

Abstract
Background
MicroRNAs (miRNAs) have been shown to function in many different cellular processes, including proliferation, apoptosis, differentiation and development. miR-181a, -181b, -181c and -181d are miR-181 members of the family, which has been rarely studied, especially uveal melanoma.
Methods
The expression level of miR-181 family in human uveal melanoma cell lines was measured via real-time PCR (RT-PCR). The function of miR-181 on cell cycle was detected through Flow Cytometry assay. Microarray assay and Bioinformatics analysis were used to find the potential target of miR-181b, and dual-luciferase reporter assays further identified the target gene.
Results
MiR-181 family members were found to be highly homologous across different species and their upregulation significantly induces UM cell cycle progression. Of the family members, miR-181b was significantly overexpressed in UM tissues and most UM cells. Bioinformatics and dual luciferase reporter assay confirmed CTDSPL as a target of miR-181b. miR-181b over-expression inhibited CTDSPL expression, which in turn led to the phosphorylation of RB and an accumulation of the downstream cell cycle effector E2F1, promoting cell cycle progression in UM cells. Knockdown CTDSPL using siRNAs showing the same effect, including increase of E2F1 and the progression of cell cycle.
Conclusions
MiR-181 family members are key negative regulators of CTDSPL-mediated cell cycle progression. These results highlight that miR-181 family members, especially miR-181b, may be useful in the development of miRNA-based therapies and may serve as novel diagnostic and therapeutic candidate for UM.
Electronic supplementary material
The online version of this article (10.1186/s13046-018-0679-5) contains supplementary material, which is available to authorized users.
Keywords: miR-181, Uveal melanoma, CTDSPL, E2F1, Cell cycle
Background
Recently, miRNAs were found to play critical roles in many different cellular processes, especially in tumor progression. There are nearly 1000 miRNAs and more than 40,000 protein-coding genes in the human genome [1, 2]. Consequently, it is more feasible to explore reliable miRNA biomarkers from genome-wide miRNA expression data than from genome-wide gene expression data. miRNAs are a class of short non-coding RNAs ranging from 19 to 25 nucleotides in length, which are transcribed as precursors and are matured to active forms by a series of enzymes, including Dicer [3]. Numerous studies have explored the instrumental roles of these small, non-coding RNA species, mostly through base-pairing to the untranslated region (UTR) of target mRNA, thus leading to its degradation and/or reduced translation [4]. Generally, an individual miRNA can regulate the expression of multiple target genes, and several miRNAs can synergistically act on one target gene, regulating cell differentiation, proliferation, mobility and apoptosis [5]. The miR-181 family contains four miRNAs (miR-181a/b/c/d). miR-181a and miR-181b are transcribed from two separated gene loci (miR-181a-1/miR-181b-1 and miR-181a-2/miR-181b-2), while miR-181c and miR-181d are transcribed from another locus [6]. It had been reported that miR-181a, -181b, -181c and -181d function differently in a tumor series. However, the homology among the miR-181 family members and the contribution of miR-181a, -181b, -181c and -181d in UM have not yet been clarified.
UM is the most frequent malignant intraocular cancer in adults, and up to 50% of UM patients are at risk of metastasis via hematogenous spread, most commonly to the liver [7]. Recently, epigenetic events mediated by miRNAs have been implicated in UM development. UM proliferation and progression are regulated by dynamic interactions between UM-specific regulators, including miRNAs, whose aberrant expression has been associated with oncogenesis and tumor suppressor activity [8]. Recent studies have implicated miRNAs in UM development. For example, miR-20a functions as an oncogenic miRNA involved in promoting cell growth in UM, and miR-454 promotes proliferation and invasion by regulating PTEN in UM [9, 10]. On the other hand, miR-32 and miR-124a both function as tumor suppressors by regulating multiple targets involved in UM development [11, 12]. Moreover, growing evidence indicates that miRNA expression can potentially be used as a biomarker for the diagnosis and prognosis of different tumors. However, the expression and function of the miR-181 family members in the pathogenesis of UM had not been established.
In the present study, the homology and function of miR-181 family members, miR-181a, -181b, -181c, and -181d, were investigated. miR-181 family members were found to be highly homologous and have the same target, CTDSPL. The CTDSPL gene contains 8 exons coding for a 4.8 kb mRNA, which has been previously denoted as HYA22 and RBSP3, is a recently identified phosphatase-like tumor suppressor gene that dephosphorylates the Rb1 serine on Ser-807 and Ser-811 [13]. The sequence analysis shows that CTDSPL belongs to a gene family of small C-terminal domain phosphatases that may control the RNA polymerase II transcription machinery [14]. Then, the pattern of miRNA expression in melanoma tissues was analyzed using microarray technology. The microarray results indicated miR-181b1 and miR-181b2 were highly expressed in melanoma tissues. Furthermore, miR-181b was found to be extremely overexpressed in most UM cells. These findings raised the possibility that miR-181b might have an important role in UM development or pathogenesis. However, the molecular basis for this phenotype has not been elucidated, and the status of the downstream targets of miR-181b in UM has not been researched. Therefore, a better understanding of the mechanisms responsible for UM and an exploration of the novel diagnostic and therapeutic strategies are crucial for achieving improved patient outcomes.
Methods
Cell culture and transfection
UM cells SP6.5, VUP, OCM1 and 92-1 were maintained in Dulbecco’s Modified Essential Medium (DMEM; Gibco, Carlsbad, CA, USA) with 10% fetal bovine serum (FBS; Gibco) OCM1a and MUM2b were maintained in Iscove’s Modified Dulbecco’s Medium (IMDM; Gibco) with 10% FBS. The normal control cells, RPE, were maintained in DMEM with 10% FBS. Cultures were maintained at 37 °C in a 5% CO2 humidified atmosphere. Cells were treated and harvested for qRT-PCR and Western blot analysis. MUM2b (3 × 105) or OCM1a (5 × 105) cells were cultured overnight in 6-well plates and transfected with 200 nM miR-NC, miR-181 family mimics, or as-miR-181 family members (GenePharma Co. Ltd., Shanghai, China) using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). Two days later, these cells were either harvested for protein and mRNA or fixed using 70% ethanol for FCM.
Cell cycle analysis
Treated UM cells along with control cells were harvested. The cells were washed twice with cold phosphate-buffered saline (PBS), fixed in 70% ethanol and stored at 4 °C overnight. The next day, the cells were washed twice with cold PBS and incubated with propidium iodide/ribonuclease staining solution (Becton Dickinson, NJ, USA) for 15 min at room temperature, following the manufacturer’s instructions. Cell cycle distribution was detected and analyzed using the FACScan instrument and CellQuest program (Becton Dickinson, NJ, USA).
Western blot analysis
After the indicated treatments, the cells were washed with PBS and lysed with ice-cold lysis buffer (RIPA; Sigma Chemical Co, MO, USA). Cell lysates were incubated at 4 °C for 50 min. After centrifugation at 12,000 g for 1 min at 4 °C, protein concentration was determined by a BCA protein assay (Bio-Rad, Hercules, CA, USA). Thirty micrograms of protein were separated on 10% SDS–PAGE and transferred to a PVDF membrane. Membranes were probed with primary antibodies against CTDSPL (Abcam, Cambridge, UK) or E2F1 (Abcam, Cambridge, UK) at 4 °C overnight. Next, the membranes were washed three times with TBS containing 0.1% Tween-20 and incubated with secondary antibody for 1 h. The PVDF membrane was washed three times with Tris-Buffered Saline Tween-20 (TBST). After washing with TBST, the bands were detected using the Odyssey Infrared imaging system (Odyssey; LI-COR, Lincoln, NE).
Dual-luciferase reporter assay
To determine the common target region of the miR-181 family in CTDSPL, a segment of wild-type and mutated 3’-UTR of the human CTDSPL cDNA was constructed. Constructs were validated by sequencing. 293 T cells were plated in 24-well flat-bottomed plates and co-transfected with the wild-type or mutated 3’-UTR of the CTDSPL reporter plasmid, pRL-TK, and miR-181 family members or miR-NC using Lipofectamine 2000 reagent. Firefly and Renilla luciferase activities were determined 24 h after transfection using the dual-luciferase reporter assay system (Promega, Madison, WI, USA). The Renilla values were normalized to firefly luciferase.
Microarray and computational analysis
Briefly, RNA from tissue samples (three melanomas and three normal tissues) was used to synthesize double-stranded cDNA, and double-stranded cDNA was labeled and hybridized to the 2.0 microRNA Expression Microarray (Affymetrix GeneChip Human Gene 2.0 ST Array, Rockville, MD, USA). Raw data were extracted as pair files using NimbleScan software (version 2.5; Roche NimbleGen, Inc., Madison, WI, USA). NimbleScan software’s implementation of RMA offers quartile normalization and background correction. Differentially expressed genes were identified through the random variance model. The AP value was calculated using the paired t-test. The threshold set for up- and downregulated genes was a fold change > 2.0 and a P-value < 0.05. Hierarchical clustering was performed based on differentially expressed miRNAs using Cluster_Treeview software from Stanford University (Palo Alto, CA, USA).
RNA extraction, reverse transcription and quantitative polymerase chain reaction (RT-qPCR)
Total RNA from UM cells was isolated using the Trizol reagent (Invitrogen, Carlsbad, CA, USA) following the protocol provided by the manufacturer. The cDNA synthesis reaction was performed according to the manufacturer’s protocol (TaKaRa Bio, Otsu, Japan). DPN1 enzyme (Sangon Biotech, Shanghai, China) was used to delete the genomic DNA from the extracted RNA, which was used to amplify the miRNAs. Screening for miRNAs was performed by qRT-PCR with the primer sets described in Table 1. PCR reactions were performed according to the manufacturer’s protocol (TaKaRa Bio) and were repeated at least three times for each sample. The miRNA loop primers were used first, and then the miRNAs PCR primers used. The relative levels of target gene miRNA transcripts to control U6 were determined by the 2-△△CT method.
Table 1.
Primers used in this study
| Primer name | Sequence (5′- 3′) |
|---|---|
| hsa-miR-181a-5p loop | GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACACTCACCG |
| hsa-miR-181a-5p F | TGCGCAACATTCAACGCTGTCG |
| hsa-miR-181a-5p R | CTCAAGTGTCGTGGAGTCGGCAA |
| hsa-miR-181b-5p loop | GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACACCCACCG |
| hsa-miR-181b-5p F | TGCGCAACATTCATTGCTGTCG |
| hsa-miR-181b-5p R | CTCAAGTGTCGTGGAGTCGGCAA |
| hsa-miR-181c-5p loop | GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACACTCACCG |
| hsa-miR-181c-5p F | TGCGCAACATTCAACCTGTCG |
| hsa-miR-181c-5p R | CTCAAGTGTCGTGGAGTCGGCAA |
| hsa-miR-181d-5p loop | GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACACCCACCG |
| hsa-miR-181d-5p F | TGCGCAACATTCATTGTTGTCG |
| hsa-miR-181d-5p R | CTCAAGTGTCGTGGAGTCGGCAA |
| U6 F | CGCTTCGGCAGCACATATAC |
| U6 R | AAATATGGAACGCTTCACGA |
| CTDSPL-F | GTGGCTGACCTCCTAGACC |
| CTDSPL-R | TTCACGTAGTTCCCACGATGA |
| GAPDH-F | GGCTGTTGTCATACTTCTCATGG |
| GAPDH-R | GGCTGTTGTCATACTTCTCATGG |
| CTDSPL-si1 | GCAGCAUCCUUAGCUCCUUTT |
| CTDSPL-si2 | UCCACCAGCUAAGUACCUUTT |
Overexpressing miR-181b plasmid construction, lentivirus package, cloning and stable transfection in UM cells
The miR-181b sequence was amplified and sequenced without mutations. Then, 293 T cells were used to package the lentivirus. 293 T cells were cultured in DMEM supplemented with 10% FBS and maintained at 37 °C at a concentration of 6 × 106 cells/ml and transfected using Lipofectamine 2000 reagent with 3 μg PL-shRNA-HSA-MIR-181b-5p, 3 μg pMD2.D, and 6.0 μg PsPax. After incubation overnight with the 293 T cells, the media was replaced with 5 mL of fresh medium. The viral supernatants were concentrated and used to obtain stably transfected miR-181b-overexpressing UM cells. Stable MUM2b and OCM1a cell lines were established by lentiviral infection and blasticidin selection. The colonies with GFP expression were selected for subsequent culture after incubation with 4 g/mL blasticidin for 3 weeks. Transduction efficiency was determined by EGFP expression and measured by qRT-PCR.
Statistical analysis
All experiments were carried out in triplicate. All statistical analyses were performed using SPSS19.0 software. The statistical analysis was performed with a double-sided Student’s t-test for comparison of two groups. All data are expressed as the mean ± standard error of the mean (SEM). Differences at P < 0.05 were considered statistically significant.
Results
miR-181 family members are highly conserved, and their upregulation promotes cell cycle progression
To explore the relationship among miR-181 family members, their sequence homology was investigated. Evolutionary conservation analysis of the miR-181 family members indicated that the sequences of miR-181a, -181b, -181c, and -181d are partly conserved in Homo sapiens, Mus musculus, Rattus norvegicus, Bos taurus and Pan troglodytes (Fig. 1a). To investigate the potential roles of miR-181 family members, miR-181 family mimics (miR-181a, -181b, -181c, and -181d) or inhibitors (as-miR-181a, -181b, -181c, and -181d) were separately transfected into MUM2b and OCM1a cells. The results demonstrated that mimics of miR-181 family members promoted cell cycle progression, while inhibitors of miR-181 family members led to cell cycle arrest (Fig. 1b-e).
Fig. 1.
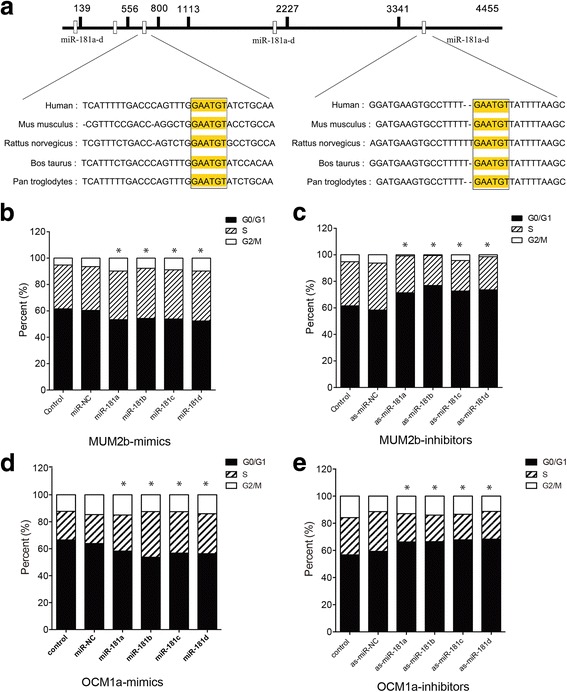
The conservation and cell cycle analysis of miR-181 family members. a Schematic of the miR-181 family putative target sites in the human 3’-UTR of CTDSPL. The sequences of the miR-181 family members are partly evolutionarily conserved in Homo sapiens, Mus musculus, Rattus norvegicus, Bos taurus and Pan troglodytes. Yellow indicates the conserved sequence. The hollow white rectangle indicates the five different gene loci of the miR-181 family members. b and (c) The cell cycle distribution was detected. The fraction of cells in G0/G1-phase was significantly decreased by 8-12%, and the periods of S-phases were significantly increased 6-15% after the mimics of miR-181 family members were transfected into MUM2b cells compared with the control and miR-NC groups (P < 0.05). In contrast, the fraction of cells in G0/G1 phase was significantly increased by 15-18%, and the period of S-phases was significantly decreased 15-20% after the inhibitors of miR-181 family members were transfected into MUM2b cells. d and (e) The fraction of cells in G0/G1-phase was significantly decreased by 10-20%, and the periods of S-phases were significantly increased 8-18% after the mimics of miR-181 family members were transfected into OCM1a cells compared with the control and miR-NC groups (P < 0.05). In contrast, the fraction of cells in G0/G1 phase was significantly increased by 12-15%, and the period of S-phases was significantly decreased 8-12% after the inhibitors of miR-181 family members were transfected into OCM1a cells
Bioinformatics and molecular biology assays confirmed CTDSPL as a target of miR-181 family members
To identify the target gene(s) of miR-181, candidate genes were identified using the miRNA target prediction database TargetScan [13] (http://www.targetscan.org/), miRanda [14] (http://www.microrna.org/) and Pictar [15] (http://pictar.mdc-berlin.de/). miR-181 family members were predicted to target CTDSPL, which had previously been denoted as RBSP3 (RB1 serine phosphatase from human chromosome 3), a key downstream mediator of cell cycle progression, and has been reported to participate in acute myeloid leukemia pathogenesis [6]. There are five predicted target sites in the 3’-UTR of CTDSPL sequence for miR-181 family members. The predicted sequences to which miR-181 binds in the 3’-UTR of CTDSPL are conserved in humans (Fig. 2a). Western blot assays further indicated that mimics of miR-181 family members led to the reduced expression of CTDSPL, while inhibitors led to the increased expression of CTDSPL in MUM2b cells (Fig. 2b-c, P < 0.05). To examine whether miR-181 family members could directly regulate CTDSPL expression, 293 T cells were transfected with a luciferase reporter construct containing the putative wild-type and mutant 3’-UTR of CTDSPL binding sites, together with one of the following miRNAs: miR-181a, -181b, -181c, -181d, miR-NC, as-miR-181a, -181b, -181c, or -181d. Compared with control cells, firefly luciferase activity was significantly decreased by nearly 3-fold after treatment with miR-181a, -181b, -181c, and -181d mimics and cotransfection with the wild-type CTDSPL gene 3’-UTR plasmid (Fig. 2d), whereas activity increased approximately 3-fold when miR-181a, -181b, -181c, or -181d inhibitors were used (Fig. 2f). Firefly luciferase activity was largely unchanged when the putative mutant of the 3’-UTR of CTDSPL binding sites was used (Fig. 2e and g). These data provide strong evidence that the miR-181 family members inhibit CTDSPL gene expression by directly binding to sites within its 3’-UTR.
Fig. 2.
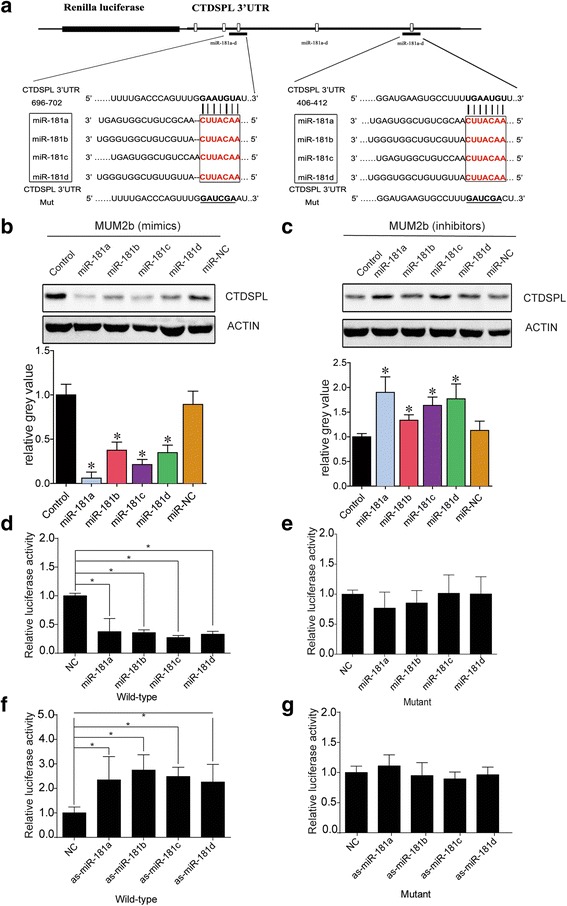
CTDSPL is a direct target of miR-181 family members. a Alignment of the seed sites in the human 3’-UTR of CTDSPL gene. The mutated 3’-UTR of CTDSPL is underlined. b and (c) MUM2b cells were transfected with 40 nM miR-NC, miR-181a, -181b, -181c, and -181d or as-miR-181a, -181b, -181c and -181d. Overexpression or knockdown of miR-181 family inhibited or enhanced CTDSPL expression, respectively. The gray level was analyzed by histogram. d and (f) Overexpression or knockdown of miR-181 expression inhibited or enhanced the Renilla luciferase activity, respectively. 293 T cells were cotransfected with 40 nM miR-NC, miR-181a, -181b, -181c, and -181d, or as-miR-181a, -181b, -181c, and -181d, and 100 ng of reporter plasmid containing the wild-type 3’-UTR of CTDSPL. After 24 h, Renilla luciferase values, normalized against firefly luciferase, were measured. e and (g) The Renilla luciferase activity was nearly unchanged after mimics and inhibitors of miR-181 family members were transfected with the mutated 3’-UTR of CTDSPL
miR-181b was extremely overexpressed in melanoma tissues and most UM cells
To investigate the expression profile of miR-181 family members in UM, microarray technology was used to detect the expression of miR-181 family members in melanoma tissues. Compared with normal samples, gene chip results showed that miR-181b1 and miR-181b2 were significantly overexpressed in melanoma tissues. miR-181a expression was also upregulated, while miR-181c and miR-181d were essentially unchanged (Fig. 3a). Next, miR-181 family members were detected in various types of UM cells, including OCM1, SP6.5, VUP, OCM1a, MUM2b and 92-1 cells. In accordance with the microarray results, miR-181b was overexpressed in OCM1, SP6.5, VUP and 92-1 cells by nearly 50-fold and was up more than 1000-fold in 92-1 cells, while miR-181b was not upregulated in OCM1a or MUM2b cells (Fig. 3b). miR-181a was also upregulated in OCM1, SP6.5, VUP and 92-1 cells 12-20-fold. The expression levels of miR-181c and miR-181d were not upregulated in most UM cell lines, except for a slight increase in miR-181c in OCM1 cells and a mild upregulation of miR-181d in OCM1a, both less than 10-fold. Additionally, there was no downregulation of any miR-181 family member (Fig. 3b) in these cell lines. These results show that miR-181b exhibited significantly higher expression in most UM cells, strongly implying a relationship between the upregulation of miR-181b and UM development and that miR-181b expression is a specific marker of UM.
Fig. 3.
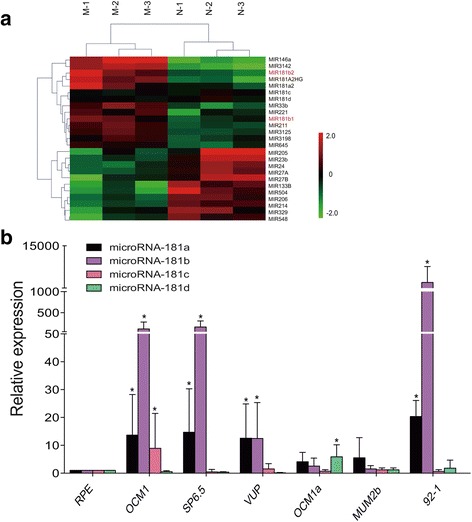
The expression profile of miR-181 in melanoma tissues and UM cells. a Hierarchical clustering analysis of miRNAs that were differentially expressed in melanoma compared with non-tumor samples. Expression values are represented in shades of red and green indicating expression above and below the median expression value across all samples (log scale 2, from − 2 to + 2), respectively. miR-181b1 and miR-181b2 were significantly upregulated in melanoma tissues. b The expression of miRNA-181a-d was measured by qRT-PCR in RPE, OCM1, SP6.5, VUP, OCM1a, MUM2b and 92-1 cells. miR-181b was overexpressed in OCM1, SP6.5, VUP and 92-1 cells by approximately 50-fold and more than 1000-fold in 92-1 cells, while miR-181b was not upregulated in OCM1a or MUM2b cells. miR-181a was upregulated in OCM1, SP6.5, VUP and 92-1 cells by approximately 12-to-20-fold. The expression levels of miR-181c and miR-181d were not upregulated in most UM cell lines, except for a slight increase in miR-181c in OCM1 cells and miR-181d in OCM1a cells, both less than 10-fold. There was no downregulation of any miR-181 family members. Triplicate assays were performed for each sample, and the relative level of each miRNA was normalized to U6 (*P < 0.05)
miR-181b overexpression promotes cell cycle progression through CTDSPL with the downstream release of E2F1 in MUM2b and OCM1a cells
To explore the function of miR-181b, we constructed a high-level expression plasmid of human miR-181b. Our earlier qRT-PCR results had demonstrated that miR-181b was highly expressed in a variety of UM cells, except MUM2b and OCM1a cells. Thus, MUM2b and OCM1a cells were stably transfected with a miR-181b overexpression plasmid (MUM2b-over-miR-181b, OCM1a-over-miR-181b), which also contained an EGFP tag. The transfection efficiency of the stably transfected MUM2b and OCM1a cells was determined by immunofluorescence and qRT–PCR (Fig. 4a-b). There were high amounts of green fluorescence in both the MUM2b-over-miR-181b and OCM1a-over-miR-181b cells. However, green fluorescence was not observed in the control cells (Fig. 4a). The qRT-PCR results showed a significantly higher expression of miR-181b with a nearly 500-fold increase in both the MUM2b-over-miR-181b and OCM1a-over-miR-181b cells (Fig. 4b, P < 0.05). Next, the cell cycle distribution was analyzed to assess whether miR-181b affected the cell cycle of MUM2b or OCM1a cells. We found that cell cycle progression was promoted in both the MUM2b-over-miR-181b and OCM1a-over-miR-181b cells. The fraction of cells in G0/G1-phase was significantly decreased by approximately 19% and 16%, while that the S-phases was significantly increased by about 10% and 18% in MUM2b-over-miR-181b and OCM1a-over-miR-181b cells, respectively, compared with the control group (Fig. 4c-d, P < 0.05). These results suggest that miR-181b might act as a regulator of UM cell cycle progression. To explore whether CTDSPL is involved in the UM cell cycle, we determined the expression level of CTDSPL protein in UM cells. A decreased expression in CTDSPL and an increased expression of E2F1 were found in MUM2b-over-miR-181b and OCM1a-over-miR-181b cells compared with the control group (Fig. 4e-f).
Fig. 4.
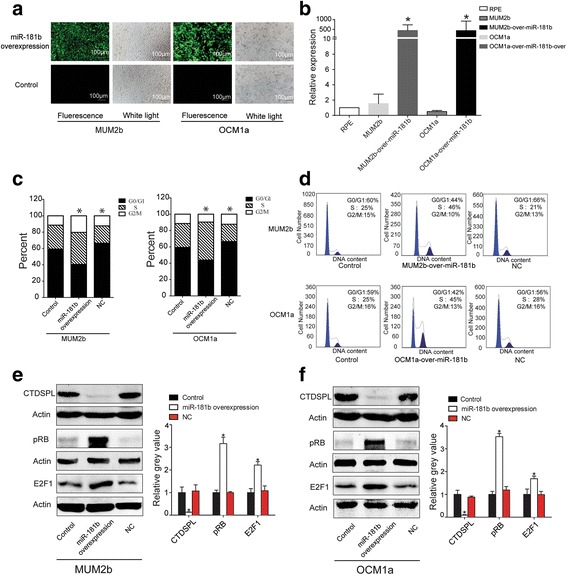
miR-181b inhibits cell cycle distribution through CTDSPL and E2F1. a The miR-181b overexpression plasmid was stably transfected into MUM2b and OCM1a cells (MUM2b-over-miR-181b, OCM1a-over-miR-181b), and the plasmid also contained the EGFP tag. Obvious green fluorescence was observed in MUM2b-over-miR-181b and OCM1a-over-miR-181b cells, but not in the control groups (original magnification 100X). b The qRT-PCR results showed significantly higher expression of miR-181b of nearly 500-fold in MUM2b-over-miR-181b and OCM1a-over-miR-181b cells. c and (d) Cell cycle progression was significantly promoted in miR-181b-stably transfected MUM2b and OCM1a cells. The G0/G1 phase was significantly decreased by approximately 19% and 16%, while that the S-phases was significantly increased by about 10% and 18% in MUM2b-over-miR-181b and OCM1a-over-miR-181b cells, respectively, compared with the control group. e CTDSPL expression was significantly inhibited in miR-181b-stably transfected MUM2b and OCM1a cells. f E2F1 expression was significantly overexpressed in miR-181b-transfected MUM2b and OCM1a cells (*P < 0.05)
Decreased CTDSPL expression promotes cell cycle progression in MUM2b and OCM1a cells
To detect the function of CTDSPL, we inhibited expression of CTDSPL in MUM2b and OCM1a cells via transfection of CTDSPL inhibitors (si-CTDSPL-1 and si-CTDSPL-2). Results of qRT-PCR demonstrated that CTDSPL expression was inhibited in UM cells after transfection of si-CTDSPL-1 and si-CTDSPL-2, respectively, approximately 65% and 75% in MUM2b cells and approximately 40% and 35% in OCM1a cells (Fig. 5a). Western blotting further confirmed that expression of CTDSPL decreased in MUM2b and OCM1a cells after transfection with si-CTDSPL-1 and si-CTDSPL-2 (Fig. 5c-d). In addition, the cell cycle G0/G1-phase proportion decreased significantly, approximately 43% and 36% for MUM2b cells and 35% and 32% for OCM1a cells after transfection with si-CTDSPL-1 and si-CTDSPL-2, compared with control group which is 65% in MUM2b cells and 64% in OCM1a cells. While the cell cycle S-phase proportion increased from 13% to 21% and 15% in MUM2b cells and from 10% to 22% and 15% in OCM1a cells after transfection with si-CTDSPL-1 and si-CTDSPL-2 seperately (Fig. 5b). Increased pRB and E2F1 expression was also confirmed after transfection with si-CTDSPL-1 and si-CTDSPL-2 (Fig. 5c-d).
Fig. 5.
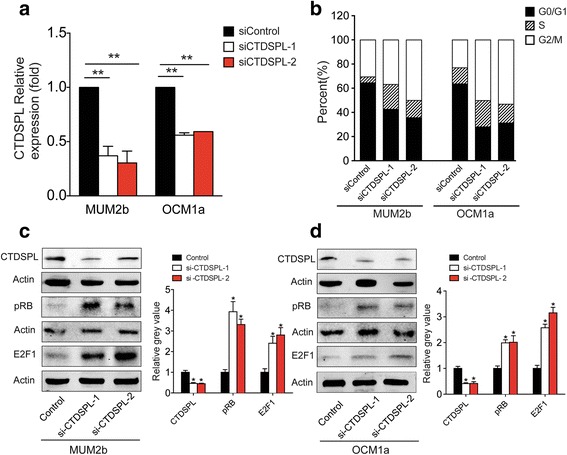
Decreased CTDSPL expression promotes cell cycle distribution through pRB and E2F1. a qRT-PCR results showed significantly decreased expression of CTDSPL after transfection with si-CTDSPL-1 and si-CTDSPL-2 in MUM2b and OCM1a cells, respectively. b The cell cycle G0/G1-phase proportion decreased significantly, approximately 43% and 36% for MUM2b cells and 35% and 32% for OCM1a cells after transfection with si-CTDSPL-1 and si-CTDSPL-2, compared with control group which is 65% in MUM2b cells and 64% in OCM1a cells. While the cell cycle S-phase proportion increased from 13% to 21% and 15% in MUM2b cells and from 10% to 22% and 15% in OCM1a cells after transfection with si-CTDSPL-1 and si-CTDSPL-2 seperately (Fig. 5b). c-d CTDSPL expression was significantly inhibited after transfection with si-CTDSPL-1 and si-CTDSPL-2 in MUM2b and OCM1a cells, whereas pRB and E2F1 expression was significantly increased in MUM2b and OCM1a cells after transfection with si-CTDSPL-1 and si-CTDSPL-2. The gray level was analyzed by histogram seperately (*P < 0.05)
miR-181 contributes to cell cycle progression via its target CTDSPL, which in turn increases expression of the cell cycle effector pRB/E2F1 in UM cells
Our work found that miR-181b is overexpressed in melanoma tissues and most UM cells and promotes cell cycle progression by repressing CTDSPL expression in UM cells. Previous studies have revealed that CTDSPL is involved in the regulation of the cell cycle through pRB-E2F1 [13, 15], which removes the phosphate group from serine 807 and 811 in its substrate, phosphorylated RB (pRB), and induces the release of E2F1 protein that then contributes to cell cycle progression [16, 17]. We thus hypothesized that miR-181b might contribute to UM pathogenesis via the CTDSPL–pRB–E2F1 pathway. The decreased CTDSPL expression along with the increased E2F1 expression in MUM2b-over-miR-181b and OCM1a-over-miR-181b cells supports our hypothesis. Collectively, these results demonstrate that miR-181b over-expression contributes to UM pathogenesis by targeting the CTDSPL–pRB–E2F1 pathway to alter cell cycle progression (Fig. 6).
Fig. 6.
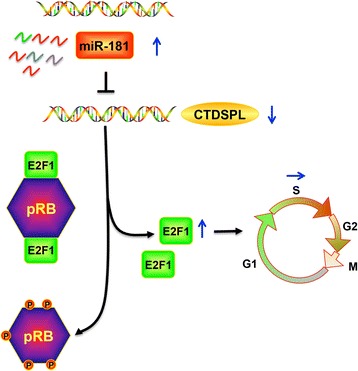
miR-181 targets CTDSPL, which modulates the cell cycle effector E2F1. Schematic representation of the pathway modulated by miR-181 in UM cells progressing through the cell cycle. miR-181 overexpression in UM cells induces progression through the G1/S transition and promotes S-phase entry. Thus, miR-181 induces cell cycle progression by repressing the downstream target CTDSPL, which in turn results in the phosphorylation of RB and an accumulation of the downstream cell cycle effector E2F1
Discussion
Recently, miRNAs have emerged as important cellular regulators that mediate cellular proliferation and progression. The miR-181 family consists of miR-181a, miR-181b, miR-181c, and miR-181d, and miR-181b, which divided into miR-181b1 and miR-181b2 and is transcribed from two separate gene loci [6]. However, there are few studies on the correlations among miR-181 family members. Here, we explored the correlations among miR-181 family members. First, we found that these miRs are highly evolutionarily conserved in different species, including Homo sapiens, Mus musculus, Rattus norvegicus, Bos taurus and Pan troglodytes (Fig. 1a), which suggests functional conservation. Cell cycle experiments demonstrated that pretreatment with miR-181 family mimics promoted cell cycle progression, while inhibitors resulted in cell cycle arrest (Fig. 1b). Similar effects usually shared the same mechanism and we know that small, non-coding RNA species mostly function through base-pairing to the untranslated region (UTR) of its target mRNA, leading to its degradation and/or reduced translation [18]. To explore the mechanism of the miR-181 family members, we performed bioinformatics analysis, and CTDSPL was identified as a candidate target gene of the miR-181 family members through three publicly available algorithms (TargetScan, miRanda and Pictar). Additionally, it had been previously reported and confirmed that miR-181a can bind to the 3′-UTR of CTDSPL mRNA. However, there are no relevant, precise studies regarding miR-181 [6]. Here, we found that all the miR-181 family members have nearly the same binding site within the 3’-UTR of CTDSPL. Additionally, we found five different binding sites (Fig. 2a). A firefly luciferase assay indicated that all miR-181 family members could inhibit CTDSPL expression through directly binding to the 3’-UTR of CTDSPL. Consequently, this work confirmed that the miR-181 family members are highly conserved and share a common function of direct binding to the 3’-UTR of CTDSPL.
The biological functions of the miR-181 family have been discussed in different tumors with different underlying biological processes. miR-181a was first identified and recognized as a contributor to hematopoietic lineage commitment and differentiation [19, 20]. In breast cancer, miR-181a could prevent and reverse drug resistance via binding to the 3’-UTR of BCRP [21]. It has been reported that miR-181a/b (miR-181a and miR-181b) suppress the translation of p300/CBP-associated factor (PCAF) mRNA, a process relevant to the epigenetic fine-tuning of epithelial inflammatory processes in liver epithelial cells [22]. There is another report that the loss of miR-181a-1/b-1 dampens the induction of experimental autoimmune encephalomyelitis and reduces basal TCR signaling in peripheral T cells and their migration from lymph nodes to pathogenic sites [23]. miR-181c inhibits glioblastoma cell invasion, migration and mesenchymal transition by targeting the TGF-beta pathway and is associated with metastatic brain cancer and high-grade osteosarcoma [24–26]. miR-181d acts as a glioma suppressor by targeting K-ras and Bcl-2 [27]. Compared with miR-181a, miR-181c and miR-181d, miR-181b was confirmed to be the most effective miRNA of the miR-181 family members [4, 6]. Studies have demonstrated that miR-181b is overexpressed in several cancers, such as colorectal cancer, acute lymphocytic leukemia (ALL), acute promyelocytic leukemia and hepatocellular carcinoma [28, 29]. Furthermore, miR-181b expression was found to be strongly associated with clinical response to S-1 in colon cancer patients [30]. On the other hand, reduced miR-181b expression has been observed in several primary human cancers, including gastric, lung, and prostate cancer, and acute myeloid leukemia and chronic lymphocytic leukemia [28, 31–33]. However, research regarding the miR-181 family is very scarce. They had been observed in glioblastoma, which could reverse mesenchymal transition by targeting KPNA4 [34]. At present, there are no investigations exploring their functions and mechanisms in UM pathogenesis and development. Here, microarray technology indicated there are different expression profiles of miR-181a, miR-181b, miR-181c, and miR-181d in UM. miR-181b1 and miR-181b2 were significantly overexpressed in melanoma tissues, while there were no significant changes in miR-181c and miR-181d. We also explored the function of these four miRNAs in UM cells. In accordance with the microarray results, we found miR-181b was extremely upregulated in most UM cell lines and that miR-181a was also upregulated in most UM cell lines. However, miR-181c and miR-181d were not upregulated in most UM cell lines, except for a slight increase of miR-181c in OCM1 and miR-181d in OCM1a. Given the different expression patterns of the miR-181 family members, we can assume that they function differently in different UM tissues and cell lines. Interestingly, there was no downregulation of any miR-181 family members in the UM cells compared with the control group. Thus, there is expression specificity of miR-181 family members in UM, but a lack of universality. In our study, miR-181b was especially interesting. miR-181b displayed the highest degree of expression in melanoma tissues and UM cell lines, and several previous studies have revealed that miR-181b is more active [4, 6] and has an intimate relationship with human malignant tumors, including hepatocellular carcinoma, colorectal gastric, lung, and prostate cancer, and ALL, acute myeloid leukemia, and chronic lymphocytic leukemia [28, 29, 31–33].
In our study, miR-181b was significantly upregulated in most UM cell lines, specifically VUP, SP6.5, OCM1 and 92-1, but was not upregulated in MUM2b or OCM1a cells. We then transfected MUM2b and OCM1a cells with a miR-181b overexpression plasmid. Compared with the control group, the cell cycle was at a later stage in miR-181b-overexpressing MUM2b and OCM1a cells. Bioinformatic analyses and dual-luciferase reporter assays demonstrated and confirmed that CTDSPL was the target of the miR-181 family members. A significant downregulation of CTDSPL and upregulation of E2F1 in MUM2b-over-miR-181b and OCM1a-over-miR-181b cells was confirmed. The underlying molecular pathway responsible for the effects of miR-181b in UM cell survival might control the G0/G1 to S phase transition through the repression of CTDSPL. CTDSPL is an important phosphatase-like tumor suppressor gene located at 3p21.3, and belongs to the small C-terminal domain phosphatase family, which modulates the RB/E2F1 signaling pathway and results in cell cycle arrest at the G1/S boundary [13, 35]. Previous studies have also reported that CTDSPL removes the phosphate group from serines 807 and 811 in its substrate, pRB, and thereby induces the formation of the RB/E2F1 complex [36, 37]. It had been reported that miR-100 regulates myeloid differentiation by targeting CTDSPL [38]. However, the involvement of CTDSPL in the regulation of cell growth in UM cells has not yet been studied. Our data demonstrated that overexpressed miR-181b knocked down CTDSPL expression and resulted in an accelerated G1/S transition in UM cells. These results indicate that overexpressed miR-181b inactivates the phosphatase CTDSPL protein and that this inactivation may be a common step that is required for UM progression.
In this work, miR-181b was found to control the G0/G1 to S phase transition by repressing CTDSPL and regulating E2F1 expression in most UM cells, except for MUM2b and OCM1a cells. This finding suggests that miR-181b expression is specifically increased or unchanged, without downregulation, in all UM cells, but there is lack of universality in UM. There were other reports that CTDSPL can directly bind to Rb just like the mechanism demonstrated by Beniaminov et al. [39, 40]. It showed a new method called surface plasmon resonance (SPR) to detect the direct interaction. However, in our co-immunoprecipitation assay, no interaction between CTDSPL and RB1 was found in OCM1a cell line (Additional file 1). Previous studies showed that cyclic phosphorylation/dephosphorylation of the pRb protein plays an important role in decreasing ppRb levels, ultimately blocks the G1/S progression. In our study, CTDSPL is another important regulator of ppRb level through phosphatase activity [14, 41, 42], showing as another parallel pathway, without relationship exists between cyclins and CTDSPL, at least in OCM1a cell line (Additional file 1).
miR-181 family members have been reported as potential therapeutic targets for myeloid dysplastic syndrome and acute myeloid leukemia [6]. Although miR-181c and -181d mimics and inhibitors could promote and inhibit CTDSPL expression through the same binding site in the CTDSPL gene, miR-181c and miR-181d were not upregulated in most UM cells except for a slight increase in miR-181c in OCM1 cells and miR-181d in OCM1a cells. This finding suggests that miR-181c and miR-181d do not play leading roles in UM cells but rather that miR-181c and 181d have support and backup functions for miR-181b, through binding to the 3’-UTR of CTDSPL and inhibiting its expression. In other words, while one of the miR-181 family members may be the primary functional miRNA in one tumor, the other miR-181 family members may assist it. Finally, miR-181 family members, and especially miR-181b overexpression, could be used as therapeutic targets for UM. Our results characterize a new role for miR-181 family members. It is regrettable that miR-181 family members were not detected in the limited UM tissues available, except via the microarray chip. Going forward, we hope to detect the expression of miR-181 in additional UM tissues. The prognostic and therapeutic value of miR-181 family members, especially miR-181b, needs to be confirmed in UM patients and other tumor types in the future. Moreover, miR-181 expression would be ideally detectable in the blood of UM patients, which would be useful in the future for UM patient diagnosis and prognosis.
Conclusions
In summary, we have presented herein the novel finding that miR-181b contributes to cell cycle progression through depressing the expression of CTDSPL, which in turn activates the downstream effector E2F1 and promotes S-phase entry. Furthermore, miR-181c and -181d might support and backup the function of miR-181b through binding to the 3’-UTR of CTDSPL and inhibit its expression in UM cells. Taken together, these results suggest that a high miR-181b expression may play an important role in UM through disrupting cell cycle control, promoting cell proliferation and consequently facilitating the development of UM via CTDSPL. This might thus represent a therapeutic target in UM. These findings constitute a comprehensive foundation for future research on the important role miR-181 in the developmental pathology of UM. The manipulation of miR-181 family members could be diagnostically and therapeutically relevant for the treatment of UM.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural Science Foundation of China grant (81602366, 81402258, 81570884), the Scientific Research Program of National Health and Family Planning Commission of China (201402014), the ShuGuang Project of Shanghai Municipal Education Commission and Shanghai Education Development Foundation (14SG18), and the Science and Technology Commission of Shanghai (17DZ2260100).
Availability of data and materials
The datasets used and analyzed during the current study are available from the corresponding authors on reasonable request.
Abbreviations
- CTDSPL
(CTD small phosphatase-like)
- miRNAs
(microRNAs)
- RBSP3
(RB1 serine phosphatase from human chromosome 3)
- RT-qPCR
(Reverse transcription quantitative polymerase chain reaction)
- UM
(Uveal melanoma)
Additional file
There are no interactions between CTDSPL and RB1, cyclins and CTDSPL. (TIF 9130 kb)
Authors’ contributions
ZLL wrote the paper; JRB and FXQ conceived and designed the study; ZLL, LF, XXF, PH, WXY and HXL did the experiments; ZH, and GSF collect and analyzed the data. XXF, HXY and LB gave important instructions to our experiment. All authors read and approved the final manuscript.
Ethics approval and consent to participate
All procedures performed in studies involving human participants were in accordance with the ethical standards of the Ethics Committee of the Institutional Ethical Review Board of Shanghai Ninth People’s Hospital. All patients studied signed an informed consent for participation.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Leilei Zhang, Xiaoyu He and Fang Li contributed equally to this report.
Xiaofang Xu, Renbing Jia and Xianqun Fan are senior authors of this report.
Electronic supplementary material
The online version of this article (10.1186/s13046-018-0679-5) contains supplementary material, which is available to authorized users.
Contributor Information
Xiaofang Xu, Email: xuxu0139@hotmail.com.
Renbing Jia, Email: jrb19760517@hotmail.com.
Xianqun Fan, Email: fanxq@sjtu.edu.cn.
References
- 1.Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F, Visone R, Iorio M, Roldo C, Ferracin M, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A. 2006;103(7):2257–2261. doi: 10.1073/pnas.0510565103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6(11):857–866. doi: 10.1038/nrc1997. [DOI] [PubMed] [Google Scholar]
- 3.Stark A, Brennecke J, Bushati N, Russell RB, Cohen SM. Animal MicroRNAs confer robustness to gene expression and have a significant impact on 3'UTR evolution. Cell. 2005;123(6):1133–1146. doi: 10.1016/j.cell.2005.11.023. [DOI] [PubMed] [Google Scholar]
- 4.Lim LP, Lau NC, Garrett-Engele P, Grimson A, Schelter JM, Castle J, Bartel DP, Linsley PS, Johnson JM. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature. 2005;433(7027):769–773. doi: 10.1038/nature03315. [DOI] [PubMed] [Google Scholar]
- 5.Davis-Dusenbery BN, Hata A. MicroRNA in cancer: the involvement of aberrant microRNA biogenesis regulatory pathways. Genes Cancer. 2010;1(11):1100–1114. doi: 10.1177/1947601910396213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Su R, Lin HS, Zhang XH, Yin XL, Ning HM, Liu B, Zhai PF, Gong JN, Shen C, Song L, et al. MiR-181 family: regulators of myeloid differentiation and acute myeloid leukemia as well as potential therapeutic targets. Oncogene. 2015;34(25):3226–3239. doi: 10.1038/onc.2014.274. [DOI] [PubMed] [Google Scholar]
- 7.Collaborative Ocular Melanoma Study Group Assessment of metastatic disease status at death in 435 patients with large choroidal melanoma in the collaborative ocular melanoma study (COMS): COMS report no. 15. Arch Ophthalmol. 2001;119(5):670–676. doi: 10.1001/archopht.119.5.670. [DOI] [PubMed] [Google Scholar]
- 8.Harbour JW. The genetics of uveal melanoma: an emerging framework for targeted therapy. Pigment Cell Melanoma Res. 2012;25(2):171–181. doi: 10.1111/j.1755-148X.2012.00979.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou J, Jiang J, Wang S, Xia X. Oncogenic role of microRNA20a in human uveal melanoma. Mol Med Rep. 2016;14(2):1560–1566. doi: 10.3892/mmr.2016.5433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sun L, Wang Q, Gao X, Shi D, Mi S, Han Q. MicroRNA-454 functions as an oncogene by regulating PTEN in uveal melanoma. FEBS Lett. 2015;589(19 Pt B):2791–2796. doi: 10.1016/j.febslet.2015.08.007. [DOI] [PubMed] [Google Scholar]
- 11.Ma YB, Song DW, Nie RH, Mu GY. MicroRNA-32 functions as a tumor suppressor and directly targets EZH2 in uveal melanoma. Genet Mol Res. 2016;15(2):gmr7935. doi: 10.4238/gmr.15027935. [DOI] [PubMed] [Google Scholar]
- 12.Chen X, He D, Dong XD, Dong F, Wang J, Wang L, Tang J, Hu DN, Yan D, Tu L. MicroRNA-124a is epigenetically regulated and acts as a tumor suppressor by controlling multiple targets in uveal melanoma. Invest Ophthalmol Vis Sci. 2013;54(3):2248–2256. doi: 10.1167/iovs.12-10977. [DOI] [PubMed] [Google Scholar]
- 13.Kashuba VI, Li J, Wang F, Senchenko VN, Protopopov A, Malyukova A, Kutsenko AS, Kadyrova E, Zabarovska VI, Muravenko OV, et al. RBSP3 (HYA22) is a tumor suppressor gene implicated in major epithelial malignancies. Proc Natl Acad Sci U S A. 2004;101(14):4906–4911. doi: 10.1073/pnas.0401238101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yeo M, Lin PS, Dahmus ME, Gill GN. A novel RNA polymerase II C-terminal domain phosphatase that preferentially dephosphorylates serine 5. J Biol Chem. 2003;278(28):26078–26085. doi: 10.1074/jbc.M301791200. [DOI] [PubMed] [Google Scholar]
- 15.Kashuba VI, Pavlova TV, Grigorieva EV, Kutsenko A, Yenamandra SP, Li J, Wang F, Protopopov AI, Zabarovska VI, Senchenko V, et al. High mutability of the tumor suppressor genes RASSF1 and RBSP3 (CTDSPL) in cancer. PLoS One. 2009;4(5):e5231. doi: 10.1371/journal.pone.0005231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liban TJ, Thwaites MJ, Dick FA, Rubin SM. Structural conservation and E2F binding specificity within the retinoblastoma pocket protein family. J Mol Biol. 2016;428(20):3960–3971. doi: 10.1016/j.jmb.2016.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fischer M, Grossmann P, Padi M, DeCaprio JA. Integration of TP53, DREAM, MMB-FOXM1 and RB-E2F target gene analyses identifies cell cycle gene regulatory networks. Nucleic Acids Res. 2016;44(13):6070–6086. doi: 10.1093/nar/gkw523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–297. doi: 10.1016/S0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 19.Chen CZ, Li L, Lodish HF, Bartel DP. MicroRNAs modulate hematopoietic lineage differentiation. Science. 2004;303(5654):83–86. doi: 10.1126/science.1091903. [DOI] [PubMed] [Google Scholar]
- 20.Li QJ, Chau J, Ebert PJ, Sylvester G, Min H, Liu G, Braich R, Manoharan M, Soutschek J, Skare P, et al. MiR-181a is an intrinsic modulator of T cell sensitivity and selection. Cell. 2007;129(1):147–161. doi: 10.1016/j.cell.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 21.Jiao X, Zhao L, Ma M, Bai X, He M, Yan Y, Wang Y, Chen Q, Zhao X, Zhou M, et al. MiR-181a enhances drug sensitivity in mitoxantone-resistant breast cancer cells by targeting breast cancer resistance protein (BCRP/ABCG2) Breast Cancer Res Treat. 2013;139(3):717–730. doi: 10.1007/s10549-013-2607-x. [DOI] [PubMed] [Google Scholar]
- 22.Zhao J, Gong AY, Zhou R, Liu J, Eischeid AN, Chen XM. Downregulation of PCAF by miR-181a/b provides feedback regulation to TNF-alpha-induced transcription of proinflammatory genes in liver epithelial cells. J Immunol. 2012;188(3):1266–1274. doi: 10.4049/jimmunol.1101976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schaffert SA, Loh C, Wang S, Arnold CP, Axtell RC, Newell EW, Nolan G, Ansel KM, Davis MM, Steinman L, et al. Mir-181a-1/b-1 modulates tolerance through opposing activities in selection and peripheral T cell function. J Immunol. 2015;195(4):1470–1479. doi: 10.4049/jimmunol.1401587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.He X, Liu Z, Peng Y, Yu C. MicroRNA-181c inhibits glioblastoma cell invasion, migration and mesenchymal transition by targeting TGF-beta pathway. Biochem Biophys Res Commun. 2016;469(4):1041–1048. doi: 10.1016/j.bbrc.2015.12.021. [DOI] [PubMed] [Google Scholar]
- 25.Tominaga N, Kosaka N, Ono M, Katsuda T, Yoshioka Y, Tamura K, Lotvall J, Nakagama H, Ochiya T. Brain metastatic cancer cells release microRNA-181c-containing extracellular vesicles capable of destructing blood-brain barrier. Nat Commun. 2015;6:6716. doi: 10.1038/ncomms7716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mori F, Sacconi A, Canu V, Ganci F, Novello M, Anelli V, Covello R, Ferraresi V, Muti P, Biagini R, et al. miR-181c associates with tumor relapse of high grade osteosarcoma. Oncotarget. 2015;6(16):13946–13961. doi: 10.18632/oncotarget.3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang XF, Shi ZM, Wang XR, Cao L, Wang YY, Zhang JX, Yin Y, Luo H, Kang CS, Liu N, et al. MiR-181d acts as a tumor suppressor in glioma by targeting K-ras and Bcl-2. J Cancer Res Clin Oncol. 2012;138(4):573–584. doi: 10.1007/s00432-011-1114-x. [DOI] [PubMed] [Google Scholar]
- 28.Schwind S, Maharry K, Radmacher MD, Mrozek K, Holland KB, Margeson D, Whitman SP, Hickey C, Becker H, Metzeler KH, et al. Prognostic significance of expression of a single microRNA, miR-181a, in cytogenetically normal acute myeloid leukemia: a cancer and leukemia group B study. J Clin Oncol. 2010;28(36):5257–5264. doi: 10.1200/JCO.2010.29.2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang B, Hsu SH, Majumder S, Kutay H, Huang W, Jacob ST, Ghoshal K. TGFbeta-mediated upregulation of hepatic miR-181b promotes hepatocarcinogenesis by targeting TIMP3. Oncogene. 2010;29(12):1787–1797. doi: 10.1038/onc.2009.468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schetter AJ, Leung SY, Sohn JJ, Zanetti KA, Bowman ED, Yanaihara N, Yuen ST, Chan TL, Kwong DL, Au GK, et al. MicroRNA expression profiles associated with prognosis and therapeutic outcome in colon adenocarcinoma. JAMA. 2008;299(4):425–436. doi: 10.1001/jama.299.4.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu X, Jia R, Zhou Y, Song X, Wang J, Qian G, Ge S, Fan X. Microarray-based analysis: identification of hypoxia-regulated microRNAs in retinoblastoma cells. Int J Oncol. 2011;38(5):1385–1393. doi: 10.3892/ijo.2011.961. [DOI] [PubMed] [Google Scholar]
- 32.Zhu W, Shan X, Wang T, Shu Y, Liu P. MiR-181b modulates multidrug resistance by targeting BCL2 in human cancer cell lines. Int J Cancer. 2010;127(11):2520–2529. doi: 10.1002/ijc.25260. [DOI] [PubMed] [Google Scholar]
- 33.Schaefer A, Jung M, Mollenkopf HJ, Wagner I, Stephan C, Jentzmik F, Miller K, Lein M, Kristiansen G, Jung K. Diagnostic and prognostic implications of microRNA profiling in prostate carcinoma. Int J Cancer. 2010;126(5):1166–1176. doi: 10.1002/ijc.24827. [DOI] [PubMed] [Google Scholar]
- 34.Wang H, Tao T, Yan W, Feng Y, Wang Y, Cai J, You Y, Jiang T, Jiang C. Upregulation of miR-181s reverses mesenchymal transition by targeting KPNA4 in glioblastoma. Sci Rep. 2015;5:13072. doi: 10.1038/srep13072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Senchenko VN, Anedchenko EA, Kondratieva TT, Krasnov GS, Dmitriev AA, Zabarovska VI, Pavlova TV, Kashuba VI, Lerman MI, Zabarovsky ER. Simultaneous down-regulation of tumor suppressor genes RBSP3/CTDSPL, NPRL2/G21 and RASSF1A in primary non-small cell lung cancer. BMC Cancer. 2010;10:75. doi: 10.1186/1471-2407-10-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ghosh A, Ghosh S, Maiti GP, Sabbir MG, Zabarovsky ER, Roy A, Roychoudhury S, Panda CK. Frequent alterations of the candidate genes hMLH1, ITGA9 and RBSP3 in early dysplastic lesions of head and neck: clinical and prognostic significance. Cancer Sci. 2010;101(6):1511–1520. doi: 10.1111/j.1349-7006.2010.01551.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Song L, Lin C, Wu Z, Gong H, Zeng Y, Wu J, Li M, Li J. MiR-18a impairs DNA damage response through downregulation of ataxia telangiectasia mutated (ATM) kinase. PLoS One. 2011;6(9):e25454. doi: 10.1371/journal.pone.0025454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zheng YS, Zhang H, Zhang XJ, Feng DD, Luo XQ, Zeng CW, Lin KY, Zhou H, Qu LH, Zhang P, et al. MiR-100 regulates cell differentiation and survival by targeting RBSP3, a phosphatase-like tumor suppressor in acute myeloid leukemia. Oncogene. 2012;31(1):80–92. doi: 10.1038/onc.2011.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Beniaminov AD, Krasnov GS, Dmitriev AA, Puzanov GA, Snopok BA, Senchenko VN, Kashuba VI. Interaction of two tumor suppressors: Phosphatase CTDSPL and Rb protein. Mol Biol. 2016;50(3):504–508. doi: 10.1134/S002689331603002X. [DOI] [PubMed] [Google Scholar]
- 40.Krasnov GS, Puzanov GA, Kudryavtseva AV, Dmitriev AA, Beniaminov AD, Kondratieva TT, Senchenko VN. Differential expression of an ensemble of the key genes involved in cell-cycle regulation in lung cancer. Mol Biol. 2017;51(5):849–856. doi: 10.1134/S0026893317050107. [DOI] [PubMed] [Google Scholar]
- 41.Wu Y, Evers BM, Zhou BP. Small C-terminal domain phosphatase enhances snail activity through dephosphorylation. J Biol Chem. 2009;284(1):640–648. doi: 10.1074/jbc.M806916200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sapkota G, Knockaert M, Alarcon C, Montalvo E, Brivanlou AH, Massague J. Dephosphorylation of the linker regions of Smad1 and Smad2/3 by small C-terminal domain phosphatases has distinct outcomes for bone morphogenetic protein and transforming growth factor-beta pathways. J Biol Chem. 2006;281(52):40412–40419. doi: 10.1074/jbc.M610172200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and analyzed during the current study are available from the corresponding authors on reasonable request.