

ABSTRACT
TG4010, a Modified Vaccinia virus Ankara (MVA) expressing human mucin1 (MUC1) has demonstrated clinical benefit for patients suffering from advanced non-small cell lung cancer (NSCLC) in combination with chemotherapy. To support its development, preclinical experiments were performed with either TG4010 or β-galactosidase-encoding MVA vector (MVA-βgal) in mice presenting tumors in the lung. Tumor growth was obtained after intravenous injection of CT26 murine colon cancer cells, engineered to express either MUC1 or βgal. Mice showed increased survival rates after repeated intravenous injections of TG4010 or MVA-βgal, compared to an empty MVA control vector. Treatment with MVA vectors led to the accumulation of CD3dimCD8dim T cells, with two subpopulations characterized as KLRG1+CD127− short-lived effector cells (SLECs), and KLRG1−CD127− early effector cells (EECs) comprising cells releasing IFNγ, Granzyme B and CD107a upon antigen-specific peptide stimulation. EECs were characterized by an up-regulation of PD-1. Tumor growth in the diseased lung correlated with the appearance of PD1+ Treg cells that partially disappeared after TG4010 treatment. At late stage of tumor development in the lung, PD-L1 was detected on CD45− tumor cells, on CD4+ cells, including Treg cells, on CD3+CD8+ and CD3dimCD8dim T lymphocytes, on NK cells, on MDSCs and on alveolar macrophages. We demonstrated that targeting the PD-1/PD-L1 pathway with blocking monoclonal antibodies several days after TG4010 treatment, at late stage of tumor development, enhanced the therapeutic protection induced by the vaccine, supporting the ongoing clinical evaluation of TG4010 immunotherapy in combination with Nivolumab.
KEYWORDS: immune checkpoint blockade, lung cancer, Modified Vaccinia virus Ankara, PD-1, PD-L1, TG4010
Introduction
Lung cancer is one of the most common malignancies worldwide, with an annual incidence of 1.61 million, and the leading cause of cancer-related deaths with 1.38 million deaths in 2008.1 Newly developed immunotherapies are challenging the current treatment paradigms. Among these, TG4010, a recombinant Modified Vaccinia virus Ankara (MVA) encoding the human mucin 1 (MUC1) tumor-associated antigen, and interleukin 2, designed to target MUC1-positive cancers. TG4010, in combination with first-line standard of care chemotherapy, improved progression-free survival in patients with advanced metastatic non-small-cell lung cancer (NSCLC) compared to chemotherapy-only treated NSCLC patients in two different randomized and controlled phase 2b clinical trials.2,3 MVA-based immunotherapy depends on the induction of a specific cellular cytotoxic response to the target antigen. In preclinical experiments, we already showed that TG4010 treatment efficacy was dependent on MVA-driven MUC1 expression, and the presence of tumor-infiltrating CD8+ T cells. This effect could be enhanced by Toll- or Rig-like receptor ligands, demonstrating the key role of the tumor microenvironment.4,5,6
Antibodies targeting the programmed cell death-1 receptor (PD-1) and its ligands (PD-L1, PD-L2) have been assessed in first and second line treatment of NSCLC. These immune checkpoint inhibitors (ICIs) have produced a rapid and durable response, particularly in patients with tumors expressing PD-L1.7,8 PD-1 is expressed on activated T cells and acts as a negative regulatory signal to limit peripheral T cell activity, und thus avoid tissue damage. Upon engagement by one of its natural ligands (PD-L1 or PD-L2), PD-1 inhibits kinases involved in T cell activation.9 Coincidentally, this pathway serves as an immune escape mechanism for tumor cells. PD-L1 expressed on tumor cells binds to PD-1-expressing T cells and leads to decreased production of effector cytokines, lower cytolytic activity, and ultimately failure to eliminate cancer cells.10-12 Since the accumulation of mutations in cancer cells leads to the expression of cancer-specific non-self-antigens,13 blockade of this pathway results in the activation of tumor-specific T cells, leading to significant anti-tumor activity.14 Despite significant improvement of clinical outcome in a fraction of the patient population, a large proportion of subjects are yet non-responders to ICIs. Possible explanations for this lack of activity are the absence of anti-tumor T cells in the so-called “immune-desert” tumors or the tumor-induced inhibition of T-cell priming and activation.15 In preclinical studies, it appeared that systemic treatment with MVA vectors augments CD8+ T cell proportions in mouse tumors and organs.4
Considering these observations, combination of TG4010 and PD1-PD-L1-targeted ICIs has been considered as a promising strategy to augment tumor-specific CD8+ T cell proportions and, activate their cytotoxic action.16 A clinical study is underway to evaluate TG4010 in combination with Nivolumab, an anti-PD-1 monoclonal antibody (NCT02823990) in patients with advanced NSCLC. The goal of the current preclinical study was to validate such combination regimens in mice presenting tumors in the lung. We could observe that systemic injection of MVA vectors led to the appearance of a defined CD3dimCD8dim T cell populations in the lung, comprising short-lived and early effector cells as defined in Plumlee et al.,17 among which antigen-specific T cells were detected. Repeated injection of MVA vaccines led to the upregulation of PD-1 on EECs. Extensive phenotyping at late tumor stage after repeated MVA vector injection also revealed a PD-1+ Treg population, and a multitude of PD-L1+ cell types. Thus, we intervened in a second step with a combination of anti-PD-1 and anti-PD-L1 to completely cover all potential binders of PD-1 and PD-L1. Such a sequential treatment with TG4010 and anti-PD-1/PD-L1 led to measurable effects on tumor growth and survival.
Results and discussion
To induce lung tumors, BALB/c mice were intravenously injected (caudal vein) with either CT26.CL25 or CT26-MUC1 cells. CT26.CL25 is a colon carcinoma cell line stably transfected with the retroviral vector LXSN that contains the lacZ gene encoding β-Galactosidase (βGal).18 CT26-MUC1 is a clonal cell line obtained after stable transfection of CT26 cells (ATCC) with an expression plasmid encoding the G418-resistance gene and the human MUC1 gene under the control of SV40 and CMV promoters, respectively. Mice developed lung tumors within a few days after injection. Tumor progression led to dyspnea and weight loss, mice having lost 10% of their body weight were sacrificed. MVA vector were intravenously injected at day 2 and 9 after tumor cells injection. Reference median survival were 16 days and 34 days, for BALB/c mice bearing respectively either CT26.CL25 or CT26-MUC1 tumors when treated with an empty MVA vector. Intravenous therapeutic treatment with 104 pfu of MVA-βGal clearly enhanced the overall survival of BALB/c mice by 18 days, while treatment of BALB/c mice injected with CT26-MUC1 cells with 5 × 107 pfu of TG4010 led to a survival increase of three days (Fig. 1A, B). Noteworthy, while TG4010 provided a modest increase in median survival, the hazard ratio (HR) was largely in favor of TG4010 compared to empty MVA (HR: 0.48; CI [0.29–0.79]). This was due to the late separation of survival curves, a phenomenon well described in cancer immunotherapy.19 The HR takes in account the overall survival curve, while calculation of the median only depends on the earlier part of the follow-up period; hence, this metric is more likely to reflect the overall effect of immunotherapeutic treatment. For instance, at 60 days, percentage survival was around 20% versus 5% in the TG4010 and control MVA treated animals, respectively.
Figure 1.
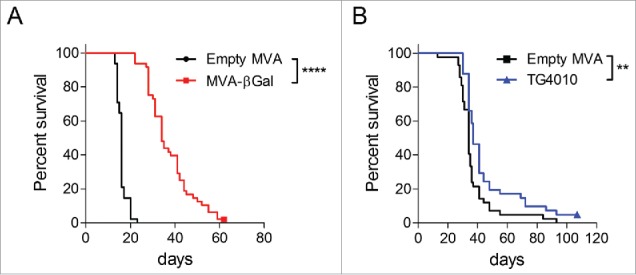
Therapeutic effects of MVA vaccines in CT26 colon cancer tumor models. BALB/c mice were IV injected (caudal vein) with either 2 × 105 CT26.CL25 (A) or CT26-MUC1 (B) cells. On day 2 and 9 after tumor challenge, mice were treated either with 1 × 104 pfu of MVA-βgal (A) or 5 × 107 pfu of TG4010 (B). An empty MVA vector was used as control in both models at the same dose. Mice were weighed twice per week and sacrificed when reaching 10% weight loss. Two to three independent experiments were carried out with groups of 10 to 15 mice per group. Overall survival rates represented as Kaplan-Meier curves were compared with a Log-rank test. Therapeutic treatment with 104 pfu MVA-βGal increased the survival by 18 days, while the treatment withTG4010 augmented the overall survival by 3 days. Animal experiments were conducted in compliance with EU directive 2010/63/EU.
Interestingly, a specific T cell subpopulation which we coined CD3dimCD8dim appeared in the lungs after repeated injection of MVA vaccines at one-week intervals (Fig. 2A). These CD127− cells were earlier characterized as either short-lived effector (SLEC) or early effector cells (EEC), depending, respectively, on the expression or not of the killer-cell lectin-like receptor G1 (KLRG1).17 In contrast to the proportion of CD3+CD8+ T cells, that was not influenced by MVA injection, the proportion of CD3dimCD8dim cells increased after repeated MVA injections (Fig. 2B, lower panel). This phenomenon was independent of the presence of growing tumors within the lungs, and did not depend on the MVA-encoded transgene, as similar infiltrations were detected after injection of either empty MVA or MVA-βGal vectors (Fig. 2B, lower panel). In mice having received the MVA-βGal vector, the proportion of antigen-specific cytotoxic CD3dimCD8dim lymphocytes triple positive for IFNγ, CD107a and Granzyme B were specifically increased as detected upon in vitro stimulation with the H-2d restricted βGal peptide T9L3 (Fig. 2B, upper panel). Within all living cells present in lung cell suspensions, such multifunctional antigen-specific cells were readily detectable only within the CD3dimCD8dim population. An increase of NKG2D-positive cells within the CD3dimCD8dim populations was observed (data not shown), most likely associated with the appearance of this marker on activated CD8 T cells.20 An implication of NKG2D+ NK-T cells cannot be ruled out. In the CT26-MUC1 model, the appearance of CD3dimCD8dim lymphocytes in the lungs of TG4010-injected mice was also observed. However, MUC1-specific cytotoxic T cells were not detected (data not shown). This observation is in accordance with the reported low immunogenicity of human MUC1 in mice and the failure to detect antigen-specific effector T cells in the periphery, even though the vaccine effect depends on the expression of MVA-encoded MUC1.4,6 The relative inefficiency in generating MUC1-specific cytotoxic T cells or their exhaustion could be one probable reason for the comparatively weaker impact of MUC1-encoding TG4010 on survival rates compared to MVA-βgal in their respective tumor models.
Figure 2.
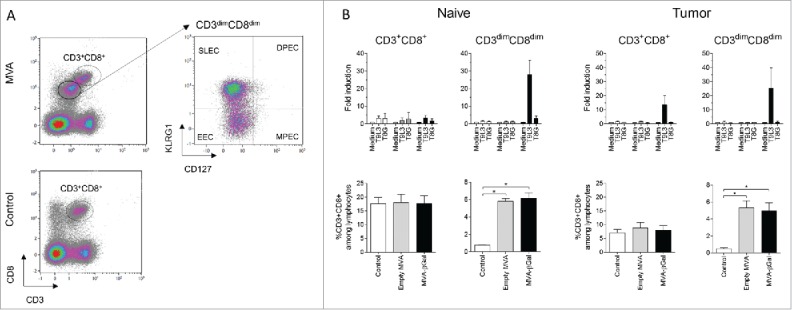
Immune cell analysis in the CT26.CL25 / MVA-βgal model. The lungs of naive or tumor-bearing mice were taken at day 14, five days after the second virus treatment. Lungs were weighed and enzymatically dissociated (Miltenyi Biotec). Single-cell suspensions were treated with fixable dead cell stain Live/Dead Aqua and labelled with anti CD3-PerCPCy5.5 (clone 145-2C11, BD Pharmingen), anti CD8-APCVio770 (clone 53–6.7, Miltenyi Biotec), anti KLRG1-PEVio770 (clone 2F1, Miltenyi Biotec) and CD127-VioBrightFITC (clone A7R34, Miltenyi Biotec). (Fig. 2A) Gating on living lymphocytes, defined a CD3dimCD8dim subpopulation in MVA-injected animals. This population was further divided into SLECs (KLRG1+CD127−) and their precursors EEC (KLRG1−CD127−). Neither memory precursor effector cells (MPECs, KLRG1−CD127+), nor double positive effector cells (DPECs, KLRG1+CD127+) were detected in the sample (right panel). (Fig. 2B) Analysis of CD3+CD8+ and CD3dimCD8dim cells. The lower panel displays the specific increase of CD3dimCD8dim T cells upon treatment with MVA in the lung from naive or tumor-bearing mice. Four independent experiments were performed and compared using the Wilcoxon-Mann-Whitney test with an adjusted p value of 0.025. The upper panel displays the results of antigen-specific T cell stimulation. Two x106 cells were ex vivo stimulated in 150 µl TexMACS medium (Miltenyi Biotec) in the presence of 1 µg anti-CD28 (abcam), and either the β-gal specific peptide T9L3 (TPHPARIGL), the control peptide T8G (TPHPARIG), or medium. Anti-CD107a-APC (clone 1D4B, eBiosciences) was added to detect degranulation events. Brefeldin A was added after one hour of incubation. After four hours, cells were analysed by flow cytometry for CD3, CD8 and KLRG1 as described before. After permeabilization (Cytofix/Cytoperm, BD Biosciences), activation was assessed by intracellular staining with anti-IFN-γ-FITC (clone XMG1.2, BD Pharmingen) or its isotype control, and anti-GranzymeB-Pacific Blue (clone GB11, BioLegend). To measure antigen-specific cytotoxic T cells, IFN-γ-secreting living CD3+CD8+ or CD3dimCD8dim cells were identified by subtracting non-specific signals observed after staining with its isotype control. The frequency fold-change of IFN-γ+CD107A+GranzymeB+ CD3dimCD8dim or CD3+CD8+ cells in the presence of the stimulatory peptide is shown in comparison to medium-stimulated control. The mean +/-SEM is calculated for three independent experiments. Fluorescence intensities were measured on a MacsQuant cytometer (Miltenyi Biotec) and data were analysed using Kaluza software (Beckman Coulter).
To augment MUC1-specific response, we tested a combination treatment of TG4010 with an anti-PD-1 in the CT26-MUC1 tumor model. Concomitant administration of anti-PD-1 with TG4010 did not increase the survival rate (data not shown). Hypothesizing that antibody injection might not coincide with the presence of potential PD-1+ target cells, we studied the phenotype of various immune cells in tumor-bearing lungs. Fig. 3A shows the percentage of SLECs and EECs within the CD3dimCD8dim population. The percentage of SLECs significantly increased after the second virus injection and remained high up to 8 days after. The percentage of EECs also raised after the first virus injection and further increased after the second injection. Fig. 3B shows that PD-1 expression on EECs was significantly higher after one injection and further increased after the second injection. Thus, repeated treatment with a viral vector augments the frequency of SLECs and EECs comprised within the CD3dimCD8dim population, and leads to up-regulation of PD-1 on EECs, representing a sign of exhaustion of this cell type.
Figure 3.
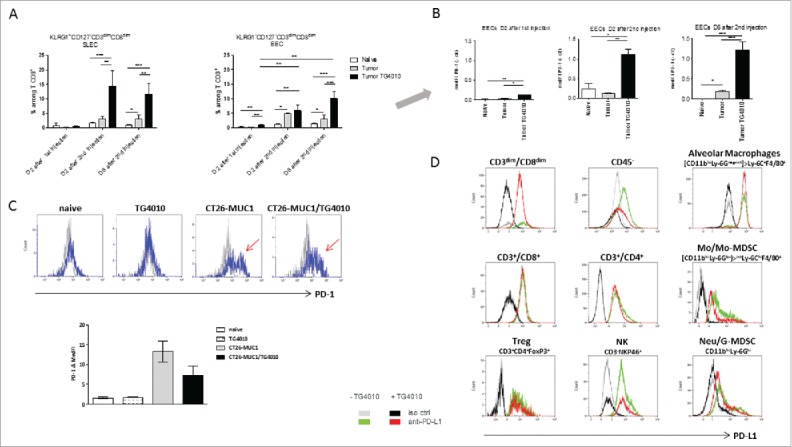
Immune cell analysis in the CT26-MUC1 model. For A and B, tumor bearing mice (four mice per group) were sacrificed two days after the first TG4010 IV injection, and 2 or 8 days after the second injection. The lungs were processed for flow cytometry as described in Fig. 2. Fig. 3A shows the percentage of CD3dimCD8dimKLRG1+CD127− SLECs, or CD3dimCD8dimKLRG1−CD127− EECs among living CD8+ T cells after one or two MVA vector injections. Fig. 3B shows the PD-1 expression on EECs as median fluorescence intensity minus control (MedFI-ctr). Antibodies used were PD-1-PE (clone HA2-7B1) and its isotype control PE-IgG2b (ES26-SE12.4), provided by Miltenyi Biotec. ANOVA model was carried out for Tumor*Time interaction, with post-hoc comparisons between samples at each time point usingTukey multiplicity adjustment (adjusted p-values are reported). For Fig. 3C and 3D, tumor-bearing mice, treated or not at day 2 and 9, with TG4010, were sacrificed at day 22 to 28 of tumor growth. Single-cell suspensions of 2 to 5 pooled lungs were firstly incubated with anti-mouse CD16/CD32 blocking antibodies (eBiosciences) and then stained using the LIVE/DEAD Fixable Near-I/R Dead Cell Stain Kit (Molecular Probes). The fluorochrome-conjugated antibodies used for cell phenotyping were: anti-CD3e-PE-Cy7 (clone 145-2C11), CD4-Alexa Fluor 700 (clone RM4-5) and Ly-6C-PE-Cy7 (clone AL-21) from BD Pharmingen, CD8a-V500 (clone 53–6.7), NKp46-BV421 (clone 29A1.4), CD11b-V450 (clone M1/70), Ly-6G-PE-CF594 (clone 1A8) and Siglec-F-PE-CF594 (clone E50-2440) from BD Horizon, and CD45-FITC (clone 30-F11), F4/80-Alexa Fluor 700 (clone BM8), PD-1-PerCP-Cy5.5 (clone 29F.1A12), PD-L1-APC (clone 10F.9G2), and respective isotype-matched controls (clones RTK2758, RTK4530) from BioLegend. Intranuclear FoxP3 was detected with the Anti-Mouse/Rat FoxP3 Staining Set (eBioscience) and an anti-FoxP3-PE-CF594 antibody (clone MF23, BD Horizon). All samples were run on a Navios flow cytometer (Beckman Coulter) and subsequently analyzed with Kaluza software (Beckman Coulter). Fig. 3C shows the development and persistence of a PD-1+ Treg cell subset in tumor-bearing lungs (blue curve, negative isotype control staining as gray curve). Bars represent means +/- SEM from 3 independent experiments. Fig. 3D: PD-L1 profiles of immune and non-immune cells in the lungs of tumor-bearing mice. Green and red distribution curves stand for PD-L1+ cells in TG4010-treated or untreated mice, respectively. Grey and black curves correspond to the respective isotype control. Lung/tumor non-immune cells were identified as living CD45− cells. Lymphoid and myeloid populations were analyzed within living CD45+ cells. The myeloid compartment was divided into CD11bhiLy-6Ghi (Neu/G-MDSCs), CD11bhiLy-6Glo-int and CD11bloLy-6Gneg-int. F4/80 and Ly-6C markers allowed further separation of Ly-6ChiF4/80+ Mo/Mo-MDSC cells within the CD11bhiLy-6Glo-int gate and Ly-6C+F4/80+ macrophages within the CD11bloLy-6Gneg-int gate. These macrophages were categorized as AM as they appeared Siglec Fhi in a separate experiment.24 Histograms are representative of the 3 experiments.
Another highly PD-1+ cell population was identified in late stage tumor-bearing lungs, among CD3+CD4+FoxP3+ Treg cells, which only partially disappeared after TG4010 treatment (Fig. 3C). These presumably immunosuppressive PD-1+ Treg cells may represent another interesting target population for PD-1/ PD-L1 inhibition.21
To identify binding partners for PD-1+ cells, we studied the PD-L1 expression in a variety of cell types in tumor-bearing lungs from mice treated or not with TG4010, taken at various time points after 20 days of CT26-MUC1 tumor growth (Fig. 3D). PD-L1 expression was observed both on CD45− cells, including the CT26-MUC1 tumor cells, and all CD45+ studied populations: CD3+CD8+, CD3dimCD8dim, CD4+, Treg and NK cells, monocyte/monocytic MDSC (Mo/Mo-MDSC) and neutrophil/Granulocytic MDSC (Neu/G-MDSC). Noteworthy, high PD-L1 expression was detected on alveolar macrophages (AM). Although there were no significant differences in the level of PD-L1 expression between MVA-treated and untreated samples, we observed a tendency of lower PD-L1 staining on CD45− cells which could be linked to a reduction of tumor load observed in preliminary histological studies on lung/tumor samples (data not shown). Low or no PD-L2 expression was observed on any of the cell types tested in the CT26-MUC1 tumor model (data not shown).
Having observed this arsenal of PD-1+ and PD-L1+ immune and lung tumor cells after TG4010 treatment and more than 20 days of tumor growth, we tested whether sequential treatment with corresponding blocking antibodies at this late time point would have a beneficial effect on survival. Groups of fifteen CT26-MUC1-injected mice, were treated with TG4010 (IV injection of 5 × 107 pfu) at day 2 and day 9, followed by intraperitoneal injection of 250 µg of anti-PD-1 and 200 µg of anti-PD-L1 antibodies at day 20, 24, 28 and 31. Two independent experiments, carried out in two different animal facilities, led to distinct survival profiles in untreated mice (Fig. 4A). A delay in tumor onset was observed in mice receiving TG4010 and anti-PD-1/PD-L1 sequential treatment versus TG4010 alone (adjusted log-rank p-values were 0.105 (experiment A) and 0.135 (experiment B)) with a median difference of 2.5 and 3 days. A stratified log-rank test was carried out by combining the two experiments to increase statistical power. Fig. 4B provides log-rank p-values and relative HR of different treatment groups in experiment A, in experiment B and in a meta-analysis of both (A+B). Significantly longer survival was found for mice receiving TG4010 and anti-PD-1/PD-L1 compared to the three other groups (for TG4010 comparison p-value = 0.024, adjusted p-value = 0.035 after Benjamini-Hochberg adjustment, HR of meta-analysis was 0.61). TG4010 was associated with a longer overall survival than buffer (adjusted p-value = 0.034) with a HR of 0.53 (95% confidence interval was [0.32,0.88]) but TG4010 and anti-PD-1/PD-L1 provided a more favorable HR of 0.32 (95% confidence interval was [0.19,0.55]). The lower HR observed with the combined therapy indicates that animals receiving the combination regimen had a lower risk of death than animals under other treatments. These data reflect the superiority of a sequential immunotherapeutic intervention over either regimen alone, while taking in account the delayed effect of immunotherapeutic treatments that may affect interpretation of median survival alone.
Figure 4.
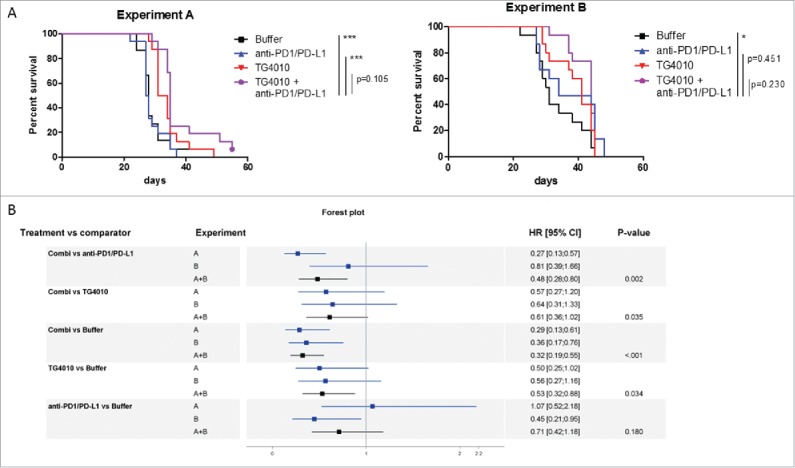
Effect of the sequential administration of anti-PD-1/anti-PD-L1 in the CT26-MUC1 model. (A) Groups of fifteen CT26-MUC1-injected mice were treated with TG4010 (IV injection of 5 × 107 pfu) at day 2 and day 9, followed by intraperitoneal injection of 250 µg of anti-PD-1 (clone RMP1.14) and 200 µg of anti-PD-L1 (clone 10F.9G2) at day 20, 24, 28 and 31. Survival was followed and criteria of sacrifice were based on weight loss. Two independent preclinical experiments (A and B) showed a trend of better overall survival for mice receiving the combination treatment. By combining the two experiments, a measurable benefit on tumor onset and survival was found for mice receiving both TG4010 and anti PD-1/PD-L1 versus monotherapy. (B) Forest plot analysis: the hazard ratio (HR) derives from the unstratified Cox proportional hazards model for experiments A and B. Meta-analysis was done for A+B with a fixed effect model by weighting estimates with the inverse of variance. The p-value was obtained using a stratified log-rank test for comparing survival curves. Animal experiments were conducted in compliance with EU directive 2010/63/EU.
In conclusion, our immunophenotyping studies suggest an immunosuppressive environment in late stage lung tumor samples. The up-regulation of PD-1 on EECs due to the TG4010 treatment, as well as the persistence of PD-1+ Treg cells, could be considered a threat for the efficiency of TG4010. EECs are crucial for T cell specific responses since they are the precursors for both effector and memory T cells.17 Up-regulation of PD-1 on EECs might prevent the proliferation of SLECs and memory precursor effector cells. Park and co-authors have identified the interaction between PD-1+ Treg cells and PD-L1 on effector T cells as one cause for potent T cell suppression in chronic viral infection in murine systems.21 In lung cancer patients, the increased frequency of Treg cells and their PD-1 expression in peripheral blood samples was discussed as a possible explanation for impaired adaptive immunity.22 In this regard, our preclinical syngeneic tumor model, even-though based on engineered murine colon cancer cells, seems to be a valuable tool to mimic certain aspects of the human disease. In addition, the triggering of an immune response in mice against human MUC1 expressed by CT26 cells is challenging due to the sequence homology between human and murine MUC1.4 Further, the serial passaging of the parental CT26 tumor in mice has most likely led to the selection of clones with lower antigen load,23 and rendered the immune stimulation against CT26-based tumors in the absence of strong antigens like for instance β-galactosidase even more difficult. Nevertheless, administration of PD-1/PD-L1 blocking agents led to measurable benefit on tumor growth and survival, suggesting that the tumor immunosuppressive action on the vaccine could be counteracted. Our observations provide a strong incentive for the combination of TG4010 with anti-PD-1/anti-PD-L1 in the clinic. The development of MVA-based immunotherapy with more immunogenic epitopes, combined with PD-1/PD-L1 blockade, may lead to further improvement of their therapeutic efficacy and holds great promise in this context of high medical need.
Abbreviations
- AM
Alveolar macrophages
- βgal
beta-galactosidase
- EEC
Early effector cells
- HR
Hazard ratio
- ICI
Immune checkpoint inhibitor
- IV
Intravenous
- KLRG1
Killer cell lectin-like receptor subfamily G member 1
- MedFI
Median Fluorescence Intensity
- MDSC
Myeloid-derived suppressor cells
- MUC1
Mucin1
- MVA
Modified Vaccinia virus Ankara
- NSCLC
Non-small cell lung cancer
- PD-1
Programmed cell death 1
- PD-L1
Programmed death-ligand 1
- pfu
plaque-forming unit
- SLEC
Short-lived effector cells
- Treg
Regulatory T cells
Disclosure of potential conflicts of interest
All authors were employees of Transgene when the work was performed. Transgene is a publicly traded French biopharmaceutical company, with Institut Mérieux as major shareholder.
Acknowledgment
We thank Caroline Tosch for her excellent assistance in multiparametric flow cytometry. We are grateful to Caroline Schenkels for editing the manuscript.
References
- 1.Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893-917. https://doi.org/ 10.1002/ijc.25516 [DOI] [PubMed] [Google Scholar]
- 2.Quoix E, Ramlau R, Westeel V, Papai Z, Madroszyk A, Riviere A, Koralewski P, Breton JL, Stoelben E, Braun D, et al.. Therapeutic vaccination with TG4010 and first-line chemotherapy in advanced non-small-cell lung cancer: A controlled phase 2B trial. Lancet Oncol. 2011;12:1125-33. https://doi.org/ 10.1016/S1470-2045(11)70259-5. PMID:22019520 [DOI] [PubMed] [Google Scholar]
- 3.Quoix E, Lena H, Losonczy G, Forget F, Chouaid C, Papai Z, Gervais R, Ottensmeier C, Szczesna A, Kazarnowicz A, et al.. TG4010 immunotherapy and first-line chemotherapy for advanced non-small-cell lung cancer (TIME): Results from the phase 2b part of a randomised, double-blind, placebo-controlled, phase 2b/3 trial. Lancet Oncol. 2016;17:212-223. https://doi.org/ 10.1016/S1470-2045(15)00483-0. PMID:26727163 [DOI] [PubMed] [Google Scholar]
- 4.Fend L, Gatard-Scheikl T, Kintz J, Gantzer M, Schaedler E, Rittner K, Cochin S, Fournel S, Préville X. Intravenous injection of MVA virus targets CD8+ lymphocytes to tumors to control tumor growth upon combinatorial treatment with a TLR9 agonist. Cancer Immunol Res. 2014;2:1163-74. https://doi.org/ 10.1158/2326-6066.CIR-14-0050. PMID:25168392 [DOI] [PubMed] [Google Scholar]
- 5.Claudepierre MC, Hortelano J, Schaedler E, Kleinpeter P, Geist M, Remy-Ziller C, Brandely R, Tosch C, Laruelle L, Jawhari A, et al.. Yeast virus-derived stimulator of the innate immune system augments the efficacy of virus vector-based immunotherapy. J Virol. 2014;88:5242-55. https://doi.org/ 10.1128/JVI.03819-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schaedler E, Remy-Ziller C, Hortelano J, Kehrer N, Claudepierre MC, Gatard T, Jakobs C, Préville X, Carpentier AF, Rittner K. Sequential administration of a MVA-based MUC1 cancer vaccine and the TLR9 ligand Litenimod (Li28) improves local immune defense against tumors. Vaccine. 2017;35:577-85. https://doi.org/ 10.1016/j.vaccine.2016.12.020. PMID:28012777 [DOI] [PubMed] [Google Scholar]
- 7.Johnson DB, Rioth MJ, Horn L. Immune checkpoint inhibitors in NSCLC. Curr Treat Options Oncol. 2014;15:658-69. https://doi.org/ 10.1007/s11864-014-0305-5. PMID:25096781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhu L, Jing S, Wang B, Wu K, Shenglin MA, Zhang S. Anti-PD-1/PD-L1 therapy as a promising option for non-small cell lung cancer: A single arm meta-analysis. Pathol Oncol Res. 2016;22(2):331-9. https://doi.org/ 10.1007/s12253-015-0011-z. PMID:26552662 [DOI] [PubMed] [Google Scholar]
- 9.Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, Fitz LJ, Malenkovich N, Okazaki T, Byrne MC, et al.. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192:1027-34. https://doi.org/ 10.1084/jem.192.7.1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K, et al.. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat Med. 2002;8:793-800. https://doi.org/ 10.1038/nm0902-1039c. PMID:12091876 [DOI] [PubMed] [Google Scholar]
- 11.Taube JM, Anders RA, Young GD, Xu H, Sharma R, McMiller TL, Chen S, Klein AP, Pardoll DM, Topalian SL, et al.. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med. 2012;4:127ra37. https://doi.org/ 10.1126/scitranslmed.3003689. PMID:22461641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blank C, Brown I, Peterson AC, Spiotto M, Iwai Y, Honjo T, Gajewski TF. PD-L1/B7H-1 inhibits the effector phase of tumor rejection by T cell receptor (TCR) transgenic CD8+ T cells. Cancer Res. 2004;64:1140-5. https://doi.org/ 10.1158/0008-5472.CAN-03-3259. PMID:14871849 [DOI] [PubMed] [Google Scholar]
- 13.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, et al.. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124-8. https://doi.org/ 10.1126/science.aaa1348. PMID:25765070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al.. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568-71. https://doi.org/ 10.1038/nature13954. PMID:25428505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. 2017;541:321-30. https://doi.org/ 10.1038/nature21349. PMID:28102259 [DOI] [PubMed] [Google Scholar]
- 16.Chen DS, Mellman I. Oncology meets immunology: The cancer-immunity cycle. Immunity. 2013;39:1-10. https://doi.org/ 10.1016/j.immuni.2013.07.012. PMID:23890059 [DOI] [PubMed] [Google Scholar]
- 17.Plumlee CR, Obar JJ, Colpitts SL, Jellison ER, Haining WN, Lefrancois L, Khanna KM. Early effector CD8 T cells display plasticity in populating the short-lived effector and memory-precursor pools following bacterial or viral infection. Sci Rep. 2015;5:12264. https://doi.org/ 10.1038/srep12264. PMID:26191658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang M, Bronte V, Chen PW, Gritz L, Panicali D, Rosenberg SA, Restifo NP. Active immunotherapy of cancer with a nonreplicating recombinant fowlpox virus encoding a model tumor-associated antigen. J Immunol. 1995;154:4685-92. PMID:7722321 [PMC free article] [PubMed] [Google Scholar]
- 19.Thoren FB, Anderson H, Strannegard O. Late divergence of survival curves in cancer immunotherapy trials: Interpretation and implications. Cancer Immunol Immunother. 2013;62:1547-51. https://doi.org/ 10.1007/s00262-013-1458-y. PMID:23979447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ehrlich LI, Ogasawara K, Hamerman JA, Takaki R, Zingoni A, Allison JP, Lanier LL. Engagement of NKG2D by cognate ligand or antibody alone is insufficient to mediate costimulation of human and mouse CD8+ T cells. J Immunol. 2005;174:1922-31. https://doi.org/ 10.4049/jimmunol.174.4.1922. PMID:15699119 [DOI] [PubMed] [Google Scholar]
- 21.Park HJ, Park JS, Jeong YH, Son J, Ban YH, Lee BH, Chen L, Chang J, Chung DH, Choi I, et al.. PD-1 upregulated on regulatory T cells during chronic virus infection enhances the suppression of CD8+ T cell immune response via the interaction with PD-L1 expressed on CD8+ T cells. J Immunol. 2015;194:5801-11. https://doi.org/ 10.4049/jimmunol.1401936. PMID:25934860 [DOI] [PubMed] [Google Scholar]
- 22.Zhong A, Pan X, Shi M. Expression of PD-1 by CD4(+)CD25(+)CD127(low) Treg cells in the peripheral blood of lung cancer patients. Onco Targets Ther. 2015;8:1831-3. https://doi.org/ 10.2147/OTT.S90538. PMID:26229490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brattain MG, Strobel-Stevens J, Fine D, Webb M, Sarrif AM. Establishment of mouse colonic carcinoma cell lines with different metastatic properties. Cancer Res. 1980;40:2142-6. PMID:6992981 [PubMed] [Google Scholar]
- 24.Misharin AV, Morales-Nebreda L, Mutlu GM, Budinger GR, Perlman H. Flow cytometric analysis of macrophages and dendritic cell subsets in the mouse lung. Am J Respir Cell Mol Biol. 2013;49:503-10. https://doi.org/ 10.1165/rcmb.2013-0086MA. PMID:23672262 [DOI] [PMC free article] [PubMed] [Google Scholar]