

Abstract
Selective potentiation of the mGlu5 subtype of metabotropic glutamate (mGlu) receptor using positive allosteric modulators (PAMs) has robust cognition-enhancing effects in rodent models that are relevant for schizophrenia. Until recently, these effects were thought to be due to potentiation of mGlu5-induced modulation of NMDA receptor (NMDAR) currents and NMDAR-dependent synaptic plasticity. However, “biased” mGlu5 PAMs that do not potentiate mGlu5 effects on NMDAR currents show efficacy that is similar to that of prototypical mGlu5 PAMs, suggesting that NMDAR-independent mechanisms must be involved in these actions. We now report that synaptic activation of mGlu5 is required for a form of long-term depression (mLTD) in mouse prefrontal cortex (PFC) that is induced by activation of M1 muscarinic acetylcholine (mAChR) receptors, which was previously thought to be independent of mGlu5 activation. Interestingly, a biased mGlu5 PAM, VU0409551, that does not potentiate mGlu5 modulation of NMDAR currents, potentiated induction of mLTD. Furthermore, coactivation of mGlu5 and M1 receptors increased GABAA-dependent inhibitory tone in the PFC pyramidal neurons, which likely contributes to the observed mLTD. Finally, systemic administration of the biased mGlu5 PAM reversed deficits in mLTD and associated cognitive deficits in a model of cortical disruption caused by repeated phencyclidine exposure that is relevant for schizophrenia and was previously shown to be responsive to selective M1 muscarinic receptor PAMs. These studies provide exciting new insights into a novel mechanism by which mGlu5 PAMs can reverse deficits in PFC function and cognition that is independent of modulation of NMDAR currents.
Keywords: positive allosteric modulators, prefrontal cortex, mGluR, muscarinic, long-term depression, schizophrenia
Graphical Abstract
INTRODUCTION
In recent years, the metabotropic glutamate (mGlu) receptor subtype 5 (mGlu5) has emerged as an exciting new target with potential for improving cognitive function and other symptoms associated with schizophrenia and other brain disorders.1–3 Multiple studies reveal that highly selective mGlu5 positive allosteric modulators (PAMs) enhance hippocampal synaptic plasticity4–9 and improve performance in a broad range of animal models of cognitive function that are dependent on intact function of the hippocampus or prefrontal cortex (PFC), and are relevant to domains of cognitive function that are disrupted in schizophrenia patients.4,10–17
Many of the physiological effects of mGlu5 activation are thought to be mediated by coupling of the receptor to Gαq GTP-binding proteins and subsequent mobilization of intra-cellular calcium, activation of protein kinase C, and associated signaling pathways.18 In addition, mGlu5 is a close signaling partner with the N-methyl D-Aspartate subtype of ionotropic glutamate receptor (NMDARs) and can potentiate NMDAR function4,10,19 by a mechanism that is thought to involve multiple adaptor proteins that are independent of Gαq signaling, including Shank, Homer, Preso 1, and Pin1.20–23 Because of the critical role of NMDARs in multiple forms of synaptic plasticity and cognitive function, it was previously thought that the cognition-enhancing effects of mGlu5 PAMs are likely mediated by potentiation of NMDAR currents. However, we recently discovered novel mGlu5 PAMs that display stimulus bias and selectively potentiate coupling of mGlu5 to Gαq, but do not do not potentiate mGlu5 effects on NMDAR currents. Interestingly, these biased mGlu5 PAMs have robust cognition-enhancing effects in unimpaired rodents,6 suggesting that some cognition-enhancing effects of mGlu5 PAMs are independent of their effects on NMDAR signaling. At present, little is known about NMDAR-independent mechanisms by which mGlu5 PAMs can enhance different domains of cognitive function. Thus, it will be critical to evaluate the effects of these novel biased mGlu5 PAMs on specific aspects of cognitive function and identified CNS circuits that may be relevant for the pathophysiology underlying schizophrenia.
We now report that VU0409551, a biased mGlu5 PAM that does not potentiate mGlu5-induced modulation of NMDAR currents, induces a complete reversal of severe deficits in recognition memory that are observed in young adult mice that were exposed to transient NMDAR blockade during juvenile development. Furthermore, VU0409551 completely reverses the deficits in a form of long-term depression (mLTD) in the prefrontal cortex (PFC) that is induced by activation of the M1 muscarinic acetylcholine receptor (mAChR) and has been shown to be closely related to deficits in recognition memory observed in this model.24 This was surprising since mGlu5 agonists do not induce LTD in PFC and mLTD is not known to involve activation of mGlu5. However, further mechanistic studies revealed that mLTD requires concurrent activation of M1 and synaptic activation of mGlu5 by stimulation of glutamatergic projections from the hippocampus. Furthermore, concurrent activation of M1 and mGlu5 induces a long-lasting increase in inhibitory transmission, suggesting a novel mechanism by which coactivation of these receptors can converge to induce mLTD. Taken together, these studies provide new insights into one mechanism by which biased mGlu5 PAMs can reverse behavioral deficits in an NMDAR hypofunction model, and provide strong evidence for an overlap of mGlu5 and M1 functionality in the PFC.
RESULTS
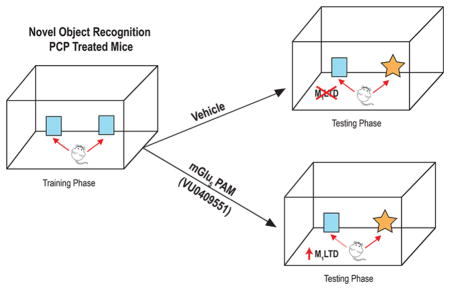
mGlu5 PAM VU0409551 Has Behavioral Efficacy and Can Rescue Muscarinic LTD Deficits in Repeated PCP Treated Mice
To test whether the mGlu5 PAM VU0409551 has behavioral efficacy in rodent models based on NMDAR hypofunction during juvenile development, we used a repeated phencyclidine (PCP) treatment mouse model of schizophrenia. 5–6 week old mice were treated with 10 mg/kg PCP for 7 days (once a day, subcutaneous) followed by a 7 day washout. On the eighth day of washout, animals were tested for recognition memory employing the novel object recognition task. As reported earlier, animals that had been transiently treated with PCP during juvenile development showed a significantly lower recognition index (−0.02 ± 0.07; Figure 1A) than did control vehicle-treated animals (0.20 ± 0.05), indicating a deficit in recognition memory (one-way analysis of variance (ANOVA): p = 0.0099; F (4, 74) = 3.59; Dunnett’s multiple comparison test: p < 0.05; q = 2.577). However, acute pretreatment of VU0409551 30 min prior to the NOR task led to a dose-dependent reversal of recognition memory deficit in the PCP-treated mice. Thus, 3 (0.08 ± 0.05) and 10 (0.17 ± 0.05) mg/kg VU0409551 showed a trend toward an increase in recognition index without a significant increase (Dunnett’s multiple comparison test: p > 0.05; q = 1.253 for 3 mg/kg and 2.401 for 10 mg/kg). However, 30 mg/kg VU0409551 (0.25 ± 0.06) significantly reversed the deficits in recognition memory in the PCP treated mice (Dunnett’s multiple comparison test: p < 0.01; q = 3.430). Thirty mg/kg VU0409551 (0.20 ± 0.07) did not significantly affect the recognition index in vehicle treated mice (Mann–Whitney test, p = 0.9477; data not shown).
Figure 1.

Biased mGlu5 PAM VU0409551 dose-dependently rescues recognition memory and mLTD deficits observed after repeated PCP exposure in juvenile mice. (A) PCP treatment (n = 17) caused a significant reduction in recognition index compared to the vehicle group (n = 12). However, acute pretreatment of VU0409551 (i.p.) prior to exposure to identical objects dose dependently enhanced recognition index in PCP treated animals. Although 3 mg/kg (n = 16) and 10 mg/kg dose (n = 17) of VU0409551 did not have a significant effect, 30 mg/kg VU0409551 (n = 17) significantly enhanced recognition memory assessed 24 h later. Data are expressed as mean ± SEM. Multiple comparisons were carried out with PCP group (white bar) as the control group. **p < 0.01 and *p < 0.05. (B) In PFC brain slices obtained from mice with repeated exposure to PCP, 50 μM CCh failed to induce mLTD (n = 7). However, pretreatment of VU0409551 (dashed line) before and during 50 μM CCh application rescued mLTD in the PCP-treated mice (n = 6) and led to robust depression in PFC fEPSP (shaded gray area). Inset shows representative fEPSP traces for each condition for baseline (black trace) and 55 min after CCh application (red trace). (C) The average magnitude of mLTD observed in the PCP-treated experimental groups was quantified and compared along with that of VU0409551/CCh group showing a significant increase in the magnitude of mLTD. This indicates that the mGlu5-selective PAM can rescue deficits in muscarinic plasticity after repeated PCP treatment. Scale bars denote 0.5 mV and 5 ms. Data are expressed as mean ± SEM. **p < 0.01.
We previously found that loss of recognition memory in this model was associated with a loss of M1-dependent mLTD induced by the cholinergic agonist carbachol (CCh) in PFC slices.24 Furthermore, M1 mAChR PAMs restored the deficit in mLTD and also restored the loss of recognition memory.24 Based on this association, we performed studies to determine whether the mGlu5-selective PAM could also restore the deficits in mLTD in PFC slices. Field excitatory post synaptic potentials (fEPSPs) were recorded from layer V of the prelimbic area of the mouse PFC in response to electrical stimulation of the superficial layer II/III. As observed previously,24 50 μM CCh induced robust mLTD in PFC slices from control mice (73.3 ± 5.4% baseline fEPSP) but failed to induce mLTD in the PCP treated mice (95.6 ± 4.5% baseline fEPSP; Figure 1B). However, acute pretreatment of the brain slices with mGlu5 PAM VU0409551 (10 μM) for 10 min followed by a 10 min coapplication of 50 μM CCh led to the induction of mLTD in fEPSPs (52.8 ± 7.3% baseline fEPSP Figure 1B and C) in slices from mice that were treated with PCP during juvenile development. Statistical analysis confirmed that the magnitude of fEPSP depression relative to the baseline was significantly different between CCh-alone and VU0409551/CCh conditions in the PCP treated mice (Mann–Whitney test, p = 0.0047; Figure 1C). Thus, the mGlu5 PAM VU0409551 can reverse both behavioral as well as physiological deficits in mLTD associated with this NMDAR hypofunction mouse model.
Muscarinic LTD is Dependent on Glutamate Release from PFC Afferents
The finding that VU0409551 reversed deficits in mLTD was somewhat surprising in light of previous studies showing that agonists of mGlu5 do not induce LTD in the PFC.25,26 Thus, while mGlu5 activation alone is not sufficient to induce PFC LTD, it is possible that synaptic activation of mGlu5 plays an important role in induction of mLTD by the cholinergic agonist. To evaluate this possibility, we performed additional studies to determine whether induction of mLTD by CCh requires activation of glutamatergic afferents and subsequent activation of mGlu5 during the period of CCh application. As observed in previous studies, bath application of 50 μM CCh for 10 min, led to an acute depression followed by mLTD of fEPSPs in the PFC (73.3 ± 5.4% baseline fEPSP) measured 55–60 min following the addition of CCh (Figure 2A). To test the hypothesis that this form of LTD required glutamatergic synaptic activity for its induction, we repeated the same experiment in the absence of electrical stimulation during CCh application. A 10 min stable baseline was recorded with a test pulse (0.05 Hz), after which stimulation was discontinued. After a 5 min period, 50 μM CCh was added for 10 min in the absence of any concurrent afferent stimulation. Stimulation was resumed and fEPSPs were recorded again 20 min after the washout of CCh. Under this condition, application of CCh failed to induce mLTD in fEPSPs measured 55–60 min following CCh add (97.1 ± 3.6% baseline fEPSP; Figure 2B and C). A statistically significant difference was observed when the magnitude of fEPSP depression during the 55–60 min period was compared between the two experimental conditions (Mann–Whitney test, p = 0.0043; Figure 2C). These results suggest that induction of mLTD by CCh is dependent on synaptic activation and requires afferent stimulation for its induction in the mouse PFC.
Figure 2.

Muscarinic LTD is dependent on glutamate release from PFC afferents. (A) Bath application of 50 μM CCh for 10 min (solid line), led to an acute depression followed by mLTD of fEPSPs measured 55–60 min following the addition of CCh (n = 6). (B) To examine whether glutamate release from PFC afferents is necessary for mLTD, electrical stimulation was suspended after 10 min of stable baseline. Following 5 min of no electrical stimulation, 50 μM CCh was added for 10 min (solid line) in the absence of any concurrent afferent stimulation. Stimulation was resumed 20 min after the washout of CCh and fEPSPs were recorded again for 40 min (n = 5). (C) Quantification of mLTD (normalized fEPSP slopes) in the different experimental groups showing a significant difference in the magnitude of mLTD observed between them. (D) Left: Schematic of viral-mediated gene transfer of ChR2 into the mouse ventral hippocampus. Right: Optically evoked fEPSPs (ofEPSPs) were recorded in layer V of the mouse prelimbic PFC using blue light (470 nM). (E) Bath application of 50 μM (in the absence of any electrical stimulations) CCh for 10 min led to a robust mLTD of ofEPSPs (n = 5) similar to electrically evoked fEPSPs. Insets in (A)–(C) show representative fEPSP/ofEPSP traces for each condition for baseline (black trace) and 55 min after CCh application (red trace). Scale bars denote 0.5 mV and 5 ms. Data are expressed as mean ± SEM. **p < 0.01.
While these results suggest that CCh-induced LTD requires concurrent activation of glutamatergic synapses during CCh application, electrical stimulation activates multiple fiber tracks, including different glutamatergic projections to the PFC as well as fibers from GABAergic and other neuronal populations. Thus, it is possible that this does not reflect a requirement for activation of glutamatergic afferents at the hippocampo-PFC synapse, which is thought to be the synapse at which mAChR activation induces mLTD.27 To more directly test the hypothesis that mLTD requires concurrent activation of the glutamatergic hippocampo-PFC synapse, we used optogenetic techniques to directly stimulate glutamatergic terminals that originate from hippocampal projections to the PFC. Thus, optically evoked fEPSPs (ofEPSPs) were recorded in layer V prelimbic PFC (Figure 2D) in slices from mice expressing ChR2 in the hippocampal terminals. Bath application of 50 μM CCh for 10 min, led to a robust mLTD of EPSPs (59.5 ± 3.9% baseline ofEPSP) measured 55–60 min following the addition of CCh (Figure 2E). This confirms that the mLTD observed using electrical stimulation reflects, at least in part, mLTD at the hippocampal-PFC synapse and indicates that electrical stimulation is not required but that optically evoked glutamate release from hippocampal terminals with concurrent addition of CCh is sufficient to induce mLTD.
Muscarinic LTD is Dependent on Activation of mGlu5 but Not NMDA Receptors
To test the hypothesis, that activation of mGlu5 is required for induction of mLTD in naive mice, we investigated the effect of the selective mGlu5 negative allosteric modulator (NAM) MPEP on mLTD. After acquiring a stable fEPSP baseline, MPEP was bath applied for 10 min followed by coapplication of CCh and MPEP for another 10 min. In the presence of MPEP (10 μM), 50 μM CCh failed to induce mLTD (95.5 ± 6.1% baseline fEPSP; Figure 3A). We next investigated the role of NMDARs in mLTD, as NMDARs are close signaling partners of mGlu5 and have been previously shown to be important in muscarinic forms of plasticity in other synapses.28,29 However, pretreatment of NMDAR antagonist AP-5 (100 μM) for 10 min followed by a 10 min coapplication with 50 μM CCh failed to block CCh induced mLTD in the PFC (71.8 ± 7.6% baseline fEPSP; Figure 3B). The magnitude of depression relative to baseline between the AP-5/CCh, MPEP/CCh and CCh-alone condition was statistically significant (one-way ANOVA; p = 0.04; F(2,11) = 4.065; Figure 3C). Moreover, posthoc multiple comparison tests revealed that the MPEP-CCh condition was significantly different from the CCh-alone condition (Dunnett’s multiple comparison test p < 0.05; q = 2.543), whereas the AP-5/CCh condition was not significantly different from the CCh/alone condition (Dunnett’s multiple comparison test p > 0.05; q = 0.165).
Figure 3.

mLTD is mGlu5 dependent but NMDA independent. (A) Bath application of the selective mGLu5 negative allosteric modulator MPEP (10 μM) for 10 min followed by coapplication of 50 μM CCh and MPEP blocked the induction of mLTD (n = 4). (B) Pharmacological blockade of NMDA receptors by AP-5 (100 μM) for 10 min followed by coapplication of 50 μM CCh failed to block mLTD (n = 4). (C) Magnitude of depression (outlined in gray) relative to baseline in AP-5/CCh, MPEP/CCh were compared with that of CCh-alone condition reported in Figure 2B and C. There was a significant difference across all three groups and posthoc analysis revealed that mLTD in the MPEP-CCh group was significantly different than CCh-alone group. (D) Pretreatment of MPEP also blocked CCh-induced mLTD of ofEPSPs in hippocampo-PFC pathway (n = 3). (E) Bar graph of ofEPSP depression relative to baseline in CCh alone (Figure 2E) and MPEP/CCh groups showing significant differences in mLTD between the 2 groups. Data are expressed as mean ± SEM. Insets in (A), (B), and (D) show representative fEPSP/ofEPSP traces for each condition for baseline (black trace) and 55 min after CCh application (red trace). Scale bars denote 0.5 mV and 5 ms. **p < 0.01 and *p < 0.05.
To further corroborate a specific role of synaptically activated mGlu5 in induction of mLTD at the hippocampo-PFC synapse, we next determined the effect of MPEP on induction of mLTD with optically evoked ofEPSPs isolated from the hippocampo-PFC synapse. After acquiring a stable ofEPSP baseline, MPEP (10 μM) was bath applied for 10 min followed by coapplication of CCh and MPEP for another 10 min. In the presence of MPEP, 50 μM CCh failed to induce LTD in ofEPSP (106.2 ± 14.2% baseline ofEPSP; Figure 3D). Statistical analysis confirmed that the magnitude of ofEPSP depression relative to baseline was significantly different between CCh-alone (Figure 2E) and MPEP/CCh conditions (Mann–Whitney test, p = 0.0318; Figure 3E). These results clearly show that mLTD at the hippocampo-PFC synapse is NMDA-independent but requires coactivation of M1 and mGlu5 receptors.
mGlu5 PAM VU0409551 Can Reduce the Threshold for Muscarinic LTD
To test whether biased mGlu5 PAMs can facilitate mLTD in untreated mice, we investigated whether allosteric potentiation of mGlu5 receptors, using the PAM VU0409551,6 can reduce the threshold for mLTD induction. Consistent with our previous results,24 a lower concentration of CCh (10uM), induces acute depression of fEPSPs but fails to induce mLTD (91.5 ± 2.5% baseline fEPSP; Figure 4A). However, pretreatment of VU0409551 (10 μM) for 10 min followed by a cotreatment of VU0409551 and 10 μM CCh, led to the induction of mLTD (66.1 ± 6.0% baseline fEPSP; Figure 4B and C). Statistical analysis revealed a significant difference between the 10 μM CCh alone condition and the VU0409551/CCh condition (Mann–Whitney test, p = 0.0177; Figure 4C). This indicates that potentiation of mGlu5 activity can reduce the effective CCh concentration required for mLTD, further emphasizing the critical role of mGlu5 receptors in mLTD.
Figure 4.

MGlu5 PAM VU0409551 can reduce the threshold of mLTD. (A) Time-course graph showing that, unlike 50 μM CCh, application of 10 μM CCh for 10 min produces negligible mLTD (shaded gray area; n = 7). (B) The biased mGlu5 PAM VU0409551 (10 μM) potentiated the threshold 10 μM CCh-mediated response as 10 min pretreatment with VU0409551 (dashed line) and 10 min coapplication of 10 μM CCh led to a robust mLTD (shaded gray area; n = 5). Inset shows representative fEPSP traces for each condition for baseline (black trace) and 55 min after CCh application (red trace). (C) Bar graph of fEPSP depression relative to baseline of the two experimental groups. There was a significant difference in the magnitude of mLTD observed with 10 μM CCh alone compared with the VU0409551/10 μM CCh condition. Scale bars denote 0.5 mV and 5 ms. Data are expressed as mean ± SEM. *p < 0.05.
Activation of mGlu5 along With CCh Application Leads to an Increased Inhibitory Tone onto Pyramidal Neurons
The finding that mLTD requires coactivation of both M1 and mGlu5, raises the question of how these receptors interact to induce mLTD. It is known that both M1 and mGlu5 activation can modulate GABAergic neurotransmission in the CNS.30,31 Furthermore, previous studies suggest that changes in inhibitory transmission play an important role in induction of LTD at the hippocampo-PFC synapse32 and in some of the behavioral deficits associated with transient exposure to NMDAR antagonists in juvenile development.33 Based on these previous findings, it is possible that the converging actions of mGlu5 and M1 on the inhibitory transmission in the PFC play an important role in induction of mLTD. To test the hypothesis that mLTD is associated with lasting changes in inhibitory transmission, spontaneous inhibitory post synaptic currents (sIPSCs) were recorded from layer V pyramidal neurons. We assessed inhibitory tone onto excitatory projection neurons of PFC, before and after application of CCh to induce mLTD. CCh (50 μM) was bath applied for 5 min after a stable baseline recording of sIPSCs. During the CCh application the superficial layer II/III was concurrently stimulated at 0.05 Hz. sIPSC frequency in a 2 min bin was calculated during baseline and then every 10 min after CCh application for 50 min, as shown in the time course (Figure 5A). Interestingly, CCh application and concurrent afferent stimulation led to a long-lasting increase in sIPSC frequency that could be observed 40–50 min following CCh application. A significant increase in the raw sIPSC frequency values was observed before and 40–50 min after CCh application (Wilcoxson matched pairs signed rank test; p = 0.0049; Figure 5A). The cumulative frequency distribution curve for inter event intervals (IEIs) for the 40–50 min post CCh time period was left shifted compared to baseline IEIs (Figure 5B). However, no change in frequency distribution for peak amplitudes was observed between baseline and 40–50 min post CCh (Figure 5C). Importantly, CCh failed to induce a long-lasting increase in sIPSC frequency when applied without concurrent stimulation of layer II/III (Wilcoxson matched pairs signed rank test; p = 0.5781; Figure 5D), which was similar to the failure in inducing mLTD with CCh-alone without concurrent stimulation. To determine whether the effect of concurrent stimulation on inhibitory tone is mediated by mGlu5, the experiments were repeated in the presence of the mGlu5 NAM MPEP (30 μM). In the presence of MPEP throughout the experiment, no long-term increase in sIPSC frequency was observed after CCh and concurrent stimulation (Figure 5E). No significant differences were observed between baseline sIPSC frequency and that observed 50 min after CCh application (Wilcoxson matched pairs signed rank test; p = 0.3125; Figure 5E). Finally, the percent change in sIPSC frequency 40–50 min following CCh add was compared between the three experimental conditions. Statistical analysis revealed that there was a significant difference between the three conditions (one-way ANOVA; p = 0.0003; F(2,21) = 12.55; Figure 5F). Moreover, posthoc multiple comparison testing revealed that the increase in sIPSC frequency observed with CCh/Stimulation condition was significantly different than the changes observed with CCh-alone (Dunnett’s multiple comparison test; p < 0.001; q = 4.252) and in the presence of MPEP (Dunnett’s multiple comparison test; p < 0.01; q = 4.036). This indicates that coactivation of mGlu5 receptors during CCh application may underlie the effects of afferent stimulation on inhibitory tone of pyramidal neurons of mouse PFC.
Figure 5.

Activation of mGlu5 with CCh leads to increased inhibitory tone. (A) Left: sIPSCs were recorded from layer V pyramidal neurons and sIPSC frequencies were normalized to baseline and displayed as a time course. After 5 min of baseline, 50 μM CCh was bath applied for 5 min with concurrent stimulation at 0.05 Hz. This led to a delayed increase in sIPSC frequency 50–60 min following addition of CCh (n = 11). Right Right top: Raw sIPSC frequency values from baseline and post CCh are plotted for each cell recorded and compared statistically showing a significant increase in frequency from baseline. Right bottom: Sample trace for a typical cell before CCh (black) and post-CCh application (red). (B,C) Cumulative percentage plots comparing pre-mLTD (black) and post-mLTD (red) for IEI (B) and amplitude (C) of sIPSCs are shown here for the data plotted in (A). Data displayed in (D) and (E) similarly to (A). (D) CCh failed to induced long-term increases in sIPSC frequency when applied without concurrent stimulation (n = 7). (E) There was no change in sIPSC frequency when 50 μM CCh was applied during concurrent stimulation in the presence of MPEP (dashed line) for the entire experiment (n = 6). (F) Graph comparing the normalized sIPSC frequency for all three conditions, shows that percent change in sIPSC frequency from baseline observed with CCh/Stimulation condition was significantly different than the changes observed with CCh-alone and in the presence of MPEP. Scale bars denote 50 pA and 2 s. Error bars represent the mean ± SEM. ***p < 0.001 and **p < 0.01.
Increased Inhibitory Tone Following CCh Application and mGlu5 Coactivation May Underlie Expression of mLTD
To evaluate the importance of changes in inhibitory transmission via GABAA receptors for mLTD expression in the PFC, we evaluated the ability of CCh to induce mLTD in the presence of the GABAA receptor antagonist bicuculline (20 μM). After a stable baseline of electrically evoked fEPSPs was acquired in the presence of continuous perfusion of 20 μM bicuculline, 50 μM CCh was applied for 10 min. In presence of bicuculline, CCh induced a significant acute depression of fEPSPs but failed to induce mLTD measured 55–60 min following CCh application (120.3 ± 4.2% baseline fEPSP; Figure 6A). The magnitude of change in fEPSP relative to baseline during the bicuculline/CCh condition was also significantly different than what was observed with CCh/Stim condition (Mann–Whitney test, p = 0.0095; Figure 6B).
Figure 6.

Increased inhibitory tone may underlie expression of mLTD. (A) Time-course graph of fEPSP recordings showing that application of 50 μM CCh in the presence of the GABAA antagonist bicuculline (20 μM) for the entire experiment fails to produce mLTD (n = 4). (B) The magnitude of mLTD (gray area of 5a) was significantly different in the presence of bicuculline compared to 50 μM CCh alone as observed in Figure 2A. Inset shows representative fEPSP traces for baseline (black trace) and 55 min after CCh application (red trace). (C) Bicuculline was added for 2 min in slices where mLTD was already induced with CCh and stimulation (n = 5). This addition was 40–50 min after the application of CCh and led to a larger change in fEPSP compared to the effect of bicuculline in slices without mLTD induction (n = 5). (D) Bar graphs plotting the percent change in fEPSP slope after bicuculline add (gray area in C) shows that the magnitude of disinhibition was significantly higher in slices with mLTD induced than in slices without any mLTD induction. Scale bars denote 0.5 mV and 5 ms. Error bars denote SEM. **p < 0.01 and *p < 0.05.
To assess the potential importance of this lasting increase in inhibitory tone in the depression of the net excitatory fEPSP output during mLTD, bicuculline (20 μM) was added for 2 min in slices where mLTD was already induced with 50 μM CCh and concurrent stimulation (40–50 min after CCh application). As a control, bicuculline was also added for 2 min in slices where no mLTD was induced but a stable baseline fEPSP was acquired (Figure 6C). The percent change in fEPSP slope observed after bicuculline add (average of 1–5 min after bicuculline add: 207.2 ± 30.7%) in slices with mLTD induced was significantly higher (Mann–Whitney test, p = 0.0317; Figure 6D) than that observed in slices without any mLTD induction (average of 1–5 min after bicuculline add: 133.4 ± 8.3%). This shows that there is greater amount of disinhibition affected by bicuculline in slices with mLTD, indicating that there is enhanced inhibitory tone during periods of mLTD. Taken together, these results provide strong evidence that mLTD is associated with a lasting increase in GABAA receptor-mediated inhibitory transmission and that this plays an important role in the expression of mLTD in mouse PFC.
DISCUSSION
Extensive previous studies suggest that cholinergic innervation of the PFC as well as glutamatergic inputs from the hippocampus to the PFC play critical roles in multiple forms of PFC-dependent cognitive function.34–37 Furthermore, mounting evidence suggests that excessive transmission at hippocampo-PFC synapses and deficits in LTD at this synapse may play important roles in the pathophysiology underlying some aspects of schizophrenia.32,38 In this study, we make the surprising finding that a previously described form of LTD that is induced by cholinergic activation of M1 mAChRs (mLTD) is dependent on synaptic activation of mGlu5 during the period of M1 receptor activation. Thus, mLTD requires coincident activation M1 and glutamatergic hippocampo-PFC synapses and is completely blocked by incubation with a selective mGlu5 NAM. Furthermore, we found that a biased mGlu5 PAM that does not potentiate mGlu5 modulation of NMDAR currents can reduce the threshold of mLTD in slices from naïve mice. One of the most exciting aspects of the current studies was the finding that the biased mGlu5 PAM completely restores deficits in mLTD, and its systemic administration reverses deficits in a measure of recognition memory that is severely impaired in mice that were transiently administered PCP during a one-week period in juvenile development. While the detailed mechanisms by which mGlu5 and M1 interact to induce mLTD and reverse deficits in this rodent model are not entirely known, our results suggest that coactivation of mGlu5 and M1 receptors converge to induce an increase in GABAergic inhibitory tone onto the pyramidal neurons of PFC. This in turn could tilt the excitatory inhibitory balance toward inhibition leading to an overall depression of fEPSPs and expression of mLTD.
The present findings are consistent with a previous report that CCh-induced LTD recorded in layer II/III pyramidal cells in the PFC also required stimulation of glutamatergic afferents,27 and provides strong evidence that this is due to a requirement for synaptic activation of mGlu5. It is interesting to note that, while activation of mGlu5 alone does not induce LTD in this particular synapse of PFC, the ability of mGlu5 to regulate LTD in this region has also been observed with LTD induced by activation of dopaminergic25 and serotonergic26 systems or in other synapses.39 Interestingly, another form of muscarinic LTD reported in layer V pyramidal neurons in the PFC was shown to be independent of concurrent afferent stimulation.40 However, this form of LTD was observed in rats rather than mice, and the studies were performed at an earlier stage of postnatal development, when mGlu5 signaling is enhanced relative to what is observed in young adult animals. This could increase tonic activation of mGlu5 in the absence of afferent stimulation. Also, the previous studies were performed at room temperature and under whole cell configuration with the membrane voltage clamped to −70 mV where there is minimal contribution of inhibitory neurotransmission.
While mGlu5 activation alone is not sufficient to induce LTD in PFC, activation of mGlu5 alone induces robust LTD in the hippocampus.25,26 Interestingly, mAChR activation can also induce hippocampal LTD, and previous studies suggest that the ability of an mGlu5 agonist to induce LTD in area CA1 of the hippocampus is lost with selective genetic deletion of the M1 mAChR in hippocampal CA3 pyramidal cells.41 Furthermore, a specific form of LTD that is induced by patterned high frequency stimulation in the hippocampus is dependent on activation of both GABAA receptors and mAChRs.42 Thus, it is possible that there are mechanistic similarities between mLTD in the PFC and the mGlu receptor LTD in the hippocampus, and that M1 and mGlu5 work in concert to induce LTD in both regions, at least under some conditions. However, there are also important differences between mGlu5 LTD in the hippocampus and mLTD in the PFC. Most importantly, LTD induced by the mGlu5 agonist DHPG at the SC-CA1 synapse of the hippocampus, is not dependent on increased inhibitory neurotransmission43,44 and is mediated by AMPA receptor endocytosis.45,46 In contrast, the mLTD we observe here appears to be at least partly mediated by an increase in inhibitory transmission. This is in stark contrast with another form of muscarinic LTD reported by others in the PFC,47 where fEPSPs were generated by stimulating layer V collaterals of PFC, instead of layer II/III. The M1 dependent LTD observed in this study was independent of GABAA receptor activation. The difference on the dependence of GABAA receptors between the two studies can be attributed to the fact that they were looking at synaptic responses largely mediated by recurrent collaterals of other excitatory inputs to PFC that are different from those recruited by stimulation of superficial layers. This indicates that there are independent mechanisms for plasticity in different synapses in the PFC.
The present findings that mLTD is accompanied by a sustained increase in inhibitory tone onto the layer V pyramidal neurons and that this form of plasticity is lost in mice that were exposed to PCP during juvenile development is especially interesting in light of previous studies on inhibitory transmission in the PFC using this model. Interestingly, transient PCP treatment at this developmental stage results in a sustained loss of GABAergic inhibition in the PFC,48,49 a selective loss of a specific subpopulation of inhibitory interneurons that express parvalbumin,50,51 and a shift in excitatory/inhibitory balance.32,52,53 This is important since loss of interneurons or inhibitory function is a hallmark in schizophrenia pathology and this interneurons dysfunction could underlie many of the cognitive symptoms of schizophrenia.54 Therefore, reversing plasticity deficits in the PFC that are dependent on inhibitory neurotransmission can be considered as a relevant mechanism for improving cognitive function.
Our previous report shows that the deficit in mLTD in PCP-treated mice cannot be explained by a simple loss of function of M1 receptors in the pyramidal neurons.24 In the light of new data reported here, it can be postulated that this deficit in mLTD may be due, at least in part, to a loss in inhibitory transmission. If so, it is possible that mGlu5 PAMs restore mLTD in PCP-treated mice by helping restore the loss in inhibitory tone observed in these mice. At present, the exact mechanism by which simultaneous activation of M1 and mGlu5 leads to a lasting increase in inhibitory tone is unknown. Both M1 and mGlu5 are expressed in interneurons and can modulate inhibitory transmission.55,30 Thus, it is possible that either M1 or mGlu5, or both of these receptors, located on interneurons are responsible for this effect. However, activation of one or both of these receptors in pyramidal neurons could also modulate inhibitory neurotransmission via a retrograde messenger system or through circuit level effects. In future studies, it will be important to systematically evaluate potential roles for both mGlu5 and M1 in specific neuronal populations in inducing this response.
Previous reports have also suggested that disruptions of PFC function in response to PCP treatment (acute or chronic) are primarily mediated by changes in the hippocampo-prefrontal pathway.32,34,56–58 This pathway has also been implicated in cognitive functions in normal human subjects that are relevant for several psychiatric disorders including schizophrenia.59 Our findings show that indeed mLTD can also be replicated in the isolated hippocampo-prefrontal pathway. However, whether the deficits observed in mLTD in the PCP treated mice are exclusive to this pathway is not known and will be an important future direction to understand the pathophysiology of PCP treatment and its relevance to schizophrenia.
Finally, while M1 and mGlu5 act in concert to induce mLTD, we previously showed that potentiation of M1 receptors by a selective M1 PAM is sufficient to rescue the mLTD and cognitive deficits of the PCP treated mice.24 Likewise, in the current studies, we found that mGlu5 potentiation using a selective mGlu5 PAM can be equally effective in rescuing mLTD and recognition memory deficits. This corroborates our hypothesis that mLTD deficits can underlie the behavioral outcome observed in this NMDAR hypofunction model and probes that can rescue mLTD can also improve the cognitive symptoms observed in this PCP treatment model. However, since recognition memory is also dependent on other cortical areas it is possible that the rescue of this phenotype are due to a potential mGlu5 and M1 interaction or mGlu5 action alone in other cortical regions as well. But in either case, these studies provide additional support to the hypothesis that mGlu5 PAMs can provide a promising therapeutic approach for the treatment of cognitive symptoms in mental disorders. Excitingly, in this study, we used VU0409551, which unlike other mGlu5 PAMs 6,9 does not potentiate coupling of mGlu5 to NMDAR function. Although this rodent PCP model was based on NMDAR hypofunction, VU0409551 showed robust in vivo efficacy in this model. This supports our hypothesis, that mGlu5 PAMs do not need to rely upon NMDAR potentiation to show efficacy in in vivo models. Moreover, this study sheds light on one potential mechanism by which biased mGlu5 PAMs that do not enhance NMDAR function can show in vivo efficacy in normal animals as well disease models.
In conclusion, in this study, we have shown that mLTD is a unique form of plasticity in the PFC that is more of a network effect. We have also provided strong evidence that mGlu5 and M1 can interact in the PFC to modulate inhibitory neuro-transmission via GABAA receptors and subsequently induce plasticity. Additionally, we have also shown that potentiation of mGlu5 alone by PAMs that do not potentiate NMDAR function can show efficacy in animal models based on NMDAR hypofunction. Finally, rescue of mLTD deffcits in the PFC can be one mechanism by which these biased mGlu5 PAMs can show behavioral efficacy in vivo.
METHODS
Animals
All animal studies were approved by the Vanderbilt University Medical Center Institutional Animal Care and Use Committee and were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Male C57BL6/J mice (Jackson laboratories) were used in electrophysiology and behavioral studies (8–9 weeks old). Animals were group housed 4–5 per cage, maintained on a 12 h light/dark cycle, and provided food and water ad libitum.
Drugs
(−)-Bicuculline methobromide, D-AP5, was purchased from Abcam bicochemicals. Carbachol (CCh) and phencyclidine (PCP) were purchased from Sigma-Aldrich. MPEP60 and VU04095516,9 were synthesized in house. All drugs used for electrophysiology experiments were diluted in artificial cerebrospinal fluid (ACSF). For behavioral studies VU0409551 and PCP were formulated in 10% Tween-80 and 0.9% Saline, respectively.
Stereotaxic Viral Injections
Mice underwent stereotaxic injections for viral-mediated gene transfer of channelrhodopsin-2 (ChR2) at 4–5 weeks of age. Dexmetometidine/ketamine (0.5:80 mg/kg, i.p.) was used to induce anesthesia. Following a craniotomy, four injections (400 nL at 100 nL/min) of AAV5-CaMKII-ChR2-EYFP (UNC Viral Vector Core) were delivered into the target regions through a pulled glass pipet. Injection site coordinates were as follows (relative to bregma): vSub [ML: ± 2.8, AP: −3.6, DV: −4.6] and vCA1 [ML: ± 3.6, AP: −3.2, DV: −3.2]. Ketoprofen (10 mg/kg, s.c.) was administered immediately after the procedure and then for at least another 3 days post procedure. Recordings were made 4–6 weeks following surgery to allow sufficient expression of ChR2 in axon terminals within the PFC.
Extracellular Field Electrophysiology
Extracellular field electro-physiology was performed as reported previously.24 In brief, 8–10 week old male C57BL6/J mice (Jackson Laboratories) were anesthetized with isofluorane, and the brains were removed and submerged in ice-cold cutting solution (in mM: 230 sucrose, 2.5 KCl, 8 MgSO4, 0.5 CaCl2, 1.25 NaH2PO4, 10 D-glucose, 26 NaHCO3). Coronal slices containing the prelimbic prefrontal cortex were cut at 400 μm and were transferred to a holding chamber containing NMDG-HEPES recovery solution (in mM: 93 NMDG, 2.5 KCl, 1.2 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 D-glucose, 5 sodium ascorbate, 2 thiourea, 3 sodium pyruvate, 10 MgSO4, 0.5 CaCl2, 12 N-acetyl-L-cysteine, pH 7.3, <310 mOsm) for 15 min at 32 °C. Slices were then transferred to a room temperature holding chamber for at least 1 h containing ACSF (in mM: 126 NaCl, 1.25 NaH2PO4, 2.5 KCl, 10 D-glucose, 26 NaHCO3, 2 CaCl2, 1 MgSO4) supplemented with 600 μM sodium ascorbate for slice viability. All buffers were continuously bubbled with 95% O2/5% CO2. Subsequently, slices were transferred to a 28–32 °C submersion recording chamber where they were perfused with ACSF at a rate of 2 mL/min. Data was analyzed offline using Clampfit 10.2 (Molecular Devices). Data were digitized using a Multiclamp 700B, Digidata 1322A, and pClamp 10 software (Molecular Devices). Paired electrical stimulus pulses (100 μs duration, every 20 s; interpulse interval of 50 ms) of the superficial layer II–III, delivered through a concentric bipolar stimulating electrode. For studies involving optical stimulation, blue light (470 nm) was delivered using a High Power LED (Thorlabs Inc. Newton, NJ), which was mounted to the epi-illumination port of an Olympus BX50WI upright microscope (Olympus). Blue light was shined onto slices through the 40× objective lens at 0.05 Hz (Maximum light intensity at the site of illumination is 2.6 mW).
Whole Cell Electrophysiology
Whole-cell patch-clamp recordings were performed using coronal PFC slices (300 μm) prepared from 8–10 week old male C57BL6/J mice (Jackson Laboratories) and a cesium methylsulfonate based intracellular solution consisting of the following (in mM) 70 CsCH3SO3, 60 CsCl, 1 MgCl2, 0.2 EGTA, 10 HEPES, 2 Mg-ATP 0.3 GTP-Na2, 10 Na2-phosphocreatine,10 TEA, and 5 QX-314. Spontaneous inhibitory postsynaptic currents (sIPSCs) were recorded at a holding potential of −70 mV in the presence of NMDAR antagonist AP-5 (100uM) and AMPA receptor antagonist CNQX (20uM). The interevent intervals (IEIs) of sIPSCs from 2 min episodes during baseline and different time points post drug application were calculated and normalized to the baseline value. These normalized values were used to generate the sIPSC timecourse graphs. Cumulative probability plots were constructed using the baseline and 50 min post CCh IEI values. The IEIs from each experiment were also expressed as frequency and the mean values from the 2 min episodes from baseline and 40–50 min post CCh were compared. All analyses were performed using Clampfit software using template matching (v10.2, Molecular Devices).
Novel Object Recognition Task
Novel object recognition task assay was performed as explained previously.24 In brief, mice were habituated in an empty dark colored plexiglass box (32 × 32 × 40 cm3) for 10 min. Approximately 24 h later, the mice were injected with vehicle (10% Tween-80), 1, 3, or 10 mg/kg of VU0409551 (intraperitoneal (i.p.); 10 mL/kg) and placed back into their home cage for 30 min. Mice were then placed back into the plexiglass box containing two identical objects and exposed to the objects for 10 min. After the exposure period, mice were placed back into their home cages. One hour later, they were returned to the plexiglass box in which one of the previously exposed (familiar) objects was replaced by a novel object. The exploration of the objects by the mice was video recorded for 10 min. A blinded observer then scored the time spent exploring the novel and familiar object. The recognition index was then calculated as [(time spent exploring novel object) − (time spent exploring familiar object)]/(total time exploring objects) and reported. The recognition index would be “0” when animals spend equal amount of time exploring each object (i.e., chance levels).
Statistical Analyses
For the novel object recognition task, groups were compared using a one-way ANOVA, followed by Dunnett’s multiple comparison tests with the PCP-vehicle mice as the control group. For the mLTD experiments where only two experimental conditions were compared, the Mann–Whitney U test was performed to calculate statistical significance. For comparisons with more than one group, a one-way ANOVA was performed followed by Dunnett’s multiple comparison tests with CCh-alone condition as control. Changes in sIPSC frequency before and after CCh addition was compared using a Wilcoxson matched pairs signed rank test. Finally, percent change in sIPSC frequency between different experimental conditions was compared using a one-way ANOVA, followed by Dunnett’s multiple comparison tests with the CCh/stimulation condition as the control group. For all statistical comparison, the critical p value was considered to be 0.05.
Acknowledgments
Funding
This work was supported by NIH R37 NS031373 (P.J.C.), R01 MH062646 (P.J.C.), and R01 MH073676 (P.J.C.).
ABBREVIATIONS
- AAV5
adeno-associated-virus serotype 5
- AMPAR
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor
- ANOVA
analysis of variance
- CaMKII
calcium calmodulin kinase II
- CCh
carbachol
- ChR2
channelrhodopsin-2
- CNS
central nervous system
- CNQX
6-cyano-7-nitroquinoxaline-2,3-dione
- DHPG
3,5-dihydroxyphenylglycine hydrate
- EYFP
enhanced yellow fluorescence protein
- fEPSP
field excitatory post synaptic potentials
- GABAA
gamma-aminobutyric acid receptor subtype A
- GTP
guanosine triphosphate
- HEPES
(4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid)
- IEI
interevent interval
- LTD
long-term depression
- M1
muscarinic receptor subtype 1
- mAchR
muscarinic acetylcholine receptor
- mGlu5
metabotropic glutamate receptor subtype 5
- mg/kg
milligrams per kilograms
- mLTD
muscarinic long-term depression
- mL/kg
milliliters per kilograms
- MPEP
2-methyl-6-(phenylethynyl)pyridine
- mV
millivolt
- mW
milliwatt
- NAM
Negative allosteric modulator
- NMDAR
N-methyl-D-Aspartate receptor
- NMDG
N-methyl-D-Glucamine
- ofEPSP
optically evoked field excitatory post synaptic potentials
- PAM
positive allosteric modulator
- PCP
phencyclidine
- PFC
prefrontal cortex
- SC-CA1
Schaffer collateral-Cornu Ammonis area 1
- sIPSC
spontaneous inhibitory post synaptic currents
- TEA
tetraethylammmonium
- vCA1
ventral Cornu Ammonis area 1
- vSub
ventral subiculum
Footnotes
Author Contributions
A.G., J.M.R., B.A.G., Z.X., C.W.L., and P.J.C. designed experiments; A.G., S.P.M., J.W.D., and M.E.J. performed experiments; A.G., S.P.M., B.A.G., C.W.L., J.M.R., and P.J.C. wrote manuscript.
Notes
The authors declare the following competing financial interest(s): Dr. Rook’s work has been funded by NIH, Alzheimer’s Drug Discovery Foundation, Harrington Discovery Institute, Thome Alzheimer’s Disease Drug Discovery Foundation, and Brain & Behavior Research Foundation. She is an inventor on patents that protect different classes of muscarinic receptor allosteric modulators. Dr. Lindsley’s work has been funded by the NIH, Johnson and Johnson, Bristol-Myers Squibb, AstraZeneca, Michael J. Fox Foundation, as well as Seaside Therapeutics. He has consulted for AbbVie and received compensation. He is an inventor on patents that protect different classes of metabotropic glutamate and muscarinic receptor allosteric modulators. Dr. Conn has been funded by NIH, AstraZeneca, Bristol-Myers Squibb, Michael J. Fox Foundation, Dystonia Medical Research Foundation, CHDI Foundation, and Thome Memorial Foundation. Over the past three years he has served on the Scientific Advisory Boards for Michael J. Fox Foundation, Stanley Center for Psychiatric Research Broad Institute (MIT/Harvard), Karuna Pharmaceuticals, Lieber Institute for Brain Development, Clinical Mechanism (POCM) and Proof of Concept (POC) Consortium, and Neurobiology Foundation for Schizophrenia and Bipolar Disorder. He is an inventor on patents that protect different classes of metabotropic glutamate and muscarinic receptor allosteric modulators. Dr. Ghoshal, Mr. Moran, Dr. Dickerson, Dr. Joffe, Dr. Grueter and Dr. Xiang declare no potential conflict of interest.
References
- 1.Conn PJ, Lindsley CW, Jones CK. Activation of metabotropic glutamate receptors as a novel approach for the treatment of schizophrenia. Trends Pharmacol Sci. 2009;30:25–31. doi: 10.1016/j.tips.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nickols HH, Conn PJ. Development of allosteric modulators of GPCRs for treatment of CNS disorders. Neurobiol Dis. 2014;61:55–71. doi: 10.1016/j.nbd.2013.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Walker AG, Conn PJ. Group I and group II metabotropic glutamate receptor allosteric modulators as novel potential antipsychotics. Curr Opin Pharmacol. 2015;20:40–45. doi: 10.1016/j.coph.2014.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ayala JE, Chen Y, Banko JL, Sheffler DJ, Williams R, Telk AN, Watson NL, Xiang Z, Zhang Y, Jones PJ, Lindsley CW, Olive MF, Conn PJ. mGluR5 positive allosteric modulators facilitate both hippocampal LTP and LTD and enhance spatial learning. Neuropsychopharmacology. 2009;34:2057–2071. doi: 10.1038/npp.2009.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Noetzel MJ, Rook JM, Vinson PN, Cho HP, Days E, Zhou Y, Rodriguez AL, Lavreysen H, Stauffer SR, Niswender CM, Xiang Z, Daniels JS, Jones CK, Lindsley CW, Weaver CD, Conn PJ. Functional impact of allosteric agonist activity of selective positive allosteric modulators of metabotropic glutamate receptor subtype 5 in regulating central nervous system function. Mol Pharmacol. 2012;81:120–133. doi: 10.1124/mol.111.075184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rook JM, Xiang Z, Lv X, Ghoshal A, Dickerson JW, Bridges TM, Johnson KA, Foster DJ, Gregory KJ, Vinson PN, Thompson AD, Byun N, Collier RL, Bubser M, Nedelcovych MT, Gould RW, Stauffer SR, Daniels JS, Niswender CM, Lavreysen H, Mackie C, Conde-Ceide S, Alcazar J, Bartolome-Nebreda JM, Macdonald GJ, Talpos JC, Steckler T, Jones CK, Lindsley CW, Conn PJ. Biased mGlu5-Positive Allosteric Modulators Provide In Vivo Efficacy without Potentiating mGlu5Modulation of NMDAR Currents. Neuron. 2015;86:1029–1040. doi: 10.1016/j.neuron.2015.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Noetzel MJ, Gregory KJ, Vinson PN, Manka JT, Stauffer SR, Lindsley CW, Niswender CM, Xiang Z, Conn PJ. A novel metabotropic glutamate receptor 5 positive allosteric modulator acts at a unique site and confers stimulus bias to mGlu5 signaling. Mol Pharmacol. 2013;83:835–847. doi: 10.1124/mol.112.082891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rook JM, Noetzel MJ, Pouliot WA, Bridges TM, Vinson PN, Cho HP, Zhou Y, Gogliotti RD, Manka JT, Gregory KJ, Stauffer SR, Dudek FE, Xiang Z, Niswender CM, Daniels JS, Jones CK, Lindsley CW, Conn PJ. Unique signaling profiles of positive allosteric modulators of metabotropic glutamate receptor subtype 5 determine differences in in vivo activity. Biol Psychiatry. 2013;73:501–509. doi: 10.1016/j.biopsych.2012.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Conde-Ceide S, Martinez-Viturro CM, Alcazar J, Garcia-Barrantes PM, Lavreysen H, Mackie C, Vinson PN, Rook JM, Bridges TM, Daniels JS, Megens A, Langlois X, Drinkenburg WH, Ahnaou A, Niswender CM, Jones CK, Macdonald GJ, Steckler T, Conn PJ, Stauffer SR, Bartolome-Nebreda JM, Lindsley CW. Discovery of VU0409551/JNJ-46778212: An mGlu5 Positive Allosteric Modulator Clinical Candidate Targeting Schizophrenia. ACS Med Chem Lett. 2015;6:716–720. doi: 10.1021/acsmedchemlett.5b00181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Homayoun H, Stefani MR, Adams BW, Tamagan GD, Moghaddam B. Functional Interaction Between NMDA and mGlu5 Receptors: Effects on Working Memory, Instrumental Learning, Motor Behaviors, and Dopamine Release. Neuropsychopharmacology. 2004;29:1259–1269. doi: 10.1038/sj.npp.1300417. [DOI] [PubMed] [Google Scholar]
- 11.Bhardwaj SK, Ryan RT, Wong TP, Srivastava LK. Loss of dysbindin-1, a risk gene for schizophrenia, leads to impaired group 1 metabotropic glutamate receptor function in mice. Front Behav Neurosci. 2015;9:72. doi: 10.3389/fnbeh.2015.00072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clifton NE, Morisot N, Girardon S, Millan MJ, Loiseau F. Enhancement of social novelty discrimination by positive allosteric modulators at metabotropic glutamate 5 receptors: adolescent administration prevents adult-onset deficits induced by neonatal treatment with phencyclidine. Psychopharmacology. 2013;225:579–594. doi: 10.1007/s00213-012-2845-3. [DOI] [PubMed] [Google Scholar]
- 13.Horio M, Fujita Y, Hashimoto K. Therapeutic effects of metabotropic glutamate receptor 5 positive allosteric modulator CDPPB on phencyclidine-induced cognitive deficits in mice. Fundam Clin Pharmacol. 2013;27:483–488. doi: 10.1111/j.1472-8206.2012.01045.x. [DOI] [PubMed] [Google Scholar]
- 14.Moghaddam B. Targeting metabotropic glutamate receptors for treatment of the cognitive symptoms of schizophrenia. Psychopharmacology. 2004;174:39–44. doi: 10.1007/s00213-004-1792-z. [DOI] [PubMed] [Google Scholar]
- 15.Stefani MR, Moghaddam B. Activation of type 5 metabotropic glutamate receptors attenuates deficits in cognitive flexibility induced by NMDA receptor blockade. Eur J Pharmacol. 2010;639:26–32. doi: 10.1016/j.ejphar.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Uslaner JM, Parmentier-Batteur S, Flick RB, Surles NO, Lam JS, McNaughton CH, Jacobson MA, Hutson PH. Dose-dependent effect of CDPPB, the mGluR5 positive allosteric modulator, on recognition memory is associated with GluR1 and CREB phosphorylation in the prefrontal cortex and hippocampus. Neuropharmacology. 2009;57:531–538. doi: 10.1016/j.neuropharm.2009.07.022. [DOI] [PubMed] [Google Scholar]
- 17.Balu DT, Li Y, Takagi S, Presti KT, Ramikie TS, Rook JM, Jones CK, Lindsley CW, Conn PJ, Bolshakov VY, Coyle JT. An mGlu5-Positive Allosteric Modulator Rescues the Neuroplasticity Deficits in a Genetic Model of NMDA Receptor Hypofunction in Schizophrenia. Neuropsychopharmacology. 2016;41:2052–2061. doi: 10.1038/npp.2016.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Niswender CM, Conn PJ. Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu Rev Pharmacol Toxicol. 2010;50:295–322. doi: 10.1146/annurev.pharmtox.011008.145533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marino MJ, Conn PJ. Direct and indirect modulation of the N-methyl D-aspartate receptor. Curr Drug Targets: CNS Neurol Disord. 2002;1:1–16. doi: 10.2174/1568007023339544. [DOI] [PubMed] [Google Scholar]
- 20.Hu JH, Yang L, Kammermeier PJ, Moore CG, Brakeman PR, Tu J, Yu S, Petralia RS, Li Z, Zhang PW, Park JM, Dong X, Xiao B, Worley PF. Preso1 dynamically regulates group I metabotropic glutamate receptors. Nat Neurosci. 2012;15:836–844. doi: 10.1038/nn.3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shepherd JD, Huganir RL. The cell biology of synaptic plasticity: AMPA receptor trafficking. Annu Rev Cell Dev Biol. 2007;23:613–643. doi: 10.1146/annurev.cellbio.23.090506.123516. [DOI] [PubMed] [Google Scholar]
- 22.Gao C, Tronson NC, Radulovic J. Modulation of behavior by scaffolding proteins of the post-synaptic density. Neurobiol Learn Mem. 2013;105:3–12. doi: 10.1016/j.nlm.2013.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Park JM, Hu JH, Milshteyn A, Zhang PW, Moore CG, Park S, Datko MC, Domingo RD, Reyes CM, Wang XJ, Etzkorn FA, Xiao B, Szumlinski KK, Kern D, Linden DJ, Worley PF. A prolyl-isomerase mediates dopamine-dependent plasticity and cocaine motor sensitization. Cell. 2013;154:637–650. doi: 10.1016/j.cell.2013.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ghoshal A, Rook JM, Dickerson JW, Roop GN, Morrison RD, Jalan-Sakrikar N, Lamsal A, Noetzel MJ, Poslusney MS, Wood MR, Melancon BJ, Stauffer SR, Xiang Z, Daniels JS, Niswender CM, Jones CK, Lindsley CW, Conn PJ. Potentiation of M1Muscarinic Receptor Reverses Plasticity Deficits and Negative and Cognitive Symptoms in a Schizophrenia Mouse Model. Neuropsychopharmacology. 2016;41:598–610. doi: 10.1038/npp.2015.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Otani S, Auclair N, Desce JM, Roisin MP, Crepel F. Dopamine receptors and groups I and II mGluRs cooperate for long-term depression induction in rat prefrontal cortex through converging postsynaptic activation of MAP kinases. J Neurosci. 1999;19:9788–9802. doi: 10.1523/JNEUROSCI.19-22-09788.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhong P, Liu W, Gu Z, Yan Z. Serotonin facilitates long-term depression induction in prefrontal cortex via p38 MAPK/Rab5-mediated enhancement of AMPA receptor internalization. J Physiol. 2008;586:4465–4479. doi: 10.1113/jphysiol.2008.155143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang L, Yuan LL. Activation of M2 muscarinic receptors leads to sustained suppression of hippocampal transmission in the medial prefrontal cortex. J Physiol. 2009;587:5139–5147. doi: 10.1113/jphysiol.2009.174821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dennis SH, Pasqui F, Colvin EM, Sanger H, Mogg AJ, Felder CC, Broad LM, Fitzjohn SM, Isaac JT, Mellor JR. Activation of Muscarinic M1 Acetylcholine Receptors Induces Long-Term Potentiation in the Hippocampus. Cerebral cortex. 2016;26:414–426. doi: 10.1093/cercor/bhv227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Sevilla DF, Buno W. The muscarinic long-term enhancement of NMDA and AMPA receptor-mediated transmission at Schaffer collateral synapses develop through different intracellular mechanisms. J Neurosci. 2010;30:11032–11042. doi: 10.1523/JNEUROSCI.1848-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yi F, Ball J, Stoll KE, Satpute VC, Mitchell SM, Pauli JL, Holloway BB, Johnston AD, Nathanson NM, Deisseroth K, Gerber DJ, Tonegawa S, Lawrence JJ. Direct excitation of parvalbumin-positive interneurons by M1 muscarinic acetylcholine receptors: roles in cellular excitability, inhibitory transmission and cognition. J Physiol. 2014;592:3463–3494. doi: 10.1113/jphysiol.2014.275453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Colavita M, Terral G, Lemercier CE, Drago F, Marsicano G, Massa F. Layer-specific potentiation of network GABAergic inhibition in the CA1 area of the hippocampus. Sci Rep. 2016;6:28454. doi: 10.1038/srep28454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thomases DR, Cass DK, Meyer JD, Caballero A, Tseng KY. Early Adolescent MK-801 Exposure Impairs the Maturation of Ventral Hippocampal Control of Basolateral Amygdala Drive in the Adult Prefrontal Cortex. J Neurosci. 2014;34:9059–9066. doi: 10.1523/JNEUROSCI.1395-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li JT, Su YA, Wang HL, Zhao YY, Liao XM, Wang XD, Si TM. Repeated Blockade of NMDA Receptors During Adolescence Impairs Reversal Learning and Disrupts GABAergic Interneurons in Rat Medial Prefrontal Cortex. Front Mol Neurosci. 2016;9:17. doi: 10.3389/fnmol.2016.00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ghoshal A, Conn PJ. The hippocampo-prefrontal pathway: a possible therapeutic target for negative and cognitive symptoms of schizophrenia. Future Neurol. 2015;10:115–128. doi: 10.2217/FNL.14.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Luchicchi A, Bloem B, Viana JN, Mansvelder HD, Role LW. Illuminating the role of cholinergic signaling in circuits of attention and emotionally salient behaviors. Front Synaptic Neurosci. 2014;6:24. doi: 10.3389/fnsyn.2014.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ragozzino ME, Kesner RP. The effects of muscarinic cholinergic receptor blockade in the rat anterior cingulate and Prelimbic/Infralimbic cortices on spatial working memory. Neurobiol Learn Mem. 1998;69:241–257. doi: 10.1006/nlme.1998.3823. [DOI] [PubMed] [Google Scholar]
- 37.Shirey JK, Brady AE, Jones PJ, Davis AA, Bridges TM, Kennedy JP, Jadhav SB, Menon UN, Xiang Z, Watson ML, Christian EP, Doherty JJ, Quirk MC, Snyder DH, Lah JJ, Levey AI, Nicolle MM, Lindsley CW, Conn PJ. A selective allosteric potentiator of the M1 muscarinic acetylcholine receptor increases activity of medial prefrontal cortical neurons and restores impairments in reversal learning. J Neurosci. 2009;29:14271–14286. doi: 10.1523/JNEUROSCI.3930-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jodo E. The role of the hippocampo-prefrontal cortex system in phencyclidine-induced psychosis: a model for schizophrenia. J Physiol. 2013;107:434–440. doi: 10.1016/j.jphysparis.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 39.Martin HG, Lassalle O, Manzoni OJ. Differential Adulthood Onset mGlu5 Signaling Saves Prefrontal Function in the Fragile X Mouse. Cereb Cortex. 2016:1–11. doi: 10.1093/cercor/bhw328. [DOI] [PubMed] [Google Scholar]
- 40.Caruana DA, Warburton EC, Bashir ZI. Induction of activity-dependent LTD requires muscarinic receptor activation in medial prefrontal cortex. J Neurosci. 2011;31:18464–18478. doi: 10.1523/JNEUROSCI.4719-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kamsler A, McHugh TJ, Gerber D, Huang SY, Tonegawa S. Presynaptic m1 muscarinic receptors are necessary for mGluR long-term depression in the hippocampus. Proc Natl Acad Sci U S A. 2010;107:1618–1623. doi: 10.1073/pnas.0912540107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhu YY, Jing L, Duan TT, Yuan Q, Cao J, Zhou QX, Xu L. Patterned high-frequency stimulation induces a form of long-term depression dependent on GABAA and mACh receptors in the hippocampus. Neuroscience. 2013;250:658–663. doi: 10.1016/j.neuroscience.2013.07.059. [DOI] [PubMed] [Google Scholar]
- 43.Palmer MJ, Irving AJ, Seabrook GR, Jane DE, Collingridge GL. The group I mGlu receptor agonist DHPG induces a novel form of LTD in the CA1 region of the hippocampus. Neuropharmacology. 1997;36:1517–1532. doi: 10.1016/s0028-3908(97)00181-0. [DOI] [PubMed] [Google Scholar]
- 44.Rohde M, Tokay T, Kohling R, Kirschstein T. GABA(A) receptor inhibition does not affect mGluR-dependent LTD at hippocampal Schaffer collateral-CA1 synapses. Neurosci Lett. 2009;467:20–25. doi: 10.1016/j.neulet.2009.09.053. [DOI] [PubMed] [Google Scholar]
- 45.Collingridge GL, Peineau S, Howland JG, Wang YT. Long-term depression in the CNS. Nat Rev Neurosci. 2010;11:459–473. doi: 10.1038/nrn2867. [DOI] [PubMed] [Google Scholar]
- 46.Gladding CM, Collett VJ, Jia Z, Bashir ZI, Collingridge GL, Molnar E. Tyrosine dephosphorylation regulates AMPAR internalisation in mGluR-LTD. Mol Cell Neurosci. 2009;40:267–279. doi: 10.1016/j.mcn.2008.10.014. [DOI] [PubMed] [Google Scholar]
- 47.Martin HG, Bernabeu A, Lassalle O, Bouille C, Beurrier C, Pelissier-Alicot AL, Manzoni OJ. Endocannabinoids Mediate Muscarinic Acetylcholine Receptor-Dependent Long-Term Depression in the Adult Medial Prefrontal Cortex. Front Cell Neurosci. 2015;9:457. doi: 10.3389/fncel.2015.00457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Amitai N, Kuczenski R, Behrens MM, Markou A. Repeated phencyclidine administration alters glutamate release and decreases GABA markers in the prefrontal cortex of rats. Neuropharmacology. 2012;62:1422–1431. doi: 10.1016/j.neuropharm.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yu B, Wang C, Liu J, Johnson KM, Gallagher JP. Adaptation to chronic PCP results in hyperfunctional NMDA and hypofunctional GABA(A) synaptic receptors. Neuroscience. 2002;113:1–10. doi: 10.1016/s0306-4522(02)00163-x. [DOI] [PubMed] [Google Scholar]
- 50.Kaalund SS, Riise J, Broberg BV, Fabricius K, Karlsen AS, Secher T, Plath N, Pakkenberg B. Differential expression of parvalbumin in neonatal phencyclidine-treated rats and socially isolated rats. J Neurochem. 2013;124:548–557. doi: 10.1111/jnc.12061. [DOI] [PubMed] [Google Scholar]
- 51.Wang CZ, Yang SF, Xia Y, Johnson KM. Postnatal phencyclidine administration selectively reduces adult cortical parvalbumin-containing interneurons. Neuropsychopharmacology. 2008;33:2442–2455. doi: 10.1038/sj.npp.1301647. [DOI] [PubMed] [Google Scholar]
- 52.du Bois TM, Deng C, Han M, Newell KA, Huang XF. Excitatory and inhibitory neurotransmission is chronically altered following perinatal NMDA receptor blockade. Eur Neuropsychopharmacol. 2009;19:256–265. doi: 10.1016/j.euroneuro.2008.12.002. [DOI] [PubMed] [Google Scholar]
- 53.Kehrer C, Maziashvili N, Dugladze T, Gloveli T. Altered Excitatory-Inhibitory Balance in the NMDA-Hypofunction Model of Schizophrenia. Front Mol Neurosci. 2008;1:6. doi: 10.3389/neuro.02.006.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Inan M, Petros TJ, Anderson SA. Losing your inhibition: linking cortical GABAergic interneurons to schizophrenia. Neurobiol Dis. 2013;53:36–48. doi: 10.1016/j.nbd.2012.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Barnes SA, Pinto-Duarte A, Kappe A, Zembrzycki A, Metzler A, Mukamel EA, Lucero J, Wang X, Sejnowski TJ, Markou A, Behrens MM. Disruption of mGluR5 in parvalbumin-positive interneurons induces core features of neuro-developmental disorders. Mol Psychiatry. 2015;20:1161–1172. doi: 10.1038/mp.2015.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Blot K, Kimura S, Bai J, Kemp A, Manahan-Vaughan D, Giros B, Tzavara E, Otani S. Modulation of hippocampus-prefrontal cortex synaptic transmission and disruption of executive cognitive functions by MK-801. Cerebral cortex. 2015;25:1348–1361. doi: 10.1093/cercor/bht329. [DOI] [PubMed] [Google Scholar]
- 57.Jodo E, Suzuki Y, Katayama T, Hoshino KY, Takeuchi S, Niwa S, Kayama Y. Activation of medial prefrontal cortex by phencyclidine is mediated via a hippocampo-prefrontal pathway. Cerebral cortex. 2005;15:663–669. doi: 10.1093/cercor/bhh168. [DOI] [PubMed] [Google Scholar]
- 58.Suzuki Y, Jodo E, Takeuchi S, Niwa S, Kayama Y. Acute administration of phencyclidine induces tonic activation of medial prefrontal cortex neurons in freely moving rats. Neuroscience. 2002;114:769–779. doi: 10.1016/s0306-4522(02)00298-1. [DOI] [PubMed] [Google Scholar]
- 59.Godsil BP, Kiss JP, Spedding M, Jay TM. The hippocampal-prefrontal pathway: the weak link in psychiatric disorders? Eur Neuropsychopharmacol. 2013;23:1165–1181. doi: 10.1016/j.euroneuro.2012.10.018. [DOI] [PubMed] [Google Scholar]
- 60.Rodriguez AL, Grier MD, Jones CK, Herman EJ, Kane AS, Smith RL, Williams R, Zhou Y, Marlo JE, Days EL, Blatt TN, Jadhav S, Menon UN, Vinson PN, Rook JM, Stauffer SR, Niswender CM, Lindsley CW, Weaver CD, Conn PJ. Discovery of novel allosteric modulators of metabotropic glutamate receptor subtype 5 reveals chemical and functional diversity and in vivo activity in rat behavioral models of anxiolytic and antipsychotic activity. Molecular pharmacology. 2010;78:1105–1123. doi: 10.1124/mol.110.067207. [DOI] [PMC free article] [PubMed] [Google Scholar]