

Abstract
Transverse-axial tubules (TATs) are commonly assumed to be sparse or absent in atrial myocytes from small animals. Atrial myocytes from rats, cats and rabbits lack TATs, which results in a characteristic “V”-shaped Ca release pattern in confocal line-scan recordings due to the delayed rise of Ca in the center of the cell. To examine TAT expression in isolated mouse atrial myocytes, we loaded them with the membrane dye Di-4-ANEPPS to label TATs. We found that >80% of atrial myocytes had identifiable TATs. Atria from male mice had a higher TAT density than female mice, and TAT density correlated with cell width. Using the fluorescent Ca indicator Fluo-4-AM and confocal imaging, we found that wild type (WT) mouse atrial myocytes generate near-synchronous Ca transients, in contrast to the “V”-shaped pattern typically reported in other small animals such as rat. In atrial-specific Na–Ca exchanger (NCX) knockout (KO) mice, which develop sinus node dysfunction and atrial hypertrophy with dilation, we found a substantial loss of atrial TATs in isolated atrial myocytes. There was a greater loss of transverse tubules compared to axial tubules, resulting in a dominance of axial tubules. Consistent with the overall loss of TATs, NCX KO atrial myocytes displayed a “V”-shaped Ca transient with slower and reduced central (CT) Ca re-lease and uptake in comparison to subsarcolemmal (SS) Ca release. We compared chemically detubulated (DT) WT cells to KO, and found similar slowing of CT Ca release and uptake. However, SS Ca transients in the WT DT cells had faster uptake kinetics than KO cells, consistent with the presence of NCX and normal sarcolemmal Ca efflux in the WT DT cells. We conclude that the remodeling of NCX KO atrial myocytes is accompanied by a loss of TATs leading to abnormal Ca release and uptake that could impact atrial contractility and rhythm.
Keywords: Transverse-axial tubules, Atrial cardiomyocytes, Sodium-calcium exchange, Excitation-contraction coupling, Atrial remodeling
1. Introduction
Transverse-axial tubules (TATs) are invaginations of surface sarcolemma in cardiomyocytes. In ventricular myocytes, many ion channels appear to be concentrated in the membranes of TATs [1,2]. Upon depolarization, the tubular structures can rapidly conduct the action potential deep into the cell interior, allowing voltage gated Na and L-type Ca channels (LCCs) to open simultaneously throughout the cell. The small flux of Ca entering the dyadic cleft triggers sarcoplasmic reticulum (SR) Ca release by ryanodine receptors (RyRs) to produce the Ca transient (CaT) [3,4]. The released Ca then binds to myofilaments and causes contraction. Therefore, TATs play a pivotal role in excitation-contraction (EC) coupling by conducting electrical signals deep into the cell interior and bringing functionally related Ca handling proteins, such as Na channels, Na–Ca exchangers (NCX), LCCs and RyRs, into close proximity to form numerous Ca release units, known as couplons, throughout the cell. The widely distributed couplons ensure homogenously distributed Ca release to provide effective and coordinated contraction. Thus a well-organized TAT structure is essential for the proper contractile behavior of all mammalian ventricular cardiomyocytes [2].
Until recently, TATs in atrial cardiomyocytes were considered to be sparse or absent, particularly in small animals. The absence of TATs is thought to be responsible for the non-uniform Ca release pattern observed in atrial cells from rats [5–8], cats [9], rabbits [10,11] and guinea pigs [12]. Without the TAT network, couplons can only be formed at the periphery or subsarcolemmal (SS) space where the LCCs on the surface membrane are found in close proximity to RyRs on the SR membrane. RyRs in the central cellular (CT) space and distant from the sarcolemma are “uncoupled” from the LCCs. This results in a characteristic “V”-shaped CaT on transverse line-scan images due to the fast Ca-induced Ca release (CICR) initiated in couplons near the cell surface, followed by the slower spread of Ca into the cell center by a cascade of RyR-mediated Ca release triggered by Ca diffusion rather than apposing LCCs [5,9,13]. In rat atrial cells, there is evidence of rudimentary [6, 14], and in some cases, well-organized TATs [15], though most cells have few TATs and the CaTs are “V”-shaped. In large animals, previous studies [16–18] have shown that atrial myocytes possess extensive TATs. The existence of a well-developed TAT network makes the CaT more synchronous in sheep atrial cells, with CT and SS Ca release occur-ring simultaneously, similar to mammalian ventricular myocytes [7,19]. Several studies in large animals have shown that atrial TAT remodeling can occur under disease conditions (such as HF, AF) [16,20,21] and impair normal EC coupling, suggesting involvement in atrial pathologies associated with reduced contractility. Similar remodeling has been identified in the ventricle [22–24].
We have previously generated an atrial specific Na–Ca exchange (NCX) knockout (KO) mouse [25,26]. These mice have profound sinus node dysfunction and slow atrial conduction. Both right and left atrial sizes are increased, consistent with atrial remodeling. In the present study, we investigated TATs and CaTs in WT and NCX KO atrial myocytes. We found that WT atrial myocytes have surprisingly extensive TATs and synchronous CaTs, whereas KO atrial myocytes lose TATs and have “V”-shaped CaTs similar to rabbit atria (which normally lack TATs). In addition, the absence of NCX impaired SS Ca extrusion. We conclude that atrial remodeling in the NCX KO mouse results in loss of TATs, which, combined with the absence of NCX, causes abnormal Ca release and uptake.
2. Methods
2.1. Ethical approval
All procedures were approved by the Institutional Animal Care and Use Committee at Cedars-Sinai Medical Center (IACUC #: 003574) and strictly conformed to the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health.
2.2. Isolation of adult atrial cardiomyocytes
In the present study, we investigated both NCX1fx/fx mice (referred to as WT) and atrial-specific NCX1 knockout (KO) mice (on a C57BL/6 background), as we described previously [25,26]. We used an established protocol [27] to isolate mouse atrial cardiomyocytes with slight modifications (details are described below). Equal numbers of male (M) and female (F) mice (12–16 wks) were deeply anesthetized with isoflurane and then subjected to thoracotomy. Hearts were quickly excised and mounted on a Langendorff apparatus. We perfused the hearts with Ca-free Tyrode’s solution containing (in mmol/l): 136 NaCl, 5.4 KCl, 10 HEPES, 1.0 MgCl2, 0.33 NaH2PO4, 10 Glucose, pH adjusted to 7.4 with NaOH for 5 min, followed by Tyrode’s containing collagenase (collagenase type II, Worthington Biochemical, NJ, 280 U/ml) and protease (0.1 mg/ml protease type XIV) (Sigma-Aldrich, St. Louis, MO) for 10–12 min. We then washed out the enzyme solution with low-calcium Tyrode’s containing 0.2 mmol/l CaCl2 and 1 mg/ml BSA for 7 min. The above steps were performed at 37 °C. After digestion, the right and left atria were carefully separated from the ventricles, placed in a petri dish containing low-calcium Tyrode’s and cut into small pieces. Single cells were dissociated by gentle trituration with a wide-bore glass pipette. We gradually increased the CaCl2 concentration in the cell suspension from 0.2 mmol/l to 1.0 mmol/l over 20 min. Cells were then stored in Tyrode’s solution with 1 mmol/l CaCl2 at room temperature and were used within 4 h. Tyrode’s solution containing 1.0 mmol/l CaCl2 served as bath solution for all experiments. Healthy cells, i.e. cells with smooth edges and clear striations, without blebs or spontaneous contractions, were randomly selected for experiments.
2.3. Transverse-axial tubule (TAT) imaging in isolated atrial myocytes and in intact, live atrium
We loaded freshly isolated atrial cells with the membrane dye, Di-4-ANEPPS (5 μmol/l; Invitrogen) and Pluronic F-127 (0.02%; Invitrogen) for 5 min at room temperature (20–22 °C). We found 5 min of incubation sufficient for clear TAT visualization, thereby avoiding longer incubations that could cause dye internalization. We used the x-y mode of a Leica TCS-SP5-II confocal microscope (Leica Microsystems Inc.; Wetzlar, Germany) to image the membrane structure with a 63× water immersion objective lens (Numerical Aperture 1.2). For Di-4-ANEPPS we set the excitation wavelength at the 488 nm line of an Argon laser and emission at 560–675 nm. We imaged the central focal plane (1024 × 1024 pixels, 0.1 μm/pixel) for each cell.
To image TATs in atrial tissue, we quickly cut off both left and right atria and immersed the tissues in dye loading solution, which contained 10 μmol/l Di-4-ANEPPS and 0.02% Pluronic F-127, for 15 min in dark at room temperature (20–22 °C). We then placed the tissue on a coverslip-bottomed microscopy petri dish and recorded Di-4-ANEPPS images as described above for isolated cells. Images were obtained from >5 randomly selected epicardial areas.
2.4. Ca imaging in atrial myocytes
To record systolic CaTs from atrial myocytes, we incubated the cells with standard bath solution containing the fluorescent Ca indicator Fluo-4-AM (5 μmol/l; Invitrogen) and Pluronic F-127 (0.02%; Invitrogen) for 20 min, followed by washout with dye-free bath solution (two 10 min washes). The loading and washout times were sufficient for de-esterification of the dye. We then placed the cells in a coverslip-bottomed imaging chamber mounted on the microscope and perfused with standard bath solution. We used the line-scan (x-t) mode of the confocal system described above. Excitation was again at 488 nm and emission was detected at 500–650 nm for Fluo-4. The scan line was positioned transversely across the width of the cell. Cells were externally paced at 1 Hz with a field stimulator (Myopacer, IonOptix, MA; bipolar, 3 ms duration, 20 V) starting 20 s prior to imaging. Spatial resolution of the line-scan Ca images was 0.1–0.2 μm per pixel and the temporal resolution was 1 ms per line (scan speed: 1000 Hz). We carried out these experiments at 20–22 °C.
2.5. Detubulation of atrial myocytes
To separate the effects of the absence of NCX versus the loss of TATs in NCX KO mouse atrial myocytes, we used detubulated (DT) atrial myocytes as control and compared the local CaTs from either the SS region where the RyRs-LCC couplings remained intact or from the CT region where tubule structures were absent and RyRs uncoupled. We adopted a well-established method [28,29] to induce acute detubulation. Briefly, we incubated freshly isolated WT atrial myocytes with 1.5 mol/l formamide dissolved in standard bath solution for 15 min at 20–22 °C. Then we rapidly washed the cells with formamide-free bath solution for another 10 min to cause “osmotic shock”, resulting in detachment of tubule structures from surface sarcolemma. We loaded the DT atrial myocytes with Di-4-ANEPPS to confirm the detubulation and with Fluo-4-AM to investigate Ca dynamics. Images were recorded as described above.
2.6. Image analysis
We used two independent methods to estimate TAT abundance. We first quantified total cell area occupied by TATs (Tarea) by binarizing the image using an auto-thresholding algorithm (Otsu thresholding [30]) embedded in ImageJ [31]. Tarea was quantified by measuring the ratio of above-threshold pixels within the boundary of the cell to the whole cell area. We also analyzed TAT abundance by skeletonizing the image in Fiji [32–34], and then calculating the ratio of skeletonized tubule length to area of the ROIs (Tskel) and TAT orientation using the “directionality analysis” function (see an example of image processing in Supplementary Fig. 1). Details (including the custom-made macro) for the latter method are described in the Supplemental materials. Skeletonization and directionality analysis allow determination of tubule orientation by designating the 0° angle of tubule structure as axially positioned and the 90° angle as transversely positioned, as described previously [35,36]. The nucleus area was excluded from the image for TAT analysis. All cell size parameters were obtained using ImageJ.
For tissue staining images, we used 2-D Fast Fourier Transform (FFT) to determine TAT network regularities [24].
2.7. Reagents
All chemicals and reagents indicated in the text were purchased from Sigma-Aldrich (St. Louis, MO) or Fisher (Fair Lawn, NJ), unless otherwise specified.
2.8. Statistics
Statistical analysis was performed using Prism 5.0 (GraphPad Software, Inc.; La Jolla, CA) software and Clampfit 10.2 (Molecular Devices, LLC.; Sunnyvale, CA). Local CaTs were derived from 1.5 μm regions at the edge and center of the transverse line-scan images to represent SS Ca release and central (CT) Ca release, respectively. The decay constant (tau) was calculated from a mono-exponential function fitted to the decay phase of the CaT (from 90% of peak to diastolic level). The fluorescence intensity (F) reflecting intracellular Ca concentration was normalized to diastolic fluorescence (F0) after background subtraction. Data are expressed as mean ± standard error of the mean (SEM), unless otherwise indicated. One-way ANOVA with Bonferroni multiple-comparison and unpaired two-tail Student’s t-test were applied as appropriate. P < 0.05 was considered statistically significant. Cell and animal numbers are indicated in the figure legends.
3. Results
3.1. The heterogeneity of TATs in mouse atria
To characterize TAT expression in mouse atria, we labeled surface and TAT membranes with the fluorescent indicator, Di-4-ANEPPS, and imaged the cells using confocal microscopy. As described in the Methods (2.6. Image analysis), we used an auto-thresholding algorithm to quantify Tarea (Fig. 1). Cells with a Tarea above 2% were considered tubulated, as previously defined [15], whereas a typical ventricular myocyte has a Tarea of ≈8% [15]. By visual inspection, we found 84% of mouse atrial cells (162 out of 193 cells, N = 8 mice) contained identifiable TATs (Fig. 1B, C) with varying degrees of TAT organization. 23% of mouse atrial cells with visibly detectable TATs displayed well organized spacing of transverse tubules (≈1.8 μm, Fig. 1B), similar to the classical organization in ventricular myocytes (Fig. 1A). However, the rest of the atrial cells possessing TATs had a disorganized TAT pattern without regular spacing, as shown by the intensity plot in the bottom panel of Fig. 1C. These results indicate that while TATs commonly appear in mouse atrial cells, they are not usually as well organized as TATs in ventricular myocytes.
Fig. 1.
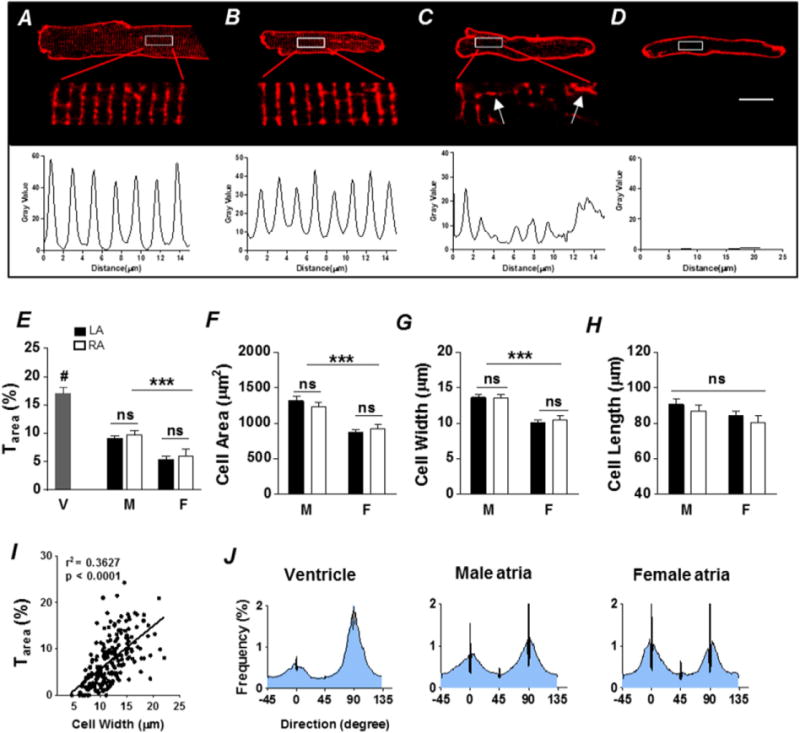
Heterogeneity of TATs in mouse atria. Di-4-ANEPPS membrane staining showing an organized TAT network in a representative ventricular myocyte (A) and three representative atrial myocytes with either well-organized TATs (B), disorganized TATs (C), or no TATs (D). Scale bar = 10 μm. Corresponding fluorescence intensity plots (shown below the images) of the enlarged TAT area show a spacing of ≈1.8 μm in ventricular myocytes (A) and organized atrial myocytes (B), while the pattern in a disorganized atrial myocyte (C) is less regular. Arrows in panel C indicate obvious axial tubules in atrial myocytes. E, summary plot of TAT density (Tarea,%) in left atria (LA, black) and right atria (RA, white) from male (M) and female (F). Ventricular (V) cells are included for comparison. ***P < 0.001, M vs. F; #P < 0.01, V vs. A, one-way ANOVA. Summary plots of cell area (F), width (G) and length (H) of left and right atrial myocytes from M vs. F. ***P < 0.001, one-way ANOVA. I, dependence of Tarea on cell width. n = 63 (M LA), 38 (M RA), 62 (F LA) and 30 (F RA) cells from 8 animals, and n = 12 cells from 3 animals for ventricle (V). Average frequency distributions of TAT orientation (J) in ventricular myocytes (left), male atrial myocytes (middle) and female atrial myocytes (right), indicate that atrial myocytes generally have a higher proportion of axial tubules (0° ± 20°) compared with ventricular myocytes. Nevertheless, male atrial cells possess prominent transverse (90° ± 20°) elements.
To investigate the distribution of TATs across atrial chambers and sexes, we compared the average TAT density (Tarea) among left and right atria from male and female mice, and found that Tarea was significantly higher in atrial cells from male (M) rather than female (F) mice (M vs. F, P < 0.05; Fig. 1E). There was no significant difference in Tarea between left and right atrium. We confirmed this finding using an image skeletonization approach to quantify Tskel (Supplementary Fig. 2). The average Tarea in male atria (A) was about half that observed in mouse ventricular cells (V) (A vs. V, P < 0.05; Fig. 1E). Male atrial cells were about 42% larger in area than female (P < 0.05, Fig. 1F) and about 33% wider than female (P < 0.05, Fig. 1G). Cell length was not significantly different among all groups (Fig. 1H). Since cell width has been implicated as a major parameter influencing TAT density in rat atrial cells [6,37], we examined the relationship between Tarea and cell width using linear regression. We found a positive correlation between cell width and Tarea (r2 = 0.36, slope = 0.94 ± 0.09, P < 0.001, Fig. 1I).
To investigate the orientation of TATs, we combined image skeletonization and directionality analysis as previously described [32, 33] (see example of image processing in Supplementary Fig. 1). Tubule fragments at 0° are parallel with the longitudinal axis of the cell, while tubules at 90° are perpendicular. We considered tubules oriented at 0° ± 20° angles as axial tubules (AT) and those oriented at 90° ± 20° angles as transverse tubules (TT), as defined previously [33]. We found that in ventricular cells, there were more transverse components than axial components. Atrial cells on average had a higher axial fraction than ventricular cells, although male atria had prominent transverse elements compared to female (Fig. 1J).
3.2. Mouse atrial myocytes generate near-synchronous Ca transients
To investigate the systolic Ca release pattern in mouse atria, we positioned the confocal scan line transversely across Fluo-4-AM loaded cells to simultaneously record peripheral and central Ca release. In almost all cases we observed near-synchronous CaTs, as evidenced by the smooth vertical appearance of the increase in fluorescence with each paced beat in the line scan as well as the superimposed global, SS and CT CaTs (see example, Fig. 2A, B). Overall, we found no difference in mean time to peak (TTP) of the global (28.3 ± 1.2 ms), SS (26.0 ± 1.2 ms) and CT (25.3 ± 1.2 ms) CaTs (n = 61 cells from N = 6 animals, P > 0.05 by one-way ANOVA with Bonferroni multiple-comparison; Fig. 2C). These results indicate that the RyRs in the periphery and the center released Ca almost simultaneously.
Fig. 2.
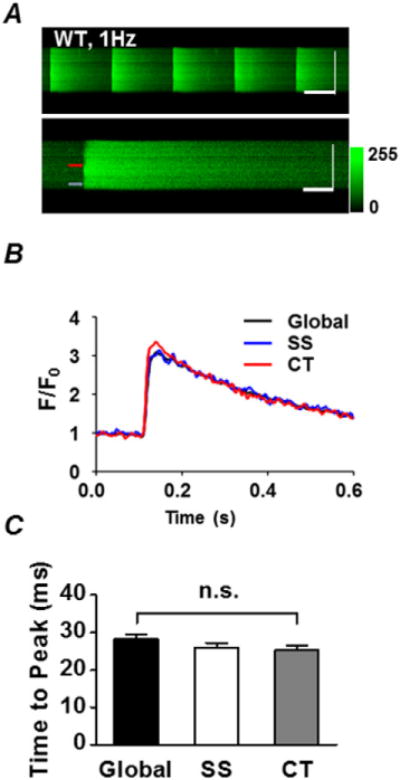
Synchronous Ca release in atrial myocytes. A, confocal line-scan images of five Ca transients (CaT) from a representative atrial myocyte loaded with Fluo-4-AM and field stimulated at 1 Hz (upper image), and a single CaT taken from the same cell but shown on an expanded time scale (lower image). Horizontal scale bars represent 500 ms (upper panel) and 100 ms (lower panel), while vertical scale bars represent 10 μm for both images. B, fluo-4 fluorescence intensity (F/F0) plots over time from the cell in A, at 1.5 μm from the subsarcolemmal space (SS, blue) or in the cell center (CT, red). Colour-coded arrows on the upper image correspond to the locations where the fluorescence was sampled for panel B. The global CaT is shown in black. Note the typical synchronous Ca release and uptake at both locations (with the global trace overlapping the SS and CT traces). C, summary plot of the time to peak (TTP) of the global, SS and CT CaT. P > 0.05, one way ANOVA with Bonferroni’s Multiple Comparison post-test, n = 61 cells from N = 6 animals. Note that there are no significant differences in TTP among the groups indicating simultaneous release.
3.3. Loss of TATs in Na–Ca exchanger (NCX) KO mice
We have shown previously that atrial-specific NCX KO mice develop sinus node dysfunction and atrial hypertrophy with dilation [25,26]. Since cardiac remodeling has been associated with loss of TATs, we tested the hypothesis that TAT density and orientation would be altered in remodeled atrial myocytes from KO mice. Atrial myocytes isolated from NCX KO mice were much larger than WT (WT vs. KO, P < 0.001; Fig. 3C– E), indicating hypertrophy at the cellular level. Despite the increased cell size, overall TAT density (Tarea) in KO was half that of WT (WT vs. KO, P < 0.001; Fig. 3B). KO cells were either completely depleted of TATs (Fig. 3A, a) or the remaining TATs were disordered (Fig. 3A, b). The frequency distribution of Tskel (the ratio of skeletonized tubule length to area) in KO cells was left shifted, indicating an increase in the fraction of non-tubulated cells (Fig. 3F, right panel). In addition, we found greater loss of transverse components (TT) in KO cells that were not completely depleted of TATs, leaving them with an axial tubule (AT) dominant TAT network (P < 0.05; Fig. 3G).
Fig. 3.
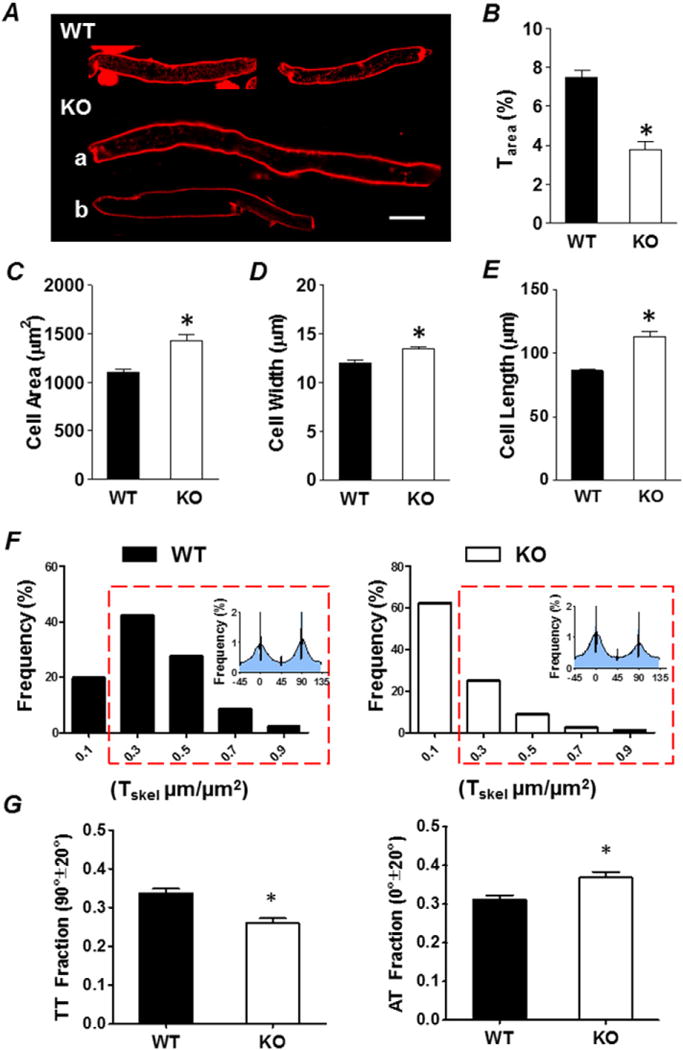
Depletion of TATs in atrial-specific NCX KO Mice. A, representative wild type (WT) and NCX knockout (KO) atrial myocytes with either decreased and deranged TATs (KO, a) or completely depleted TATs (KO, b). Scale bar = 20 μm. Summary plots of Tarea (B), cell area (C), cell width (D) and cell length (E), WT (black) vs. KO (white). *P < 0.05, unpaired Student’s t-test, n = 193 cells from 8 animals for WT, n = 156 cells from 6 animals for KO. F, frequency distributions of TAT abundance displayed as Tskel (μm/μm2) calculated from skeletonization/directionality analysis show WT (black) have more tubulated cells than KO (white). The red box indicates Tskel values with a density indicating tubulated cells. G, summary plots show the small fraction of KO cells that remain tubulated are remodeled and have decreased transverse components (TT) and increased axial components (AT). *P < 0.05, unpaired Student’s t-test, n = 153 cells from 8 animals for WT, n = 58 tubulated cells from 6 animals for KO. Tubulated cells defined as those with Tskel > 0.2 μm/μm2.
To confirm that the cell isolation process did not alter the tubule morphology, we labeled intact tissue with Di-4-ANEPPS for confocal imaging (Fig. 4). The preservation of TATs in WT tissue and the lack of TATs in KO was visually obvious. To quantify the presence of TATs, we applied the Fast Fourier Transform (FFT) function in ImageJ. When the TAT network is intact, the FFT image (lower right-hand corners of Fig. 4A and B) shows a prominent 2nd harmonic signal (indicated by the yellow arrows in Fig. 4A) representing the repetitive pattern of TATs. Consistent with our results in isolated atrial myocytes, we found a dramatic loss of TATs in KO atrial tissue compared to WT. In WT tissue, the majority of ROIs (>80%, Fig. 4C) have significant 2nd harmonic signals on their corresponding FFT images, whereas most ROIs (>90%, Fig. 4C) from KO mice lack 2nd harmonic signals (WT, n = 105 ROIs from 3 animals, vs. KO, n = 95 ROIs from 3 animals, P < 0.05; Fig. 4C).
Fig. 4.
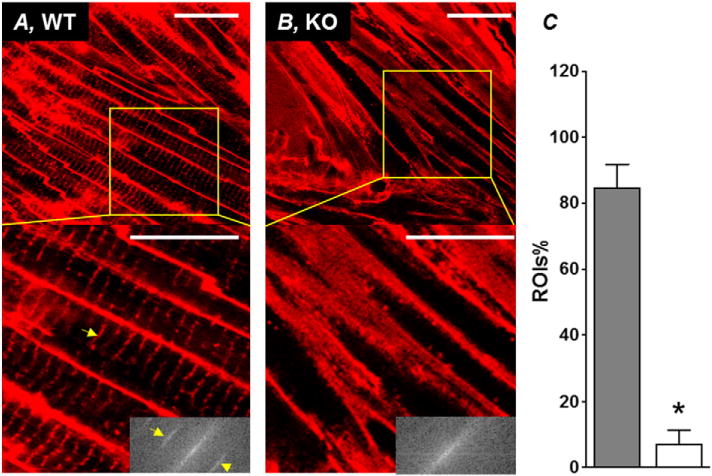
In situ confocal imaging of TATs in intact atrium from WT and KO mouse. Representative in situ confocal images of atrial myocytes from WT (A) and KO (B) mouse atria stained with the lipophilic membrane marker Di-4-ANEPPS. Scale bar = 25 μm. Insets at bottom right of each enlarged area from WT and KO show the 2-D Fast Fourier Transform (FFT) image of the region of interest bounded by the yellow box. Yellow arrow heads point to the 2nd harmonic signal indicative of a repetitive pattern (regular transverse-tubule structure) retrieved from the images. C, summary plot showing the percentage of 25 μm × 25 μm ROIs (% ROIs) with a 2nd harmonic signal. *P < 0.05, unpaired Student’s t-test, WT (n = 105 ROIs from 3 animals) vs. KO (n = 95 ROIs from 3 animals).
3.4. “V”-shaped Ca transients in NCX KO atrial myocytes
Next we investigated the hypothesis that loss of TATs in NCX KO atrial myocytes would alter the spread of the CaT. In confocal line-scan recordings, we found that KO cells frequently exhibited non-uniform “V”-shaped CaTs (Fig. 5A) during external pacing, consistent with the dramatic loss of TATs in KO cells. Similar loss of TATs has been observed by other groups in both diseased atrial [16,20] and ventricular [38] cardiomyocytes. SS CaTs displayed a more rapid time-to-peak and faster decay phase compared to the slower CT CaTs (Fig. 5A, right panel). There was no difference in the SR Ca content of WT vs. KO atrial myocytes, measured as the fluo-4 fluorescence peak upon rapid application of 5 mM caffeine (WT peak F/Fo: 3.6 ± 0.3, n = 10; KO peak F/Fo: 3.6 ± 0.8, n = 7; P > 0.05, unpaired Student t-test).
Fig. 5.
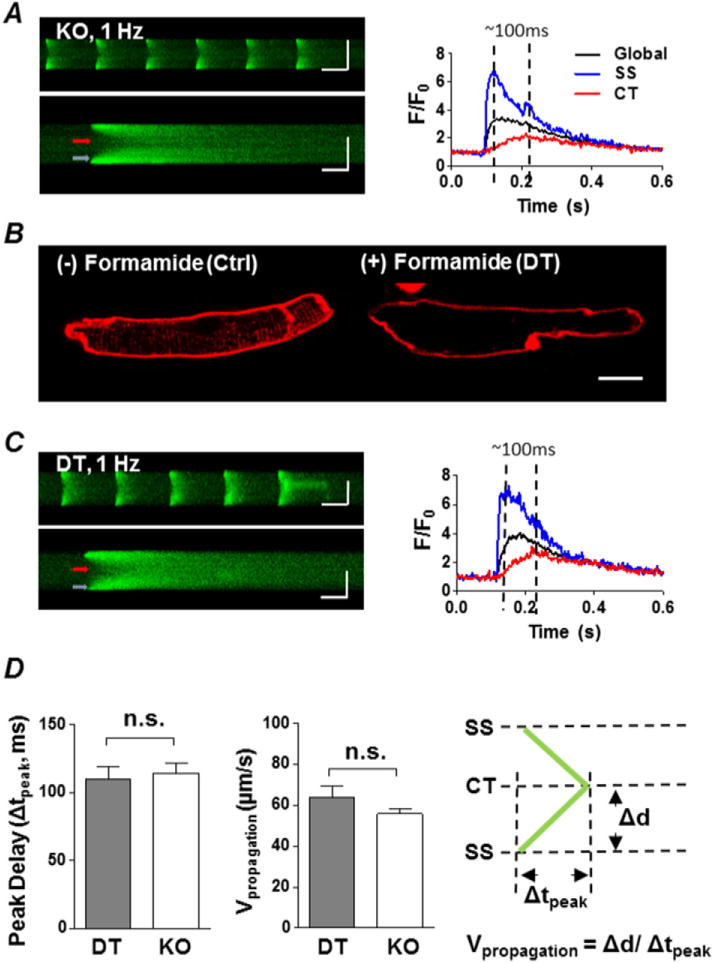
KO atrial myocytes have a similar centripetal propagation process as detubulated (DT) atrial myocytes. A, left, representative transverse confocal line-scan image of a series of electrically stimulated CaTs (upper panel) and a single transversal CaT recording (lower panel) of a KO atrial myocyte. Colour coded arrows indicate locations used to plot SS (blue) and CT (red) fluorescence intensity over time (right panel). Global CaT is shown in black. Horizontal scale bars represent 500 ms (upper panel) and 100 ms (lower panel), while vertical scale bars represent 10 μm for both images. B, Di-4-ANEPPS staining shows one normal atrial myocyte before formamide incubation (Ctrl, left) and one detubulated cell after formamide incubation (DT, right). Scale bar = 10 μm. C, similar to A but in a DT myocyte. D, summary plot of mean time difference (Δtpeak) between the peak of CT CaTs and the peak of SS CaTs during pacing, and the centripetal propagating velocity (Vpropagtion) in DT vs. KO. Vpropagtion was calculated as propagating distance divided by peak delay (right panel). P > 0.05, DT (n = 9 cells from 3 animals) vs. KO (n = 27 cells from 6 animals), unpaired Student’s t-test.
To separate the effects of TAT loss versus absence of NCX on the CaT, we compared NCX KO atrial myocytes with chemically detubulated WT atrial myocytes. We used formamide to induce detubulation [25,26] in WT atrial myocytes isolated from male mice. We chose male mice for these experiments since they have higher TAT density than females. Formamide effectively detubulated WT cells (Fig. 5B). We then recorded confocal line-scan images of CaTs during external pacing. We found that CaTs in the formamide-detubulated WT atrial myocytes (Fig. 5C) resembled the “V”-shaped transients recorded in KO myocytes (Fig. 5A), with initial release of Ca from the SS region, spreading to the center.
We next compared the propagation of the CaT from the SS to the CT in KO and DT cells. For this analysis, we deliberately selected KO cells that had no TATs, similar to DT cells that had no TATs, in order to focus on SS vs. CT Ca dynamics. We calculated the velocity of propagation (Vpropagation) by dividing the distance between the SS and CT of the cell (Δd) by the delay between the peak of the SS CaT and the peak of the CT CaT (Δtpeak, see dashed lines on the right side of Fig. 5A and C, both ~100 ms). There was no difference in the peak delay (Δtpeak) and the propagation velocity between DT and KO, suggesting that NCX has no effect on centripetal Vpropagation.
DT and KO each had similar SS and CT CaT rise-time kinetics and amplitudes. In both cell types, CT CaT amplitude (Fig. 6A) and TTP (Fig. 6B) were smaller and longer than SS CaT. However, the decay phase of the SS CaT in the KO was much slower than in DT, even though there was no difference in the kinetics of CT CaT re-uptake (Fig. 6C). This slowed SS uptake is consistent with reduced extrusion of SS Ca in the KO caused by the absence of NCX.
Fig. 6.
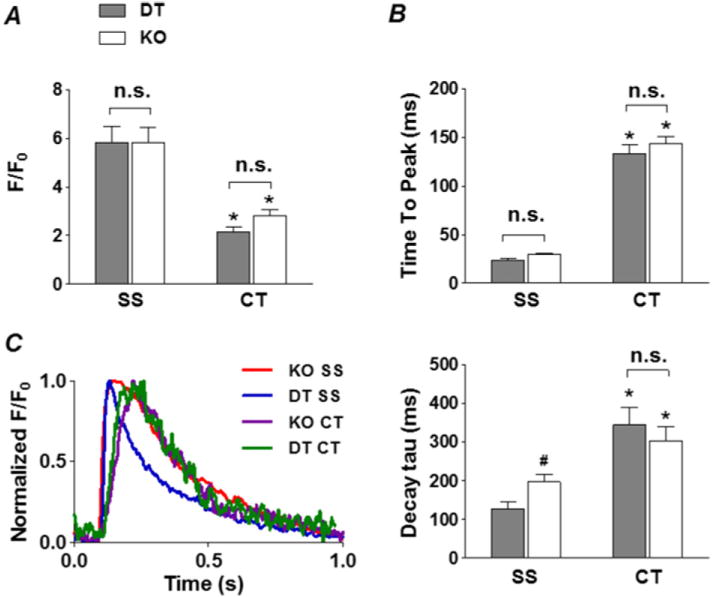
Effect of NCX KO on Ca uptake. Summary plots of the amplitude (F/F0, A), time to peak (B) and local CaTs from SS and CT regions in DT (grey) and KO (white) atrial myocytes. C, left, shows normalized, field stimulation-induced intracellular CaTs from representative DT and NCX KO myocytes at the SS regions (DT, blue; NCX KO, red), as well as the CT regions (DT, green; NCX KO, purple); right, summary plot of the local CaT decay tau from SS and CT regions in DT (grey) and KO (white) atrial myocytes. *P < 0.05, CT vs. SS, #P < 0.05, KO vs. DT, unpaired Student’s t-test, n = 9 cells from 3 animals for DT, n = 27 cells from 6 animals for KO. Note the slowed decay in the SS region of NCX KO compared to DT.
4. Discussion
Transverse-axial tubules (TATs) in cardiomyocytes serve as extensions of surface sarcolemma and help to facilitate EC coupling by transmitting membrane voltage changes deep into the cell interior to allow for synchronous depolarization and intracellular Ca release. Ventricular cardiomyocytes require a well-developed TAT structure to ensure homogeneous systolic Ca release so that ventricular contraction will be efficient for optimal pump function. In contrast, atria contribute less to cardiac output under basal conditions and atrial cells reportedly have fewer TATs than their ventricular counterparts. Numerous studies have been conducted in small laboratory animals like rabbits, cats and guinea pigs, and one of the commonly reported findings is that their atrial cells are practically devoid of TATs [10,12,13]. Unlike small species, larger animals, like sheep, cow, horse, pig and human, all have been found to exhibit varying but generally abundant TATs [16,17,20].
The relationship between TATs and the CaT is complex. Junctions between the SR and the sarcolemmal (SL) membranes allow LCCs and RyRs to come into close proximity for efficient EC coupling. In ventricular myocytes, this occurs along transverse-tubules which dive deep into the cell interior, allowing for synchronous Ca release. In contrast to ventricular myocytes, the CaTs in atrial myocytes are often reported to be “V”-shaped, where Ca release begins at the cell periphery (SS) and then spreads via CICR to the center (CT). Despite the common association of atrial myocytes with “V”-shaped Ca release, several studies have revealed extensive tubular networks and synchronous Ca release in the atria from large animals [16]. A recent study has shown that axial tubules (AT) are also present in mouse atria [33].
In the present study, we characterized TATs and CaTs in WT and NCX KO atrial myocytes. Not only are TATs present in the vast majority of WT cells, but also there is a significant sex difference in TAT occurrence, with more TATs present in male atria compared to female. Our results show that synchronous systolic Ca release occurs in most WT mouse atrial myocytes, which contrasts with other small mammals like cat, rat and guinea pigs. We compared our findings in WT with the atrial-specific NCX KO mouse. The KO mouse has sinus node dysfunction, slow atrial conduction and pronounced atrial remodeling featuring hypertrophy and dilation. Compared to WT, TATs appear to be rare in atrial-specific NCX KO mice, consistent with the structural remodeling observed in these cells. The rarity of TATs in the NCX KO corresponds with a lack of synchronous Ca release, similar to chemically detubulated cells, but the absence of NCX further reduces the rate of Ca efflux from the SS region (compared to DT).
4.1. The prevalence of TATs in mouse atria and its dependence on cell width and sex
Previous studies have investigated TATs in mouse atria. Using electron microscopy, Forbes et al. [39] found primitive TATs in mouse, though the physiological consequences were not investigated. More recently, Greiser et al. [10] and Brandenburg et al. [33] both identified complex TATs in mouse atria, but the heterogeneity across atria and sex was not characterized. We used laser scanning confocal microscopy to study TAT structure in live cells from mouse atria. Not only did we find that TATs are prevalent, which contrasts with other small animals such as cats, rats, rabbits and guinea pigs [6,8–12,40], but also there is a significant population of cells exhibiting a well-organized TAT net-work similar to ventricular cells (Fig. 1B).
TAT expression was more pronounced in wider atrial myocytes (Fig. 1I), consistent with prior studies in other species such as rat, sheep and pig [14–16,37]. Indeed, the male mouse atrial cells we examined had a cell width close to that reported in some large animals where TATs are prevalent [16]. Frisk et al. [15] reported regional differences of TAT density in pig atria, with right atrium having more TATs than the left atrium and epicardium having more TATs than the endocardium. Notably, the right atrial cells and epicardial cells were significantly wider than the left atrial cells and endocardial cells, respectively, consistent with the density of TATs correlating with cell width as we described in mouse (Fig. 1I). It has been suggested that the existence of TATs in wider cells might facilitate transmission of the action potential more efficiently to the cell interior [15], ensuring more synchronous Ca release [17]. It has also been proposed that heart rate might affect the density and complexity of t-tubules in ventricular cells [2]. For example, mouse ventricular myocytes have a more extensive t-tubule network and much higher basal heart rate than pig ventricular cells [19]. This heart rate dependence could also be the case for atrial myocytes.
Inter-chamber differences of TAT density and organization have been reported in other animals like rats and pigs [6,15]. However, we did not find significant differences between TATs in left and right atria in mouse. On the other hand, we identified a prominent sex difference in atrial cell TAT expression. Male mice exhibit significantly higher TAT density than female, possibly because their cells are wider. Nevertheless, we cannot exclude the possibility that sex hormones may directly or indirectly affect TAT expression. For example, it has been shown in rat skeletal muscle that testosterone can increase t-tubule units [41]. Furthermore, many studies have reported sex differences in Ca cycling and contractility in ventricular cardiomyocytes using both animal models and human specimens [42–44]. Since TAT complexity is correlated with EC coupling efficiency in cardiac cells [45], it is possible that differences in Ca cycling are partly caused by the variability in TAT development across sexes. Studies relating sex hormones to cardiac t-tubule development are lacking. Understanding the dependence of t-tubule development and density on sex may have clinical implications for understanding different manifestations of atrial diseases in men and women.
4.2. TATs facilitate synchronous Ca release in mouse atria
Using line-scan confocal imaging in Fluo-4-loaded myocytes, we found that CaTs are usually synchronous in mouse atrial myocytes, consistent with RyRs in close apposition to LCCs in both the periphery and center due to the presence of TATs. This contrasts with the “V”-shaped Ca release and spatial Ca gradient, where the peak Ca at CT is ~30 ms to ~80 ms delayed than the SS region [5,9,13], typically reported in other small animals such as rats [6] and cats [9].
Our observations of synchronized CaTs and abundant TAT network are consistent with the recent findings of Brandenburg et al. [33], who found that the axial tubules in mouse atrial myocytes were often coupled with hyper-phosphorylated RyR2 clusters resulting in faster Ca release at axial tubule locations residing in the cell center.
4.3. The remodeling of TATs in atrial-specific NCX KO mice
Whereas ventricular t-tubule remodeling and its impact on Ca dynamics has been extensively studied in various animal [22,38,46] and human models of heart failure [47,48], atrial TAT remodeling has not been studied in depth, perhaps because atrial myocytes are often assumed to have few if any TATs.
We found substantially reduced TAT density and altered tubule orientation in intact tissue and atrial myocytes isolated from atrial-specific NCX KO mice (Figs. 3 and 4). Consistent with the loss of TATs, CaTs from these cells were initiated at the cell periphery and then propagated to the center (Fig. 5), similar to what has been described in most rat myocytes [14]. Atrial-specific NCX KO mice exhibit dilated/ hypertrophied atria featuring sinus node exit block and slow atrial conduction [25,26]. Blood clots frequently occur in their atria suggesting reduced or absent atrial contraction [25]. We speculate that the lack of TATs and corresponding loss of synchronous Ca release contribute to these electrical and mechanical abnormalities. Although we believe we are the first to describe a loss of TATs in remodeled mouse atria, re-modeling of the atrial TAT system has been reported in several large animal models of atrial fibrillation and heart failure characterized by atrial dilation and hypertrophy, including sheep [16,20] and dog [21]. These changes have been linked to the loss of synchronous systolic Ca release, similar to what we found in the NCX KO atrial myocytes. In contrast, Brandenburg et al. [33] described an adaptive proliferation of axial tubules in mouse atria after 2 weeks of transaortic constriction (TAC) and consequently, an increase in AT/TT ratio. These proliferative ATs were shown to help maintain intracellular Ca signaling in the context of reduced RyR density and were considered to be a compensatory adaptation during TAC-induced hypertrophy. Despite dramatic loss of TATs in the majority of NCX KO cells, we too observed an increase in AT components and reduction in TT components (a greater AT/TT ratio) in cells that were not completely depleted of TATs (Fig. 3G). The discrepancies in these studies are probably due to different stages of disease or simply model dependent.
The cause of the TAT remodeling in the NCX KO is unknown. Several different proteins have been identified as important modulators of TAT development in ventricular cardiomyocytes. For example, BIN1 (bridging integrator 1) has been shown to play a pivotal role in t-tubule development and in trafficking Ca channels to the sarcolemmal membrane [49,50]. BIN1 levels in the plasma have been shown to be associated with heart function in patients [51] and the t-tubule network was disrupted in BIN1 knockout mice [52]. A small, stretch-sensitive molecule, telethonin, also known as Tcap, has also been identified as an important factor in t-tubule formation [53]. Reynolds et al. [54] found junctophilin-2 (JP-2) to be necessary for t-tubule maturation during mouse heart development and the defects of JP-2 can lead to t-tubule remodeling in ventricular myocytes [55]. A recent study suggested that NCX was causally linked to the spatial localization of JP-2 and organization of t-tubules in pressure-overloaded hearts [56], possibly due to abnormal JP-2 proteolysis through Ca-mediated calpain activation as previously described [57,58]. Whether the loss of TATs is a concomitant phenomenon of cellular remodeling or a direct consequence from the intracellular Ca elevation due to the absence of NCX in the KO cells remains to be established.
4.4. The impact of TATs depletion and the absence of NCX on the Ca transients in NCX KO atrial myocytes
Loss of t-tubules in ventricular myocytes can compromise the synchrony of SR Ca release and lead to a reduced amplitude of the global CaT [14,59,60]. Thus it is not surprising that a similar change in Ca release occurs in atrial myocytes after loss of t-tubules. We used formamide detubulation to examine the effect of TAT loss in WT atrial myocytes, and to compare with NCX KO myocytes. Removal of TATs by formamide was associated with a major decrease in the spatial uniformity of the CaT release phase. Detubulated cells generated “V”-shaped CaTs with the Ca rise initiated at the cell periphery and then propagating into the cell center with a peak delay (~100 ms) in the same range we found in NCX KO cells. The changes we observed in both KO and DT atrial myocytes were reminiscent of the effect of detubulation on ventricular myocytes [14,28]. Thus, we conclude that depletion of TATs in mouse atrial cells impairs the EC coupling process. In rat atrial myocytes, Brette et al. [28] and Smyrnias et al. [14] found a negligible change after form-amide detubulation, consistent with the paucity of TATs in control rat atrial myocytes.
In NCX KO myocytes, we found that loss of TATs corresponded to a slowing and lower amplitude of central Ca release, similar to WT cells that were detubulated by formamide (DT cells) (Fig. 5). The peak delay, propagation velocity (Vpropagation), amplitude and upstroke kinetics of CaTs from SS and CT were not different, indicating that propagation from SS to CT is not altered in KO (Fig. 5D). However, SS Ca decay rate was much slower in the NCX KO cells compared with DT cells (Fig. 6), which is consistent with lack of NCX and therefore reduced extrusion of Ca from the SS space. Increased NCX expression in an AF sheep model has been associated with abbreviated atrial SS Ca release, mirroring our findings in NCX KO [20]. We then examined the kinetics of propagation of Ca from the periphery to the center of the cell in the NCX KO and in DT cells (Fig. 5D). The average delay between SS and CT (~100 ms) and Vpropagation (~50 μm/s) was slower than what has been reported in other non-tubulated atrial cells like cat and rat [5,61] but similar to detubulated ventricular myocytes [60]. Likewise, Dibb et al. [16] examined TAT structure and Ca release properties in atrial myocytes from the heart failure sheep model. They found dramatic loss of t-tubules associated with occurrence of “V”-shaped CaTs on line-scan images. Propagation parameters were similar to what we found in NCX KO and DT, with a peak delay from SS to CT of ~150 ms. Collectively, these data show that in cells where TATs are normally abundant, their loss can play an important role in pathological changes that impair normal EC coupling. On the other hand, the absence of NCX mostly affects the Ca extrusion at junctional sites, which is usually the SS region in the context of TAT depletion.
5. Implications
The presence of heterogeneous TATs in atrial myocytes has significant implications for understanding the regulation of contractility and arrhythmogenesis in atria. Disruptions of tubule structure may contribute to the formation of pro-arrhythmic substrates and contractile insufficiency during disease. The observation of a disrupted TAT network in the NCX KO mouse suggests that depressed Ca efflux promotes TAT re-modeling through a Ca-sensitive process.
6. Conclusion
We conclude that mouse atrial myocytes commonly express TATs and, unlike other small animals including rat, Ca release often occurs rapidly and simultaneously in the periphery and the center of the cells. TATs are more extensive in male mice, and in wider atrial myocytes. Finally, dramatic loss of TATs and dyssynchronous Ca release in NCX KO mouse atrial myocytes is a concomitant pathological event with the atrial hypertrophy/dilation we have described previously [25, 26], which compromises the synchrony of Ca dynamics and contractility.
Supplementary Material
Acknowledgments
Sources of funding
This work was supported by the Chinese Scholarship Council (X.Y.) and the National Institutes of Health R01HL048509 (J.I.G., K.D.P), and by the Dorothy and E. Philip Lyon Chair in Laser Research from the Cedars-Sinai Research Institute (J.I.G.).
Abbreviations
- AM
atrial myocytes
- BIN1
bridging integrator 1
- CaT
Ca transient
- CT
central cellular
- CICR
Ca-induced Ca release
- EC
excitation-contraction
- FFT
Fast Fourier Transform
- jSR
junctional SR
- JHP2
junctophilin-2
- KO
knockout
- LA
left atrium
- LCC
L-type calcium channel
- NCX
Na-Ca exchanger
- RA
right atrium
- ROI
region of interest
- RyR
ryanodine receptor
- SR
sarcoplasmic reticulum
- SS
sub-sarcolemmal
- TAT
transverse-axial tubule
- TTP
time to peak
- VM
ventricular myocytes
- WT
wild type
Appendix A. Supplementary data
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.yjmcc.2017.05.008.
Footnotes
Disclosures
None.
References
- 1.Song LS, Guatimosim S, Gomez-Viquez L, Sobie EA, Ziman A, Hartmann H, et al. Calcium biology of the transverse tubules in heart. Ann N Y Acad Sci. 2005;1047:99–111. doi: 10.1196/annals.1341.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brette F, Orchard C. T-tubule function in mammalian cardiac myocytes. Circ Res. 2003;92:1182–1192. doi: 10.1161/01.RES.0000074908.17214.FD. [DOI] [PubMed] [Google Scholar]
- 3.Fabiato A. Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum. Am J Phys. 1983;245:C1–14. doi: 10.1152/ajpcell.1983.245.1.C1. [DOI] [PubMed] [Google Scholar]
- 4.London B, Krueger JW. Contraction in voltage-clamped, internally perfused single heart cells. J Gen Physiol. 1986;88:475–505. doi: 10.1085/jgp.88.4.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Woo SH, Cleemann L, Morad M. Ca2+ current-gated focal and local Ca2+ release in rat atrial myocytes: evidence from rapid 2-D confocal imaging. J Physiol. 2002;543:439–453. doi: 10.1113/jphysiol.2002.024190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kirk MM, Izu LT, Chen-Izu Y, McCulle SL, Wier WG, Balke CW, et al. Role of the transverse-axial tubule system in generating calcium sparks and calcium transients in rat atrial myocytes. J Physiol. 2003;547:441–451. doi: 10.1113/jphysiol.2002.034355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Walden AP, Dibb KM, Trafford AW. Differences in intracellular calcium homeo-stasis between atrial and ventricular myocytes. J Mol Cell Cardiol. 2009;46:463–473. doi: 10.1016/j.yjmcc.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 8.Mackenzie L, Bootman MD, Berridge MJ, Lipp P. Predetermined recruitment of calcium release sites underlies excitation-contraction coupling in rat atrial myocytes. J Physiol. 2001;530:417–429. doi: 10.1111/j.1469-7793.2001.0417k.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huser J, Lipsius SL, Blatter LA. Calcium gradients during excitation-contraction coupling in cat atrial myocytes. J Physiol. 1996;494(Pt 3):641–651. doi: 10.1113/jphysiol.1996.sp021521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Greiser M, Kerfant BG, Williams GS, Voigt N, Harks E, Dibb KM, et al. Tachycardia-induced silencing of subcellular Ca2+ signaling in atrial myocytes. J Clin Invest. 2014;124:4759–4772. doi: 10.1172/JCI70102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hohendanner F, Walther S, Maxwell JT, Kettlewell S, Awad S, Smith GL, et al. Ino-sitol-1,4,5-trisphosphate induced Ca2+ release and excitation-contraction coupling in atrial myocytes from normal and failing hearts. J Physiol. 2015;593:1459–1477. doi: 10.1113/jphysiol.2014.283226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Berlin JR. Spatiotemporal changes of Ca2+ during electrically evoked contractions in atrial and ventricular cells. Am J Phys. 1995;269:H1165–H1170. doi: 10.1152/ajpheart.1995.269.3.H1165. [DOI] [PubMed] [Google Scholar]
- 13.Kockskamper J, Sheehan KA, Bare DJ, Lipsius SL, Mignery GA, Blatter LA. Activation and propagation of Ca(2+) release during excitation-contraction coupling in atrial myocytes. Biophys J. 2001;81:2590–2605. doi: 10.1016/S0006-3495(01)75903-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smyrnias I, Mair W, Harzheim D, Walker SA, Roderick HL, Bootman MD. Comparison of the T-tubule system in adult rat ventricular and atrial myocytes, and its role in excitation-contraction coupling and inotropic stimulation. Cell Calcium. 2010;47:210–223. doi: 10.1016/j.ceca.2009.10.001. [DOI] [PubMed] [Google Scholar]
- 15.Frisk M, Koivumaki JT, Norseng PA, Maleckar MM, Sejersted OM, Louch WE. Variable t-tubule organization and Ca2+ homeostasis across the atria. Am J Physiol Heart Circ Physiol. 2014;307:H609–H620. doi: 10.1152/ajpheart.00295.2014. [DOI] [PubMed] [Google Scholar]
- 16.Dibb KM, Clarke JD, Horn MA, Richards MA, Graham HK, Eisner DA, et al. Characterization of an extensive transverse tubular network in sheep atrial myocytes and its depletion in heart failure. Circ Heart Fail. 2009;2:482–489. doi: 10.1161/CIRCHEARTFAILURE.109.852228. [DOI] [PubMed] [Google Scholar]
- 17.Richards MA, Clarke JD, Saravanan P, Voigt N, Dobrev D, Eisner DA, et al. Trans-verse tubules are a common feature in large mammalian atrial myocytes including human. Am J Physiol Heart Circ Physiol. 2011;301:H1996–H2005. doi: 10.1152/ajpheart.00284.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arora R, Aistrup GL, Supple S, Frank C, Singh J, Tai S, et al. Regional distribution of T-tubule density in left and right atria in dogs. Heart Rhythm. 2016;14:273–281. doi: 10.1016/j.hrthm.2016.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heinzel FR, Bito V, Volders PG, Antoons G, Mubagwa K, Sipido KR. Spatial and temporal inhomogeneities during Ca2+ release from the sarcoplasmic reticulum in pig ventricular myocytes. Circ Res. 2002;91:1023–1030. doi: 10.1161/01.res.0000045940.67060.dd. [DOI] [PubMed] [Google Scholar]
- 20.Lenaerts I, Bito V, Heinzel FR, Driesen RB, Holemans P, D’Hooge J, et al. Ultra-structural and functional remodeling of the coupling between Ca2+ influx and sar-coplasmic reticulum Ca2+ release in right atrial myocytes from experimental persistent atrial fibrillation. Circ Res. 2009;105:876–885. doi: 10.1161/CIRCRESAHA.109.206276. [DOI] [PubMed] [Google Scholar]
- 21.Wakili R, Yeh YH, Yan Qi X, Greiser M, Chartier D, Nishida K, et al. Multiple po-tential molecular contributors to atrial hypocontractility caused by atrial tachycardia remodeling in dogs. Circ Arrhythm Electrophysiol. 2010;3:530–541. doi: 10.1161/CIRCEP.109.933036. [DOI] [PubMed] [Google Scholar]
- 22.He J, Conklin MW, Foell JD, Wolff MR, Haworth RA, Coronado R, et al. Reduction in density of transverse tubules and L-type Ca(2+) channels in canine tachycardia-induced heart failure. Cardiovasc Res. 2001;49:298–307. doi: 10.1016/s0008-6363(00)00256-x. [DOI] [PubMed] [Google Scholar]
- 23.Hong TT, Smyth JW, Chu KY, Vogan JM, Fong TS, Jensen BC, et al. BIN1 is reduced and Cav 1.2 trafficking is impaired in human failing cardiomyocytes. Heart Rhythm. 2012;9:812–820. doi: 10.1016/j.hrthm.2011.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wei S, Guo A, Chen B, Kutschke W, Xie YP, Zimmerman K, et al. T-tubule remodeling during transition from hypertrophy to heart failure. Circ Res. 2010;107:520–531. doi: 10.1161/CIRCRESAHA.109.212324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Groenke S, Larson ED, Alber S, Zhang R, Lamp ST, Ren X, et al. Complete atrial-specific knockout of sodium-calcium exchange eliminates sinoatrial node pacemaker activity. PLoS One. 2013;8:e81633. doi: 10.1371/journal.pone.0081633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Torrente AG, Zhang R, Zaini A, Giani JF, Kang J, Lamp ST, et al. Burst pacemaker activity of the sinoatrial node in sodium-calcium exchanger knockout mice. Proc Natl Acad Sci U S A. 2015;112:9769–9774. doi: 10.1073/pnas.1505670112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Neco P, Rose B, Huynh N, Zhang R, Bridge JH, Philipson KD, et al. Sodium-calcium exchange is essential for effective triggering of calcium release in mouse heart. Biophys J. 2010;99:755–764. doi: 10.1016/j.bpj.2010.04.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brette F, Komukai K, Orchard CH. Validation of formamide as a detubulation agent in isolated rat cardiac cells. Am J Physiol Heart Circ Physiol. 2002;283:H1720–H1728. doi: 10.1152/ajpheart.00347.2002. [DOI] [PubMed] [Google Scholar]
- 29.Kawai M, Hussain M, Orchard CH. Excitation-contraction coupling in rat ventricular myocytes after formamide-induced detubulation. Am J Phys. 1999;277:H603–H609. doi: 10.1152/ajpheart.1999.277.2.H603. [DOI] [PubMed] [Google Scholar]
- 30.Otsu N. A threshold selection method from gray-level histograms. IEEE Trans Sys Man Cyber. 1979;9:62–66. [Google Scholar]
- 31.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to Image J: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wagner E, Brandenburg S, Kohl T, Lehnart SE. Analysis of tubular membrane net-works in cardiac myocytes from atria and ventricles. J Vis Exp. 2014:e51823. doi: 10.3791/51823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brandenburg S, Kohl T, Williams GS, Gusev K, Wagner E, Rog-Zielinska EA, et al. Axial tubule junctions control rapid calcium signaling in atria. J Clin Invest. 2016;126:3999–4015. doi: 10.1172/JCI88241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wagner E, Lauterbach MA, Kohl T, Westphal V, Williams GS, Steinbrecher JH, et al. Stimulated emission depletion live-cell super-resolution imaging shows pro-liferative remodeling of T-tubule membrane structures after myocardial infarction. Circ Res. 2012;111:402–414. doi: 10.1161/CIRCRESAHA.112.274530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Crossman DJ, Young AA, Ruygrok PN, Nason GP, Baddelely D, Soeller C, et al. T-tubule disease: relationship between t-tubule organization and regional contractile performance in human dilated cardiomyopathy. J Mol Cell Cardiol. 2015;84:170–178. doi: 10.1016/j.yjmcc.2015.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Glukhov AV, Balycheva M, Sanchez-Alonso JL, Ilkan Z, Alvarez-Laviada A, Bhogal N, et al. Direct evidence for microdomain-specific localization and remodeling of functional L-type calcium channels in rat and human atrial myocytes. Circulation. 2015;132:2372–2384. doi: 10.1161/CIRCULATIONAHA.115.018131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Louch WE, Mork HK, Sexton J, Stromme TA, Laake P, Sjaastad I, et al. T-tubule disorganization and reduced synchrony of Ca2+ release in murine cardiomyocytes following myocardial infarction. J Physiol. 2006;574:519–533. doi: 10.1113/jphysiol.2006.107227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Forbes MS, Hawkey LA, Sperelakis N. The transverse-axial tubular system (TATS) of mouse myocardium: its morphology in the developing and adult animal. Am J Anat. 1984;170:143–162. doi: 10.1002/aja.1001700203. [DOI] [PubMed] [Google Scholar]
- 40.Woo SH, Cleemann L, Morad M. Diversity of atrial local Ca2+ signalling: evidence from 2-D confocal imaging in Ca2+−buffered rat atrial myocytes. J Physiol. 2005;567:905–921. doi: 10.1113/jphysiol.2005.092270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ustunel I, Akkoyunlu G, Demir R. The effect of testosterone on gastrocnemius muscle fibres in growing and adult male and female rats: a histochemical, morphometric and ultrastructural study. Anat Histol Embryol. 2003;32:70–79. doi: 10.1046/j.1439-0264.2003.00441.x. [DOI] [PubMed] [Google Scholar]
- 42.Curl CL, Wendt IR, Kotsanas G. Effects of gender on intracellular [Ca2+] in rat cardiac myocytes. Pflugers Arch. 2000;441:709–716. doi: 10.1007/s004240000473. [DOI] [PubMed] [Google Scholar]
- 43.Fischer TH, Herting J, Eiringhaus J, Pabel S, Hartmann NH, Ellenberger D, et al. Sex-dependent alterations of Ca2+ cycling in human cardiac hypertrophy and heart failure. Europace. 2015 doi: 10.1093/europace/euv313. [DOI] [PubMed] [Google Scholar]
- 44.Parks RJ, Howlett SE. Sex differences in mechanisms of cardiac excitation-contraction coupling. Pflugers Arch. 2013;465:747–763. doi: 10.1007/s00424-013-1233-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Torres NS, Sachse FB, Izu LT, Goldhaber JI, Spitzer KW, Bridge JH. A modified local control model for Ca2+ transients in cardiomyocytes: junctional flux is ac-companied by release from adjacent non-junctional RyRs. J Mol Cell Cardiol. 2014;68:1–11. doi: 10.1016/j.yjmcc.2013.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Song LS, Sobie EA, McCulle S, Lederer WJ, Balke CW, Cheng H. Orphaned ryanodine receptors in the failing heart. Proc Natl Acad Sci U S A. 2006;103:4305–4310. doi: 10.1073/pnas.0509324103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Louch WE, Bito V, Heinzel FR, Macianskiene R, Vanhaecke J, Flameng W, et al. Reduced synchrony of Ca2+ release with loss of T-tubules-a comparison to Ca2+ release in human failing cardiomyocytes. Cardiovasc Res. 2004;62:63–73. doi: 10.1016/j.cardiores.2003.12.031. [DOI] [PubMed] [Google Scholar]
- 48.Zhang HB, Li RC, Xu M, Xu SM, Lai YS, Wu HD, et al. Ultrastructural uncoupling between T-tubules and sarcoplasmic reticulum in human heart failure. Cardiovasc Res. 2013;98:269–276. doi: 10.1093/cvr/cvt030. [DOI] [PubMed] [Google Scholar]
- 49.Hong TT, Smyth JW, Gao D, Chu KY, Vogan JM, Fong TS, et al. BIN1 localizes the L-type calcium channel to cardiac T-tubules. PLoS Biol. 2010;8:e1000312. doi: 10.1371/journal.pbio.1000312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee E, Marcucci M, Daniell L, Pypaert M, Weisz OA, Ochoa GC, et al. Amphiphysin 2 (Bin1) and T-tubule biogenesis in muscle. Science. 2002;297:1193–1196. doi: 10.1126/science.1071362. [DOI] [PubMed] [Google Scholar]
- 51.Hong TT, Cogswell R, James CA, Kang G, Pullinger CR, Malloy MJ, et al. Plasma BIN1 correlates with heart failure and predicts arrhythmia in patients with arrhythmogenic right ventricular cardiomyopathy. Heart Rhythm. 2012;9:961–967. doi: 10.1016/j.hrthm.2012.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hong T, Yang H, Zhang SS, Cho HC, Kalashnikova M, Sun B, et al. Cardiac BIN1 folds T-tubule membrane, controlling ion flux and limiting arrhythmia. Nat Med. 2014;20:624–632. doi: 10.1038/nm.3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ibrahim M, Siedlecka U, Buyandelger B, Harada M, Rao C, Moshkov A, et al. A critical role for Telethonin in regulating t-tubule structure and function in the mammalian heart. Hum Mol Genet. 2013;22:372–383. doi: 10.1093/hmg/dds434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Reynolds JO, Chiang DY, Wang W, Beavers DL, Dixit SS, Skapura DG, et al. Junctophilin-2 is necessary for T-tubule maturation during mouse heart development. Cardiovasc Res. 2013;100:44–53. doi: 10.1093/cvr/cvt133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang C, Chen B, Guo A, Zhu Y, Miller JD, Gao S, et al. Microtubule-mediated defects in junctophilin-2 trafficking contribute to myocyte transverse-tubule remodeling and Ca2+ handling dysfunction in heart failure. Circulation. 2014;129:1742–1750. doi: 10.1161/CIRCULATIONAHA.113.008452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ujihara Y, Mohri S, Katanosaka Y. Effects of induced Na+/Ca2+ exchanger over-expression on the spatial distribution of L-type Ca2+ channels and junctophilin-2 in pressure-overloaded hearts. Biochem Biophys Res Commun. 2016;480:564–569. doi: 10.1016/j.bbrc.2016.10.090. [DOI] [PubMed] [Google Scholar]
- 57.Wu CY, Chen B, Jiang YP, Jia Z, Martin DW, Liu S, et al. Calpain-dependent cleav-age of junctophilin-2 and T-tubule remodeling in a mouse model of reversible heart failure. J Am Heart Assoc. 2014;3:e000527. doi: 10.1161/JAHA.113.000527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Guo A, Hall D, Zhang C, Peng T, Miller JD, Kutschke W, et al. Molecular determinants of calpain-dependent cleavage of Junctophilin-2 protein in cardiomyocytes. J Biol Chem. 2015;290:17946–17955. doi: 10.1074/jbc.M115.652396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brette F, Salle L, Orchard CH. Differential modulation of L-type Ca2+ current by SR Ca2+ release at the T-tubules and surface membrane of rat ventricular myocytes. Circ Res. 2004;95:e1–e7. doi: 10.1161/01.RES.0000135547.53927.F6. [DOI] [PubMed] [Google Scholar]
- 60.Ferrantini C, Coppini R, Sacconi L, Tosi B, Zhang ML, Wang GL, et al. Impact of detubulation on force and kinetics of cardiac muscle contraction. J Gen Physiol. 2014;143:783–797. doi: 10.1085/jgp.201311125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Blatter LA, Kockskamper J, Sheehan KA, Zima AV, Huser J, Lipsius SL. Local calcium gradients during excitation-contraction coupling and alternans in atrial myocytes. J Physiol. 2003;546:19–31. doi: 10.1113/jphysiol.2002.025239. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.