

Abstract
During a 2013 cruise in the Southern Ocean we collected specimens of the octocoral Plumarella delicatissima between 800 and 950 m depth. Five new furanocembranoid diterpenes, keikipukalides A–E (1–5), the known diterpene pukalide aldehyde (6), and the known norditerpenoid ineleganolide (7) were isolated from the coral. These Plumarella terpenes lack mammalian cytotoxicity, while 2–7 display activity against Leishmania donovani between 1.9 and 12 μM. Structure elucidation was facilitated by one- and two-dimensional NMR spectroscopy and mass spectrometry, and keikipukalides A and E were confirmed by X-ray crystallography.
Octocorals belong to the Cnidarian order Alcyonacea and are prolific sources of bioactive natural products,1 including neurotoxic venoms used to paralyze vertebrate prey2 and potent small-molecule toxins that are most often terpenoids.3 Octocoral diterpenes exhibit potent environmental toxicity, including the protection of coral eggs during annual spawning events, inhibiting fouling by microbes and algae, and ichthyotoxicity, and contribute to the destruction of coral reefs.4 Biomedical studies have followed such ecological observations and found many of the same diterpenes cytotoxic to mammalian cells,3 although the most notable biomedical advancement was the introduction of the diterpene pseudopterosin for commercial use in anti-inflammatory and wound-healing applications.5
Octocorals range in distribution from shallow water
coral reefs
to the depths of the abyssal plane. Shallow water species dominate
the alcyonaceans with nearly 4000 species described in the World Record
of Marine Species (WoRMS) database6 compared
to just over 600 (15%) species that are recorded in the Deep Sea Octocorals
Online database.7 Natural product reports
from the two groups indicate the deep water species are less studied:
roughly 750 alcyonacean compounds are cataloged in MarinLit,8 but only 28 (3.5%) compounds have been described
from 12 deep water species.9−12 The difficulty of accessing deep sea habitats has
clearly hampered research of species hiding there, yet bioactivity
profiles of, for example, shagenes11 and
cristaxenicin A,13 suggest the deep water
species are an understudied drug discovery resource. We collected
the deep water alcyonacean Plumarella delicatissima from the “Plateau of Fascination”, approximately 240
km NE of Stanley, Falkland Islands (Islas Malvinas), in the Southern
Ocean. While a Plumarella sp. from the northwest
Pacific Ocean near the Kuril Islands was reported to produce the diterpene
plumarellide,14 a rearranged furanocembranoid,
our Southern Ocean species has yielded five new furanocembranoid diterpenes,
keikipukalides A–E15 (1–5), along with the known diterpene pukalide
aldehyde16 (6) and norditerpenoid
ineleganolide17 (7). This
family of diterpenes displays activity against the leishmaniasis parasite, Leishmania donovani, ranging from 1.9 to 12 μM with
no corresponding mammalian cytotoxicity below 50 μM.
Results and Discussion
P. delicatissima was abundant on the Plateau of Fascination (S 50°8.453′ W 55°28.106′) near the Falkland Islands in the Southern Ocean. The specimens were collected in April 2013 from 800 to 950 m depth, immediately frozen, and stored for chemical analyses. Frozen coral was subsequently freeze-dried and extracted using a Soxhlet apparatus with refluxing CH2Cl2. The 1H NMR spectrum of the organic extract contained signals indicative of terpenes; therefore it was dried onto silica gel and fractionated by normal-phase MPLC. Normal-phase semipreparative HPLC was used for the initial purification, although some metabolites required an additional purification step using reversed-phase analytical HPLC, yielding keikipukalides A (1) (12 mg), B (2) (5 mg), C (3) (3 mg), D (4) (18 mg), and E (5) (48 mg), pukalide aldehyde (6) (4 mg), and ineleganolide (7) (3 mg).
Keikipukalide A (1) was isolated as a crystalline solid that analyzed for C20H20O6 by high-resolution EIMS. The 1H and 13C NMR data (Table 1) corroborated the proton and carbon counts established by EIMS and further identified aldehyde, ester, and four olefinic functional groups, accounting for six of keikipukalide A’s 11 unsaturations, making it a five-ring structure. The HMBC spectrum facilitated locating the aldehyde group on an olefin (Figure 1), with correlations of δH 9.89 (H-18) to δC 105.6 (C-5) and 124.3 (C-4). A singlet proton, δH 6.47, correlating by HSQC to C-5, extended the spin system by observation of HMBC correlations to two additional olefinic carbons at δC 149.2 (C-6) and 162.2 (C-3). H-5 also displayed a long-range COSY correlation to an oxymethine singlet at δH 4.01 (H-7) that could be shown to be part of a trisubstituted oxirane based on its chemical shift and that of the adjacent carbon, C-8, δC 56.9. HMBC correlations of H3-19 (δH 1.23 singlet) and H2-9 established their position around the oxirane group, at C-7 (δC 54.6) and C-8, respectively. Further, a butenolide, characterized by HMBC correlations of H-11 (δH 4.09) to oxymethines C-10 (δC 75.3) and C-12 (δC 58.0), and a lactone-type carbonyl, C-20 (δC 169.0), joined the trisubstituted oxirane at C-9 based on HMBC correlations of H-9a (δH 2.09) to C-10 and C-11. Extending the system, an olefinic proton, δH 5.14 on C-13 (δC 115.6), displayed an HMBC correlation to C-20, anchoring the butenolide to a C4 substituent established by COSY as the C-13 to C-2 segment of keikipukalide A. HMBC correlations of C-2 (δC 31.0) to C-3 and C-4 formulated a C14-macrocycle. An isopropylene substituent at C-1 (δC 47.1) was established by HMBC correlations among C-15, -16, and -17 (δC 143.5, 113.6, and 23.2, respectively) and by correlation of their respective protons (δH 4.56 and 4.97 (H2-16) and 1.70 (H3-17)) back to C-1. Two unsaturations remained, and two oxygen atoms were unaccounted. Chemical shifts of C-3 to C-6 are well suited to assignment as a trisubstituted furan, as are C-11 and -12 suited to an oxirane, establishing the furanocembranoid planar structure (Figure 1) of keikipukalide A.
Table 1. NMR Data for Keikipukalide A (1).
| position | 13C,b type | 1Ha (J in Hz) | HMBCa |
|---|---|---|---|
| 1 | 47.1, CH | 3.47, m | 3, 14 |
| 2a | 31.0, CH2 | 3.31, m | 1, 3, 4, 14, 15 |
| 2b | 3.42, m | 1, 15 | |
| 3 | 162.2, C | ||
| 4 | 124.3, C | ||
| 5 | 105.6, CH | 6.47, s | 3, 4, 6 |
| 6 | 149.2, C | ||
| 7 | 54.6, CH | 4.01, s | 5, 6, 8 |
| 8 | 56.9, C | ||
| 9a | 40.4, CH2 | 2.09, dd (15.6, 1.8) | 8, 10, 11, 19 |
| 9b | 2.58, dd (15.6, 4.2) | 7, 8 | |
| 10 | 75.3, CH | 4.78, dd (4.0, 2.7) | |
| 11 | 64.1, CH | 4.09, s | 10, 12, 20 |
| 12 | 58.0, C | ||
| 13 | 115.6, CH | 5.14, dd (16.3, 1.5) | 1, 14, 20 |
| 14 | 145.8, CH | 7.41, dd (16.3, 4.4) | 15 |
| 15 | 143.5, C | ||
| 16a | 113.6, CH2 | 4.56, s | 1, 17 |
| 16b | 4.97, s | ||
| 17 | 23.2, CH3 | 1.70, s | 1, 15, 16 |
| 18 | 184.3, CH | 9.89, s | 4, 5 |
| 19 | 21.3, CH3 | 1.23, s | 7, 8, 9 |
| 20 | 169.0, C |
CDCl3, 400 MHz.
CDCl3, 100 MHz, multiplicity determined by HMQC.
Figure 1.
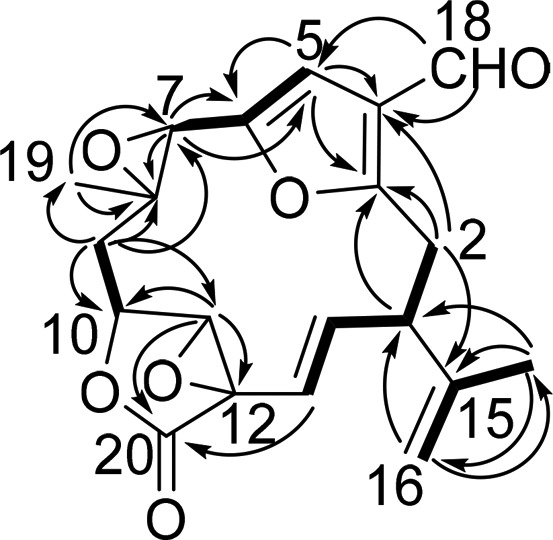
Key COSY (bold lines) and HMBC (→) correlations establishing the planar structure of keikipukalide A (1).
Stereochemical evaluation of keikipukalide A suffered from the flexibility of the macrocycle but was informed by through-bond or through-space correlations near rigid portions of the cycle. That H-10 and H-11 lack coupling established a 90°, trans, relationship between them and fixes the epoxide oxygen and C-10 substituent on opposite faces of the butenolide, as previously seen with pukalide and its derivatives.18 The C-13/C-14 olefin could similarly be defined as trans based on the large 3J13,14 coupling (16.3 Hz). The proximities of substituents around the C-7/C-8 oxirane are suited to NOE analysis, which demonstrated H-9a and H-7 to occupy the same face of the three-membered ring, defining a trans-epoxide. However, the relationship among the disparate centers required X-ray analysis for definitive assignment, and keikipukalide A (1) provided suitable crystals. The crystallographic metadata (Flack parameter 0.17(10)), Bijvoet-pair analysis, and Bayesian statistics method (P2 (true) = 1|P3 (true) = 0.981|P3 (false) = 0.4 × 10–17|P3(rac-twin) = 0.019 (see Supporting Information Table S2)) assigned the configuration depicted in Figure 2 as the absolute configuration of keikipukalide A.
Figure 2.
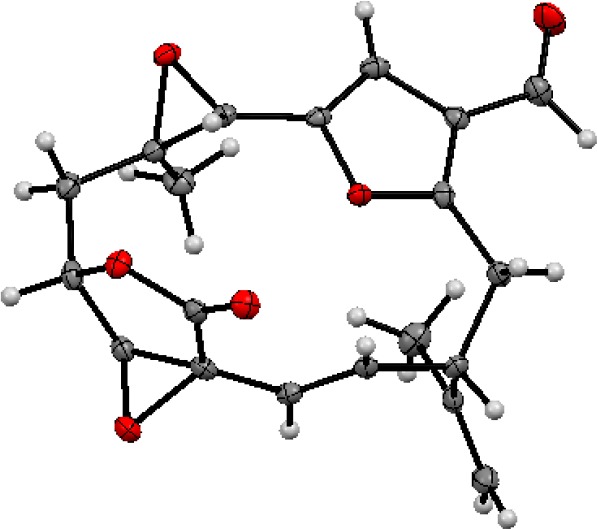
Asymmetric unit of keikipukalide A (1). Anisotropic displacement parameters are drawn at 50% probability.
Keikipukalide B (2) was isolated as an oil with a formula of C24H26O9 based on mass spectrometric analysis corroborated by 1H and 13C NMR data (Tables 2 and 3). A major feature of the 1H NMR spectrum that deviated from that of keikipukalide A (1) was the presence of two acetate methyl resonances (δH 2.11 and 2.12). However, the entire northern portion, from C-1 to C-10, was established by analysis of COSY and HMBC spectra (Figure 3) to match that of keikipukalide A. That the keikipukalide B C-11 was not part of an oxirane, as found in 1, was established not only by the chemical shift (δC 72.9) but by a correlation of H-11 (δH 5.65) to an acetoxy carbonyl (δC 170.6). The butenolide was found to bear, in addition to the C-11 acetoxy group, an exocyclic olefin at C-12 based on H-11 correlations in the HMBC spectrum to two olefinic carbons, a quaternary carbon at δC 127.4 (C-12) and a methine at δC 145.6 (C-13). COSY and HMBC both extended the macrocycle from C-13 back to C-1 via an oxymethine at δC 70.6 (C-14), which, like C-11, displayed an HMBC correlation to an acetoxy carbonyl (δC 169.4), defining the position of the second acetate group and completing the planar structure of keikipukalide B.
Table 2. 1H NMR Data for Keikipukalides B–E (2–5).
| position | 2as | 3a | 4a | 5b |
|---|---|---|---|---|
| 1 | 3.18, t (4.4) | 3.01 m | 3.19, m | 4.07, dt (9.9, 5.9) |
| 2a | 3.45, d (4.4) | 3.49, dd (15.7, 4.4) | 3.37, dd (14.2, 5.5) | 3.00, d (4.7) |
| 2b | 3.12, dd (15.7, 5.3) | 2.99, dd (14.3, 10.4) | 2.99 (s) | |
| 3 | ||||
| 4 | ||||
| 5 | 6.46, s | 6.46, s | 6.62, s | 6.44, s |
| 6 | ||||
| 7 | 4.03, s | 4.02, s | 5.71, s | 4.12, s |
| 8 | ||||
| 9a | 1.97, dd (15.3, 3.4) | 1.94, dd (15.2, 3.1) | 2.14, dd (8.9, 4.1) | 2.22, dd (15.0, 3.3) |
| 9b | 2.61, dd (15.3, 3.4) | 2.60, dd (15.2, 3.6) | 2.53, ddd (15.0, 6.4, 3.7) | |
| 10 | 4.70, t (3.3) | 4.66,t (3.4) | 4.62, t (4.7) | 5.25, dt (3.6, 1.7) |
| 11 | 5.65, s | 5.62, s | 3.77, br s | 7.31, d (1.3) |
| 12 | ||||
| 13 | 6.54, d (9.1) | 6.69, dd (11.6, 2.8) | 5.72, d (15.9) | 5.85, d (6.2) |
| 14a | 6.97, d (9.1) | 4.23, dd (17.3, 11.7) | 5.57, dd (15.9, 10.0) | 1.26, br d (15.0) |
| 14b | 2.75, ddd (17.9, 9.0, 2.2) | 2.58, ddd (15.0, 6.4, 3.4) | ||
| 15 | ||||
| 16a | 4.34, s | 4.53, s | 4.84, s | 4.92, s |
| 16b | 4.96, s | 4.93, s | 4.90, s | 5.03, s |
| 17 | 1.82, s | 1.78, s | 1.82, s | 1.91, s |
| 18 | 9.86, s | 9.90, s | 9.87, s | 9.86, s |
| 19 | 1.17, s | 1.20, s | 1.47, s | 0.98, s |
| 20 | ||||
| 21 | ||||
| 22 | 2.12, s | 2.10, s | 2.21, s | 2.01, s |
| 23 | ||||
| 24 | 2.11, s |
Recorded in CDCl3 (400 MHz).
Recorded in CDCl3 (500 MHz).
Table 3. 13C NMR Data for Keikipukalides B–E (2–5).
| position | 2b | 3a | 4a | 5b |
|---|---|---|---|---|
| 1 | 47.4, CH | 40.8, CH | 49.2, CH | 36.7, CH |
| 2 | 28.7, CH2 | 30.7, CH2 | 31.2, CH2 | 33.1, CH2 |
| 3 | 161.6, C | 162.4, C | 163.0, C | 162.3, C |
| 4 | 123.4, C | 123.3, C | 124.5, C | 123.2, C |
| 5 | 104.7, CH | 105.1, CH | 106.2, CH | 104.5, CH |
| 6 | 149.5, C | 149.7, C | 150.3, C | 149.8, C |
| 7 | 54.7, CH | 54.4, CH | 75.4, CH | 54.8, CH |
| 8 | 56.3, C | 56.8, C | 74.3, C | 56.8, C |
| 9 | 40.1, CH2 | 40.4, CH2 | 37.9, CH2 | 39.9, CH2 |
| 10 | 81.0, CH | 80.8, CH | 75.8, CH | 77.7, CH |
| 11 | 72.9, CH | 73.9, CH | 67.7, CH | 151.1, CH |
| 12 | 127.4, C | 124.1, C | 58.2, C | 134.8, C |
| 13 | 145.6, CH | 151.6, CH | 121.1, CH | 68.6, CH |
| 14 | 70.6, CH | 30.4, CH2 | 134.4, CH | 35.7, CH2 |
| 15 | 143.1, C | 144.8, C | 144.6, C | 148.5, C |
| 16 | 114.8, CH2 | 113.0, CH2 | 111.2, CH2 | 111.3, CH2 |
| 17 | 24.7, CH3 | 23.7, CH3 | 21.4, CH3 | 20.7, CH3 |
| 18 | 184.2, CH | 184.4, CH | 184.3,CH | 184.4, CH |
| 19 | 20.7, CH3 | 20.99, CH3c | 31.3, CH3 | 19.7, CH3 |
| 20 | 166.6, C | 168.2, C | 171.6, C | 170.3, C |
| 21 | 170.6, C | 170.6, C | 169.0, C | 170.5, C |
| 22 | 21.2, CH3c | 21.03, CH3c | 20.9, CH3 | 20.6, CH3 |
| 23 | 169.4, C | |||
| 24 | 21.0, CH3c |
Recorded in CDCl3 (100 MHz).
Recorded in CDCl3 (125 MHz). Multiplicity determined by HSQC (2 and 5) or HMQC (3 and 4).
Interchangeable.
Figure 3.

Key COSY (bold lines) and HMBC (→) correlations establishing the planar structure of keikipukalides B (2), C (3), D (4), and E (5).
The stereochemical assignment of keikipukalide B (2) is informed by comparison to chemical shifts and coupling constants of keikipukalide A (1). The C-7/C-8 epoxide bears the same configuration in 1 and 2, based not only on the close correspondence of both the 1H and 13C chemical shifts but also on a NOESY correlation in both between H-9b and H3-19. Both 1 and 2, like pukalide,18 lack coupling between H-10 and H-11, indicative of the 90° dihedral angle between them. The configuration of the Δ12 olefin was assigned as Z based on observation of a NOESY correlation between H-11 and H-13, analogous to that observed in the assignment of the same partial structure in an unnamed furanocembranoid from Sinularia polydactyla.18
The C-1/C-14 relationship in keikipukalide B (2) was established taking account of both NOESY and J coupling data. With the X-ray conformation of keikipukalide A (1) (Figure 2) as a starting point, modifying the conformation to introduce the functionalization of keikipukalide B yields a new MM2-minimized conformation (Figure 4) that agrees with observed spectroscopic data, including proximity of H-11/H-13 and H-14/H2-2 that would result in nuclear Overhauser enhancement, and a dihedral angle between H-1 and H-14 resulting in negligible coupling (3J1,14 ≈ 0).
Figure 4.
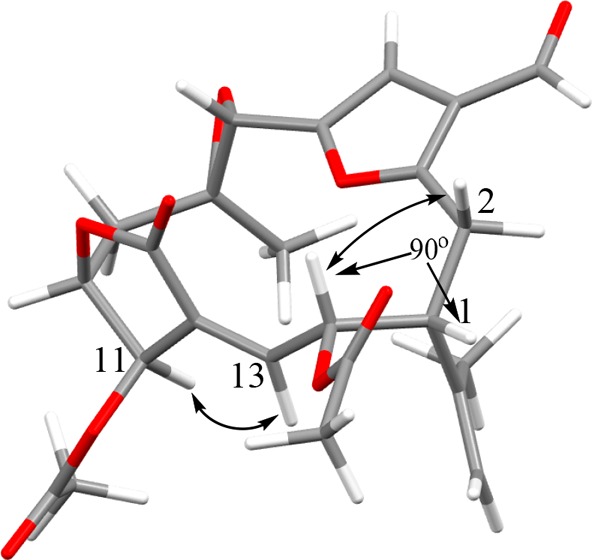
Relative configuration of keikipukalide B (2) C-1 and C-14 based on 3J1,14 ≈ 0 and NOESY (↔) correlations.
Keikipukalide C (3) was isolated as a white semisolid and determined to have the formula C22H24O7 based on mass spectrometric and 1H and 13C NMR data (Tables 2 and 3). The planar structure of keikipukalide C was found by COSY and HMBC (Figure 3) to be identical to that of keikipukalide B (2) except for C-14, where a methylene (δC 30.4; δH 2.75 and 4.23) replaced the oxymethine of keikipukalide B. The configuration similarly matched 2, including the trans epoxide at C-7/C-8, a trans relationship between H-10 and H-11 (3J10,11 = 0), and NOESY correlations between H-13 and H-11, establishing Δ12 as Z.
Keikipukalide D (4), an amorphous solid, was found to have a formula of C22H24O8 from mass spectrometric and NMR data. Keikipukalide D shared more spectroscopic features with keikipukalide A (1) than any of the other keikipukalides, with proton and carbon shifts, COSY correlations, and HMBC correlations establishing structural identity except for C-7 and C-8 (Figure 3). Keikipukalide D, therefore, differs from keikipukalide A by the presence of an acetoxy function and concomitant addition of acetic acid to the molecular formula. For oxymethine C-7 (δC 75.4; δH 5.71), the deshielded shifts are indicative of epoxide ring-opening, relative to 1, and C-8 (δC 74.3) matched the effect, reflective of a diol-type arrangement of C-7 and C-8. H-7 displayed an HMBC correlation to an ester-type carbon (δC 169.0), identifying the location of the acetate group and securing the planar structure depicted in 4. The configuration of 4 could be derived from comparison of NOESY correlations to those observed for 1. As suggested by the X-ray structure of keikipukalide A, the proximity of H-11 to H3-19 facilitated NOESY correlation between the two, and the same correlation, albeit weaker, could be observed in keikipukalide D. Further, H3-19 in keikipukalide D demonstrated a NOESY correlation to H-7, while H2-9 did not, placing H-7, H-11, and H3-19 on the same face of the macrocycle.
Crystalline keikipukalide E (5) analyzed for C22H24O7 by HRESIMS, making 5 isomeric with keikipukalide C (3). COSY and HMBC NMR spectra established a macrolide skeleton (Figure 3) matching pukalide aldehyde,19 but identified position C-13 (δC 68.6; δH 5.85) as an oxymethine rather than the methylene group found in pukalide aldehyde. Crystals of keikipukalide E were submitted to X-ray diffraction studies, resulting in full stereochemical assignment (Figure 5). The crystallographic metadata (Flack parameter 0.02(8)), Bijvoet-pair analysis, and Bayesian statistics method (P2 (true) = 1|P3 (true) = 1|P3 (false) = 0.9 × 10–38|P3 (rac-twin) = 0.9 × 10–09 (Table S4)) facilitated assignment of the absolute configuration of keikipukalide E as shown.
Figure 5.
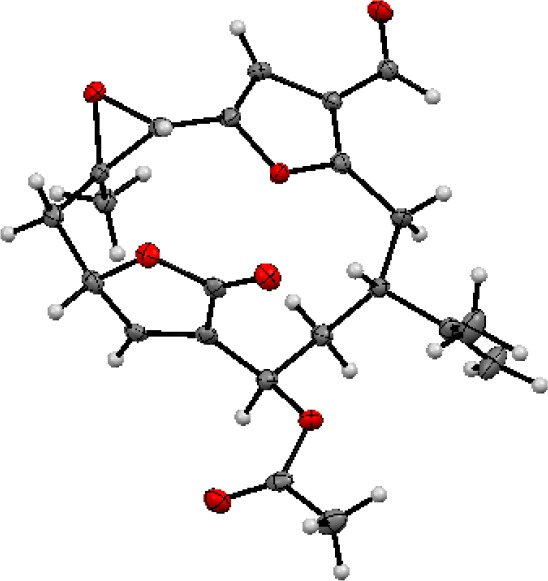
Asymmetric unit of keikipukalide E (5). Anisotropic displacement parameters are drawn at 50% probability.
The seven terpenoids isolated from P. delicatissima were evaluated against a number of infectious disease targets. Leishmania donovani was quite sensitive, displaying an IC50 of 1.9–12 μM for compounds 2–7 (Table 4) in the infected macrophage assay, compared to the IC50 of 6.2 for miltefosine, a drug currently used for the treatment of leishmaniasis. No mammalian cytotoxicity was detected in the compounds below 50 μM. However, testing against a number of other infectious diseases, including Naegleria fowleri, the ESKAPE20 panel of drug-resistant bacteria, and Clostridium difficile, found none of these pathogens susceptible to the compounds. Although this seeming specificity for L. donovani is promising for this scaffold, most of these compounds contain the neurotoxic pharmacophore of lophotoxin,16 which may prove to be a liability.
Table 4. Bioactivity of Plumarella Terpenes (IC50, μM)a.
| Leishmania donovani | A549 cytotoxicity | |
|---|---|---|
| keikipukalide A (1) | >28 | >50 |
| keikipukalide B (2) | 8.5 | >50 |
| keikipukalide C (3) | 8.8 | >50 |
| keikipukalide D (4) | 12 | >50 |
| keikipukalide E (5) | 8.8 | >50 |
| pukalide aldehyde (6) | 1.9 | >50 |
| ineleganolide (7) | 4.4 | >50 |
| miltefosine (control) | 6.2 | not determined |
The > symbol indicates the sample was inactive at the highest concentration tested.
Plumarella belongs in the family Primnoidae (suborder Calcaxonia), while nearly all similar furanocembranoid scaffolds have been reported from the Alcyoniidae or Gorgoniidae (suborder Alcyoniina or Holaxonia, respectively). However, briarane diterpenes have been isolated from Ellisellidae, which, like the Primnoidae, is in the suborder Calcaxonia. Octocoral phylogeny is in a state of flux after the integration of molecular data, and further investigations into the origin of these metabolites to possibly define a pattern between this and other coral genera could give chemotaxonomic or evolutionary insight into octocoral species around the globe.
Experimental Section
General Experimental Procedures
Optical rotations were measured using an AutoPol IV polarimeter at 589 nm. UV absorptions were measured by an Agilent Cary 60 UV–vis spectrophotometer; IR spectra were recorded with an Agilent Cary FTIR 630 spectrometer and a PerkinElmer Spectrum Two equipped with a UATR (single reflection diamond) sample introduction system. NMR spectra were recorded at 298 K on Varian Inova 400, Varian Direct Drive 500, or Varian Inova 600 MHz NMR spectrometers. Chemical shifts are reported with the use of the residual CDCl3 signals (δH 7.27 ppm; δC 77.0 ppm) as internal standards for 1H and 13C NMR spectra, respectively. The 1H and 13C NMR assignments were supported by COSY, HSQC/HMQC, HMBC, and NOESY experiments. The high-resolution electrospray ionization mass spectra were performed on an Agilent 6230 TOF LC/MS. Low-resolution electrospray ionization mass spectra were obtained on an Agilent 6120 Quadrupole LC/MS, and high-resolution electron ionization mass spectra were obtained on an Agilent 7890 GC/7200 MS QToF. Semipreparative and analytical HPLC was performed on a Shimadzu LC-20 AT system equipped with an evaporative light-scattering detector (ELSD) and an ultraviolet detector using a Luna silica column (5 μm, 250 × 10 mm) and a YMC C-18 column (10 μm, 150 × 4 mm). MPLC was performed on a Teledyne Isco CombiFlash Rf 200i equipped with an ELSD using a RediSep Rf silica 120 g flash column, and commercial silica gel 230–400 mesh was used to load samples.
Biological Material
Plumarella delicatissima is a delicate feather-like coral that was collected at the Plateau of Fascination NE of the Falkland Islands (Islas Malvinas) (S 50°8.453′ W 55°28.106′) during the austral autumn in late April 2013. The specimens were collected from 800 to 950 m depth, frozen immediately upon collection, and maintained at −80 °C until extraction. Subsamples from two specimens were extracted for DNA and had a portion of their mitochondrial genome amplified to confirm their identification. We used the primers ND42599F/mut3458R21,22 to produce msh1 (muts homologue) amplicons and outsourced these for sequencing. The resulting bidirectional sequences (MG603067, MG603068) were compared to other available Plumarella sequences in GenBank using a maximum likelihood analysis in RAxML.23,24 The tree topology was assessed using 10 runs of 100 thorough bootstrap replicates and shows our specimens fall into a clade with other P. delicatissima specimens (Figure S31).
Isolation of the Keikipukalides
Frozen P. delicatissima was freeze-dried, and 435.2 g of dry weight was extracted in refluxing CH2Cl2 using a Soxhlet apparatus. After partitioning between H2O and CH2Cl2, 10.8 g of the concentrated CH2Cl2 partition fraction was resuspended in CH2Cl2 and dried onto silica gel for fractionation by MPLC on a Teledyne CombiFlash fitted with UV and ELS detection. Fractions F–H, eluting from MPLC using EtOAc/n-hexane (1:1) to EtOAc/n-hexane (8:1), displayed NMR signatures of diterpene metabolites and were selected for purification using NP and RP HPLC with UV and ELSD detection. Semipreparative normal-phase gradient HPLC on a Shimadzu 10A instrument, using n-hexanes to EtOAc/n-hexanes (3:1) over a 50 min gradient, yielded keikipukalides A and C from MPLC fraction F, keikipukalides B and D and pukalide aldehyde from MPLC fraction G, and keikipukalide E from MPLC fraction H. Keikipukalides A and B needed no further purification, while the others were further purified using RP HPLC in H2O and MeCN, yielding keikipukalides A (1, 12 mg), B (2, 5 mg), C (3, 3 mg), D (4, 18 mg), and E (5, 48 mg), pukalide aldehyde (6, 4 mg), and ineleganolide (7, 3 mg).
Keikipukalide A (1):
clear crystals, [α]24D −104 (c 0.11, CHCl3); UV (MeCN) λmax (log ε) 273 nm (3.8); IR ν (thin film) 2936, 1778, 1677, 1566, 1391, 1350, 1089, 1048, 739 cm–1; 1H and 13C NMR data, Table 1; LRESIMS m/z 357.1 [M + H]+; 70 eV HREIMS m/z 356.1244 [M]+ (calcd for C20H20O6, 356.1260).
Keikipukalide B (2):
clear oil, [α]24D −32.7 (c 0.11, CHCl3); UV (MeCN) λmax (log ε) 275 nm (3.5); IR ν (thin film) 2936, 1741, 1677, 1566, 1424, 1372, 1223, 1085, 1022, 739 cm–1; 1H NMR data, Table 2; 13C NMR data, Table 3; LRESIMS m/z 459.2 [M + H]+; 70 eV HREIMS m/z 338.1141 [M – 2 HOAc]+ (calcd for C20H18O5, 338.1154).
Keikipukalide C (3):
white powder, [α]24D −38.7 (c 0.11, CHCl3); UV (MeCN) λmax (log ε) 262 nm (3.9); IR ν (thin film) 3458, 3059, 2973, 2936, 1756, 1681, 1566, 1383, 1271, 1089, 1052, 1026, 899, 735, 705 cm–1; 1H NMR data, see Table 2; 13C NMR data, see Table 3; LRESIMS m/z 401.2 [M + H]+; 70 eV HREIMS m/z 340.1307 [M – HOAc]+ (calcd for C20H20O5, 340.1311).
Keikipukalide D (4):
amorphous solid, [α]24D +64.8 (c 0.11, CHCl3); UV (MeCN) λmax (log ε) 269 nm (3.7); IR ν (thin film) 3361, 3063, 2977, 2936, 1785, 1748, 1677, 1566, 1376, 1223, 1044, 914, 735, 705 cm–1; 1H NMR data, see Table 2; 13C NMR data, see Table 3; LRESIMS m/z 417.2 [M + H]+; 70 eV HREIMS m/z 356.1247 [M – HOAc]+ (calcd for C20H20O6, 356.1260).
Keikipukalide E (5):
crystalline solid, [α]24D +24.3 (c 0.11, CHCl3); UV (EtOH) λmax (log ε) 270 nm (3.1); IR ν (thin film) 3050, 2940, 2900, 1740, 1580, 1440, 1390, 1350, 1330, 1220, 1140, 1000, 880, 780 cm–1; 1H NMR data, see Table 2; 13C NMR data, see Table 3; HRESIMS m/z 401.1576 [M + H]+ (calcd for C22H25O7, 401.1600).
X-ray Crystallography
Crystals of 1 and 5 were obtained from EtOAc/hexane and MeCN, respectively. The X-ray diffraction data for 1 and 5 were measured on a Bruker D8 Venture PHOTON 100 CMOS system equipped with a Cu Kα INCOATEC ImuS microfocus source (λ = 1.541 78 Å). Data integration and reduction were performed using SaintPlus 6.01.25 Absorption correction was performed by the multiscan method implemented in SADABS.26 Space groups were determined using XPREP implemented in APEX3.27 Structures were solved using SHELXT and refined using SHELXL-201428−30 (full-matrix least-squares on F2) through the OLEX2 interface program.31 All non-hydrogen atoms were refined anisotropically. Hydrogen atoms of −CH, −CH2, and −CH3 groups were placed in geometrically calculated positions and were included in the refinement process using a riding model with isotropic thermal parameters Uiso(H) = 1.2(1.5)Ueq(−CH, −CH2, (−CH3)). The absolute configuration for each compound has been established based on the Flack parameter value and verified additionally with Bijvoet-pair analysis and Bayesian statistics methods.32,33 P2 ≈ 1 for all cases and is the probability that the current model is correct assuming two possibilities: one out of two enantiomers present. Crystal data and refinement conditions are shown in Tables S1–S4.
Crystallographic data for the structures reported in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication nos. CCDC 1570913 for 1 and CCDC 1570914 for 5. Copies of the data can be obtained, free of charge, on application to the Director, CCDC, 12 Union Road, Cambridge CB2 1EZ, UK [tel: (+44) 1223-336-408; fax: (+44) 1223-336-033; or e-mail: deposit@ccdc.cam.ac.uk).
Crystallographic data for keikipukalide A (1):
C20H20O6, M = 356.36, crystal size 0.14 × 0.07 × 0.02 mm3, orthorhombic, a = 6.74700(10) Å, b = 13.2449(3) Å, c = 18.8700(4) Å, α = β = γ = 90°, V = 1686.29(6) Å3, T = 100 K, space group P212121, Z = 4, Dcalcd = 1.404 g/cm3, μ = 0.862 mm–1, F(000) = 752.0, 3552 independent reflections (Rint = 0.0710, Rsigma = 0.0356). The final R1 = 0.0352 (I ≥ 2σ(I)), wR2 = 0.0698 (I ≥ 2σ(I)), R1 = 0.0433 (all data), wR2 = 0.0731 (all data), S = 1.048. The Flack parameter was 0.17(10).
Crystallographic data for keikipukalide E (5):
C22H24O7, M = 400.41, crystal size 0.31 × 0.12 × 0.03 mm3, orthorhombic, a = 8.7671(2) Å, b = 11.4162(3) Å, c = 19.4875(4) Å, α = β = γ = 90°, V = 1950.44(8) Å3, T = 100 K, space group P212121, Z = 4, Dcalcd = 1.364 g/cm3, μ = 0.845 mm–1, F(000) = 848.0, 4102 independent reflections (Rint = 0.0664, Rsigma = 0.0323). The final R1 = 0.0344 (I ≥ 2σ(I)), wR2 = 0.0782 (I ≥ 2σ(I)), R1 = 0.0401 (all data), wR2 = 0.0812 (all data), S = 1.031. The Flack parameter was 0.02(8).
Biological Assays
Leishmania donovani Cell Line
A total of 130 L. donovani axenic amastigotes were cultured in RPMI 1640 at a pH of 5.5 with 7.5 g/L Hepes (Invitrogen Corp.), 5.86 g/L 2-(N-morpholino)ethanesulfonic acid (MES, Sigma-Aldrich), 2 g/L sodium bicarbonate (Fisher Scientific), 10 mg/L hemin (Sigma-Aldrich), 100 μM xanthine (Sigma-Aldrich), 40 mg/L Tween-80 (Sigma-Aldrich), 1% penicillin–streptomycin, 5 g/L trypton-peptone (BD Bioscience), and 20% 16 h heat-inactivated fetal bovine serum (FBS). L. donovani was incubated at 37 °C. All culturing was done using nonvented 25 cm2 tissue culture flasks (Corning).
Infected Macrophage Assay
In a 384-well plate (CellCarrier-384 black, optically clear bottom, tissue culture treated, sterile), 2000 J774A.1 cells were seeded. L. donovani axenic amastigotes (MHOM/SD/75/1246/130 cell line) were then added to the plate at a ratio of 10:1 and incubated at 37 °C, 5% CO2 for 24 h. The excess extracellular amastigotes were then washed away using prewarmed media. Compounds were prepared in a 384-well plate (Thermo Scientific Nunc 384-well clear polystyrene plates (nontreated surfaces)) with a starting concentration of 10 μg/mL and serially diluted at 1:2. Drugs were then added to the assay plate and incubated at 37 °C, 5% CO2 for 72 h. Cells were then fixed with 2% paraformaldehyde (Alfa Aesar paraformaldehyde, 16% /v aqueous solution, MeOH-free) and incubated for 15 min at rt, then stained with 5 μM Draq5 (Thermo Scientific DRAQ5 fluorescent probe) and incubated for 5 min at rt. A PerkinElmer Operetta (high-content imager) was used to capture images for each well and find macrophage and amastigote nuclei within macrophage cytoplasm using Harmoney software that counts the number of amastigotes per 500 macrophages in each well and generates IC50 values.
Cytotoxicity Assay
Cytotoxicity was determined by using the CellTiter 96 AQueous One Solution cell proliferation assay (Promega) on A549, human lung carcinoma, cells. A549 cells were seeded at a concentration of 1.6 × 104 cells/mL (1440 cell/well) in a 96-well tissue culture plate (Corning), in the presence of serially diluted pure compounds previously identified to be active. Positive control wells contained cells and media, and negative control wells contained media alone. Cells were grown in F12K medium supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin (all supplied from Fisher Scientific). The final inhibitor concentration started at 50 μg/mL and was diluted in doubling dilutions to determine cytotoxicity. The total volume of each well was 100 μL, and plates were incubated at 37 °C, 5% CO2 for 72 h. Each well received 20 μL of MTS (Promega) 4 h before the final time-point. Inhibition of A549 growth was assessed at the 72 h time-point measuring the OD values determined at 490 nm using a SpectraMax i3X (Molecular Devices). Curve fitting using nonlinear regression was carried out using DataAspects Plate Manager analysis software to obtain IC50 values.
Acknowledgments
We would like to thank the scientists and crew aboard the Nathaniel B. Palmer that aided our research efforts during the 2013 research cruise. For the crystallographic data, we thank Dr. L. Wojtas and W. Gao for their assistance. Funding for this project was provided by grants ANT-1043749 (N.G.W.), ANT-0838776, and PLR-1341339 (to B.J.B.), from the Antarctic Organisms and Ecosystems program of the National Science Foundation, by grant AI103673 (to B.J.B. and D.E.K.) from the National Institute of Allergy and Infectious Diseases, and a by a Center of Excellence award from the State of Florida to support the Center for Drug Discovery and Innovation, whose facilities made much of the chemical analysis possible. G. W. Rouse, Scripps Institution of Oceanography, was instrumental in the logistical and conceptual implementation of the field work. We are grateful for screening against ESKAPE pathogens in the lab of Dr. L. Shaw, against Clostridium difficile in the lab of Dr. X. Sun, and against Naegleria fowleri by Dr. C. Rice in the Kyle lab.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jnatprod.7b00732.
Author Contributions
∥ S. A. L. Thomas and J. L. von Salm contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Blunt J. W.; Copp B. C.; Keyzers R. A.; Munro M. H. G.; Prinsep M. R. Nat. Prod. Rep. 2017, 34, 235–294. 10.1039/C6NP00124F. [DOI] [PubMed] [Google Scholar]
- Jouiaei M.; Yanagihara A. A.; Madio B.; Nevalainen T. J.; Alewood P. F.; Fry B. G. Toxins 2015, 7, 2251–2271. 10.3390/toxins7062251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roethle P. A.; Trauner D. Nat. Prod. Rep. 2008, 25, 298–317. 10.1039/b705660p. [DOI] [PubMed] [Google Scholar]
- Coll J. C. Chem. Rev. 1992, 92, 613–631. 10.1021/cr00012a006. [DOI] [Google Scholar]
- Martins A.; Vieira H.; Gaspar H.; Santos S. Mar. Drugs 2014, 12, 1066–1101. 10.3390/md12021066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WoRMS, World Register of Marine Species. www.marinespecies.org/aphia.php?p=taxlist&pid=1365&rComp=%3E%3D&tRank=220 (accessed 17 May 2017).
- Deep Sea Octocorals Online. deepseaoctocoral.myspecies.info/search/site/alcyonacea (accessed 17 May 2017).
- MarinLit, a database of the marine natural products literature. http://pubs.rsc.org/marinlit/searchresult/compounds?FullText=True&Page=tax&Taxonomy=%7ctxc000000000006%7ctxo000000000008%7c%7c%7c%7cfalse (accessed 17 May 2017).
- Skropeta D. Nat. Prod. Rep. 2008, 25, 1131–1166. 10.1039/b808743a. [DOI] [PubMed] [Google Scholar]
- Skropeta D.; Wei L. Nat. Prod. Rep. 2014, 31, 999–1025. 10.1039/C3NP70118B. [DOI] [PubMed] [Google Scholar]
- von Salm J. L.; Wilson N. G.; Vesely B. A.; Kyle D. E.; Cuce J.; Baker B. J. Org. Lett. 2014, 16, 2630–3. 10.1021/ol500792x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iken K. B.; Baker B. J. J. Nat. Prod. 2003, 66, 888–890. 10.1021/np030051k. [DOI] [PubMed] [Google Scholar]
- Ishigami S. T.; Goto Y.; Inoue N.; Kawazu S.; Matsumoto Y.; Imahara Y.; Tarumi M.; Nakai H.; Fusetani N.; Nakao Y. J. Org. Chem. 2012, 77, 10962–10966. 10.1021/jo302109g. [DOI] [PubMed] [Google Scholar]
- Stonik V. A.; Kapustina I. I.; Kalinovsky A. I.; Dmitrenok P. S.; Grebnev B. B. Tetrahedron Lett. 2002, 43, 315–317. 10.1016/S0040-4039(01)02114-1. [DOI] [Google Scholar]
- The pukalides were originally isolated from a Hawaiian soft coral collected near the popular tourist attraction known as the Blow Hole:Missakian M. G.; Burreson B. J.; Scheuer P. J. Tetrahedron 1975, 31, 2513–2515. 10.1016/0040-4020(75)80262-6. [DOI] [Google Scholar]; Puka is the Hawaiian word for hole. We use the Hawaiian word for child, keiki, to distinguish our pukalide derivatives.
- Abramson S. N.; Trischman J. A.; Tapiolas D. M.; Harold E. E.; Fenical W.; Taylor P. J. Med. Chem. 1991, 34, 1798–1804. 10.1021/jm00110a007. [DOI] [PubMed] [Google Scholar]
- Duh C.-Y.; Wang S.-K.; Chia M.-C.; Chiang M. Y. Tetrahedron Lett. 1999, 40, 6033–6035. 10.1016/S0040-4039(99)01194-6. [DOI] [Google Scholar]
- Bowden B. F.; Coll J. C.; Wright A. D. Aust. J. Chem. 1989, 42, 757–763. 10.1071/CH9890757. [DOI] [Google Scholar]
- Bandurraga M. M.Natural Product Studies of Selected East Pacific Gorgonians. Ph.D. Dissertation, University of California, San Diego, 1982. [Google Scholar]
- Rice L. B. J. Infect. Dis. 2008, 197, 1079–1081. 10.1086/533452. [DOI] [PubMed] [Google Scholar]
- France S. C.; Hoover L. L. Bull. Biol. Soc. Wash. 2001, 10, 110–118. [Google Scholar]
- France S. C.; Hoover L. L. Hydrobiologia 2002, 471, 149–155. 10.1023/A:1016517724749. [DOI] [Google Scholar]
- Stamatakis A. Bioinformatics 2006, 22, 2688–2690. 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- Silvestro D.; Michalak I. Org. Divers. Evol. 2012, 12, 335–337. 10.1007/s13127-011-0056-0. [DOI] [Google Scholar]
- Bruker. SAINT, 8.35A; Bruker AXS Inc.: Madison, WI, USA, 2016.
- Sheldrick G. M.SADABS, Program for Empirical Absorption Correction; University of Gottingen, 1996.
- Bruker. APEX3 2015.9; Bruker AXS Inc: Madison, WI, USA, 2016.
- Sheldrick G. M. Acta Crystallogr. 2015, C71, 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldrick G. M. Acta Crystallogr. 1990, A46. [Google Scholar]
- Sheldrick G. M. Acta Crystallogr., Sect. A: Found. Crystallogr. 2008, A64, 112–122. 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- Dolomanov O. V.; Bourhis L. J.; Gildea R. J.; Howard J. A. K.; Puschmann H. J. Appl. Crystallogr. 2009, 42, 339–341. 10.1107/S0021889808042726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooft R. W. W.; Straver L. H.; Spek A. L. J. Appl. Crystallogr. 2008, 41, 96–103. 10.1107/S0021889807059870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spek A. L.Acta Crystallogr., Sect. D: Biol. Crystallogr. 2009, D65, 148–155 10.1107/S090744490804362X. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.