple monomer-dimer equilibrium models, with close overlays and low root-mean-squared deviations between the raw data (black traces) and fitted curves (red traces) and small random residuals (Fig. 3E and F and Table 1). This analysis revealed that Bcc miniTRIM5α dimerized with a KD of 1.6 μM (Table 1), which is consistent with our previous measurements (35), whereas the Bcc miniTRIMCyp affinity was 10-fold lower (KD of 18.1 μM). Our centrifugation data were therefore in excellent agreement with the SEC-MALS data and confirmed that the B-box 2 domain of TRIMCyp self-associates more weakly than the equivalent domain of TRIM5α.
FIG 3.

Velocity sedimentation analyses of miniTRIM proteins. (A and B) Model-free van Holde-Weischet analysis of the indicated RBcc (A) and Bcc (B) miniTRIMs. (C and D) Size distribution plots derived from PCSA and Monte Carlo analysis of the RBcc (C) and Bcc (D) data sets revealed two major populations corresponding to monomer and dimer. Minor, higher-order species (asterisks) are also present in the RBcc plots. (E and F) Finite element fitting of the Bcc scans. (Upper) Raw scans (black curves) with overlaid fits (red curves). Only a subset out of more than 300 scans are shown, and they are spaced for clarity. (Lower) Residual differences between the real and simulated data.
TABLE 1.
Summary of finite element fitting results for the Bcc miniTRIM sedimentation data
| Construct | Fit RMSD | KD (μM) | Loading concn (μM) | Partial specific vol (ml/g) | Frictional ratio, f/f0 |
Sedimentation coefficient (Svedberg units) |
||
|---|---|---|---|---|---|---|---|---|
| Monomer | Dimer | Monomer | Dimer | |||||
| miniTRIMCyp | 0.0034 | 18.14 | 63.32 | 0.7336 | 1.36 | 1.56 | 1.67 | 2.32 |
| miniTRIM5α | 0.0042 | 1.55 | 88.36 | 0.7444 | 1.23 | 1.30 | 1.78 | 2.68 |
Contribution of the RING domain to miniTRIM oligomerization.
Unlike the miniTRIM5α protein, the RING domain of miniTRIMCyp appeared to contribute significantly to self-association. We compared the relative importance of the RING domains by mutating an arginine residue (Arg120 in TRIMCyp and Arg121 in TRIM5α) that is critical for B-box–B-box interactions (26–28, 35, 36, 54). By SEC-MALS, the RBcc miniTRIM5α R121E mutant eluted primarily as a monomer (Fig. 2C, red), demonstrating that dimerization of miniTRIM5α occurred principally through the B-box 2 domain, as observed previously (35). In contrast, the elution profile of the RBcc miniTRIMCyp R120E mutant was similar to that of the wild-type (WT) construct, i.e., the relative fractions of monomer and dimer remained similar (compare red and blue curves in Fig. 2A). Thus, the RING domain of TRIMCyp dimerized more readily than the RING domain of TRIM5α, whereas the reverse was true of the corresponding B-box 2 domains.
RING domain dimerization is required for enzymatic activity of TRIM proteins, because the dimer facilitates interactions with ubiquitin (Ub)-loaded E2 enzymes and reorganization of the E2 active site, thereby positioning the ubiquitin moiety for efficient conjugation to acceptor lysines (41, 55, 56). The TRIM5α RING domain dimerizes inefficiently, and B-box–B-box interactions therefore are apparently required for the formation of the RING dimer in the context of the full-length TRIM5α (41). In contrast, our experiments described above suggested that efficient TRIMCyp RING dimerization reduces the need for B-box-mediated self-association. To test these models directly, we compared the relative B-box dependence of TRIM5α and TRIMCyp RING ubiquitination activities in vitro. The intrinsic ability of RING E3 ligases to stimulate ubiquitination can be measured by a “ubiquitin discharge” assay, in which ubiquitin is transferred from an activated E2∼Ub complex onto free lysine (57) (Fig. 4). We found that, as expected, the isolated TRIM5α RING domain discharged ubiquitin at a significantly lower rate than the RBcc miniTRIM5α construct that contained both the RING and B-box 2 domains (Fig. 4B, compare orange and blue curves). This result confirmed that TRIM5α RING dimerization is promoted by the B-box and that the B-box therefore is required for optimal ubiquitin transfer. In contrast, the isolated TRIMCyp RING and RBcc miniTRIMCyp proteins stimulated ubiquitin discharge to equivalent degrees in this assay (Fig. 4D). This equivalence is consistent with the SEC-MALS data showing that the RBcc miniTRIMCyp dimerizes efficiently, even with a mutated B-box 2 domain that is impaired in self-association (Fig. 2A). Thus, as expected, the in vitro E3 ligase activity of miniTRIM5α was more dependent on the presence of its B-box than was miniTRIMCyp.
FIG 4.

Comparison of in vitro ubiquitin discharge activities of RING and miniTRIM RBcc constructs. (A and C) Representative Coomassie-stained gels showing release of free UbcH5b (E2) and free ubiquitin (Ub) on incubation of the E2∼Ub complex with excess free lysine, in the absence of E3 (boxed in black) or in the presence of the indicated RING (orange) and miniTRIM proteins (blue). (B and D) Plot of relative densitometry values of the E2∼Ub bands during the course of the discharge assays. Experiments were performed in triplicate, and error bars indicate one standard deviation from the average values. No E3 control, black; RING domains alone, orange; miniTRIM proteins, blue.
Bcc miniTRIMCyp crystallizes as a trimer.
Our previous crystallization screens of miniTRIM5α yielded a large number of hits, consistent with its high propensity to oligomerize (35). In contrast, extensive screening of miniTRIMCyp with various precipitants failed to yield crystals, consistent with the weaker self-association propensity of the TRIMCyp B-box 2 domain. However, we were able to crystallize a Bcc miniTRIMCyp construct that contained two engineered cysteines in the coiled-coil hairpin linker (indicated by asterisks in Fig. 1C). These cysteines were designed to stabilize the hairpin by forming a disulfide bond, but instead they promoted crystallization by coordinating an extra zinc atom that facilitated crystal lattice formation. Importantly, this crystal contact did not perturb the tertiary folds of the B-box and coiled-coil domains and was well removed from the symmetric B-box–B-box contacts. The structure was refined to R/Rfree values of 0.23/0.29 at 2.3-Å resolution (Fig. 5A). Statistics for the Bcc miniTRIMCyp crystal structure are summarized in Table 2.
FIG 5.
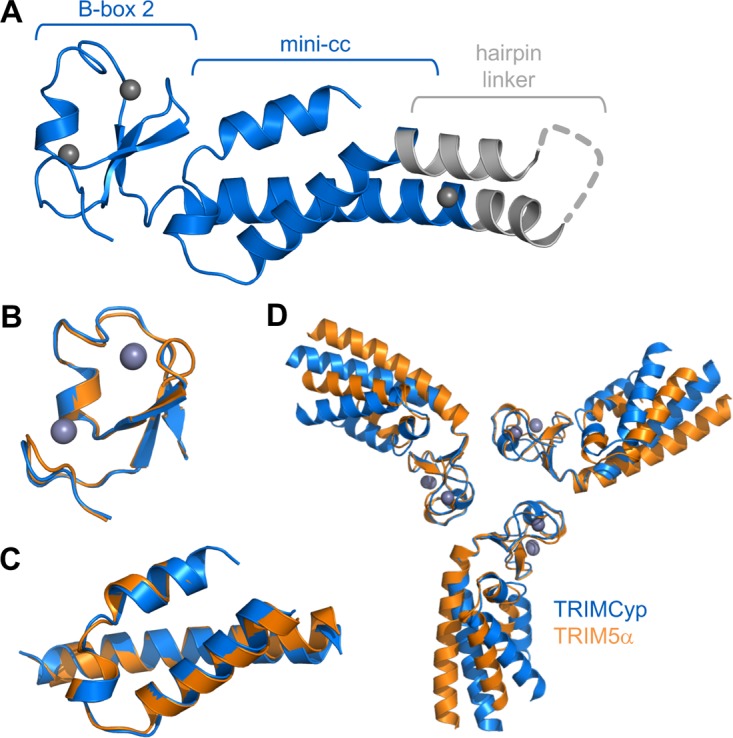
MiniTRIMCyp and miniTRIM5α Bcc constructs have similar structures. (A) Crystal structure of Bcc miniTRIMCyp. TRIMCyp-derived regions are colored blue, and the exogenous hairpin linker is gray. Unmodeled residues are represented as a gray dashed line. Zinc atoms are shown as charcoal gray spheres. (B) Superposition of the TRIMCyp (blue) and TRIM5α (orange) B-box 2 domains. (C) Superposition of the coiled-coil domains. (D) Superposition of the B-box 2 domains as trimeric units.
TABLE 2.
Structure statistics
| Parameter | Value |
|---|---|
| Data collection statistics | |
| Beamline | APS 22ID |
| Wavelength (Å) | 1.000 |
| Space group | I23 |
| Unit cell | a = b = c = 104.6 Å, α = β = γ = 90° |
| Resolution range, Å | 50–2.30 (2.38–2.30)a |
| Rsym/Rmeas/Rpim | 0.13 (1.00)/0.14 (1.00)/0.05 (0.68) |
| Mean I/σ<I> | 12.3 (1.1) |
| Completeness, % | 99.6 (100.0) |
| Avg redundancy | 10.6 (8.5) |
| Mosaicity range, ° | 0.47–0.90 |
| Wilson B-factor, Å2 | 74.2 |
| Refinement statistics | |
| Resolution range | 27.9–2.31 (2.45–2.31) |
| No. of unique reflections | 8,544 (1,281) |
| No. of reflections in free set | 862 (144) |
| Rwork | 0.23 (0.31) |
| Rfree | 0.29 (0.35) |
| No. of nonhydrogen atoms | |
| Protein and zinc | 985 |
| Solvent | 25 |
| Avg B-factor, Å2 | |
| Protein and zinc | 90.3 |
| Solvent | 86.9 |
| Coordinate deviations | |
| Bond lengths, Å | 0.008 |
| Bond angles, ° | 0.375 |
| Ramachandran plot | |
| Favored, % | 99 |
| Outliers, % | 0 |
| MolProbity clash score | 0.51 |
| PDB entry | 5VA4 |
Values in parentheses are for the highest-resolution shell.
Consistent with their high degree of sequence similarity, the miniTRIMCyp and miniTRIM5α monomers were very similar and superimposed with root-mean-square deviations of 0.44 Å (B-box 2 residues) (Fig. 5B) and 0.52 Å (coiled-coil residues) (Fig. 5C). Although miniTRIMCyp monomers and dimers predominated in solution at micromolar concentrations, the Bcc protein crystallized as a symmetric, triskelion-shaped trimer in which the B-box 2 domains form a trimeric vertex and the coiled coils emanate as spokes (Fig. 5D). This structure is very similar to the trimer observed previously for miniTRIM5α (Fig. 5D and compare Fig. 6A and B). Both trimers are stabilized by three layers of interactions, with a central hydrophobic layer centered around a ring of three aromatic residues (Trp116 in TRIMCyp and Trp117 in TRIM5α) sandwiched between two layers of polar interactions (Fig. 6, central panels). The interfacial residues are identical between the miniTRIMCyp and miniTRIM5α trimers, with the exception of Val104 in TRIMCyp, which is Leu105 in TRIM5α. We hypothesized that this single amino acid change is sufficient to explain the differences in the self-association propensities of the different miniTRIMs. This simple model was not supported by mutational and oligomerization analyses, however, because the profiles of both the V104L miniTRIMCyp and L105V miniTRIM5α mutants were similar to their corresponding wild-type constructs in SEC-MALS experiments (Fig. 2B and D, green curves).
FIG 6.

Comparison of the miniTRIMCyp trimer (A) and the miniTRIM5α trimer (33) (B). The protein chains are rendered as ribbons, with each subunit in a different color. The exogenous linker regions are colored gray. Zinc atoms are shown as charcoal gray spheres. Boxed panels show details of the three layers of interactions. Sidechains that mediate trimerization contacts are shown as sticks and labeled. Hydrogen bonds are shown as dashed lines.
The TRIMCyp and TRIM5α B-box vertices superimposed very well as trimeric units (0.75 Å over equivalent Cα atoms) (Fig. 5D). However, the trajectories of the coiled-coil spokes differed significantly between the two structures. We observed similar variations in interdomain orientations within the ensemble of our previous TRIM5α structures and proposed that these variations represent a flexion mechanism that allows the hexagonal lattice to accommodate the surface curvature of retroviral capsids (35). In the present case, the miniTRIMCyp trimer structure contains only a single molecule per asymmetric unit, but we presume that this protein also can exhibit similar coiled-coil flexion.
Purified TRIMCyp and HIV-1 CA proteins coassemble in vitro.
Our finding that the TRIMCyp B-box 2 domain can associate into trimers supports the idea that both forms of TRIM5 make similar hexagonal lattices that facilitate capsid binding. Indeed, pure recombinant TRIM5α and TRIMCyp proteins both can form flat hexagonal lattices in vitro under either spontaneous assembly conditions or atop templating two-dimensional HIV-1 CA crystals (37). Real retroviral capsids present curved binding surfaces, however, so we also tested the ability of TRIMCyp to form hexagonal lattices on the surface of HIV-1 CA tubes using a CA/TRIM coassembly assay (35, 37). Purified HIV-1 CA and TRIMCyp were coincubated under conditions in which neither protein assembled alone. The samples then were visualized by negative-stain electron microscopy, which revealed the presence of assembled CA tubes that were extensively decorated with TRIMCyp (Fig. 7). The negative-stain images of TRIMCyp-decorated tubes appeared very similar to equivalent negative-stain images of TRIM5α-decorated tubes that we previously described (35, 37). Repeating units were observed on the exterior tube surfaces (white arrows), consistent with an ordered, lattice-like arrangement of TRIMCyp, and in some cases apparent hexagonal shapes could be discerned (red arrowheads). These results, taken together with those of previous studies (37), indicate that like TRIM5α, TRIMCyp can form a deformable hexagonal lattice that could surround a viral capsid.
FIG 7.
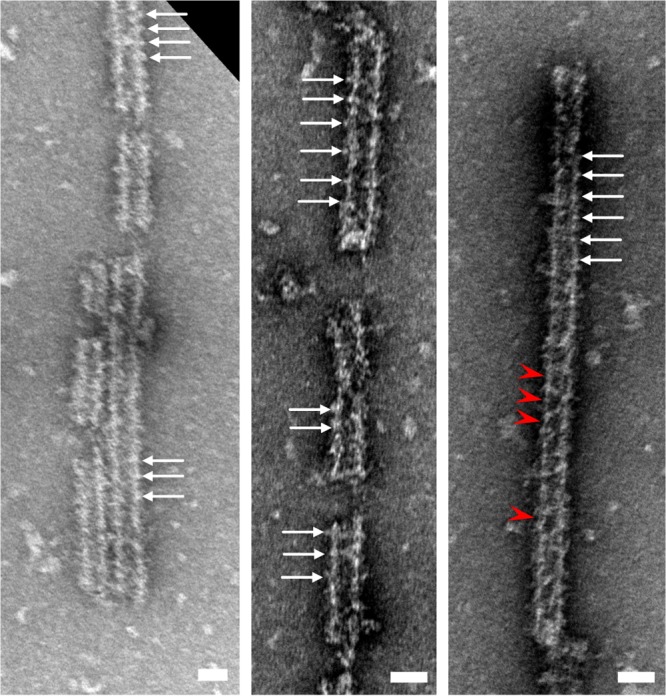
Negative-stain electron micrographs of in vitro-assembled complexes of purified TRIMCyp and HIV-1 CA proteins. White arrows denote regularly spaced surface features indicating that the TRIM proteins are assembled around the CA tubes. Red arrowheads indicate apparent rings. Scale bars, 50 nm.
Restriction activities of TRIMCyp B-box 2 mutants.
We next tested whether TRIMCyp restriction of HIV-1 replication is dependent on the B-box–B-box interactions we observed in vitro. For these experiments, HeLa cells were transduced with a lentiviral vector that expressed owl monkey TRIMCyp and then were infected with increasing levels of a single-cycle reporter HIV-green fluorescent protein (GFP) virus. The TRIMCyp transcripts also encoded DsRed under the control of an internal ribosome entry site, which allowed us to quantify viral infectivity selectively in cells that expressed TRIMCyp. Consistent with previous studies (51), we found that TRIMCyp proteins that harbored the W116A, W116E, R120A, and R120E mutations were just as restrictive as the wild-type protein, implying that TRIMCyp restriction was independent of B-box 2-mediated interactions under these relatively high protein expression conditions (Fig. 8A). This result was in stark contrast to TRIM5α, where the equivalent mutations have been shown to severely disrupt or completely abolish restriction activity (26, 27, 35). It was also noteworthy that while wild-type TRIMCyp restricted well at low expression levels, the steady-state protein levels of all of the restricting TRIMCyp mutants were significantly elevated (by at least 10-fold) relative to wild-type protein (compare lanes 2 to 5 to lane 1 in the immunoblot shown in Fig. 8A). Similar increases in steady-state protein levels were previously observed for analogous TRIM5α constructs and were attributed to accelerated turnover of assembled (versus free) TRIM5 subunits (35, 51).
FIG 8.

Restriction activities of FLAG/Strep-tagged owl monkey TRIMCyp proteins expressed in HeLa cells. (A) GFP-labeled HIV-1 vector (HIV-GFP) efficiently transduced cells that did not express exogenous TRIMCyp (no TRIM), whereas transduction was significantly inhibited in cells that expressed wild-type (WT) TRIMCyp or the indicated B-box 2 domain mutants. (B) Cells were transduced with HIV-GFP (MOI, 4) in the presence of the indicated concentrations of CsA. (C) Restriction activities of mutant and WT omTRIM-rhSPRY, a chimeric protein that contained the RBCC domains of owl monkey TRIMCyp and the SPRY domain of rhesus TRIM5α. Native rhesus TRIM5α (rhTRIM5α) was used as a positive control for restriction. Western blots (anti-FLAG) in all panels indicate TRIM protein expression levels, with GAPDH used as a loading control. Note that steady-state levels of the wild-type proteins were very low, as observed previously for TRIM5α (35, 51). Experiments were performed three times independently, with similar results.
We reasoned that the strong restriction and apparent B-box independence of TRIMCyp might reflect the relatively high intrinsic affinity of the cyclophilin domain for individual HIV-1 CA subunits (and perhaps compounded further by the elevated mutant protein levels). We therefore repeated the restriction assays with a single dose of HIV-GFP but in the presence of increasing concentrations (0 to 5 μM) of cyclosporine (CsA), which binds cyclophilin A with high affinity and competitively inhibits CA binding (58). As expected, the restriction activity in cells that expressed wild-type TRIMCyp decreased with increasing CsA levels, with full restriction at 0 μM CsA and no restriction at 5 μM (Fig. 8B, filled squares, red). Importantly, under these more stringent conditions, the TRIMCyp B-box 2 mutants lost activity at lower CsA concentrations than wild-type TRIMCyp, with complete loss of restriction occurring at ∼2.5 μM CsA for the mutants (Fig. 8B, blue curves). This enhanced restriction attenuation was particularly notable because the mutant TRIMCyp protein levels still expressed at much higher levels than wild-type TRIMCyp (Fig. 8B, immunoblot). In control experiments, cells that did not express TRIMCyp were infected equally well by HIV-GFP irrespective of CsA concentrations (Fig. 8B, filled diamonds, green). These experiments demonstrated that TRIMCyp restriction becomes B-box dependent under conditions where strong capsid binding is limiting.
The TRIMCyp RBCC motif supports capsid recognition and restriction by the SPRY domain.
To confirm that the TRIMCyp RBCC motif can undergo higher-order assembly in cells, we generated and analyzed a chimeric protein (called omTRIM-rhSPRY) that contained the tripartite motif of owl monkey TRIMCyp (residues 1 to 299) and the SPRY domain of rhesus TRIM5α (residues 301 to 495). This chimeric protein restricted HIV vector transduction (Fig. 8C, closed squares, red), albeit less efficiently than native rhesus TRIM5α (open squares, red). We speculate that the weaker restriction activity of omTRIM-rhSPRY reflects the weaker self-association propensity of the TRIMCyp B-box 2 domain and/or suboptimal packing of the TRIM5α SPRY domain against the TRIMCyp coiled-coil domain. Regardless, we found that the chimera clearly required homomeric B-box interactions for restriction, because the omTRIM-rhSPRY W116A, W116E, R120A, and R120E mutants all failed to restrict (Fig. 8C, blue curves), again despite substantially elevated expression levels (Fig. 8C, immunoblot). These results indicated that the TRIMCyp RBCC motif can provide sufficient avidity gains to support productive capsid recognition by the SPRY domain in cells.
DISCUSSION
Because the cyclophilin domain has a substantially higher affinity for the HIV-1 CA protein than the SPRY domain, it had been thought that productive capsid recognition by TRIMCyp did not require the affinity amplification mechanism used by TRIM5α. However, the significant sequence similarity between the TRIM5α and TRIMCyp tripartite motifs suggested to us that these proteins share the ability to assemble into hexagonal lattices. Our previous (37) and current studies support this hypothesis. Specifically, we find that the TRIMCyp B-box 2 domain can both dimerize and trimerize and that the trimer facilitates assembly in vitro, much as we and others have previously shown for TRIM5α (35, 36, 54). Importantly, the owl monkey TRIMCyp RBCC domains can functionally replace the equivalent region of rhesus TRIM5α, and the restriction activity of the chimeric construct is disrupted by the same B-box 2 mutations that disrupt the activity of native TRIM5α (Fig. 8C). Since the SPRY domain requires higher-order assembly to bind and restrict retroviral capsids efficiently (11, 26, 27, 35), we conclude that the TRIMCyp B-box 2 domain also mediates higher-order assembly of native TRIMCyp in cells. Moreover, we have shown that this ability to self-assemble plays an important role in restriction when the TRIMCyp-CA interaction is limiting (Fig. 8B).
How might TRIMCyp assembly facilitate restriction? In the case of TRIM5α, hexagonal lattice formation not only facilitates capsid binding but also promotes the effector functions of the RING domain, because E3 ligase activation requires RING dimerization (41). Higher-order assembly of TRIM5α therefore serves to couple the essential step of capsid recognition with subsequent, ubiquitin-dependent steps of restriction. Although many studies have shown that loss of viral infectivity caused by TRIM5 proteins is only partially RING domain dependent (3, 50, 59–62), there is also good evidence that both TRIM5-mediated inhibition of reverse transcription and accelerated capsid dissociation are, indeed, ubiquitin-dependent aspects of the restriction mechanism (40, 42, 43, 50, 63). We propose that higher-order assembly of TRIMCyp serves the same function of coupling capsid binding and ubiquitination. The resulting avidity effects are likely to significantly contribute to recognition efficiency, particularly under conditions where TRIMCyp concentrations are limiting, as might be expected at early time points of a natural infection. Interestingly, we observed that TRIMCyp RING activation is less dependent on its B-box than TRIM5α, at least in vitro. We speculate that the greater self-association propensity of the TRIMCyp RING compensates for its weaker B-box and still allows for efficient enzymatic activation in cells.
Comparison of the underlying energetics for TRIM5α versus TRIMCyp proteins in capsid binding and lattice formation reveals an interesting reciprocal relationship that can be explained by evolutionary pressure to recognize and signal the presence of an invading retroviral capsid efficiently yet avoid signaling in the absence of an invader. Specifically, we observe an inverse correlation between the intrinsic CA-binding affinities of the TRIM5 C-terminal domains and the self-association affinities of their corresponding B-box 2 domains. We therefore suggest that TRIM5 self-assembly is “tuned” to be as weak as possible, perhaps because higher-order assembly in the absence of a capsid template is likely to be harmful to the cell. In this scenario, relatively strong B-box–B-box interactions would favor capsid recognition but also promote spontaneous assembly of TRIM5 subunits that are not bound to retroviral capsids, which could then lead to spontaneous polyubiquitination and innate immune signaling in the absence of infection. Our observations also support the idea that cells turn over these spontaneously assembled TRIM5 proteins much more rapidly than nonassembled TRIM5 subunits, because TRIM5 B-box mutants that lack assembly activity consistently accumulate to much higher cellular concentrations than their wild-type counterparts (35, 51, and this study). Efficient disposal of such assemblies may have evolved to prevent the deleterious effects of nonvirally triggered signaling. Recent studies have indicated that TRIM5 assemblies are turned over by autophagy (64, 65), which we speculate is operative only when large, repeating arrays of TRIM5 subunits are recognized by the autophagic initiators.
In summary, our studies show that TRIM5α and TRIMCyp share the inherent capacity to form higher-order assemblies through their tripartite or RBCC motifs. We therefore conclude that the hexagonal lattice model of retroviral pattern recognition applies to both forms of TRIM5.
MATERIALS AND METHODS
MiniTRIMCyp construction, expression, and purification.
Synthetic DNA (GenScript, Inc.) encoding the protein sequence shown in Fig. 1C was subcloned into a pET (Novagen) plasmid with a His6-SUMO leader sequence. From this plasmid, the RING-less construct was generated by using QuikChange mutagenesis (Agilent) to remove TRIMCyp residues 1 to 87. All other mutations were also introduced by this method.
MiniTRIMCyp constructs were expressed and purified by following the same method as that described previously for miniTRIM5α (35), with a few modifications. E. coli cells were lysed in 2× lysis buffer [100 mM Tris, pH 8.5, 200 mM NaCl, 10% glycerol, 1% Triton X-100, 2 mM Tris(2-carboxyethyl)phosphine (TCEP), 2 mM phenylmethylsulfonyl fluoride]. After lysis using a microfluidizer, an equal volume of ice-cold water was added to bring the lysis buffer to 1×. The protein was purified by affinity chromatography using nickel agarose as before, except that buffers contained 200 mM NaCl instead of LiCl. The His6-SUMO tag was cleaved with Ulp1 protease and removed by incubation with nickel agarose. The unbound fractions containing untagged miniTRIMCyp in 1× sample buffer (25 mM Tris, pH 8, 100 mM NaCl, 2.5% glycerol, 1 mM TCEP) were applied to HyperD anion exchange columns (Pall Lifesciences). In order to prevent miniTRIM5 from binding to the HyperD matrix, the columns were preequilibrated with 2× sample buffer. Protein in the flowthrough was concentrated and purified further by gel filtration on a Superdex 75 10/300 GL column (GE Healthcare) in running buffer (10 mM Tris, pH 8, 100 mM NaCl, 1 mM TCEP). Major peak fractions were collected and used without freezing. Typical yields were ∼0.5 mg/liter culture (RBcc constructs) and ∼1.2 mg/liter (Bcc).
SEC-MALS.
Experiments using size exclusion with multiangle light scattering (SEC-MALS) were carried out on a Dionex UltiMate3000 high-performance liquid chromatography (HPLC) system (ThermoFisher) equipped with a UV detector and a Superdex 200 HR 10/300 GL column (GE Healthcare). Mass measurements were made with an in-line miniDAWN TREOS static light scattering detector (Wyatt Technology) and Optilab T-rEX differential refractometer (Wyatt Technology). Sample volumes of 100 μl at 100 μM concentration were injected and then developed at a flow rate of 0.4 ml/min in 10 mM Tris, pH 8, 100 mM NaCl, 1 mM TCEP.
Velocity sedimentation analytical ultracentrifugation.
MiniTRIMs were prepared for analytical ultracentrifugation at 12 to 100 μM protein concentrations in 10 mM N-cyclohexyl-2-aminoethanesulfonic acid (CHES), pH 9, 100 mM NaCl, 1 mM TCEP. Samples (420 μl) were spun at 42,000 rpm and 20°C in a Beckman Optima XLA-1 centrifuge equipped with an eight-hole An50-Ti rotor. About 300 scans were collected for each sample. Noise-filtered data were analyzed graphically using the van Holde-Weischet extrapolation method (66). Results were further analyzed by performing a parametrically constrained grid search procedure (PCSA) using the NNLS algorithm in Ultrascan III with 100 Monte Carlo iterations (67, 68). Direct fitting of the raw Bcc data was performed using the adaptive space-time finite element method (69), assuming a reversible monomer-dimer equilibrium and holding the monomer and dimer molecular weights constant. Ninety-six Monte Carlo iterations of the fits were performed to generate statistics (68), which are summarized in Table 1.
Ubiquitin discharge assay.
E1, E2, and ubiquitin proteins either were obtained commercially (Enzo, UBPBio, and Sigma) or were purified in house. UbcH5b∼Ub conjugates were generated in a solution containing 120 μM E2, 0.2 μM human E1, 120 μM Ub, 3 mM ATP in reaction buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 5 mM MgCl2, 0.5 mM TCEP, 0.1% Triton X-100). After incubation at 37°C for 1 h, unreacted ATP was removed by addition of 4.5 U/ml apyrase (NEB). Discharge reactions were prepared by mixing the E2∼Ub conjugate solution with an equal volume of reaction buffer solution containing 10 μM E3 (RING or miniTRIM) and 60 mM l-lysine, followed by incubation at room temperature. Aliquots were collected at the indicated time points, quenched in nonreducing SDS loading buffer, and analyzed by SDS-PAGE. Coomassie-stained gels were scanned on an Odyssey scanner (LiCor Biosciences), and intensities of the E2∼Ub bands were quantified. Experiments were performed three times each with freshly purified miniTRIM and RING proteins.
Structure determination.
MiniTRIMCyp Bcc WT and the RL→CC hairpin linker mutant proteins were concentrated to 5.0 mg/ml for crystallization screening. Only two conditions yielded crystals of the double-cysteine mutant, which turned out to be the same crystal form. Crystals for data collection were obtained in hanging drops by mixing equal volumes of protein solution and precipitant (17% polyethylene glycol 3350, 0.4 M sodium-potassium tartrate). Crystals were cryoprotected with 25% ethylene glycol in well solution. Diffraction data were collected at beamline 22ID of the Advanced Photon Source and processed with HKL2000 (70). Phases were determined by molecular replacement using PHASER software (71) and miniTRIM5α (Protein Data Bank [PDB] accession number 5EIU) (35) as a search model. Modeling, refinement, and validation were carried out using the tools in Coot (72) and PHENIX software suite version 1.10.1-2155 (73). Restraints for secondary structure and zinc ion coordination were used during refinement. Structure statistics are summarized in Table 2.
Purification and assembly of full-length TRIMCyp.
Full-length TRIMCyp was expressed in insect cells and purified as described previously (37). Two point mutations in the linker 2 region (K283D/Q287D) were included to minimize proteolytic degradation during expression. The purified protein in 20 mM CHES, pH 10, 0.1 μM ZnCl2, 1 mM TCEP was concentrated to 3 mg/ml and used as soon as possible after purification.
TRIMCyp/HIV-1 CA coassemblies were produced by mixing purified HIV-1 CA (∼600 μM final concentration) and TRIMCyp (∼1 to 5 μM) in assembly buffer (20 mM Tris, pH 8, 50 mM NaCl) and incubating at 37°C for 1 h and then at room temperature overnight. Assembled particles were applied on carbon-coated grids, stained with uranyl acetate, and visualized by electron microscopy.
HIV restriction assays.
Single-cycle replication assays utilized HIV-GFP vectors and were performed as described previously for TRIM5α (35). The lentiviral vector CSII-IDR2-owlTRIMCyp-FOS (plasmid 79067; Addgene) was used to express TRIMCyp with a C-terminal FLAG-One-Strep (FOS) tag followed by an internal ribosome entry sequence and DsRed. For restriction assays in the presence of CsA (C3662; Sigma) in dimethyl sulfoxide (DMSO), HeLa cells first were treated with inhibitor for 2 h prior to transduction with HIV-GFP. CsA treatment was maintained for 72 h after transduction, after which GFP expression was quantified by flow cytometry. Expression levels were determined by immunoblotting of whole-cell lysates with anti-FLAG M2 (1:1,000; F1804; Sigma) and anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (1:10,000; MAB374; EMD Millipore) to control for loading. All restriction assays were repeated three times.
The omTRIM-rhSPRY chimera was generated by overlap extension PCR. TRIMCyp residues 1 to 299 were amplified with primer 1 (GTACTAGTGCTAGCCACCAT GGCTTCCAGAATCCTGGT) and primer 2 (GTAGCCAGTGTCACATCAACCCAGTAGCGTTGGACTTCTG), while TRIM5α residues 301 to 495 were amplified with primer 3 (GTAGCCAGTGTCACATCAACCCAGTAGCGTTGGACTTCTG) and primer 4 (GAAGCTTGAGCTCGAGTTATTTTTCG, which is complementary to the FOS tag). These PCR products were combined and amplified with primers 1 and 4 to generate full-length omTRIM-rhSPRY, which then was cloned into the CSII-IDR2 vector using NheI and XhoI restriction sites. The W116A, W116E, R120A, and R120E mutations were introduced by using QuikChange mutagenesis (Agilent).
Accession number(s).
Coordinates and structure factors were deposited in PDB under accession number 5VA4.
ACKNOWLEDGMENTS
We thank members of our laboratories for technical advice and experimental support.
This study was supported by NIH grants R01-GM112508 (O.P.) and P50-GM082545 (B.K.G.-P., D.N.I., and W.I.S.). J.M.W. was supported by a postdoctoral NIH fellowship (F32-GM115007).
REFERENCES
- 1.Grütter MG, Luban J. 2012. TRIM5 structure, HIV-1 capsid recognition, and innate immune signaling. Curr Opin Virol 2:142–150. doi: 10.1016/j.coviro.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sanz-Ramos M, Stoye JP. 2013. Capsid-binding retrovirus restriction factors: discovery, restriction specificity and implications for the development of novel therapeutics. J Gen Virol 94:2587–2598. doi: 10.1099/vir.0.058180-0. [DOI] [PubMed] [Google Scholar]
- 3.Stremlau M, Owens CM, Perron MJ, Kiessling M, Autissier P, Sodroski J. 2004. The cytoplasmic body component TRIM5α restricts HIV-1 infection in Old World monkeys. Nature 427:848–853. doi: 10.1038/nature02343. [DOI] [PubMed] [Google Scholar]
- 4.Sayah DM, Sokolskaja E, Berthoux L, Luban J. 2004. Cyclophilin A retrotransposition into TRIM5 explains owl monkey resistance to HIV-1. Nature 430:569–573. doi: 10.1038/nature02777. [DOI] [PubMed] [Google Scholar]
- 5.Nisole S, Lynch C, Stoye JP, Yap MW. 2004. A Trim5-cyclophilin A fusion protein found in owl monkey kidney cells can restrict HIV-1. Proc Natl Acad Sci U S A 101:13324–13328. doi: 10.1073/pnas.0404640101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sawyer SL, Wu LI, Emerman M, Malik HS. 2005. Positive selection of primate TRIM5α identifies a critical species-specific retroviral restriction domain. Proc Natl Acad Sci U S A 102:2832–2837. doi: 10.1073/pnas.0409853102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sebastian S, Luban J. 2005. TRIM5α selectively binds a restriction-sensitive retroviral capsid. Retrovirology 2:40. doi: 10.1186/1742-4690-2-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stremlau M, Perron M, Welikala S, Sodroski J. 2005. Species-specific variation in the B30.2(SPRY) domain of TRIM5α determines the potency of human immunodeficiency virus restriction. J Virol 79:3139–3145. doi: 10.1128/JVI.79.5.3139-3145.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yap MW, Nisole S, Stoye JP. 2005. A single amino acid change in the SPRY domain of human Trim5α leads to HIV-1 restriction. Curr Biol 15:73–78. doi: 10.1016/j.cub.2004.12.042. [DOI] [PubMed] [Google Scholar]
- 10.Li Y, Li X, Stremlau M, Lee M, Sodroski J. 2006. Removal of arginine 332 allows human TRIM5α to bind human immunodeficiency virus capsids and to restrict infection. J Virol 80:6738–6744. doi: 10.1128/JVI.00270-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stremlau M, Perron M, Lee M, Li Y, Song B, Javanbakht H, Diaz-Griffero F, Anderson DJ, Sundquist WI, Sodroski J. 2006. Specific recognition and accelerated uncoating of retroviral capsids by the TRIM5α restriction factor. Proc Natl Acad Sci U S A 103:5514–5519. doi: 10.1073/pnas.0509996103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Virgen CA, Kratovac Z, Bieniasz PD, Hatziioannou T. 2008. Independent genesis of chimeric TRIM5-cyclophilin proteins in two primate species. Proc Natl Acad Sci U S A 105:3563–3568. doi: 10.1073/pnas.0709258105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Biris N, Yang Y, Taylor AB, Tomashevski A, Guo M, Hart PJ, Diaz-Griffero F, Ivanov DN. 2012. Structure of the rhesus monkey TRIM5α PRYSPRY domain, the HIV capsid recognition module. Proc Natl Acad Sci U S A 109:13278–13283. doi: 10.1073/pnas.1203536109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang H, Ji X, Zhao G, Ning J, Zhao Q, Aiken C, Gronenborn AM, Zhang P, Xiong Y. 2012. Structural insight into HIV-1 capsid recognition by rhesus TRIM5α. Proc Natl Acad Sci U S A 109:18372–18377. doi: 10.1073/pnas.1210903109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hatziioannou T, Cowan S, Von Schwedler UK, Sundquist WI, Bieniasz PD. 2004. Species-specific tropism determinants in the human immunodeficiency virus type 1 capsid. J Virol 78:6005–6012. doi: 10.1128/JVI.78.11.6005-6012.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Owens CM, Song B, Perron MJ, Yang PC, Stremlau M, Sodroski J. 2004. Binding and susceptibility to postentry restriction factors in monkey cells are specified by distinct regions of the human immunodeficiency virus type 1 capsid. J Virol 78:5423–5437. doi: 10.1128/JVI.78.10.5423-5437.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Song H, Nakayama EE, Yokoyama M, Sato H, Levy JA, Shioda T. 2007. A single amino acid of the human immunodeficiency virus type 2 capsid affects its replication in the presence of cynomolgus monkey and human TRIM5αs. J Virol 81:7280–7285. doi: 10.1128/JVI.00406-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mortuza GB, Dodding MP, Goldstone DC, Haire LF, Stoye JP, Taylor IA. 2008. Structure of B-MLV capsid amino-terminal domain reveals key features of viral tropism, Gag assembly and core formation. J Mol Biol 376:1493–1508. doi: 10.1016/j.jmb.2007.12.043. [DOI] [PubMed] [Google Scholar]
- 19.Kono K, Song H, Yokoyama M, Sato H, Shioda T, Nakayama EE. 2010. Multiple sites in the N-terminal half of simian immunodeficiency virus capsid protein contribute to evasion from rhesus monkey TRIM5α-mediated restriction. Retrovirology 7:72. doi: 10.1186/1742-4690-7-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuroishi A, Bozek K, Shioda T, Nakayama EE. 2010. A single amino acid substitution of the human immunodeficiency virus type 1 capsid protein affects viral sensitivity to TRIM5 α. Retrovirology 7:58. doi: 10.1186/1742-4690-7-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ohkura S, Goldstone DC, Yap MW, Holden-Dye K, Taylor IA, Stoye JP. 2011. Novel escape mutants suggest an extensive TRIM5α binding site spanning the entire outer surface of the murine leukemia virus capsid protein. PLoS Pathog 7:e1002011. doi: 10.1371/journal.ppat.1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McCarthy KR, Schmidt AG, Kirmaier A, Wyand AL, Newman RM, Johnson WE. 2013. Gain-of-sensitivity mutations in a Trim5-resistant primary isolate of pathogenic SIV identify two independent conserved determinants of Trim5α specificity. PLoS Pathog 9:e1003352. doi: 10.1371/journal.ppat.1003352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ohkura S, Stoye JP. 2013. A comparison of murine leukemia viruses that escape from human and rhesus macaque TRIM5αs. J Virol 87:6455–6468. doi: 10.1128/JVI.03425-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Biris N, Tomashevski A, Bhattacharya A, Diaz-Griffero F, Ivanov DN. 2013. Rhesus monkey TRIM5α SPRY domain recognizes multiple epitopes that span several capsid monomers on the surface of the HIV-1 mature viral core. J Mol Biol 425:5032–5044. doi: 10.1016/j.jmb.2013.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kovalskyy DB, Ivanov DN. 2014. Recognition of the HIV capsid by the TRIM5α restriction factor is mediated by a subset of pre-existing conformations of the TRIM5alpha SPRY domain. Biochemistry 53:1466–1476. doi: 10.1021/bi4014962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li X, Sodroski J. 2008. The TRIM5α B-box 2 domain promotes cooperative binding to the retroviral capsid by mediating higher-order self-association. J Virol 82:11495–11502. doi: 10.1128/JVI.01548-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Diaz-Griffero F, Qin XR, Hayashi F, Kigawa T, Finzi A, Sarnak Z, Lienlaf M, Yokoyama S, Sodroski J. 2009. A B-box 2 surface patch important for TRIM5α self-association, capsid binding avidity, and retrovirus restriction. J Virol 83:10737–10751. doi: 10.1128/JVI.01307-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ganser-Pornillos BK, Chandrasekaran V, Pornillos O, Sodroski JG, Sundquist WI, Yeager M. 2011. Hexagonal assembly of a restricting TRIM5α protein. Proc Natl Acad Sci U S A 108:534–539. doi: 10.1073/pnas.1013426108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sanchez JG, Okreglicka K, Chandrasekaran V, Welker JM, Sundquist WI, Pornillos O. 2014. The tripartite motif coiled-coil is an elongated antiparallel hairpin dimer. Proc Natl Acad Sci U S A 111:2494–2499. doi: 10.1073/pnas.1318962111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goldstone DC, Walker PA, Calder LJ, Coombs PJ, Kirkpatrick J, Ball NJ, Hilditch L, Yap MW, Rosenthal PB, Stoye JP, Taylor IA. 2014. Structural studies of postentry restriction factors reveal antiparallel dimers that enable avid binding to the HIV-1 capsid lattice. Proc Natl Acad Sci U S A 111:9609–9614. doi: 10.1073/pnas.1402448111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roganowicz MD, Komurlu S, Mukherjee S, Plewka J, Alam SL, Skorupka KA, Wan Y, Dawidowski D, Cafiso DS, Ganser-Pornillos BK, Campbell EM, Pornillos O. 2017. TRIM5α SPRY/coiled-coil interactions optimize avid retroviral capsid recognition. PLoS Pathog 13:e1006686. doi: 10.1371/journal.ppat.1006686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kar AK, Diaz-Griffero F, Li Y, Li X, Sodroski J. 2008. Biochemical and biophysical characterization of a chimeric TRIM21-TRIM5α protein. J Virol 82:11669–11681. doi: 10.1128/JVI.01559-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Langelier CR, Sandrin V, Eckert DM, Christensen DE, Chandrasekaran V, Alam SL, Aiken C, Olsen JC, Kar AK, Sodroski JG, Sundquist WI. 2008. Biochemical characterization of a recombinant TRIM5α protein that restricts human immunodeficiency virus type 1 replication. J Virol 82:11682–11694. doi: 10.1128/JVI.01562-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao G, Ke D, Vu T, Ahn J, Shah VB, Yang R, Aiken C, Charlton LM, Gronenborn AM, Zhang P. 2011. Rhesus TRIM5α disrupts the HIV-1 capsid at the inter-hexamer interfaces. PLoS Pathog 7:e1002009. doi: 10.1371/journal.ppat.1002009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wagner JM, Roganowicz MD, Skorupka K, Alam SL, Christensen D, Doss G, Wan Y, Frank GA, Ganser-Pornillos BK, Sundquist WI, Pornillos O. 2016. Mechanism of B-box 2 domain-mediated higher-order assembly of the retroviral restriction factor TRIM5α. eLife 5:e16309. doi: 10.7554/eLife.16309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Keown JR, Yang JX, Douglas J, Goldstone DC. 2016. Characterisation of assembly and ubiquitylation by the RBCC motif of Trim5α. Sci Rep 6:26837. doi: 10.1038/srep26837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li YL, Chandrasekaran V, Carter SD, Woodward CL, Christensen DE, Dryden KA, Pornillos O, Yeager M, Ganser-Pornillos BK, Jensen GJ, Sundquist WI. 2016. Primate TRIM5 proteins form hexagonal nets on HIV-1 capsids. eLife 5:e16269. doi: 10.7554/eLife.16269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li X, Yeung DF, Fiegen AM, Sodroski J. 2011. Determinants of the higher order association of the restriction factor TRIM5α and other tripartite motif (TRIM) proteins. J Biol Chem 286:27959–27970. doi: 10.1074/jbc.M111.260406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pertel T, Hausmann S, Morger D, Zuger S, Guerra J, Lascano J, Reinhard C, Santoni FA, Uchil PD, Chatel L, Bisiaux A, Albert ML, Strambio-De-Castillia C, Mothes W, Pizzato M, Grütter MG, Luban J. 2011. TRIM5 is an innate immune sensor for the retrovirus capsid lattice. Nature 472:361–365. doi: 10.1038/nature09976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Roa A, Hayashi F, Yang Y, Lienlaf M, Zhou J, Shi J, Watanabe S, Kigawa T, Yokoyama S, Aiken C, Diaz-Griffero F. 2012. RING domain mutations uncouple TRIM5α restriction of HIV-1 from inhibition of reverse transcription and acceleration of uncoating. J Virol 86:1717–1727. doi: 10.1128/JVI.05811-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yudina Z, Roa A, Johnson R, Biris N, de Souza Aranha Vieira DA, Tsiperson V, Reszka N, Taylor AB, Hart PJ, Demeler B, Diaz-Griffero F, Ivanov DN. 2015. RING dimerization links higher-order assembly of TRIM5α to synthesis of K63-linked polyubiquitin. Cell Rep 12:788–797. doi: 10.1016/j.celrep.2015.06.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fletcher AJ, Christensen DE, Nelson C, Tan CP, Schaller T, Lehner PJ, Sundquist WI, Towers GJ. 2015. TRIM5α requires Ube2W to anchor Lys63-linked ubiquitin chains and restrict reverse transcription. EMBO J 34:2078–2095. doi: 10.15252/embj.201490361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Campbell EM, Weingart J, Sette P, Opp S, Sastri J, O'Connor SK, Talley S, Diaz-Griffero F, Hirsch V, Bouamr F. 2015. TRIM5α-mediated ubiquitin chain conjugation is required for inhibition of HIV-1 reverse transcription and capsid destabilization. J Virol 90:1849–1857. doi: 10.1128/JVI.01948-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Caines ME, Bichel K, Price AJ, McEwan WA, Towers GJ, Willett BJ, Freund SM, James LC. 2012. Diverse HIV viruses are targeted by a conformationally dynamic antiviral. Nat Struct Mol Biol 19:411–416. doi: 10.1038/nsmb.2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gamble TR, Vajdos FF, Yoo S, Worthylake DK, Houseweart M, Sundquist WI, Hill CP. 1996. Crystal structure of human cyclophilin A bound to the amino-terminal domain of HIV-1 capsid. Cell 87:1285–1294. doi: 10.1016/S0092-8674(00)81823-1. [DOI] [PubMed] [Google Scholar]
- 46.Price AJ, Marzetta F, Lammers M, Ylinen LM, Schaller T, Wilson SJ, Towers GJ, James LC. 2009. Active site remodeling switches HIV specificity of antiretroviral TRIMCyp. Nat Struct Mol Biol 16:1036–1042. doi: 10.1038/nsmb.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vajdos FF, Yoo S, Houseweart M, Sundquist WI, Hill CP. 1997. Crystal structure of cyclophilin A complexed with a binding site peptide from the HIV-1 capsid protein. Protein Sci 6:2297–2307. doi: 10.1002/pro.5560061103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yoo S, Myszka DG, Yeh C, McMurray M, Hill CP, Sundquist WI. 1997. Molecular recognition in the HIV-1 capsid/cyclophilin A complex. J Mol Biol 269:780–795. doi: 10.1006/jmbi.1997.1051. [DOI] [PubMed] [Google Scholar]
- 49.Diaz-Griffero F, Vandegraaff N, Li Y, McGee-Estrada K, Stremlau M, Welikala S, Si Z, Engelman A, Sodroski J. 2006. Requirements for capsid-binding and an effector function in TRIMCyp-mediated restriction of HIV-1. Virology 351:404–419. doi: 10.1016/j.virol.2006.03.023. [DOI] [PubMed] [Google Scholar]
- 50.Diaz-Griffero F, Kar A, Lee M, Stremlau M, Poeschla E, Sodroski J. 2007. Comparative requirements for the restriction of retrovirus infection by TRIM5α and TRIMCyp. Virology 369:400–410. doi: 10.1016/j.virol.2007.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Diaz-Griffero F, Kar A, Perron M, Xiang SH, Javanbakht H, Li X, Sodroski J. 2007. Modulation of retroviral restriction and proteasome inhibitor-resistant turnover by changes in the TRIM5α B-box 2 domain. J Virol 81:10362–10378. doi: 10.1128/JVI.00703-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Javanbakht H, Diaz-Griffero F, Yuan W, Yeung DF, Li X, Song B, Sodroski J. 2007. The ability of multimerized cyclophilin A to restrict retrovirus infection. Virology 367:19–29. doi: 10.1016/j.virol.2007.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yap MW, Mortuza GB, Taylor IA, Stoye JP. 2007. The design of artificial retroviral restriction factors. Virology 365:302–314. doi: 10.1016/j.virol.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 54.Keown JR, Goldstone DC. 2016. Crystal structure of the Trim5α Bbox2 domain from rhesus macaques describes a plastic oligomerisation interface. J Struct Biol 195:282–285. doi: 10.1016/j.jsb.2016.07.004. [DOI] [PubMed] [Google Scholar]
- 55.Sanchez JG, Chiang JJ, Sparrer KM, Alam SL, Chi M, Roganowicz MD, Sankaran B, Gack MU, Pornillos O. 2016. Mechanism of TRIM25 catalytic activation in the antiviral RIG-I pathway. Cell Rep 16:1315–1325. doi: 10.1016/j.celrep.2016.06.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Koliopoulos MG, Esposito D, Christodoulou E, Taylor IA, Rittinger K. 2016. Functional role of TRIM E3 ligase oligomerization and regulation of catalytic activity. EMBO J 35:1204–1218. doi: 10.15252/embj.201593741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wenzel DM, Lissounov A, Brzovic PS, Klevit RE. 2011. UBCH7 reactivity profile reveals parkin and HHARI to be RING/HECT hybrids. Nature 474:105–108. doi: 10.1038/nature09966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Luban J, Bossolt KL, Franke EK, Kalpana GV, Goff SP. 1993. Human immunodeficiency virus type 1 Gag protein binds to cyclophilins A and B. Cell 73:1067–1078. doi: 10.1016/0092-8674(93)90637-6. [DOI] [PubMed] [Google Scholar]
- 59.Javanbakht H, Diaz-Griffero F, Stremlau M, Si Z, Sodroski J. 2005. The contribution of RING and B-box 2 domains to retroviral restriction mediated by monkey TRIM5α. J Biol Chem 280:26933–26940. doi: 10.1074/jbc.M502145200. [DOI] [PubMed] [Google Scholar]
- 60.Perez-Caballero D, Hatziioannou T, Zhang F, Cowan S, Bieniasz PD. 2005. Restriction of human immunodeficiency virus type 1 by TRIM-CypA occurs with rapid kinetics and independently of cytoplasmic bodies, ubiquitin, and proteasome activity. J Virol 79:15567–15572. doi: 10.1128/JVI.79.24.15567-15572.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Maegawa H, Miyamoto T, Sakuragi J, Shioda T, Nakayama EE. 2010. Contribution of RING domain to retrovirus restriction by TRIM5α depends on combination of host and virus. Virology 399:212–220. doi: 10.1016/j.virol.2010.01.003. [DOI] [PubMed] [Google Scholar]
- 62.Kim J, Tipper C, Sodroski J. 2011. Role of TRIM5α RING domain E3 ubiquitin ligase activity in capsid disassembly, reverse transcription blockade, and restriction of simian immunodeficiency virus. J Virol 85:8116–8132. doi: 10.1128/JVI.00341-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Campbell EM, Perez O, Anderson JL, Hope TJ. 2008. Visualization of a proteasome-independent intermediate during restriction of HIV-1 by rhesus TRIM5α. J Cell Biol 180:549–561. doi: 10.1083/jcb.200706154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mandell MA, Kimura T, Jain A, Johansen T, Deretic V. 2014. TRIM proteins regulate autophagy: TRIM5 is a selective autophagy receptor mediating HIV-1 restriction. Autophagy 10:2387–2388. doi: 10.4161/15548627.2014.984278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Imam S, Talley S, Nelson RS, Dharan A, O'Connor C, Hope TJ, Campbell EM. 2016. TRIM5α degradation via autophagy is not required for retroviral restriction. J Virol 90:3400–3410. doi: 10.1128/JVI.03033-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Demeler B, van Holde KE. 2004. Sedimentation velocity analysis of highly heterogeneous systems. Anal Biochem 335:279–288. doi: 10.1016/j.ab.2004.08.039. [DOI] [PubMed] [Google Scholar]
- 67.Gorbet G, Devlin T, Hernandez Uribe BI, Demeler AK, Lindsey ZL, Ganji S, Breton S, Weise-Cross L, Lafer EM, Brookes EH, Demeler B. 2014. A parametrically constrained optimization method for fitting sedimentation velocity experiments. Biophys J 106:1741–1750. doi: 10.1016/j.bpj.2014.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Demeler B, Brookes E. 2008. Monte Carlo analysis of sedimentation experiments. Colloid Polym Sci 286:129–137. doi: 10.1007/s00396-007-1699-4. [DOI] [Google Scholar]
- 69.Cao W, Demeler B. 2008. Modeling analytical ultracentrifugation experiments with an adaptive space-time finite element solution for multicomponent reacting systems. Biophys J 95:54–65. doi: 10.1529/biophysj.107.123950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Otwinowski Z, Minor W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 71.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. 2007. Phaser crystallographic software. J Appl Crystallogr 40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]