

Abstract
Background
Glioblastomas are the most aggressive type of brain tumor. A successful treatment should aim at halting tumor growth and protecting neuronal cells to prevent functional deficits and cognitive deterioration. Here, we exploited a Rho GTPase-activating bacterial protein toxin, cytotoxic necrotizing factor 1 (CNF1), to interfere with glioma cell growth in vitro and vivo. We also investigated whether this toxin spares neuron structure and function in peritumoral areas.
Methods
We performed a microarray transcriptomic and in-depth proteomic analysis to characterize the molecular changes triggered by CNF1 in glioma cells. We also examined tumor cell senescence and growth in vehicle- and CNF1-treated glioma-bearing mice. Electrophysiological and morphological techniques were used to investigate neuronal alterations in peritumoral cortical areas.
Results
Administration of CNF1 triggered molecular and morphological hallmarks of senescence in mouse and human glioma cells in vitro. CNF1 treatment in vivo induced glioma cell senescence and potently reduced tumor volumes. In peritumoral areas of glioma-bearing mice, neurons showed a shrunken dendritic arbor and severe functional alterations such as increased spontaneous activity and reduced visual responsiveness. CNF1 treatment enhanced dendritic length and improved several physiological properties of pyramidal neurons, demonstrating functional preservation of the cortical network.
Conclusions
Our findings demonstrate that CNF1 reduces glioma volume while at the same time maintaining the physiological and structural properties of peritumoral neurons. These data indicate a promising strategy for the development of more effective antiglioma therapies.
Keywords: cytotoxic necrotizing factor 1, dendritic structure, evoked potentials, senescence, visual cortex
Glioblastoma (GBM) is an aggressive form of brain tumor typically associated with a poor prognosis.1 Survival rates and quality of life of the patients have scarcely improved in the last years. Thus, it is urgent to find innovative approaches for GBM treatment. The development of novel therapies requires a better understanding of the biology of glioma cells and their interactions with resident brain cells. In particular, glioma cells are known to release high amounts of glutamate, leading to overexcitation of peritumoral neurons and epileptic seizures,2 with consequent neuronal death by excitotoxicity. This neuronal loss may facilitate glioma invasion3 and underlie cognitive impairments in patients.4,5 A successful brain tumor treatment should therefore aim at protecting neuronal cells to prevent functional deficits and cognitive deterioration, which have a strong impact on patients’ quality of life.
Currently, very little information is available about the functional status of peritumoral tissue at the border of the glioma mass. Previous studies have reported accumulation of glutamate6 as well as excitatory actions of gamma-aminobutyric acid (GABA),7 which may explain the occurrence of epileptic seizures. However, how tumor growth reverberates on the activity of single cells and the function of neuronal networks remain completely uncharted. Gathering this information is critical from at least 2 points of view. First, neuronal activity in peritumoral areas can potently influence brain tumor growth via the release of glioma mitogens.8 Second, novel antiglioma therapies should ideally aim at blocking tumor growth without noxious side effects on neurons. Indeed, an important advancement in preclinical tests of antineoplastic drugs would be the incorporation of physiological assays measuring neuronal function at the glioma border.
In this manuscript, we have exploited glioma cell transplantation into the mouse visual cortex to obtain quantitative measures of neuronal responsiveness at the border of the tumor. The visual cortex is a suitable model system, since it is particularly easy to identify alterations in the response to physiological stimuli.9
To halt glioma growth and preserve peritumoral regions, we used a Rho GTPase-activating protein, the bacterial toxin cytotoxic necrotizing factor 1 (CNF1). This toxin induces multinucleation of glioma cells10 while exerting a “plasticizing” effect on neurons, with enhanced dendritic branching and long-term potentiation.11,12 CNF1 enters cells close to the site of delivery and catalyzes the deamidation of a single glutamine residue in Rho GTPases (Rho, Rac, and Cdc42), thus impairing hydrolysis of guanosine triphosphate (GTP) and leading to their permanent activation.13,14 This leads to a double effect: (i) actin stabilization, blockade of cytodieresis, and eventually cell death in proliferating glioma cells10 and (ii) promotion of neuron health and plasticity.12 We therefore tested whether CNF1 treatment may reduce tumor growth and at the same time preserve the functionality and structure of the neurons surrounding the glioma.
Materials and Methods
For detailed experimental procedures, see the Supplementary Material.
Cell Cultures
GL261 cells were grown according to American Type Culture Collection protocols. Primary human glioblastoma cells were collected from 2 subjects as described in our previous publication.10 The study was approved by the Human Ethics Committee of the University of Pisa and Pisa Hospital. Cells were treated with CNF1 (3 nM) and incubated for 48 h before β-galactosidase measurements. The percentage of positive cells was determined after counting 3 random fields.
Microarray Analysis and Real-Time PCR
After RNA extraction, gene expression profiling was performed using a standard 2-color protocol by Agilent. Complementary RNA was hybridized to an Agilent 4 × 44k whole mouse genome microarray (G4122F) using the facility at the European Brain Research Institute (Rome, Italy). Quantitative real-time (RT) PCR reactions were performed using the SYBR PCR Mastermix (Applied Biosystems) on a StepOnePlus Real-Time PCR System (Applied Biosystems).
Proteomics Analysis
The proteomics experiment was performed as described before.15 Briefly, cells were lysed in a urea-based lysis buffer and digested with Lys-C and trypsin. Peptide labeling was performed with Tandem Mass Tag 6-plex labeling, and fractionation was performed using high pH reverse phase fractionation on a BRAVO AssayMAP (Agilent Technologies). Nano-scale liquid chromatography–tandem mass spectrometry analysis was performed on a 50 cm Easyspray column and an Orbitrap Fusion for MS3 analysis (Thermo Fisher Scientific). Data were analyzed using MaxQuant16 and Database for Annotation, Visualization and Integrated Discovery (DAVID)17 software.
Animals and Tumor Induction
Adult (age >postnatal day 60) C57BL/6J and Thy1-GFP mice were used. All experimental procedures conformed to the European Communities Council Directive #86/609/EEC and were approved by the Italian Ministry of Health. To induce glioma formation, C57BL/6 and Thy1-GFP mice received a stereotaxically guided injection of 40 000 GL261 cells (20 000 cells/µL phosphate buffered saline solution) into the visual cortex (2 mm lateral to the midline and in correspondence with lambda). Five days after injection of GL261 cells (tumor induction), mice were divided into 2 groups. The first group received CNF1 injection, the second, named “vehicle,” Tris–HCl buffer injection (glioma control condition).
Immunohistochemistry
Glioma volumes were measured in serial cortical sections using Stereo Investigator software (MicroBrightField). To quantify density of glial cells in the peritumoral areas, we used primary antibodies directed against glial fibrillary acidic protein (GFAP) (1:500; Dako) and Iba-1 (1:500; Wako).12 Positive cells were then counted in regions adjacent to the tumor.
Assessment of Senescence In vivo
Coronal sections were stained for senescence-associated β-galactosidase (SA β-gal) and coverslipped. Images of the tumor border and surrounding neural tissue were acquired with a Zeiss microscope and Axiovision software. Mean signal intensity was measured with Metamorph software.
Electrophysiological In vivo Recordings
Recordings were performed as described previously.9,18 Mice were anesthetized with urethane (150 mg/mL/kg), and a tungsten electrode was inserted into the visual cortex to record visual evoked potentials (VEPs) and single unit spiking activity. VEPs were recorded in response to a horizontal square wave grating of different spatial frequency and contrast. We measured either the peak-to-trough amplitude of the major negative component (transient VEPs) or the amplitude of the second harmonic of the Fourier transform computed from the recorded signal (steady-state). For spiking activity, the visual stimulus consisted of a light bar drifting in the central part of the visual field. Spikes were discriminated from background by a voltage threshold, as described.9,18 Peak responses were determined from peristimulus time histograms.
Neuronal Morphology
To investigate morphological alterations in peritumoral neurons, we transplanted glioma cells into the visual cortex of Thy1-GFP mice.19 Pyramidal neurons 100–300 µm away from the tumor borders were 3D reconstructed with Neurolucida software (MicroBrightField). Total dendritic length and dendritic branch number were quantified for each reconstructed neuron with Neurolucida Explorer software (MicroBrightField). For each neuron, the dendritic tree complexity was quantified using Sholl analysis.
Statistical Analysis
Differences between 2 groups were assessed with a t-test, or a Mann–Whitney rank sum test for datasets not normally distributed. Differences among 3 or more groups were evaluated with ANOVA followed by either Holm–Sidak or Dunn's test. For Sholl analysis and contrast gain curves, we carried out a 2-way repeated measures ANOVA.
Results
Molecular Changes Triggered by CNF1 in Glioma Cells
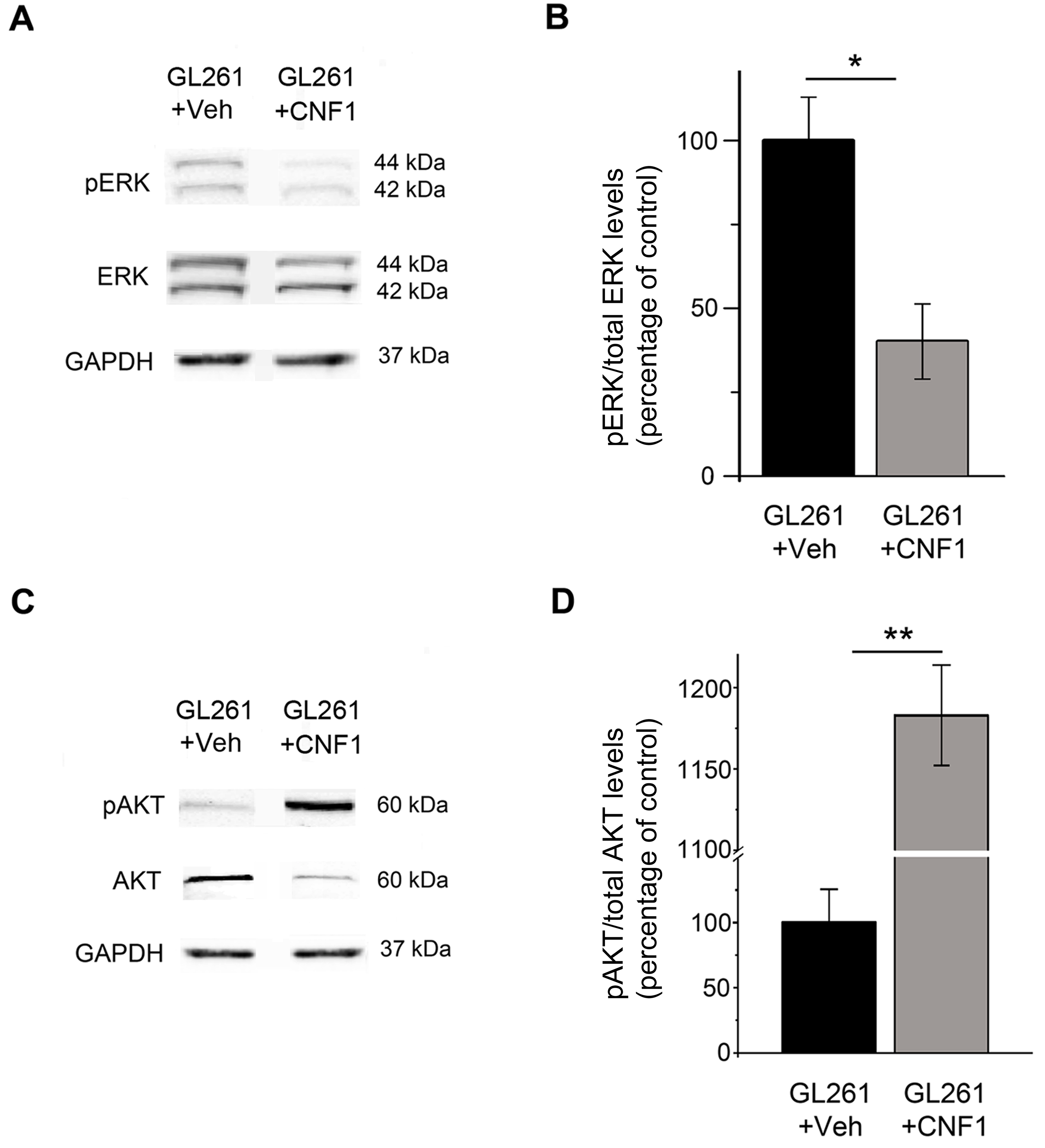
We have previously demonstrated that CNF1 induces multinucleation and eventually death of glioma cells.10 To fully describe the molecular profile induced by CNF1 in glioma cells, GL261 cells were treated with CNF1 or vehicle followed by microarray transcriptomic and in-depth proteomic analysis at 48 h (Fig. 1A–C). The expression profile of 27 122 transcripts and 4246 proteins was analyzed. Using a fold change cutoff of 2, we found a total of 1711 transcripts that were downregulated, while another 1318 were upregulated. Due to the very strict constraints in the proteomic analysis, a lower number of regulated proteins (129 proteins upregulated and 67 downregulated) was reported when compared with the microarray data. In order to get an overview of CNF1-altered processes/pathways, we performed functional enrichment analysis on both the transcriptomics and proteomics, using the bioinformatics tools WebGestalt20,21 and DAVID.17,22 The most significantly enriched transcripts (threshold value, P < .05) in the network were associated with cell cycle/senescence, DNA replication, and mitogen-activated protein kinase signaling (Fig. 1A). In CNF1-treated samples, we found a significant downregulation of receptors for growth factors (such as epidermal growth factor [EGF] and platelet-derived growth factor [PDGF]) involved in glioma progression (Fig. 1B). There was also a consistent and marked upregulation of senescence-associated genes such as Cdkn1a/p21 and uridine phosphorylase 1 (Upp1) (Fig. 1B). Accordingly, the proteomic screening showed a strong upregulation of p21 and Upp1 protein levels (Fig. 1C). Western blot was performed to address some of the intracellular signaling pathways highlighted by the microarray analysis. We found that CNF1 treatment induced a clear reduction of phosphorylated Erk while the fraction of phosphorylated Akt was increased (Supplementary Fig. 1), consistent with a previous report.23
Fig. 1.

Transcriptomic and proteomic profile of CNF1-treated glioma cells. Genes and proteins differentially expressed in CNF1- vs vehicle-treated glioma cells, 48 h after treatment. (A) The most significant gene categories are displayed at the top of the graph downward (P-value, red line) and gene numbers are arranged horizontally by the length of the bars (gene counts, blue bars). (B) Microarray heat map of selected genes differentially expressed at 48 h in CNF1-treated GL261 cells. Gene expression is represented by the intensity of colors (green: lowest expression; red: higher expression). Note significant upregulation of senescence-associated genes such as p21 and Upp1, and downregulation of several growth factor receptors (EGF and PDGF receptors) in CNF1-treated samples. (C) Volcano plot of all quantified proteins (4246 total). The triplicate average ratio CNF1 vs control was plotted against their t-test statistical log P-values. Significantly regulated proteins (total of 196 proteins, permutation corrected false discovery rate < 0.001) were colored in red and green for up- and downregulation, respectively. (D) Quantitative RT-PCRs showing the relative expressions of the senescence markers p21 (Cdkn1a, cyclin-dependent kinase inhibitor 1) and p16 (Cdkn2a, cyclin-dependent kinase inhibitor 2), and the negative p21 regulator FoxG1 (Forkhead box protein G1) in GL261 cells. (E, F) Increased expression of p21 in human glioma cells: U87 cell line (E) and patient-derived GBM cell cultures (F). Data are mean ± SEM. *P < .05, **P < .01, ***P < .001 (t-test).
We next compared the effects of CNF1 in murine GL261 and human glioma cells (U87 glioma cell line and patient-derived GBM cell cultures). In a first set of experiments, we used quantitative RT-PCR to examine the expression of genes related to senescence. In GL261 cells, we found that the expression of senescence-associated genes p16 and p21 was upregulated, while the Forkhead box protein G1 gene,24 a negative transcriptional regulator of p21, was strongly downregulated (t-test, P < .05; Fig. 1D). A similar upregulation of p21 was detected in CNF1-treated U87 and patient-derived cells (t-test, P < .05; Fig. 1E and F). Second, we examined the senescence phenotype at the morphological level by performing staining for SA β-gal activity in GL261 cells and human glioma cells treated with CNF1 (3 nM) or vehicle for 48 h. GL261 cells exposed to CNF1 exhibited a significant increase in SA β-gal activity compared with control cells (t-test, P < .01; Fig. 2A). As expected by the upregulation of p21, U87 and GBM-derived cells also stained strongly positive for SA β-gal following CNF1 treatment (t-test, P < .01; Fig. 2B and C). Altogether, these data indicate that CNF1 triggers molecular and morphological hallmarks of senescence in GL261 and human glioma cells, including patient-derived cells.
Fig. 2.

CNF1 induces senescence of glioma cells. (A–C) Percentage of SA β-gal positive GL261 (A), U87 (B), and GBM-derived glioma cells (C) 48 h after CNF1 treatment. Data are mean ± SEM. **P < .01 (t-test). (D, E) Representative coronal brain sections from glioma-bearing mice treated with either vehicle (D) or CNF1 (E). Note very intense SA β-gal staining (blue) in the CNF1-treated animal. For both images, the glioma mass is on the right while the intact peritumoral cortex is on the left. Scale bar = 200 µm. (F) Quantification of SA β-gal staining in vehicle- and CNF1-treated mice. The horizontal lines in the box denote the 25th, 50th, and 75th percentile values. The error bars denote the 5th and 95th percentile values, and the square symbol represents the mean of the column of data. SA β-gal staining is more intense in CNF1-treated mice (Mann–Whitney rank sum test, ***P < .001).
CNF1 Induces Senescence of Glioma Cells and Reduces Tumoral Volume In vivo
To evaluate the effects of CNF1 on glioma cells in vivo, GL261 cells were transplanted into the primary visual cortex of adult mice, and CNF1 (or vehicle) was delivered into the transplant site 5 days later. The neuroanatomical analysis was performed at day 21. We first stained coronal sections for SA β-gal activity to check activation of the senescence pathway following CNF1 treatment in vivo. A robust staining for SA β-gal was apparent in CNF1- but not vehicle-treated glioma-bearing animals, especially at the border of the tumor mass (Fig. 2D and E). The quantitative analysis demonstrated a significant difference in SA β-gal labeling between CNF1 and vehicle treatment (t-test, P < .001; Fig. 2F).
A stereological analysis was also performed to quantify the tumoral volume in vehicle- and CNF1-treated mice. We found that CNF1-treated animals exhibited markedly reduced tumor volumes compared with controls (Mann–Whitney rank sum test, P < .05; Fig. 3A and B). To investigate whether CNF1 could affect recruitment of astroglial and microglial cells at the border of the tumor, we performed immunostainings for Iba-1 and GFAP 21 days following glioma cell transplant (Fig. 3B). GFAP-positive astrocytes in peritumoral borders have typical thick processes that allow their identification with respect to GFAP-stained glioma cells within the tumor mass (Fig. 3B, insets). The quantitative analysis demonstrated an increased density of astrocytes and microglia in CNF1-treated mice (Mann–Whitney rank sum test, P < .001; Fig. 3C, D), indicating that CNF1 treatment recruits glial cells in proximity to the tumor.
Fig. 3.

CNF1 reduces tumor volume in vivo. (A) Reduced tumoral volume in CNF1-treated animals. Volumes were measured 21 d after GL261 injection in vehicle- (Veh, n = 10) and CNF1-treated animals (n = 17; Mann–Whitney rank sum test, P < .05). Horizontal lines in the box chart denote the 25th, 50th, and 75th percentile values. Error bars denote the 5th and 95th percentile values, while the square indicates the mean of the data. *P < .05. (B) Representative Hoechst-stained coronal sections through the visual cortex from mice inoculated with GL261 cells and treated either with vehicle (top) or CNF1 (bottom). Note the bigger tumoral mass in the vehicle mouse. Dorsal is up and lateral is to the right. The box on the left shows GFAP labeling within the tumor mass, while the boxes at right show GFAP-positive astrocytes and Iba-1–stained cells. Scale bar = 500 µm (10 µm for insets). (C) Quantification of GFAP-positive cells in the visual cortex surrounding the tumoral mass. Number of cells per boxes counted in vehicle- (Veh, gray box chart, number of boxes counted = 65) and CNF1-treated mice (blue box chart, number of boxes counted = 135; Mann–Whitney rank sum test, ***P < .001). (D) Quantification of Iba-1–positive cells in the visual cortex surrounding the tumoral mass. Number of cells per boxes counted in vehicle- (Veh, gray box chart, number of boxes counted = 67) and CNF1-treated mice (blue box chart, number of boxes counted = 120; Mann−Whitney rank sum test, ***P < .001).
Sparing of Neuronal Responses in CNF1-Treated Glioma-Bearing Mice
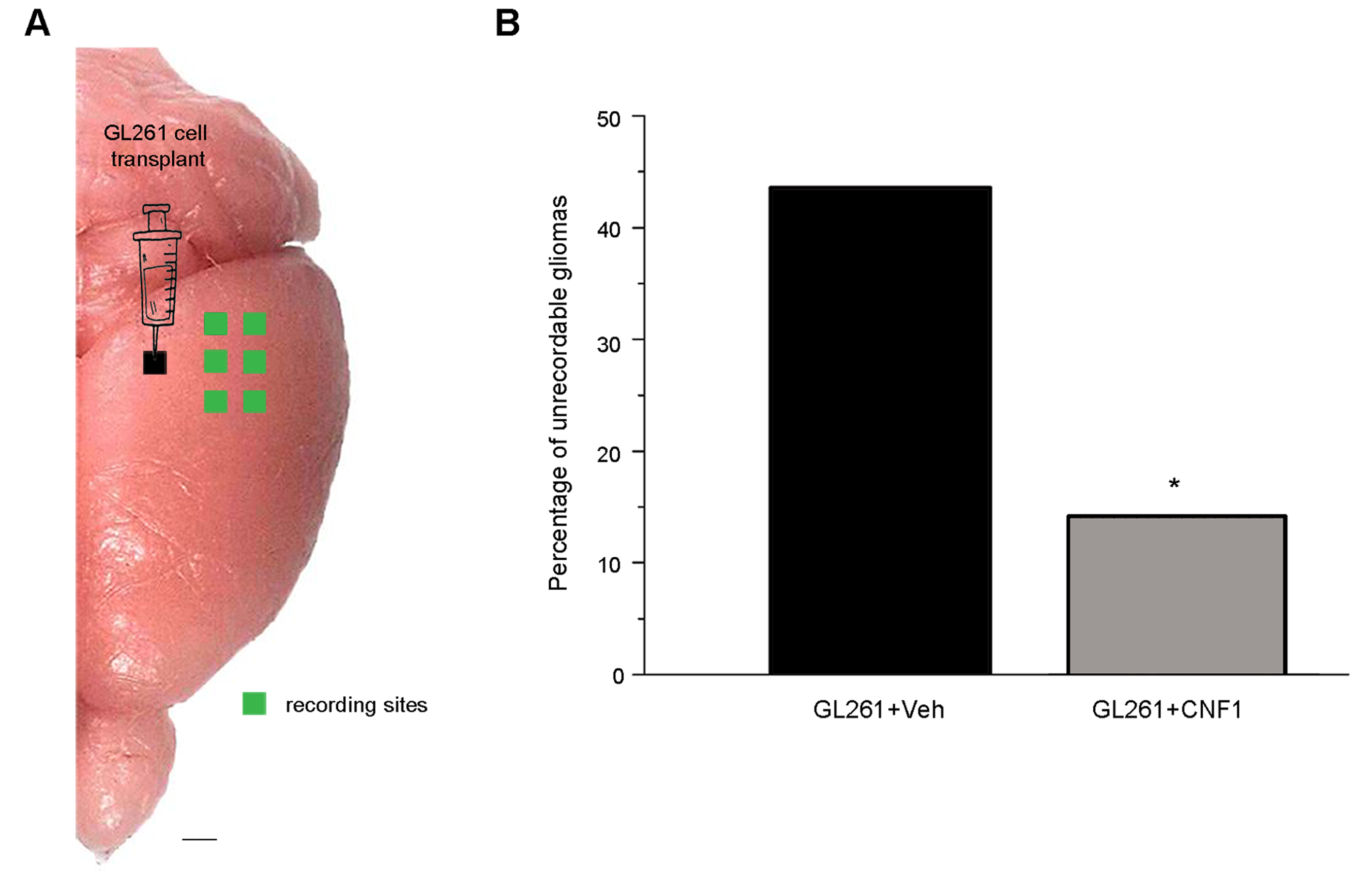
To evaluate functional changes induced by GL261 injection, we performed extracellular recordings of local field potentials and spiking activity from the primary visual cortex of anesthetized mice 21 days after glioma cell inoculation. A schematic of the transplant and recording sites is provided in Supplementary Fig. 2A. In keeping with the neuroanatomical data, we found different extents of tumor growth in vehicle- and CNF1-treated mice prepared for electrophysiological experiments. Indeed, more than 40% of vehicle-treated glioma-bearing mice could not be recorded, since the tumoral mass had completely covered the cortical surface in correspondence with the planned recording sites (Supplementary Fig. 2B). In contrast, this occurred in only about 14% of glioma-bearing mice treated with CNF1 (z-test, P < .05).
We initially used VEP recordings to compare visual responsiveness of naïve animals and glioma-bearing mice treated with either CNF1 or vehicle. Representative VEP waveforms are shown in Fig. 4A. We found a significant reduction of absolute VEP amplitudes in vehicle-treated glioma mice (1-way ANOVA followed by Holm–Sidak test, P < .05; Fig. 4A). This reduction was completely counteracted by CNF1 treatment (Fig. 4A). Latency of visual drive (i.e., of the major positive VEP peak) was in the order of 100 ms and was unaffected by experimental treatment (1-way ANOVA, P = .175; Fig. 4B). We then evaluated visual responses at different contrast levels and found that in vehicle-treated glioma-bearing mice, VEP amplitudes were scaled down across a range of contrasts compared with naïve and CNF1-treated mice (2-way repeated measures ANOVA followed by Holm–Sidak test, P < .05; Fig. 4C). Importantly, CNF1-treated animals showed a normal contrast gain function, with VEP amplitudes that were superimposable to those of naïve mice (P > .05; Fig. 4C).
Fig. 4.

Preservation of VEP responses in CNF1-treated glioma-bearing mice. (A) Transient VEP amplitude in naïve, vehicle (Veh), and CNF1 glioma-bearing mice. Representative waveforms from each group are shown on top. Responses in vehicle-treated mice (n = 7) are consistently lower compared with CNF1-treated glioma mice (n = 6) and naïve animals (n = 6; 1-way ANOVA followed by Holm–Sidak test, P < .05). Data are mean ± SEM. *P < .05. (B) Latency of transient VEPs in naïve, vehicle-, and CNF1-injected glioma-bearing mice. There are no statistical differences among groups (1-way ANOVA, P = .175). Data are mean ± SEM. (C) Amplitude of steady-state VEPs is plotted as a function of variable contrast (10%, 20%, 30%, 90%) in control and GL261-injected mice. Noise level (dotted line) represents the response to a blank stimulus (0% contrast). VEP responses are strongly reduced in vehicle glioma-bearing mice with respect to both naïve and CNF1-infused animals (2-way repeated measures ANOVA followed by Holm–Sidak test). Naïve, n = 6; vehicle-treated glioma-bearing mice, n = 7; CNF1-treated glioma-bearing mice, n = 6. Data are mean ± SEM. *P < .05, **P < .01.
To further probe functional changes induced by GL261 cell injection and the maintenance of neuronal responses in the peritumoral area of CNF1-treated animals, we also recorded extracellular spiking activity. Analysis of spontaneous discharges (i.e., without visual stimuli) revealed a profound enhancement of baseline firing in both groups of glioma-bearing mice (1-way ANOVA on ranks followed by Dunn's test, P < .01; Fig. 5A), indicating hyperexcitability of peritumoral regions. We then quantified the responsiveness of cortical neurons to visual stimulation as the peak-to-baseline ratio, that is, the peak discharge evoked by visual stimulation divided by spontaneous activity.9,18 We found a profound depression of responsivity in vehicle-infused glioma-bearing mice compared with naïve and CNF1-treated animals (1-way ANOVA on ranks followed by Dunn's test, P < .01 for both comparisons; Fig. 5B). Visual responsiveness in CNF1-treated mice was intermediate between vehicle-treated glioma-bearing and naïve mice (P < .01; Fig. 5B). To investigate whether the dampened peak-to-baseline ratio in neurons from glioma-bearing mice may be ascribed to a reduced reliability of visual responses, we counted failures (the percentage of stimulus presentations unable to modify spontaneous activity) in all experimental groups.9 We detected an abnormally high failure rate (about 30%) in control glioma-bearing mice, which was completely recovered by CNF1 delivery (1-way ANOVA followed by Holm–Sidak test, vehicle vs naïve and CNF1, P < .05; CNF1 vs naïve, P = .73; Fig. 5C). Moreover, vehicle-infused glioma-bearing mice showed an increase in neuronal receptive field size in comparison with naïve and CNF1-treated groups (1-way ANOVA on ranks followed by Dunn's test, P = .011; Fig. 5D).
Fig. 5.

Sparing of functional properties of cortical units by CNF1 treatment. (A) Spontaneous firing of neurons in naïve (n = 11, 164 cells recorded), vehicle-injected (Veh; n = 8, 121 cells recorded), and CNF1-injected (n = 9, 111 cells recorded) glioma-bearing mice. Both glioma groups differ from naïve (ANOVA on ranks, post hoc Dunn's test, **P < .01). (B) Neuronal responsivity (peak firing evoked by visual stimulation divided by spontaneous activity) in naïve, vehicle, and CNF1 glioma-bearing mice. Compared with naïve mice, glioma-bearing animals display a lower responsivity which is partially counteracted by CNF1 (1-way ANOVA on ranks, post hoc Dunn's test, glioma-bearing vehicle vs CNF1, **P < .01). (C) Percentage of failures (lack of response to a light bar drifting into the receptive field) of cortical units. Note the higher percentage of failures in vehicle animals with glioma (1-way ANOVA, post hoc Holm–Sidak test, *P < .05). (D) Box charts showing neuronal receptive field size in naïve animals, and vehicle- and CNF1-treated glioma-bearing mice. Note the increase in receptive field size for vehicle glioma-bearing mice compared with naïve and CNF1-treated group (1-way ANOVA on ranks, post hoc Dunn's test, **P < .01).
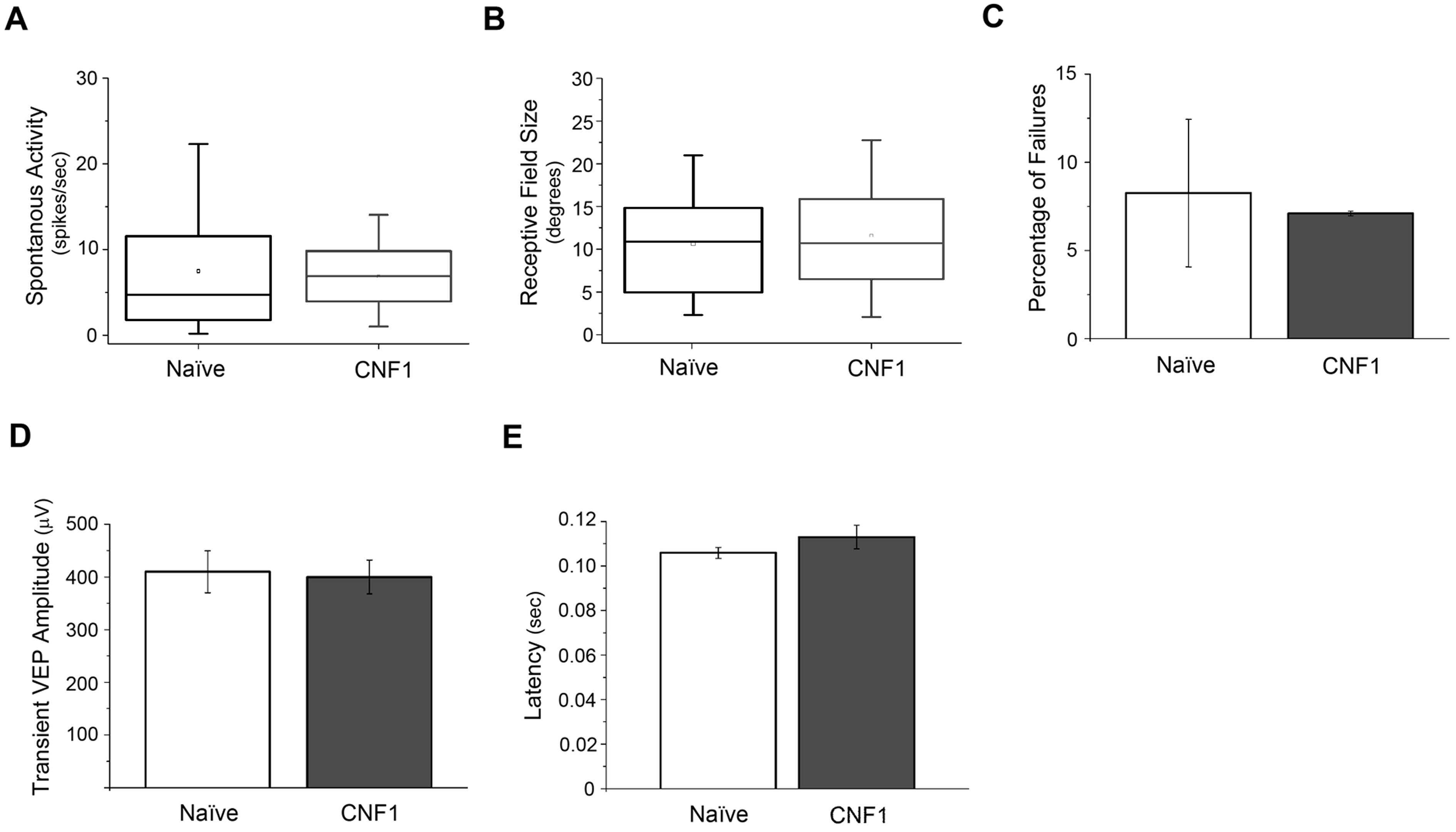
To define more precisely how CNF1 treatment per se affects normal cortical function, we conducted control experiments to measure neuronal properties in CNF1-injected naïve mice (n = 4). Consistent with our previous report in rats,12 we found no alterations in cell physiology (t-test and Mann–Whitney rank sum test, P > .05; Supplementary Fig. 3). We noted a small, nonsignificant increase in spontaneous activity (Supplementary Fig. 3A) which may be linked to the enhanced density of dendritic spines in CNF1-treated neurons.12
Morphological Preservation of Peritumoral Neurons by CNF1
To investigate alterations of neuronal structure in peritumoral areas, we transplanted GL261 cells in Thy1-GFP mice expressing GFP in a subset of layer V pyramidal neurons (Fig. 6A). Two weeks after transplant, we reconstructed GFP-stained neurons in visual cortex (Fig. 6B) and quantified total dendritic length and complexity in peritumoral neurons (cell somas were located 100–300 µm from the tumor border). Overall, neurons in peritumoral regions had a shrunken appearance and reduced branching (Fig. 6B). Total dendritic length was strongly reduced in vehicle-infused glioma-bearing mice, and CNF1 treatment partially recovered this phenotype (Mann–Whitney rank sum test, P < .05; Fig. 6C). We also examined dendrite complexity by Sholl analysis. The number of intersections between dendrites and Sholl circles was significantly reduced in vehicle-treated glioma-bearing mice (Fig. 6D). Dendritic branching was significantly preserved by CNF1 at distances ≤30 µm from the cell soma (2-way repeated measures ANOVA followed by Holm–Sidak test, P < .05; Fig. 6D). Altogether, these data demonstrate that CNF1 treatment protects the dendritic architecture of peritumoral neurons, indicating a morphological correlate of the maintained visual responsivity.
Fig. 6.

CNF1 counteracts the impaired dendritic branching in glioma-bearing mice. (A) Representative Thy1-GFP neurons (green) in a Hoechst-stained coronal section through the mouse primary visual cortex. Tumor borders are indicated by the dotted line. Dorsal is up and lateral is to the right. Scale bar = 500 µm. (B) Neurolucida tracings of representative Thy1-GFP pyramidal neurons in naïve, vehicle-, and CNF1-treated glioma-bearing animals. Scale bar = 10 µm. (C) Box charts showing total dendritic length for neurons from glioma-bearing mice. The horizontal dotted line represents the median value observed in naïve mice for comparison. Dendritic length is significantly increased in CNF1- vs vehicle-treated mice (Mann–Whitney rank sum test, *P < .05). (D) Sholl analysis of layer V pyramidal neurons in naïve, vehicle-, and CNF1-treated glioma-bearing mice. CNF1 treatment maintains a higher number of intersections close to the cell soma (2-way repeated measures ANOVA followed by Holm–Sidak test, 10-20-30 µm from soma, CNF1 vs vehicle, *P < .05). Data are mean ± SEM.
Discussion
We previously demonstrated that intracerebral CNF1 delivery increases survival of glioma-bearing mice,10 but the molecular pathways set in motion by CNF1 remain only partly understood. To characterize in detail the effect of CNF1 on glioma cells, we carried out a full transcriptomic and proteomic analysis. These data are important for the definition of CNF1 mechanisms of action.25–28 First, we found a significant downregulation of several trophic factors and receptors involved in glioma progression, which may be linked to the antitumoral properties of CNF1. Second, we found evidence for activation of the senescence pathway in CNF1-treated cells, with activation of SA β-gal. SA β-gal activity is a widely used and reliable biomarker in studies of cellular senescence in culture and in vivo, even if it is not completely specific.29 The activation of senescence was supported by enhanced expression of p21/p16 and was consistently observed in murine and human cell lines. To link these data in cell cultures with the in vivo condition, we evaluated staining for SA β-gal in tumors treated with CNF1. Consistently, the results indicated a robust and massive induction of cellular senescence (see Fig. 2D–F). These senescence-promoting effects are likely one of the key mechanisms involved in the reduced tumor growth observed following intracerebral CNF1 treatment (see Fig. 3A).
In addition, we used glioma cell injections into the mouse visual cortex to precisely characterize functional and structural alterations in pyramidal neurons located in peritumoral areas. One of the most dramatic physiological alterations detected in our recordings was the increased baseline firing of cortical neurons, which may represent the cellular substrate underlying enhanced propensity to spontaneous seizures in peritumoral areas.7,30 Higher firing rates may depend on glutamate release from the tumor2 as well as on alterations of chloride homeostasis in neurons, resulting in a net depolarizing efflux of chloride-negative ions upon GABA-A receptor opening.7,31 Changes in the excitation/inhibition ratio may also be at the basis of the increased receptive field size of cortical neurons.32 Our data indicate reduced visual responses in the peritumoral regions, as shown by a dampened VEP amplitude. This may be due to 2 distinct, nonmutually exclusive possibilities: a damage to afferent thalamocortical fibers secondary to tumor growth in the white matter, or a reduced recruitment of cortical neurons because of alterations in intracortical connectivity. Visually evoked firing activity was also downregulated, and this was linked to a higher incidence of failures in responding to the visual stimuli. It is possible that the higher baseline activity occludes response to physiological stimuli, as we recently observed in the visual cortex rendered epileptic by tetanus neurotoxin.9 Altogether, these data provide a set of quantitative parameters to measure dysfunction in peritumoral regions during glioma growth and to assess the effect of neuroprotective strategies. These parameters can also be employed to measure precisely the impact of radiotherapy, chemotherapy, and anti-epileptic drugs on neuronal function in glioma-bearing animals. The inclusion of physiological endpoints in preclinical glioma studies is highly warranted in order to deepen our understanding and treatment of neuronal dysfunction related to cancer and cancer therapies.
Importantly, CNF1 injection appears to rescue, at least in part, the pathological phenotypes of peritumoral neurons. In our electrophysiological experiments, we found that CNF1 preserves VEP amplitudes, enhances the reliability of visually evoked discharges, and maintains a normal contrast response curve and receptive field size in cortical neurons. It is worth noting that the GL261 model represents a rather focal glioma model; in more diffuse/infiltrating tumors with less distinct borders, the electrophysiological effects of the tumor and the impact of CNF1 therapy may be different. The physiological differences in vehicle- versus CNF1-treated glioma-bearing mice cannot be ascribed to the reduced tumor growth, that is, to the sampling of cortical regions more distant from the tumor border in CNF1-injected mice. Indeed, we discarded more than 40% of vehicle-treated mice (Supplementary Fig. 2B) and made sure that physiological data were collected from a subgroup of vehicle- and CNF1-treated animals with similar tumor volumes (see Supplementary material). In addition, enhancement of spontaneous activity was noted in both vehicle- and CNF1-treated mice, likely reflecting similar exposure to glutamate and other neuroexcitants released from the nearby glioma cells. These data strongly suggest that CNF1 maintains functional responses via a direct action on neurons, independently of the reduced tumoral growth. In keeping with this scenario, CNF1 administration to brain slices was found to enhance excitatory synaptic transmission and long-term potentiation.11
At the level of fine neuronal structure, glioma growth potently impacted on the dendritic arbors of peritumoral neurons, which were dramatically reduced in length and complexity. To our knowledge, this is the first report of neurite alterations in glioma. Importantly, CNF1 improved dendritic length and increased dendritic complexity, at least close to the cell soma (see Fig. 6). This indicates selective preservation of primary dendrites with concomitant loss of higher-order branches in CNF1-treated cortical neurons. This morphological rescue is in line with the improved neuronal responses, indicating that CNF1 maintains neuron health and function.
In summary, we have shown that CNF1 treatment results in reduced glioma volume while at the same time maintaining the physiological and structural properties of peritumoral neurons. These data therefore suggest a promising strategy for the development of a more effective antiglioma therapy. Our physiological approach is very relevant, since neuronal activity in the vicinity of the tumor can influence glioma growth and progression.8 It is worth noting that tissue preservation per se does not predict sparing at the functional level.33,34 We introduce here an electrophysiology-based approach to test the effectiveness of anticancer therapies by measuring neuronal function and neuroprotection at the glioma border.
Supplementary Material
Supplementary material is available online at Neuro-Oncology (http://neuro-oncology.oxfordjournals.org/).
Funding
This work was supported by AIRC (Italian Association for Cancer Research) grant #IG13252, Fondazione Pisa grant #158/2011, CNR InterOmics project, and CNR NanoMax project.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank Enrico Pracucci for contributing to data collection for Fig. 3, and Francesca Biondi for excellent animal care. Riccardo Vannozzi and Nicola Benedetto (Neurosurgery, Pisa Hospital) provided the surgical samples of human GBM.
Conflict of interest statement. The authors have nothing to disclose.
References
- 1. Johnson DR, O'Neill BP. Glioblastoma survival in the United States before and during the temozolomide era. J Neurooncol. 2012;107(2):359–364. [DOI] [PubMed] [Google Scholar]
- 2. Buckingham SC, Campbell SL, Haas BR et al. Glutamate release by primary brain tumors induces epileptic activity. Nat Med. 2011;17(10):1269–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sontheimer H. A role for glutamate in growth and invasion of primary brain tumors. J Neurochem. 2008;105(2):287–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brown PD, Jensen AW, Felten SJ et al. Detrimental effects of tumor progression on cognitive function of patients with high-grade glioma. J Clin Oncol. 2006;24(34):5427–5433. [DOI] [PubMed] [Google Scholar]
- 5. Hilverda K, Bosma I, Heimans JJ et al. Cognitive functioning in glioblastoma patients during radiotherapy and temozolomide treatment: initial findings. J Neurooncol. 2010;97(1):89–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Marcus HJ, Carpenter KL, Price SJ et al. In vivo assessment of high-grade glioma biochemistry using microdialysis: a study of energy-related molecules, growth factors and cytokines. J Neurooncol. 2010;97(1):11–23. [DOI] [PubMed] [Google Scholar]
- 7. Pallud J, Le Van Quyen M, Bielle F et al. Cortical GABAergic excitation contributes to epileptic activities around human glioma. Sci Transl Med. 2014;6(244):244ra289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Venkatesh HS, Johung TB, Caretti V et al. Neuronal activity promotes glioma growth through neuroligin-3 secretion. Cell. 2015;161(4):803–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vannini E, Restani L, Pietrasanta M et al. Altered sensory processing and dendritic remodeling in hyperexcitable visual cortical networks. Brain Struct Funct. 2015. [DOI] [PubMed] [Google Scholar]
- 10. Vannini E, Panighini A, Cerri C et al. The bacterial protein toxin, cytotoxic necrotizing factor 1 (CNF1) provides long-term survival in a murine glioma model. BMC Cancer. 2014;14:449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Diana G, Valentini G, Travaglione S et al. Enhancement of learning and memory after activation of cerebral Rho GTPases. Proc Natl Acad Sci U S A. 2007;104(2):636–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cerri C, Fabbri A, Vannini E et al. Activation of Rho GTPases triggers structural remodeling and functional plasticity in the adult rat visual cortex. J Neurosci. 2011;31(42):15163–15172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Flatau G, Lemichez E, Gauthier M et al. Toxin-induced activation of the G protein p21 Rho by deamidation of glutamine. Nature. 1997;387(6634):729–733. [DOI] [PubMed] [Google Scholar]
- 14. Schmidt G, Sehr P, Wilm M et al. Gln 63 of Rho is deamidated by Escherichia coli cytotoxic necrotizing factor-1. Nature. 1997;387(6634):725–729. [DOI] [PubMed] [Google Scholar]
- 15. de Graaf EL, Kaplon J, Mohammed S et al. Signal transduction reaction monitoring deciphers site-specific PI3K-mTOR/MAPK pathway dynamics in oncogene-induced senescence. J Proteome Res. 2015;14(7):2906–2914. [DOI] [PubMed] [Google Scholar]
- 16. Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26(12):1367–1372. [DOI] [PubMed] [Google Scholar]
- 17. Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. [DOI] [PubMed] [Google Scholar]
- 18. Restani L, Cerri C, Pietrasanta M et al. Functional masking of deprived eye responses by callosal input during ocular dominance plasticity. Neuron. 2009;64(5):707–718. [DOI] [PubMed] [Google Scholar]
- 19. Feng G, Mellor RH, Bernstein M et al. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28(1):41–51. [DOI] [PubMed] [Google Scholar]
- 20. Wang J, Duncan D, Shi Z et al. WEB-based GEne SeT AnaLysis Toolkit (WebGestalt): update 2013. Nucleic Acids Res. 2013;41:(Web Server issue)W77–W83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang B, Kirov S, Snoddy J. WebGestalt: an integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res. 2005;33:(Web Server issue)W741–W748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Miraglia AG, Travaglione S, Meschini S et al. Cytotoxic necrotizing factor 1 prevents apoptosis via the Akt/IkappaB kinase pathway: role of nuclear factor-kappaB and Bcl-2. Mol Biol Cell. 2007;18(7):2735–2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pancrazi L, Di Benedetto G, Colombaioni L et al. Foxg1 localizes to mitochondria and coordinates cell differentiation and bioenergetics. Proc Natl Acad Sci U S A. 2015;112(45):13910–13915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Travaglione S, Loizzo S, Rizza T et al. Enhancement of mitochondrial ATP production by the Escherichia coli cytotoxic necrotizing factor 1. FEBS J. 2014;281(15):3473–3488. [DOI] [PubMed] [Google Scholar]
- 26. Piteau M, Papatheodorou P, Schwan C et al. Lu/BCAM adhesion glycoprotein is a receptor for Escherichia coli Cytotoxic Necrotizing Factor 1 (CNF1). PLoS Pathog. 2014;10(1):e1003884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Augspach A, List JH, Wolf P et al. Activation of RhoA,B,C by yersinia cytotoxic necrotizing factor (CNFy) induces apoptosis in LNCaP prostate cancer cells. Toxins (Basel). 2013;5(11):2241–2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Loizzo S, Rimondini R, Travaglione S et al. CNF1 increases brain energy level, counteracts neuroinflammatory markers and rescues cognitive deficits in a murine model of Alzheimer's disease. PLoS One. 2013;8(5):e65898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Debacq-Chainiaux F, Erusalimsky JD, Campisi J et al. Protocols to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat Protoc. 2009;4(12):1798–1806. [DOI] [PubMed] [Google Scholar]
- 30. Armstrong TS, Grant R, Gilbert MR et al. Epilepsy in glioma patients: mechanisms, management, and impact of anticonvulsant therapy. Neuro Oncol. 2016;18(6):779–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Campbell SL, Robel S, Cuddapah VA et al. GABAergic disinhibition and impaired KCC2 cotransporter activity underlie tumor-associated epilepsy. Glia. 2015;63(1):23–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liu BH, Li P, Sun YJ et al. Intervening inhibition underlies simple-cell receptive field structure in visual cortex. Nat Neurosci. 2010;13(1):89–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dumas TC, Sapolsky RM. Gene therapy against neurological insults: sparing neurons versus sparing function. Trends Neurosci. 2001;24(12):695–700. [DOI] [PubMed] [Google Scholar]
- 34. Caleo M, Medini P, von Bartheld CS et al. Provision of brain-derived neurotrophic factor via anterograde transport from the eye preserves the physiological responses of axotomized geniculate neurons. J Neurosci. 2003;23(1):287–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.