

ABSTRACT
There is increasing evidence to suggest that antibodies directed toward influenza A virus (IAV) neuraminidase (NA) are an important correlate of protection against influenza in humans. Moreover, the potential of NA-specific antibodies to provide broader protection than conventional hemagglutinin (HA) antibodies has been recognized. Here, we describe the isolation of two monoclonal antibodies, N1-7D3 and N1-C4, directed toward the N1 NA. N1-7D3 binds to a conserved linear epitope in the membrane-distal, carboxy-terminal part of the NA and reacted with the NA of seasonal H1N1 isolates ranging from 1977 to 2007 and the 2009 H1N1pdm virus, as well as A/Vietnam/1194/04 (H5N1). However, N1-7D3 lacked NA inhibition (NI) activity and the ability to protect BALB/c mice against a lethal challenge with a range of H1N1 viruses. Conversely, N1-C4 bound to a conformational epitope that is conserved between two influenza virus subtypes, 2009 H1N1pdm and H5N1 IAV, and displayed potent in vitro antiviral activity mediating both NI and plaque size reduction. Moreover, N1-C4 could provide heterosubtypic protection in BALB/c mice against a lethal challenge with 2009 H1N1pdm or H5N1 virus. Glutamic acid residue 311 in the NA was found to be critical for the NA binding and antiviral activity of monoclonal antibody N1-C4. Our data provide further evidence for cross-protective epitopes within the N1 subtype and highlight the potential of NA as an important target for vaccine and therapeutic approaches.
IMPORTANCE Influenza remains a worldwide burden on public health. As such, the development of novel vaccines and therapeutics against influenza virus is crucial. Human challenge studies have recently highlighted the importance of antibodies directed toward the viral neuraminidase (NA) as an important correlate of reduced influenza-associated disease severity. Furthermore, there is evidence that anti-NA antibodies can provide broader protection than antibodies toward the viral hemagglutinin. Here, we describe the isolation and detailed characterization of two N1 NA-specific monoclonal antibodies. One of these monoclonal antibodies broadly binds N1-type NAs, and the second displays NA inhibition and in vitro and in vivo antiviral activity against 2009 H1N1pdm and H5N1 influenza viruses. These two new anti-NA antibodies contribute to our understanding of the antigenic properties and protective potential of the influenza virus NA antigen.
KEYWORDS: influenza A virus, neuraminidase, monoclonal antibody, therapeutic
INTRODUCTION
Human influenza is an important respiratory disease that is caused by influenza A and B viruses. These viruses display two glycoproteins on their envelopes: hemagglutinin (HA) and neuraminidase (NA), a tetrameric receptor-destroying enzyme. Both HA and NA play pivotal roles in the replication of influenza A and B viruses. HA binds to sialic acid on the surfaces of cells to initiate entry, while the NA enzymatic activity cleaves off sialic acid, allowing release of the virus from the cell surface and preventing aggregation of the virus (1). Further, NA activity contributes to virion entry by the removal of sialic acid from decoy receptors present within the airways and by contributing to HA-mediated membrane fusion (2–7).
Antibodies directed to NA can reduce viral replication and the formation of lung lesions in the mouse and ferret models (8–10) by aggregating the virus at the surfaces of cells (11) and promoting complement defenses (12). Analysis of human serum antibody responses to influenza vaccination has shown that NA-inhibiting (NI) antibodies correlate with protection independently of hemagglutination-inhibiting (HI) and microneutralization titers (13). Furthermore, in a controlled human influenza virus challenge with the 2009 H1N1pdm virus, the levels of preexisting NI antibodies correlated significantly better than HI antibodies with a reduced disease severity score, decreased symptoms, and even a reduction in the duration of virus shedding (14).
NA also induces broadly heterologous reactive antibodies. A ferret study demonstrated that vaccination with soluble recombinant tetrameric NA (rNA) derived from the 2009 H1N1pdm virus could induce antibodies that inhibited the NA activity of rNA derived from a 2007 seasonal H1N1 and an avian H5N1 isolate (8). Similar protective responses in ferrets were induced by an adjuvanted 2006-2007 influenza trivalent inactivated vaccine against challenge with an avian H5N1 virus, which was mainly attributable to NI antibodies induced by the N1 component of the vaccine (15). Further, vaccination with A/PR8/1934 rNA significantly protected mice from a homologous infection, but also from challenge with H5N1 and A(H1N1)pdm09. Protection was correlated with the presence of NI antibodies (16). In human serum samples, NI antibodies against avian H5N1 have also been detected, even though this virus has never been widespread in humans (17). Moreover, a recent study by Rajendran et al. identified a number of age groups that had NI antibodies to H1N1, H3N2, and influenza B viruses that they had never been exposed to previously (18). These data, among others, are evidence of the presence of cross-reactive epitopes that span the N1 subtype, including those of different N1 virus clades.
Compared to the numerous HA-specific monoclonal antibodies that have been described, there is still a paucity of well-defined NA-specific monoclonal antibodies. A study by Wan et al. described a number of cross-reactive NA epitopes present within the 1918 H1N1, seasonal H1N1, A(H1N1)pdm09, and H5N1 viruses (19). Other work has also identified additional cross-reactive NA epitopes within the H5N1 clades and between H5N1 and H1N1 viruses (20). Moreover, the Li group isolated a rabbit monoclonal antibody, named HCA-2, by immunization with a peptide (ILRTQESEC; amino acids 222 to 230) that corresponds to a highly conserved sequence within the NA enzymatic pocket. HCA-2 could bind to and inhibit NA of influenza A and B viruses and, when given at a high dose, partially protected against influenza A and B virus challenges in mice (5, 21, 22).
In this study, we isolated two mouse monoclonal antibodies, N1-7D3 and N1-C4, that can bind to NAs of multiple N1 subtype viruses. Monoclonal antibody N1-7D3 binds to a highly conserved epitope in the carboxy terminus of N1 NA but could not mediate NI or plaque size reduction or protect BALB/c mice from H1N1 or H5N1 virus challenge. N1-C4 could bind to and mediate NI and plaque size reduction of 2009 H1N1pdm and H5N1 viruses. Furthermore, this monoclonal antibody displayed therapeutic and prophylactic protection in BALB/c mice.
RESULTS
Isolation of mouse monoclonal antibodies that bind to N1 NA.
To generate N1-specific monoclonal antibodies, mice were infected and vaccinated consecutively as outlined in Materials and Methods. Splenocytes were isolated and fused to the immortal cell line SP2/0 to generate hybridomas. After several rounds of selection and screening, we selected two monoclonal antibodies of interest: N1-7D3 and N1-C4. Initially, the monoclonal antibodies were tested for their antibody isotype by enzyme-linked immunosorbent assay (ELISA). Both N1-7D3 and N1-C4 could be detected by an anti-mouse IgG1 antibody but not by anti-mouse antibodies specific for IgG2a or IgG2b (data not shown).
Previous work has shown that several conserved epitopes exist within NA that allow broad cross-reactivity within a subtype and, exceptionally, between subtypes (19, 21, 22). To examine if monoclonal antibodies N1-7D3 and N1-C4 could bind to a broad range of N1 NAs, the ability to bind to rNA and to the surfaces of influenza A virus (IAV)-infected cells was assessed by ELISA and by flow cytometry detection, respectively. N1-7D3 displayed the broadest range of reactivity, capable of binding to rNAs derived from USSR/77, NC/99, Sing/86, Bris/07, and Bel/09 (Fig. 1A, i), whereas N1-C4 bound only to rNA from the A(H1N1)pdm09 virus (Fig. 1A, ii). We also evaluated if the monoclonal antibodies could bind to other NAs from different subtypes that are relevant to human disease. We found that both N1-7D3 (Fig. 1B, i) and N1-C4 (Fig. 1B, ii) could not bind to rNA from an H3N2 virus from 1975, to X-47, or to the H7N9 strain A/Anhui/1/2013. An anti-streptavidin antibody that targets the purification tag of the recombinant protein confirmed that coating levels were equivalent for all three rNAs (data not shown). The same pattern was observed when binding to the surfaces of N1 subtype-infected MDCK and HEK293T cells was assessed by flow cytometry. N1-7D3 bound to all H1N1 IAVs tested, whereas N1-C4 showed negligible binding to cells that had been infected with seasonal H1N1 viruses but could bind to A(H1N1)pdm09 (Fig. 1C and D and data not shown). Moreover, we tested if the monoclonal antibodies could also bind to the H5N1 strain NIBRG-14, a PR8/34 reassortant virus with the NA and HA (lacking the polybasic cleavage site) segments of A/Vietnam/1194/2004. Both monoclonal antibodies bound to NIBRG-14-infected MDCK and HEK293T cells, as detected by flow cytometry (Fig. 1C and D and data not shown). Taken together, these data show that N1-7D3 can bind to NAs from H1N1 IAV (seasonal and pandemic strains) and H5N1 IAVs, whereas N1-C4 has a narrower range of reactivity and binds to NAs of A(H1N1)pdm09 IAV and H5N1 IAV.
FIG 1.
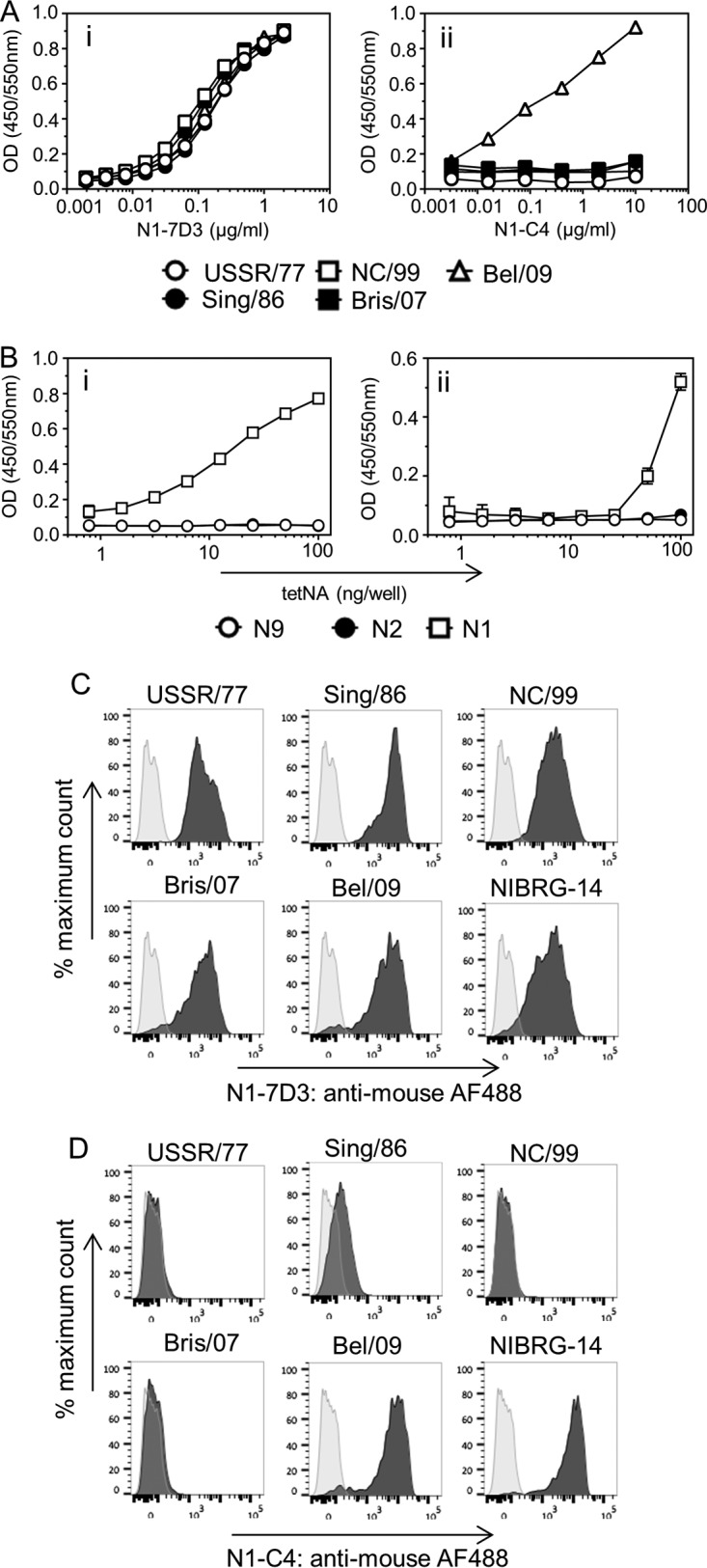
(A) Binding of monoclonal antibody N1-7D3 and N1-C4 to soluble rNA. Wells of 96-well flat-bottom ELISA plates were coated with 0.5 μg/ml of recombinant soluble NA from USSR/77, Sing/86, NC/99, Bris/07, or Bel/09 in sodium carbonate buffer. After overnight incubation and blocking with 5 mg/ml BSA in PBS, N1-7D3 (i) or N1-C4 (ii) was applied in serial dilutions for at least 1 h at room temperature in 2 mg/ml BSA in PBST plus 5 mM CaCl2. Binding of the monoclonal antibody to rNA was detected by anti-mouse antibody conjugated to HRP. The data represent results from two independent experiments. OD, optical density. (B) N1-7D3 and N1-C4 do not bind to soluble rNA derived from the N2 or N9 subtype. Ninety-six-well ELISA plates were coated with recombinant NA from X47 or A/Anuhi/1/2013 at increasing concentrations overnight. After blocking, N1-7D3 (i) or N1-C4 (ii) was applied at 5 μg/ml. Binding was detected by anti-mouse antibody conjugated to HRP. The data represent the averages of three replicates (±1 standard deviation [SD]). (C and D) Binding of N1-7D3 (C) or N1-C4 (D) to infected MDCK cells. Confluent monolayers of MDCK cells were infected with USSR/77, Sing/86, NC/99, Bris/07, Bel/09, or NIBRG-14 at an MOI of 1. Uninfected cells were taken along as a negative control. Sixteen hours postinfection, the cells were collected from the tissue culture plastic using enzyme-free cell dissociation solution. Infected and uninfected MDCK cells were stained with 10 μg/ml monoclonal antibody N1-7D3 (C) or N1-C4 (D) in 2 mg/ml BSA in PBS containing 2 mM EDTA at 4°C for 30 min. The cells were washed and subsequently stained with goat anti-mouse IgG conjugated to AF488, and fixable viability dye was added. The cells were fixed with 4% PFA for 1 h on ice, and binding of anti-NA monoclonal antibodies was determined by flow cytometry. Shown are histograms of cells in the AF488 channel with dead cells and debris gated out. Infected cells are shown in dark-gray histograms and uninfected cells in light gray.
Monoclonal antibody N1-C4, but not monoclonal antibody N1-7D3, has NA inhibition activity and reduces the plaque size of N1 IAVs.
Antibodies directed toward the NA of influenza virus have been shown to mediate a variety of antiviral in vitro effects against influenza virus, including NI, neutralization of viral infectivity, reduction of plaque size, and even blocking of HA-induced agglutination of red blood cells (23–25). N1-7D3 could not inhibit the release of sialic acid from fetuin by the NA of any of the seasonal H1N1, pandemic H1N1, or H5N1 viruses tested (Fig. 2A, i). Conversely, monoclonal antibody N1-C4 displayed dose-dependent NI activity against Bel/09 and NIBRG-14, with 50% inhibitory concentrations (IC50s) of 0.6 ± 0.07 μg/ml and 1.7 ± 0.57 μg/ml, respectively (Fig. 2A, i and ii). However, monoclonal antibody N1-C4 could not block hydrolysis of the small substrate MUNANA by NA, suggesting that N1-C4 does not have an allosteric effect on the enzyme active site or shields the active site.
FIG 2.

(A) Monoclonal antibody N1-C4 inhibits IAV NA activity. (i) USSR/77, Sing/86, NC/99, Bris/07, Bel/09, or NIBRG-14 was incubated with 1 (black bars), 5 (white bars), or 10 (gray bars) μg/ml of N1-7D3 or N1-C4, and NA activity was determined at 18 h postincubation on fetuin as described in Materials and Methods. The data are mean percentages of virus NA activity (plus 1 SD) from triplicate wells and are representative of 2 independent experiments. (ii) IC50 doses of Bel/09, NIBRG-14, and a NIBRG-14 oseltamivir-resistant variant (NIBRG-14R) were determined by nonlinear regression analysis. The data are means (plus 1 SD) of the results of experiments performed in triplicate. (B) Ability of monoclonal antibody N1-C4 to restrict viral growth in MDCK cells. Bel/09 (100 PFU) or NIBRG-14 (50 PFU) was added to MDCK cells for 1 h, and the cells were subsequently washed thoroughly with serum-free medium. Avicel (0.6%) containing 2 μg/ml trypsin with N1-C4 (20 μg/ml or 5 μg/ml) or without N1-C4 was overlaid. The plaques were immunostained (i), and their sizes were determined (ii) as described in Materials and Methods. The immunostaining is representative of one well of the assay performed in triplicate. The plaque sizes are averages of the three wells plus 1 SD. ***, P < 0.001; one-way ANOVA followed by Tukey's postanalysis.
Oseltamivir, an NA inhibitor, is the most widely used influenza antiviral medication in public health. While recent seasonal H3N2, A(H1N1)pdm09, and influenza B virus remain sensitive to oseltamivir treatment, this was not always the case. Prior to 2009, circulating seasonal H1N1 IAVs were resistant to oseltamivir, and resistance has also been detected in the past in H5N1 viruses (26). It was therefore important to test if N1-C4 could inhibit the NA activity of oseltamivir-resistant viruses. We tested a NIBRG-14 oseltamivir-resistant strain that carries an H274Y (N2 numbering) mutation that is well known to lead to resistance (27). N1-C4 could inhibit the NA activity of the oseltamivir-resistant virus (IC50, 1.2 ± 0.24 μg/ml) to the same degree as the wild-type (WT) virus (IC50, 1.7 ± 0.57 μg/ml) (Fig. 2A).
Next, the ability of N1-C4 to restrict viral growth in vitro was determined. Bel/09 and NIBRG-14 were added to a monolayer of MDCK cells for 1 h to allow binding and endocytosis. Unbound virus was subsequently washed away, and an Avicel overlay was added with 20 or 5 μg/ml of N1-C4 or without N1-C4. Both concentrations of N1-C4 significantly reduced the plaque size of the two viruses compared to an untreated control (P < 0.001; one-way analysis of variance [ANOVA]) (Fig. 2B). Finally, we tested if N1-C4 could inhibit agglutination of red blood cells by Bel/09 and NIBRG-14 virions, as previous studies have shown that antibodies directed to NA could hinder access of HA to sialic acid (24, 25). Bel/09 and NIBRG-14 agglutination was unaffected by the addition of N1-C4, even at the highest concentration tested (20 μg/ml) (data not shown). The in vitro antiviral properties of N1-7D3 were also tested against USSR/77, Sing/86, NC/99, Bris/07, Bel/09, and NIBRG-14. However, this monoclonal antibody could not reduce plaque size (data not shown).
Identification of the binding sites that determine recognition by monoclonal antibody N1-7D3 or N1-C4.
Compared to HA, there are far fewer reports on the antigenic properties of NA as defined by monoclonal antibodies. As such, we aimed to define the binding sites and critical residues of N1-7D3 and N1-C4 on NA. Initially, to determine if the monoclonal antibodies bound to a linear or conformational epitope, Bel/09 rNA was resolved under denaturing and reducing conditions by SDS-PAGE, and subsequently, a Western blot was performed that was probed with N1-7D3 or N1-C4. N1-7D3 could bind to Bel/09 rNA in the Western blot, suggesting that N1-7D3 recognizes a linear epitope, whereas N1-C4 failed to do so and likely recognizes a conformational epitope in NA (Fig. 3A).
FIG 3.

(A) N1-7D3 binds to a linear epitope, but N1-C4 binds to a conformational epitope. Recombinant soluble NA derived from Bel/09 was resolved on SDS-10% PAGE under denaturing and reducing conditions. The gel was subsequently transferred to nitrocellulose, and binding of N1-7D3 (left) or N1-C4 (right) was detected with goat anti-mouse conjugated to HRP by Western blotting. One microgram of monoclonal antibody N1-7D3 or N1-C4 was run and blotted concurrently as a positive control for binding of secondary antibody and a control for reducing conditions. (B) Binding of N1-7D3 to overlapping peptides derived from Cal/09 H1N1 IAV. An ELISA plate was coated with 115 overlapping peptides representing the full-length NA of Cal/09 A(H1N1)pdm09 IAV in sodium carbonate buffer overnight at 37°C. The plate was blocked with 5 mg/ml BSA in PBS for 1 h at room temperature and washed with PBST. N1-7D3 (2 μg/ml) was applied for 2 h, and binding of the monoclonal antibody to the peptides was detected by ELISA with goat anti-mouse IgG conjugated to HRP. (C) Overlapping peptide sequence responsible for the binding of N1-7D3 modeled on the N1 protein. Depicted is the N1 NA tetramer viewed from the side (left) or the top (right) (Protein Data Bank [PDB] 2HTY) with the active site (red) and the peptide sequence involved in the binding site of N1-7D3 (blue) indicated.
As N1-7D3 likely recognized a linear epitope, we utilized a tiled peptide array derived from A/California/04/2009 A(H1N1)pdm09 NA to identify the binding site. N1-7D3 bound to a peptide sequence at the C terminus of the NA protein (F457 to K469) (Fig. 3B). This sequence is present in all H1N1 and H5N1 viruses tested in this study and is highly conserved among N1 influenza viruses in nature, where the majority of these residues, except E462 (42% conservation), were more than 97% conserved across N1 NAs from human, avian, and swine viruses (Table 1). The epitope is modeled on an NA tetramer (Fig. 3C, highlighted in blue). As expected, N1-C4 did not bind to any of the overlapping peptides in the peptide array (data not shown).
TABLE 1.
N1-7D3 epitope percent residue conservation in human, avian, and swine N1 viruses
| Virusa | % conservation at residueb: |
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| W457 | P458 | D459 | G460 | A461 | E462 | D462 | L463 | P464 | F465 | T466 | I467 | D468 | K469 | |
| Human H1N1 pre-2009 | 100 | 100 | 100 | 99.5 | 99.9 | 98.8 | 1.2 | 99.8 | 99.9 | 99.6 | 98.4 | 99.7 | 99.8 | 99.8 |
| A(H1N1)pdm09 | 99.8 | 99.8 | 99.9 | 100 | 100 | 99.7 | 0.04 | 99.6 | 99.9 | 99.9 | 98.5 | 97.8 | 99.8 | 98.4 |
| Avian H*N1 | 99.8 | 99.7 | 99.7 | 99.5 | 99.8 | 99.5 | 0.43 | 97.5 | 100 | 99.7 | 98.8 | 99.2 | 99.7 | 99.3 |
| Swine H*N1 | 99.9 | 99.9 | 99.8 | 100 | 99.2 | 42.4 | 54.5 | 99.7 | 100 | 99.8 | 92.3 | 96.8 | 99.4 | 99.6 |
To determine the likely binding site of N1-C4 on Bel/09 NA, an escape mutant approach was used. Sequence analysis of NAs cloned from viruses that could escape from the N1-C4 plaque size reduction effect identified several mutations, alone or in combination, that were possibly involved in escape from N1-C4 (Table 2). To confirm which mutations were responsible for N1-C4 escape, site-directed mutagenesis was performed, and the corresponding Bel/09 NA mutant viruses were rescued by reverse genetics (RG) on the PR8 background (as described in Materials and Methods). Four single-point-mutant viruses were generated: RG Bel/09K260E, RG Bel/09S266A, RG Bel/09E311K, and RG Bel/09S364G. The plaque sizes of RG Bel/09K260E and RG Bel/09S364G viruses were still inhibited by N1-C4 (Fig. 4A). N1-C4 failed to reduce the plaque size of RG Bel/09E311K, indicating that residue E311 is critical for the antiviral activity of N1-C4 against Bel/09 NA (Fig. 4A). Additionally, the ability of N1-C4 to reduce the plaque size of RG Bel/09S266A was diminished in comparison to the RG-WT virus (Fig. 4A). Therefore, we suggest that residue S266 is part of the footprint of N1-C4 but only partially contributes to N1-C4 binding. Of interest, E311 is more than 98% conserved in A(H1N1)pdm09 and avian N1s. In contrast, E311 is rarely observed in human pre-2009 H1N1 viruses (0.2%), where D311 is the dominant residue (99.7% conservation) (Table 3), aligning with the inability of N1-C4 to bind to pre-2009 IAVs. Furthermore, 40% of swine N1 viruses contain E311 (Table 3), suggesting that some swine N1s may also be sensitive to the antiviral activities of N1-C4. Finally, to address if there was a fitness cost associated with mutation of S266 or E311 in the NA, viruses were compared to wild-type virus in a multicycle growth curve in MDCK cells. The introduction of A266 or K311 to the NA did not affect the ability of these viruses to replicate over time compared to the wild type, indicating no loss in fitness (data not shown).
TABLE 2.
Residues identified in escape mutant selectiona
| Escape mutant | Mutation |
|---|---|
| 1 | E311D |
| 2 | E311K |
| 3 | K260E/E311K |
| 4 | S266A/S364G |
| 5 | S266A/E311K |
Bel/09 was selected for resistance against N1-C4. Resistant viruses were identified via escape in a plaque reduction assay where the NA was cloned into pBluescript and sequenced to determine mutations.
FIG 4.
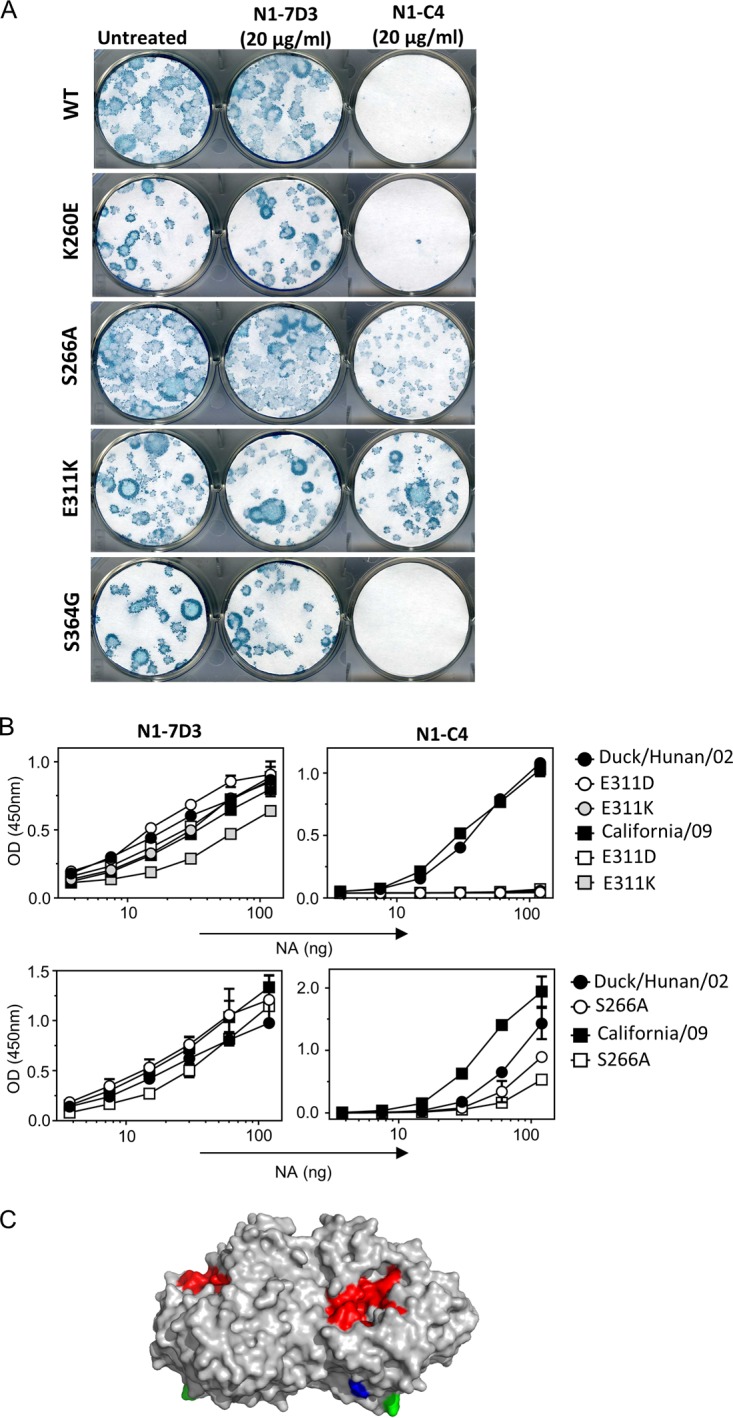
E311 is critical for the binding of N1-C4 to N1 NA. (A) N1-C4 does not reduce the plaque size of the RG Bel/09 E311K mutant. RG Bel/09 WT or NA mutants (100 PFU) were added to MDCK cells for 1 h, and the cells were subsequently washed thoroughly with serum-free medium. Avicel (0.6%) containing 2 μg/ml trypsin without added monoclonal antibody (untreated) or with either N1-7D3 or N1-C4 (20 μg/ml) was overlaid. The plaques were immunostained with mouse Bel/09 postchallenge serum and anti-mouse HRP. TrueBlue substrate was used to visualize the plaques. (B) E311 and S266 are important for N1-C4 binding to A(H1N1)pdm09 and H5N1 NA. Recombinant WT and mutant NAs were produced as described in Materials and Methods and titrated in 2-fold dilutions at a starting concentration of 120 ng/well. An ELISA was used to test the binding of N1-7D3 (1 μg/ml) (left) and N1-C4 (2 μg/ml) (right). (C) S266 and E311 modeled on the N1 protein. Depicted is the N1 NA from the side perspective (PDB 3NSS) with the active site in red and residues S266 and E311 highlighted in green and blue, respectively.
TABLE 3.
N1-C4 potential binding site percent residue conservation in human, avian and swine N1 viruses
Next, we wanted to define if E311 and S266 were also involved in the binding of N1-C4 to H5N1 viruses. The mutations S266A, E311K, and E311D were therefore introduced into a vector expressing the soluble recombinant NA from A/Duck/Hunan/02 (H5N1) and A/California/04/09 [A(H1N1)pdm09], and the binding of N1-7D3 and N1-C4 was assessed in an ELISA. D311 was included, as it was the residue that was most frequently found in N1-C4-resistant human H1N1 isolates. N1-7D3 bound to all soluble NAs with similar efficiencies, indicating the ELISA plates were coated with comparable levels of NA. Conversely, the binding of N1-C4 was completely abolished when D311 or K311 was introduced into rNAs from A/Duck/Hunan/02 and A/California/04/09, confirming that E311 is important for recognition of both H5N1 and A(H1N1)pdm09 viruses by N1-C4 (Fig. 4B). The introduction of the S266A mutation significantly reduced the binding of N1-C4 to rNAs from both A/Duck/Hunan/02 and A/California/04/09, in line with the plaque reduction results (Fig. 4). The positions of residues E311 and S266 in the crystal structure of H1N1pdm NA is depicted in Fig. 4C and indicates that they are in close proximity to each other. Taken together, these data show that residues E311 and S266 play a role in the binding of N1-C4 to A(H1N1)pdm09 NA and avian N1 NA.
Monoclonal antibody N1-C4, but not N1-7D3, protects mice from lethal infection with IAV and reduces viral loads in lungs.
NA antibodies generated by vaccination or natural infection correlate with protection against influenza in humans (14, 17). Further, therapeutic treatment with NA targeting monoclonal antibodies has been shown to ameliorate disease in the mouse model of influenza virus infection (19, 23, 28). First, we tested the ability of N1-7D3 to protect against morbidity and mortality induced by N1 IAV infection. Mice were intraperitoneally (i.p.) administered 100 μg of N1-7D3 and challenged 24 h later with 4 50% lethal doses (LD50) of either Sing/86, NC/99, Bris/07, Bel/09, or NIBRG-14. Pretreatment of mice with N1-7D3 did not alter weight loss (data not shown) or survival outcomes (Fig. 5). Additionally, preincubation of N1-7D3 with 2 LD50 of Bel/09 or Sing/86 prior to infection did not protect mice from mortality (data not shown).
FIG 5.
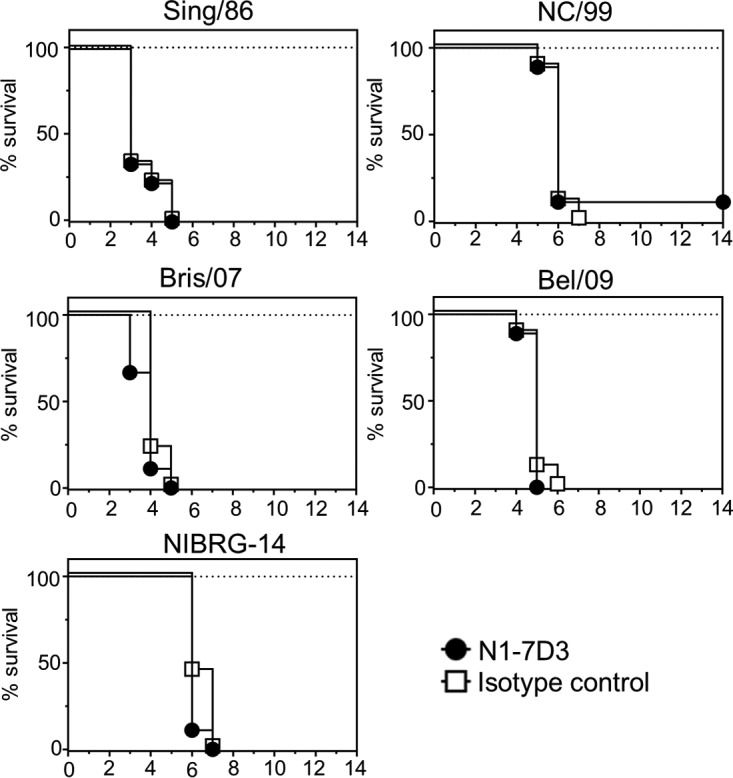
N1-7D3 does not protect against lethal infection with H1N1 and H5N1 viruses. Nine mice per group were treated with 100 μg of N1-7D3 (black circles) or an IgG1 isotype control (white squares) via the i.p. route. Twenty-four hours later, the mice were challenged intranasally with 4 LD50 of either Sing/86, NC/99, Bris/07, Bel/09, or NIBRG-14 and monitored for survival. Mice that had lost ≥25% of their original body weight were sacrificed. Survival was assessed using the log rank test.
N1-C4 monoclonal antibody displayed a more discrete range of binding activity than N1-7D3. As such, only the ability of N1-C4 to protect against Bel/09 and NIBRG-14 infection was tested. Mice were treated via the intranasal route one day prior to infection with 20 μg of N1-C4 or isotype control and challenged the following day with 1 or 4 LD50 of Bel/09 or NIBRG-14. N1-C4 significantly protected mice compared to isotype-treated controls (Fig. 6). Bel/09-challenged mice infected with 1 or 4 LD50 displayed no significant body weight loss, and all the mice survived infection at both low (Fig. 6A, i) and high (Fig. 6B, i) doses when treated with N1-C4. NIBRG-14-infected mice, pretreated with N1-C4, displayed transient weight loss at 1 or 4 LD50, with the majority of the mice recovering fully from the infection, which corresponded to 100% survival after 1 LD50 (Fig. 6A, ii) and 84% survival after infection with 4 LD50 (Fig. 6B, ii).
FIG 6.
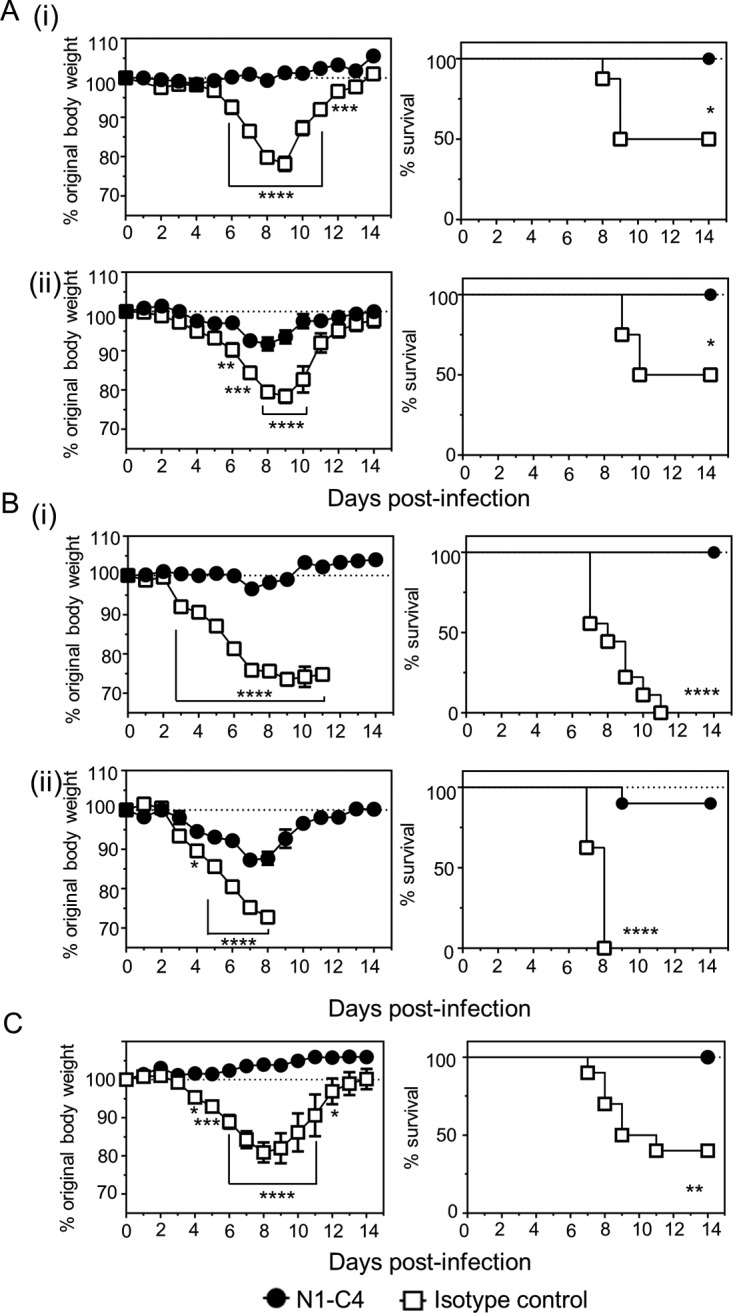
Treatment of mice with N1-C4 protects against lethal infection with H1N1 or H5N1 IAV. (A and B) BALB/c mice were treated with 20 μg of N1-C4 or an isotype control via the intranasal route under isoflurane sedation. The following day, the mice were infected with 1 LD50 (A) or 4 LD50 (B) of Bel/09 (i) or NIBRG-14 (ii) and monitored daily for weight loss (left) and survival (right). (C) Therapeutic treatment of mice with N1-C4 ameliorates disease induced by Bel/09. BALB/c mice were infected via the intranasal route with 1 LD50 of Bel09. Monoclonal antibody N1-C4 (200 μg) was administered by the i.p. route on days 1, 3, and 5 postinfection. A group of mice received an isotype control. The mice were monitored daily, and any mice that had lost ≥25% of their original body weight were euthanized. The weight loss data represent the mean percentages (± standard errors of the mean [SEM]) of original body weight over time (n = 8), and the survival data are shown as percent survival over time (n = 8). Weight loss over time was analyzed by two-way ANOVA with Bonferroni correction, and survival proportions were assessed using a two-tailed log rank (Mantel-Cox) test. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Next, we tested if monoclonal antibody N1-C4 could also therapeutically protect mice from disease induced by Bel/09 challenge (Fig. 6C). Groups of mice were infected with 1 LD50 of Bel/09 and treated i.p. with 200 μg of N1-C4 or an isotype control antibody on days 1, 3, and 5 postinfection. Mice treated with the isotype control lost significantly more weight on days 4 to 11 postinfection than N1-C4-treated mice. Moreover, 100% of the mice treated with N1-C4 survived infection compared to 40% survival of isotype control-treated mice (P < 0.01; log rank test).
To assess if monoclonal antibody N1-C4 could reduce lung viral loads, mice were prophylactically treated intranasally (i.n.) with 20 μg N1-C4 or isotype control, and lung samples were isolated at days 3 and 6 after infection. Compared to the isotype control group, treatment of mice 1 day prior to infection with monoclonal antibody N1-C4 significantly reduced the level of virus in the lung homogenates of mice that had been infected with 0.01 LD50 Bel/09 (at day 3 [P < 0.0001; Student's t test] and day 6 [P < 0.01; Student's t test] postinfection) (Fig. 7A). To rule out a possible in vitro viral-titer-reducing effect due to residual N1-C4 in the lung samples, RNA was extracted from the lung homogenates and reverse transcription-quantitative PCR (RT-qPCR) was performed on the matrix (M) gene (Fig. 7B). Confirming the TCID50 results, on both day 3 and day 6 there was a significant decrease in viral M RNA levels when mice were treated with N1-C4 versus isotype-treated animals (P < 0.0001 on day3; P < 0.01 on day 6; Student's t test). Taken together, these data show that monoclonal antibody N1-C4 can significantly reduce disease and control mortality caused by H1N1pdm or an H5N1 infection in mice.
FIG 7.
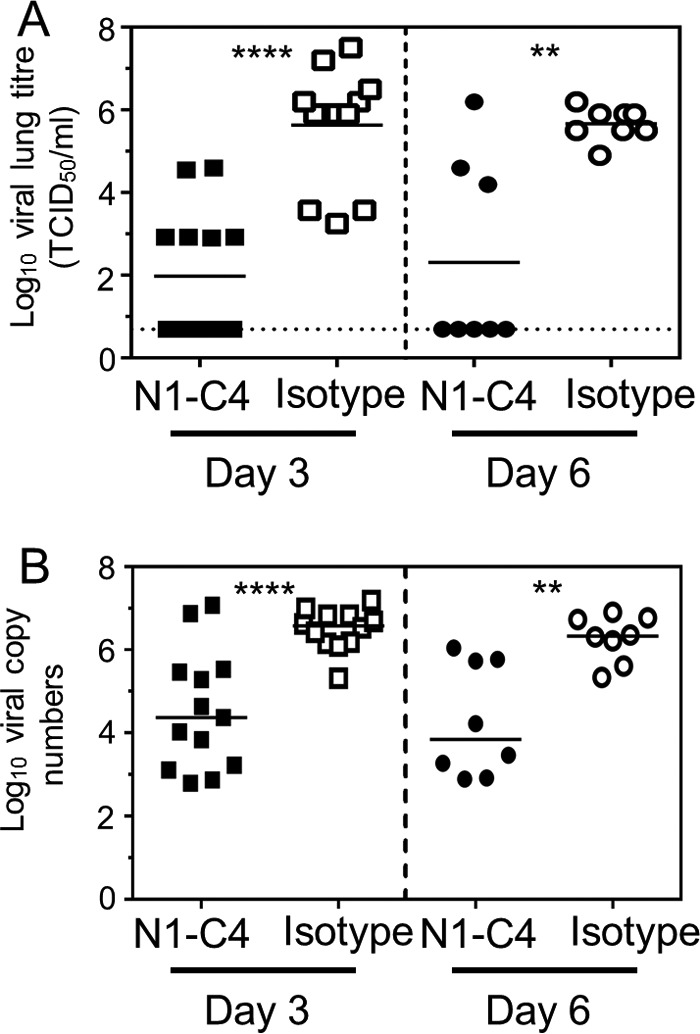
Administration of N1-C4 monoclonal antibody reduces viral loads within the lungs of Bel09-infected mice. One day prior to infection with 0.1 LD50 of Bel/09, groups of mice were administered 20 μg of N1-C4 monoclonal antibody or isotype control (Isotype) via the intranasal route. On days 3 and 6 postinfection, lungs were harvested and lung homogenates were assessed for viral titers by TCID50 (A) and RT-qPCR (B) on the viral matrix gene as outlined in Materials and Methods. Data for individual mice are shown. The horizontal lines represent the means (n = 11, pooled from three independent experiments). The dotted line indicates the detection limit for the TCID50. **, P < 0.01; ****, P < 0.0001; Student's t test.
DISCUSSION
With the prevalence of resistance to licensed influenza antiviral drugs increasing, it is becoming more important to develop new therapeutics and prophylactics to control infection. Monoclonal antibodies targeting conserved regions in HA are being developed to try to control influenza virus infection (reviewed in reference 29), and other monoclonal antibodies against a wide range of infectious diseases are licensed or in clinical development (reviewed in reference 30). Monoclonal antibodies toward the NAs of H7N9 (31), H10N8 (32), H1N1 (21, 28), H5N1 (20), and H3N2 (21) can ameliorate influenza virus infection within various animal models. Here, we characterize two N1 NA-specific monoclonal antibodies, one of which is able to inhibit NA enzymatic activity, has in vivo antiviral activity, and mediates heterosubtypic protective immunity, both prophylactic and therapeutic, against H1N1 and H5N1 infection in mice.
Wan et al. have previously described a set of N1 monoclonal antibodies raised to Bris/07 and classified them into distinct groups depending on which N1 viruses they could bind to (19). Based on this nomenclature, N1-7D3 would fit into group B, as it binds to a range of N1 viruses. N1-C4, however, does not fit into any of the groups identified by Wan and colleagues. Recent studies from the same group have addressed A(H1N1)pdm09-specific monoclonal antibodies. The studies highlight NA monoclonal antibodies as potent therapeutics against influenza virus infection in the mouse model (23, 28). Importantly, N1-C4, to our knowledge, is distinct from other published N1-specific monoclonal antibodies, displaying potent heterosubtypic antiviral activity against A(H1N1)09pdm and H5N1 viruses. NA residue E311 was found to be critical for the binding of monoclonal antibody N1-C4 to both A(H1N1)pdm09 and H5N1 IAVs. Several NA residues have been identified in the past as parts of cross-reactive sites between A(H1N1)09pdm and H5N1s. From the study by Wan et al., conserved residues 273, 338, and 339 were present in the NAs of 1918 H1N1, seasonal H1N1, A(H1N1)pdm09, and H5N1 viruses (19). Serological evidence also indicates that there are cross-protective serum antibodies in humans directed against NAs of H5N1 and human H1N1 viruses. NI antibodies against H5N1 can be detected, even though the virus is not widespread in the human population (17). Moreover, a number of animal studies show protection from H5N1 by vaccination with human N1 NA in a variety of vaccination platforms (reviewed in reference 33). The binding sites of both N1-C4 and N1-7D3 showed a high degree of conservation in circulating N1 human and avian IAVs and are promising targets for further drug development. Interestingly, NA from circulating H1N1 and H3N2 undergoes slower antigenic drift than HA but continuous genetic evolution, akin to HA (34). It remains to be seen, though, if considerably more immune pressure is exerted on influenza A and B viruses by increasing NA-based immune protection by vaccination or antivirals, if the rate of antigenic evolution of NA would increase to a level similar to that observed for HA.
N1-C4 could block the NA activity of A(H1N1)pdm09 and H5N1 viruses, although the critical residue for antibody binding is situated some distance from the active site of NA. The A(H1N1)pdm09-specific monoclonal antibody CD6 has been crystalized in complex with NA. Crystallization studies revealed a unique binding pattern where the monoclonal antibody recognized a quaternary epitope in NA that spanned two monomers of the NA tetramer. Upon binding, the amino acids of the variable domains of the antibody light chain are in close proximity to the active site. The authors concluded that CD6 could hinder NA function by either preventing access of multimeric sialic acid substrates to the enzymatic site by its size or by cross-linking adjacent NA tetramers (23). It is plausible that N1-C4 blocks NA activity through a similar mechanism of steric hindrance, given that N1-C4 did not block the access of the small substrate MUNANA to the active site. Further studies are required to confirm this.
While it is clear that the main antiviral activity of inhibition by N1-C4 is primarily by blocking the NA enzymatic activity and, presumably, preventing viral release from the cell surface, it has also been suggested that NA-specific monoclonal antibodies can mediate antibody-dependent cellular cytotoxicity (ADCC) (35, 36) and complement activation (12). It is known that different antibody subclasses can mediate ADCC to varying degrees. N1-C4 and N1-7D3 are both of the IgG1 isotype, which in mice only weakly mediates ADCC and complement-dependent cytotoxicity (CDC), whereas IgG2a and IgG2b are much stronger mediators of these effector mechanisms (37, 38). Furthermore, our data suggest that for a mouse IgG1 subclass antibody, anti-neuraminidase activity is key to protection in vivo. Subclass switch variants, i.e., with the same variable domains expressed on a different subclass Fc chain backbone, are frequently used to modulate such Fc receptor-dependent mechanisms (reviewed in reference 30). As N1-7D3 displayed a broad range of binding to H1N1 IAVs, it would be interesting to see if switching the constant part of its heavy chain to IgG2a or IgG2b would lead to enhanced ADCC or CDC, resulting in protection against influenza A virus infection. Studies have recently shown that the epitope specificity can also play a role in determining ADCC activation. Different nonneutralizing monoclonal antibodies directed to HA or NA could mediate ADCC to varying degrees, and together, NA and stem monoclonal antibodies potently activated ADCC (36). More studies of what governs the engagement of IgG Fc receptors (FcyRs) and activation of ADCC for NA monoclonal antibodies and the cooperative effect with other antibodies is clearly warranted.
To generate the monoclonal antibodies, we used an infection followed by an extensive vaccination schedule in the presence of incomplete Freund's adjuvant (IFA) with the goal of promoting high levels of cross-reactive N1 antibodies. Anti-NA antibodies, however, can be induced in mice by immunization with NA purified from IAV in the absence of adjuvants (39). Furthermore, trivalent inactivated vaccines, which are not adjuvanted, are also known to induce detectable levels of NA responses in humans (40).
NA immune responses have often been neglected in past studies and are commonly overshadowed by the response directed to HA. However, recently, NA has been gaining more interest, and the importance of NA antibodies in humans is becoming more evident (14). Here, we expand on recent studies examining NA monoclonal antibodies directed toward the N1 subtype and highlight NA as an appropriate target for monoclonal antibody antiviral therapies and as a vaccine candidate.
MATERIALS AND METHODS
Viruses.
The IAVs used in this study included the H5N1 IAV strain NIBRG-14, obtained from the United Kingdom national Institute for Biological Standards and Control, a center of the Health Protection Agency. NIBRG-14 is a 6:2 reverse genetics-derived reassortant expressing the NA and HA segments of A/Vietnam/1194/2004 and the other 6 genes from A/Puerto Rico/8/34 (PR8/34). The H1N1 strains used in this study were A/USSR/90/1977 (USSR/77), provided by Guus Rimmelzwaan (Erasmus University Medical Centre, Rotterdam, Netherlands), and A/Singapore/6/1989 (Sing/86), A/Brisbane/59/2007 (Bris/07), and A/New Caledonia/20/1999 (NC/99), provided by Alan Hay (Medical Research Council National Institute for Medical Research, United Kingdom). The A(H1N1)pdm09 virus A/Belgium/1/2009 (Bel/09) was obtained from Isabelle Thomas, Scientific Institute of Public Health, Brussels, Belgium. All the viruses used in this study were adapted to mice by serial passage in mouse lungs, as described previously (41), unless otherwise stated. For simplicity, the above-mentioned abbreviations for viruses are used for the mouse-adapted viruses. A previously described oseltamivir-resistant variant of NIBRG-14 was also used (27). The viruses were propagated in MDCK cells in serum-free medium in the presence of tosylsulfonyl phenylalanyl chloromethyl ketone (TPCK)-trypsin (Sigma-Aldrich), and the median tissue culture infective dose (TCID50) and LD50 of the viruses in BALB/c mice were calculated by the method of Reed and Muench (42). Standard H1N1 numbering is used throughout this study unless otherwise indicated.
Monoclonal antibody N1-C4-resistant mutants of Bel/09 were selected as described previously (27). Briefly, a clonal stock of Bel/09 was diluted 10-fold starting from a multiplicity of infection (MOI) of 10 and used to infect MDCK cells in the presence of 10 μg/ml (final concentration) monoclonal antibody N1-C4 and TPCK-trypsin. Three days postinfection, virus in the supernatant of the highest dilution displaying cytopathic effect (CPE) was harvested and used for a subsequent round of selection. After 2 selection rounds, virus was harvested and tested for escape from N1-C4 in a plaque reduction assay. Escape mutants were plaque picked based on plaque size and grown at small scale on MDCK monolayers. Viral RNA was isolated from the MDCK supernatant using the Nucleospin RNA virus kit (Macherey-Nagel) according to the manufacturer's instructions. cDNA was synthesized with a gene-specific primer (43) using the Transcriptor First Strand cDNA synthesis kit (Roche). NA segments were amplified with a gene-specific forward primer and a universal reverse primer (43, 44) using a Phusion High Fidelity DNA polymerase kit (Finnzymes). PCR products were purified using the Isolate II PCR and gel kit (Bioline) and cloned in the pBluescript II Phagemid KS+ vector (Agilent Technologies) by blunt-end ligation using T4 ligase (Fermentas). Candidate positive colonies were selected by blue-white screening, and the extracted plasmid DNA was sequenced to identify mutations in NA.
RG viruses were generated by the eight-plasmid RG technique as described previously (45). Site-directed mutagenesis using a quick-change kit (Stratagene) was used to introduce nucleic acid mutations K260E, S266A, E311K, and S364K in the NA of Bel/09. The viruses generated were 6:2 reassortants consisting of 6 genes from PR8 in conjunction with wild-type HA and NA from Bel/09 or mutant NAs.
Generation of monoclonal antibodies directed toward N1 NA.
To generate NA-specific monoclonal antibodies, mice were first infected via the i.n. route with 0.2 LD50 of Bel/09. Three weeks later, the mice were boosted by intramuscular injection of 300 hemagglutination units of Bel/09. A third immunization, again 3 weeks later, given subcutaneously, was with 1 μg of purified soluble recombinant tetrameric Bel/09 NA (46) in IFA (Sigma-Aldrich). Finally, 3 weeks later, the mice were boosted via the i.p. route with 5 μg of IFA-adjuvanted A/Crested Eagle/Belgium/01/04 recombinant NA produced in insect cells (27). Four days after the final immunization, splenocytes were isolated and pelleted with SP2/0-Ag14 cells in a 5:1 ratio and resuspended with 1 ml polyethylene glycol (PEG) 1500. The cells were washed and incubated in RPMI medium plus 10% fetal calf serum (FCS), 0.4 mM sodium pyruvate, nonessential amino acids, 100 U/ml penicillin, 0.1 mg/ml streptomycin, and 10% BM Condimed H1 (Roche) for 5 h in a tissue culture flask at 37°C. Hybridomas were selected for by growth in hypoxanthine-aminopterin-thymidine medium. Supernatants from candidate hybridomas were screened for binding in an ELISA to MDCK cells infected at an MOI of 1 with USSR/77, Sing/86, NC/99, Bris/07, or Bel/09, and positive clones were further subcloned. Positive hybridoma clones were grown in bulk in roller bottles, and cell-free supernatant was collected and purified using a HiTrap MabSelect SuRE column (GE Healthcare, Life Sciences).
Production and purification of soluble rNA.
Recombinant tetrameric NAs were produced essentially as described previously for Bel/09 rNA (46). In brief, the stalk of the NAs was replaced by a tetramerization coiled coil, and secretion was facilitated by an N-terminal CD5-derived secretion signal. A Strep-tag was cloned between the secretion signal and the coiled coil for purification (47). The coding sequences of the NAs from USSR/77, Sing/86, NC/99, and Bris/07 were isolated by RT-PCR of RNA isolated from MDCK cell-grown virus and cloned into pEF. rNA was affinity trapped from the supernatant of transiently transfected HEK293T cells using a StrepTrap HP column and eluted with 2.5 mM desthiobiotin in phosphate-buffered saline (PBS), followed by size exclusion chromatography in PBS using an AKTAexplorer purification system (GE Healthcare Life Sciences).
ELISA for antibody binding.
Ninety-six-well MaxiSorp plates (Nunc) were coated overnight with NA (1 μg/ml) or individual peptides from the peptide array derived from A/California/04/2009 (H1N1)pdm09 neuraminidase protein (NR-18975; obtained through BEI Resources, NIAID, NIH) in sodium bicarbonate buffer. The plates were blocked for at least 1 h at room temperature with 5% skim milk, and subsequently, hybridoma supernatant (neat) or purified monoclonal antibody (increasing concentrations) in PBS plus 2.5% skim milk and 0.05% Tween 20 (Sigma-Aldrich) were added for 2 h at room temperature. The plates were washed with PBS plus 0.1% Tween 20 (PBST), and horseradish peroxidase (HRP)-coupled goat anti-mouse total IgG, IgG1, IgG2a, or IgG2b antibody (Southern Biotech) was added. For detection, 3,3′,5,5′-tetramethylbenzidine (TMB) substrate (Pharmingen BD) was added, the reaction was stopped by using 1 M H2SO4, and absorbance was read at 450 and 655 nm.
Flow cytometry analysis of monoclonal antibodies binding to influenza virus-infected cells.
MDCK and HEK293T cells in 6-well plates were infected with IAV at an MOI of 1 for 16 h. The cells were released from the tissue culture plates using enzyme-free dissociation buffer (Gibco) and stained with monoclonal antibody N1-7D3 or N1-C4 at 10 μg/ml in PBS containing 2 mg/ml bovine serum albumin (BSA) and 2 mM EDTA for 30 min on ice. Subsequently, a goat anti-mouse IgG conjugated to Alexa Fluor 488 (AF488) and the fixable viability dye eFluor 506 (eBiosciences) was added according to the manufacturer's instructions. The cells were fixed with 4% paraformaldehyde (PFA) on ice, and binding was determined by flow cytometry analysis on an LSRII (BD Biosciences).
NA and NA inhibition assays.
The ability of monoclonal antibodies to inhibit the activity of the viral NA was measured using the standard enzyme-linked lectin assay (ELLA) essentially as described previously (48). Briefly, dilutions of monoclonal antibody were incubated with IAV at a predetermined concentration of virus to give 90% maximal NA activity for 30 min at 37°C in PBS supplemented with 10 mg/ml BSA, 1 mM CaCl2, 0.5 mM MgCl2, and 0.5% Tween 20. Dilutions were added to PBST-washed fetuin (Sigma-Aldrich; 5 μg/ml)-coated wells of a 96-well MaxiSorp plate (Nunc) and incubated for 18 h at 37°C. HRP-coupled peanut agglutinin (PNA) (Sigma-Aldrich; 2.5 μg/ml) was used to detect galactose residues exposed after removal of sialic acid from fetuin. The IC50 was calculated by nonlinear regression analysis (GraphPad Prism).
Hemagglutination and HI assays.
Hemagglutination and HI tests were performed in a round-bottom 96-well microtiter plate at room temperature using 1% (vol/vol) chicken erythrocytes in PBS with 4 hemagglutinating units (HAU) of virus according to the WHO manual for influenza research (49).
Plaque and plaque size reduction assays.
Confluent monolayers of MDCK cells in 6-well plates were infected with 50 to 100 PFU of virus for 1 h at 37°C. The cells were washed thoroughly and overlaid with 0.6% Avicel RC-591 (FMC Biopolymer) alone or with monoclonal antibodies supplemented with 2 μg/ml TPCK-treated trypsin (Sigma). The cells were incubated for 3 to 5 days at 37°C and 5% CO2, the Avicel was removed, and the cells were fixed with 4% PFA for 15 min. Subsequently, plaques were stained with postchallenge mouse serum directed to the specific virus, followed by goat anti-mouse HRP-linked antibody (GE Healthcare). After washing, TrueBlue peroxidase substrate (KPL) was used to visualize the plaques. The wells were also scanned, and image analysis for plaque size was performed using Volocity 3D image analysis software (PerkinElmer).
Binding to soluble recombinant wild-type and mutant NAs.
Human codon-optimized NA ectodomains of A/duck/Hunan/795/2002 (GenBank accession no. BAM85820.1; amino acids 62 to 469) and of A/California/04/2009 (GenBank accession no. ACP41107.1; amino acids 42 to 469) were cloned into a pFRT expression plasmid (Thermo Fisher Scientific). The soluble NA-encoding sequences were preceded by sequences coding for an N-terminal signal sequence derived from Gaussia luciferase, a double Strep-tag for affinity purification (One-STrEP; IBA GmbH), and a Staphylothermus marinus tetrabrachion tetramerization domain, as described previously (50, 51). Mutations of interest were introduced into the corresponding NA genes using a Q5 site-directed mutagenesis kit (New England BioLabs) and confirmed by sequencing. NA proteins were expressed by transfection of HEK293T cells and purified from the cell culture supernatants using Strep-tactin beads (IBA) as described previously (50, 51). The concentration of soluble NA was quantified by comparative Coomassie gel staining using a BSA standard. These purified proteins were used for the subsequent ELISA. Wells of 96-well plates were coated with 2-fold serially diluted purified NA proteins (120 ng to 1.8 ng). The plates were blocked overnight with PBST containing 3% BSA. N1-7D3 or N1-C4 was added and incubated at room temperature for 1 h, followed by the addition of rabbit anti-mouse IgG-HRP (Dako; 1:2,500 dilution). Antibody binding was determined by using TMB substrate.
Treatment and infection of mice.
All mouse experimentation complied with national (Belgian laws 14/08/1986 and 22/12/20333; Belgian Royal Decree 06/04/2010) and European (European Union [EU] directives 2010/63/EU and 86/609EEG) animal regulations. Experiments were approved by the ethics committee of Ghent University, Faculty of Sciences (no. 2014-068), and efforts were made to avoid or diminish suffering of the animals.
Six- to 8-week-old female BALB/c mice (Charles River) were housed under specific-pathogen-free conditions with food and water ad libitum. The mice were treated with monoclonal antibody via the i.p. or the i.n. route under mild anesthesia 1 day prior to infection for prophylactic experiments. For therapeutic experiments, mice were initially infected with influenza virus, followed by 200 μg monoclonal antibody i.p. on days 1, 3, and 5. Influenza virus infections were performed under isoflurane anesthesia, and a total of 50 μl was instilled equally across the nostrils of the mouse. After infection, the body weight of the mice was determined daily. Animals that had lost ≥25% of their original body weight were humanely euthanized by cervical dislocation. On various days postinfection, mice were sacrificed and lungs were collected as described previously (52).
TCID50.
TCID50 assays were used to determine the amount of infectious virus in clarified lung homogenates or cell culture supernatant. Briefly, MDCK cells cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FCS, nonessential amino acids, 2 mM l-glutamine, and 0.4 mM sodium pyruvate were seeded in 96-well plates to reach confluence overnight at 37°C in 5% CO2. The cells were then washed in serum-free medium and incubated with 10-fold dilutions of virus samples in serum-free DMEM containing 1 μg/ml TPCK-treated trypsin (Sigma). After 7 days, the presence of virus in the wells was determined by agglutination of chicken erythrocytes. TCID50 values were calculated by the method of Reed and Muench (42).
RT-qPCR for detection of vRNA.
RNA was extracted from clarified lung homogenates using the Nucleospin RNA virus kit (Macherey-Nagel). Reverse transcription was performed using a Transcriptor first-strand cDNA synthesis kit (Roche) with 2 μl of extracted RNA and a primer complementary to the conserved 12 nucleotides (nt) of the 3′ terminus of the viral RNA (vRNA) (Uni12 primer) for vRNA. Levels of the M gene were determined by RT-qPCR using TaqMan chemistry. The primers and probes for the IAV M gene were as follows: forward primer, 5′-GAC CRA TCY TGT CAC CTC TGA C-3′; reverse primer, 5′-AGGG CAT TYT GGA CAA AKC GTC TA-3′; and probe, 5′-FAM-TGC AGT CCT CGC TCA CTG GGC ACG-black hole quencher 1 (BHQ1) (53) LightCycler 480 master mix for real-time PCR was used (Roche) on an LightCycler 480/1536 instrument (Roche) using a standard program. M vRNA copy numbers were calculated by generating a standard curve using serial dilutions of plasmid containing DNA for the IAV M gene.
Statistical analysis.
For comparison of two sets of values, Student's t test (two-tailed; two-sample equal variance) was used. When comparing three or more sets of values, the data were analyzed by one-way ANOVA followed by post hoc analysis using Tukey's multiple-comparison test. For changes over time, two-way ANOVA with Bonferroni correction was used. A log rank test was applied to assess survival significance. P values of <0.05 were considered significant.
ACKNOWLEDGMENTS
This work was funded by Sanofi Pasteur. K.R. was supported by EC-FP7 project FLUNIVAC.
We thank Amanda Gonçalves from the VIB Bio Imaging core facility for her expertise and processing of the plaque size images.
REFERENCES
- 1.Palese P, Tobita K, Ueda M, Compans RW. 1974. Characterization of temperature sensitive influenza virus mutants defective in neuraminidase. Virology 61:397–410. doi: 10.1016/0042-6822(74)90276-1. [DOI] [PubMed] [Google Scholar]
- 2.Job ER, Bottazzi B, Gilbertson B, Edenborough KM, Brown LE, Mantovani A, Brooks AG, Reading PC. 2013. Serum amyloid P is a sialylated glycoprotein inhibitor of influenza A viruses. PLoS One 8:e59623. doi: 10.1371/journal.pone.0059623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cohen M, Zhang X-Q, Senaati HP, Chen H-W, Varki NM, Schooley RT, Gagneux P. 2013. Influenza A penetrates host mucus by cleaving sialic acids with neuraminidase. Virol J 10:321. doi: 10.1186/1743-422X-10-321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang X, Steukers L, Forier K, Xiong R, Braeckmans K, Van Reeth K, Nauwynck H. 2014. A beneficiary role for neuraminidase in influenza virus penetration through the respiratory mucus. PLoS One 9:e110026. doi: 10.1371/journal.pone.0110026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gravel C, Li C, Wang J, Hashem AM, Jaentschke B, Xu K, Lorbetskie B, Gingras G, Aubin Y, Van Domselaar G, Girard M, He R, Li X. 2010. Qualitative and quantitative analyses of virtually all subtypes of influenza A and B viral neuraminidases using antibodies targeting the universally conserved sequences. Vaccine 28:5774–5784. doi: 10.1016/j.vaccine.2010.06.075. [DOI] [PubMed] [Google Scholar]
- 6.Su B, Wurtzer S, Rameix-Welti M-A, Dwyer D, van der Werf S, Naffakh N, Clavel F, Labrosse B. 2009. Enhancement of the influenza A hemagglutinin (HA)-mediated cell-cell fusion and virus entry by the viral neuraminidase (NA). PLoS One 4:e8495. doi: 10.1371/journal.pone.0008495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matrosovich MN, Matrosovich TY, Gray T, Roberts NA, Klenk H-D. 2004. Neuraminidase is important for the initiation of influenza virus infection in human airway epithelium. J Virol 78:12665–12667. doi: 10.1128/JVI.78.22.12665-12667.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bosch BJ, Bodewes R, de Vries RP, Kreijtz JHCM, Bartelink W, van Amerongen G, Rimmelzwaan GF, de Haan CAM, Osterhaus ADME, Rottier PJM. 2010. Recombinant soluble, multimeric HA and NA exhibit distinctive types of protection against pandemic swine-origin 2009 A(H1N1) influenza virus infection in ferrets. J Virol 84:10366–10374. doi: 10.1128/JVI.01035-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schulman JL, Khakpour M, Kilbourne ED. 1968. Protective effects of specific immunity to viral neuraminidase on influenza virus infection of mice. J Virol 2:778–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Webster RG, Laver WG. 1967. Preparation and properties of antibody directed specifically against the neuraminidase of influenza virus. J Immunol 99:49–55. [PubMed] [Google Scholar]
- 11.Seto JT, Chang FS. 1969. Functional significance of sialidase during influenza virus multiplication: an electron microscope study. J Virol 4:58–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Holmes KT, Hampson AW, Raison RL, Webster RG, O'Sullivan WJ, Mountford CE. 1982. A comparison of two anti-neuraminidase monoclonal antibodies by complement activation. Eur J Immunol 12:523–526. doi: 10.1002/eji.1830120614. [DOI] [PubMed] [Google Scholar]
- 13.Monto AS, Petrie JG, Cross RT, Johnson E, Liu M, Zhong W, Levine M, Katz JM, Ohmit SE. 2015. Antibody to influenza virus neuraminidase: an independent correlate of protection. J Infect Dis 212:1191–1199. doi: 10.1093/infdis/jiv195. [DOI] [PubMed] [Google Scholar]
- 14.Memoli MJ, Shaw PA, Han A, Czajkowski L, Reed S, Athota R, Bristol T, Fargis S, Risos K, Powers JH, Davey RT, Taubenberger JK. 2016. Evaluation of antihemagglutinin and antineuraminidase antibodies as correlates of protection in an influenza A/H1N1 virus healthy human challenge model. mBio 7:e00417-. doi: 10.1128/mBio.00417-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rockman S, Brown LE, Barr IG, Gilbertson B, Lowther S, Kachurin A, Kachurina O, Klippel J, Bodle J, Pearse M, Middleton D. 2013. Neuraminidase-inhibiting antibody is a correlate of cross-protection against lethal H5N1 influenza virus in ferrets immunized with seasonal influenza vaccine. J Virol 87:3053–3061. doi: 10.1128/JVI.02434-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wohlbold TJ, Nachbagauer R, Xu H, Tan GS, Hirsh A, Brokstad KA, Cox RJ, Palese P, Krammer F. 2015. Vaccination with adjuvanted recombinant neuraminidase induces broad heterologous, but not heterosubtypic, cross-protection against influenza virus infection in mice. mBio 6:e02556. doi: 10.1128/mBio.02556-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sandbulte MR, Jimenez GS, Boon ACM, Smith LR, Treanor JJ, Webby RJ. 2007. Cross-reactive neuraminidase antibodies afford partial protection against H5N1 in mice and are present in unexposed humans. PLoS Med 4:e59. doi: 10.1371/journal.pmed.0040059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rajendran M, Nachbagauer R, Ermler ME, Bunduc P, Amanat F, Izikson R, Cox M, Palese P, Eichelberger M, Krammer F. 2017. Analysis of anti-influenza virus neuraminidase antibodies in children, adults, and the elderly by ELISA and enzyme inhibition: evidence for original antigenic sin. mBio 8:e02281-. doi: 10.1128/mBio.02281-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wan H, Gao J, Xu K, Chen H, Couzens LK, Rivers KH, Easterbrook JD, Yang K, Zhong L, Rajabi M, Ye J, Sultana I, Wan X-F, Liu X, Perez DR, Taubenberger JK, Eichelberger MC. 2013. Molecular basis for broad neuraminidase immunity: conserved epitopes in seasonal and pandemic H1N1 as well as H5N1 influenza viruses. J Virol 87:9290–9300. doi: 10.1128/JVI.01203-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shoji Y, Chichester JA, Palmer GA, Farrance CE, Stevens R, Stewart M, Goldschmidt L, Deyde V, Gubareva L, Klimov A, Mett V, Yusibov V. 2011. An influenza N1 neuraminidase-specific monoclonal antibody with broad neuraminidase inhibition activity against H5N1 HPAI viruses. Hum Vaccin 7:199–204. doi: 10.4161/hv.7.0.14595. [DOI] [PubMed] [Google Scholar]
- 21.Doyle TM, Hashem AM, Li C, Van Domselaar G, Larocque L, Wang J, Smith D, Cyr T, Farnsworth A, He R, Hurt AC, Brown EG, Li X. 2013. Universal anti-neuraminidase antibody inhibiting all influenza A subtypes. Antiviral Res 100:567–574. doi: 10.1016/j.antiviral.2013.09.018. [DOI] [PubMed] [Google Scholar]
- 22.Doyle TM, Jaentschke B, Van Domselaar G, Hashem AM, Farnsworth A, Forbes NE, Li C, Wang J, He R, Brown EG, Li X. 2013. The universal epitope of influenza A viral neuraminidase fundamentally contributes to enzyme activity and viral replication. J Biol Chem 288:18283–18289. doi: 10.1074/jbc.M113.468884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wan H, Yang H, Shore DA, Garten RJ, Couzens L, Gao J, Jiang L, Carney PJ, Villanueva J, Stevens J, Eichelberger MC. 2015. Structural characterization of a protective epitope spanning A(H1N1)pdm09 influenza virus neuraminidase monomers. Nat Commun 6:6114. doi: 10.1038/ncomms7114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Halbherr TH, Ricklin M, Locher S, Rentsch MB, Summerfield A, Zimmer GSJL. 2015. Biological and protective properties of immune sera directed to the influenza virus neuraminidase. J Virol 89:1550–1563. doi: 10.1128/JVI.02949-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Webster RG, Brown LE, Laver WG. 1984. Antigenic and biological characterization of influenza virus neuraminidase (N2) with monoclonal antibodies. Virology 135:30–42. doi: 10.1016/0042-6822(84)90114-4. [DOI] [PubMed] [Google Scholar]
- 26.Smith JR. 2010. Oseltamivir in human avian influenza infection. J Antimicrob Chemother 65:ii25–ii33. doi: 10.1093/jac/dkq013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cardoso FM, Ibanez LI, Van den Hoecke S, De Baets S, Smet A, Roose K, Schepens B, Descamps FJ, Fiers W, Muyldermans S, Depicker A, Saelens X. 2014. Single-domain antibodies targeting neuraminidase protect against an H5N1 influenza virus challenge. J Virol 88:8278–8296. doi: 10.1128/JVI.03178-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jiang L, Fantoni G, Couzens L, Gao J, Plant E, Ye Z, Eichelberger MC, Wan H. 2015. Comparative efficacy of monoclonal antibodies that bind to different epitopes of the 2009 pandemic H1N1 influenza virus neuraminidase. J Virol 90:117–128. doi: 10.1128/JVI.01756-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Krammer F, Palese P. 2015. Advances in the development of influenza virus vaccines. Nat Rev Drug Discov 14:167–182. doi: 10.1038/nrd4529. [DOI] [PubMed] [Google Scholar]
- 30.Irani V, Guy AJ, Andrew D, Beeson JG, Richards JS. 2015. Molecular properties of human IgG subclasses and their implications for designing therapeutic monoclonal antibodies against infectious diseases. Mol Immunol 67:171–182. doi: 10.1016/j.molimm.2015.03.255. [DOI] [PubMed] [Google Scholar]
- 31.Wilson JR, Guo Z, Reber A, Kamal RP, Music N, Gansebom S, Bai Y, Levine M, Carney P, Tzeng W-P, Stevens J, York IA. 2016. An influenza A virus (H7N9) anti-neuraminidase monoclonal antibody with prophylactic and therapeutic activity in vivo. Antiviral Res 135:48–55. doi: 10.1016/j.antiviral.2016.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wohlbold TJ, Hirsh A, Krammer F. 2015. An H10N8 influenza virus vaccine strain and mouse challenge model based on the human isolate A/Jiangxi-Donghu/346/13. Vaccine 33:1102–1106. doi: 10.1016/j.vaccine.2015.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wohlbold T, Krammer F. 2014. In the shadow of hemagglutinin: a growing interest in influenza viral neuraminidase and its role as a vaccine antigen. Viruses 6:2465–2494. doi: 10.3390/v6062465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sandbulte MR, Westgeest KB, Gao J, Xu X, Klimov AI, Russell CA, Burke DF, Smith DJ, Fouchier RAM, Eichelberger MC. 2011. Discordant antigenic drift of neuraminidase and hemagglutinin in H1N1 and H3N2 influenza viruses. Proc Natl Acad Sci U S A 108:20748–20753. doi: 10.1073/pnas.1113801108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.DiLillo DJ, Palese P, Wilson PC, Ravetch JV. 2016. Broadly neutralizing anti-influenza antibodies require Fc receptor engagement for in vivo protection. J Clin Invest 126:605–610. doi: 10.1172/JCI84428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.He W, Tan GS, Mullarkey CE, Lee AJ, Lam MMW, Krammer F, Henry C, Wilson PC, Ashkar AA, Palese P, Miller MS. 2016. Epitope specificity plays a critical role in regulating antibody-dependent cell-mediated cytotoxicity against influenza A virus. Proc Natl Acad Sci U S A 113:11931–11936. doi: 10.1073/pnas.1609316113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Van den Hoecke S, Ehrhardt K, Kolpe A, El Bakkouri K, Deng L, Grootaert H, Schoonooghe S, Smet A, Bentahir M, Roose K, Schotsaert M, Schepens B, Callewaert N, Nimmerjahn F, Staeheli P, Hengel H, Saelens X. 2017. Hierarchical and redundant roles of activating FcγRs in protection against influenza disease by M2e-specific IgG1 and IgG2a antibodies. J Virol 91:e02500-. doi: 10.1128/JVI.02500-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bruhns P. 2012. Properties of mouse and human IgG receptors and their contribution to disease models. Blood 119:5640–5649. doi: 10.1182/blood-2012-01-380121. [DOI] [PubMed] [Google Scholar]
- 39.Johansson BE, Bucher DJ, Kilbourne ED. 1989. Purified influenza virus hemagglutinin and neuraminidase are equivalent in stimulation of antibody response but induce contrasting types of immunity to infection. J Virol 63:1239–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Powers DC, Kilbourne ED, Johansson BE. 1996. Neuraminidase-specific antibody responses to inactivated influenza virus vaccine in young and elderly adults. Clin Diagn Lab Immunol 3:511–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Neirynck S, Deroo T, Saelens X, Vanlandschoot P, Jou WM, Fiers W. 1999. A universal influenza A vaccine based on the extracellular domain of the M2 protein. Nat Med 5:1157–1163. doi: 10.1038/13484. [DOI] [PubMed] [Google Scholar]
- 42.Reed LJ, Muench H. 1938. A simple method of estimating fifty per cent endpoints. Am J Epidemiol 27:493–497. doi: 10.1093/oxfordjournals.aje.a118408. [DOI] [Google Scholar]
- 43.Stech J, Stech O, Herwig A, Altmeppen H, Hundt J, Gohrbandt S, Kreibich A, Weber S, Klenk HD, Mettenleiter TC. 2008. Rapid and reliable universal cloning of influenza A virus genes by target-primed plasmid amplification. Nucleic Acids Res 36:e139. doi: 10.1093/nar/gkn646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Neumann G, Watanabe T, Ito H, Watanabe S, Goto H, Gao P, Hughes M, Perez DR, Donis R, Hoffmann E, Hobom G, Kawaoka Y. 1999. Generation of influenza A viruses entirely from cloned cDNAs. Proc Natl Acad Sci U S A 96:9345–9350. doi: 10.1073/pnas.96.16.9345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hoffmann E, Krauss S, Perez D, Webby R, Webster RG. 2002. Eight-plasmid system for rapid generation of influenza virus vaccines. Vaccine 20:3165–3170. doi: 10.1016/S0264-410X(02)00268-2. [DOI] [PubMed] [Google Scholar]
- 46.Schotsaert M, Ysenbaert T, Smet A, Schepens B, Vanderschaeghe D, Stegalkina S, Vogel TU, Callewaert N, Fiers W, Saelens X. 2016. Long-lasting cross-protection against influenza A by neuraminidase and M2e-based immunization strategies. Sci Rep 6:24402. doi: 10.1038/srep24402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schmidt PM, Attwood RM, Mohr PG, Barrett SA, McKimm-Breschkin JL. 2011. A generic system for the expression and purification of soluble and stable influenza neuraminidase. PLoS One 6:e16284. doi: 10.1371/journal.pone.0016284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Couzens L, Gao J, Westgeest K, Sandbulte M, Lugovtsev V, Fouchier R, Eichelberger M. 2014. An optimized enzyme-linked lectin assay to measure influenza A virus neuraminidase inhibition antibody titers in human sera. J Virol Methods 210:7–14. doi: 10.1016/j.jviromet.2014.09.003. [DOI] [PubMed] [Google Scholar]
- 49.WHO Global Influenza Surveillance Network. 2011. Manual for the laboratory diagnosis and virological surveillance of influenza. WHO, Geneva, Switzerland. [Google Scholar]
- 50.Dai M, McBride R, Dortmans JCFM, Peng W, Bakkers MJG, de Groot RJ, van Kuppeveld FJM, Paulson JC, de Vries E, de Haan CAM. 2017. Mutation of the second sialic acid-binding site, resulting in reduced neuraminidase activity, preceded the emergence of H7N9 influenza A virus. J Virol 91:e00049-. doi: 10.1128/JVI.00049-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dai M, Guo H, Dortmans JCFM, Dekkers J, Nordholm J, Daniels R, van Kuppeveld FJM, de Vries E, de Haan CAM. 2016. Identification of residues that affect oligomerization and/or enzymatic activity of influenza virus H5N1 neuraminidase proteins. J Virol 90:9457–9470. doi: 10.1128/JVI.01346-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.De Baets S, Verhelst J, Van den Hoecke S, Smet A, Schotsaert M, Job ER, Roose K, Schepens B, Fiers W, Saelens X. 2015. A GFP expressing influenza A virus to report in vivo tropism and protection by a matrix protein 2 ectodomain-specific monoclonal antibody. PLoS One 10:e0121491. doi: 10.1371/journal.pone.0121491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.WHO. 2009. CDC protocol of real-time RT-PCR for swine influenza A(H1N1). WHO, Geneva, Switzerland. [Google Scholar]
- 54.Li W, Cowley A, Uludag M, Gur T, McWilliam H, Squizzato S, Park YM, Buso N, Lopez R. 2015. The EMBL-EBI bioinformatics web and programmatic tools framework. Nucleic Acids Res 43(W1):W580–W584. doi: 10.1093/nar/gkv279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ashkenazy H, Abadi S, Martz E, Chay O, Mayrose I, Pupko T, Ben-Tal N. 2016. ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res 44(W1):W344–W350. doi: 10.1093/nar/gkw408. [DOI] [PMC free article] [PubMed] [Google Scholar]