

Abstract
Objective
Human cognition has long been known to be under substantial genetic control. With the complete mapping of the human genome, genome-wide association studies (GWAS) for many complex traits have proliferated; however, the highly polygenic nature of intelligence has made the identification of the precise genes that influence both global and specific cognitive abilities more difficult than anticipated.
Methods
Here, we review the latest developments in the genomics of cognition, including a discussion of methodological advances in the genetic analysis of complex traits, and shared genetic contributions to cognitive abilities and neuropsychiatric disorders.
Results
A wealth of twin and family studies have provided compelling evidence for a strong heritable component of both global and specific cognitive abilities, and for the existence of ‘generalist genes’ responsible for a large portion of the variance in diverse cognitive abilities. Increasingly sophisticated analytic tools and ever-larger sample sizes are now facilitating the identification of specific genetic and molecular underpinnings of cognitive abilities, leading to optimism regarding possibilities for novel treatments for illnesses related to cognitive function.
Conclusions
We conclude with a set of future directions for the field, which will further accelerate discoveries regarding the biological pathways relevant to cognitive abilities. These, in turn, may be further interrogated in order to link biological mechanisms to behavior.
Keywords: g, polygenic risk, memory, genome-wide association, heritability, schizophrenia
In 1869, Sir Francis Galton proclaimed that “man’s natural [intellectual] abilities are derived by inheritance, under exactly the same limitations as are the form and physical features of the whole organic world” (Galton, 1869). While this hypothesis was quite controversial at the time, nearly 150 years later the notion that cognitive abilities are under strong genetic influence is well accepted. Indeed, while it is clear that non-genetic factors play a role in individual differences in intellectual ability (e.g., socioeconomic status (E Turkheimer, Haley, Waldron, D’Onofrio, & Gottesman, 2003)), and that methods for estimating genetic influence on intelligence may be biased (Devlin, Daniels, & Roeder, 1997), there is nonetheless overwhelming support for the notion that intelligence and other cognitive abilities are highly heritable (Deary, Johnson, & Houlihan, 2009; Finkel, Pedersen, McGue, & McClearn, 1995; R. Plomin & Spinath, 2004). Yet despite high heritability estimates, identifying the precise genes that influence cognitive ability has proven difficult. In the past 25 years, a number of genes have been identified that, when deleted or substantially mutated, are associated with marked cognitive disability (Bear, Huber, & Warren, 2004; Peters et al., 2005; Ramakers, 2002; Rousseau et al., 1991). However, it is still unclear if more typical variation in these genes influences individual cognitive differences within the normative range of function. Furthermore, although genome-wide searches are beginning to identify genetic loci associated with individual differences in cognitive performance (e.g., (Davies et al., 2016; Trampush et al., 2017)), the specific genes influencing these traits remain largely unknown. Yet, there is undeniable progress in unraveling the molecular genetics underpinnings of cognitive ability, spurred by large-scale data collection projects, meta analytic methods and decreasing costs of genotyping and sequencing.
In this review, we will summarize the historical context of research on the genetic basis of cognitive abilities. Long before technologies were developed to identify the contribution of specific relevant genes, behavioral genetics studies of twins provided clear evidence of a genetic basis of cognitive traits (IQ in particular; (Nisbett et al., 2012; Robert Plomin, DeFries, Knopik, & Neiderhiser, 2013)). These studies paved the way for molecular genetics studies that have taken place largely over the past decade, in ever-increasing sample sizes. We review currently published genome-wide discovery efforts. We discuss analytic developments in the field that have allowed us to advance understanding of genetic contributions to global and unique cognitive abilities, in the context of both normal variation and disease populations. We conclude with a set of key future directions for the field that we believe will help propel this vital area of research forward.
Cognition & Behavior Genetics
Heritability provides an estimate of the extent of genetic control over a trait in a specific population (Falconer & Mackay, 1996), defined as the trait variance due to additive genetic factors (σ2a) divided by total variance: h2 = σ2a/(σ2a+σ2e). Heritability estimates (h2) range between 0, indicating no genetic contribution to trait variance, to 1, suggesting that the trait is completely under genetic control. While documenting that there is a significant heritable component is a critical first step before a measure should be used in genetic analyses, the statistic is limited in a number of important ways (see below). Heritability is traditionally assessed through twin, family, and/or pedigree studies, in which the relationship between genetic proximity and similarity in cognitive performance can index the degree to which a trait is associated with genetic or environmental factors (Almasy & Blangero, 2001; Falconer & Mackay, 1996; R. Plomin & Kosslyn, 2001). More recently, efforts to estimate heritability in unrelated individuals, or more accurately the proportion of variance explained by common genetic variation, have been developed (Yang et al., 2010), as discussed in more detail below.
One important qualification is that heritability estimates reflect the magnitude of the overall genetic effect on a trait, but do not indicate the total number of genes that may be implicated, nor the relative contributions of those genes (Almasy, 2003). Thus, while higher heritability estimates would suggest that a trait is under stronger genetic influence, it does not inform us about the genetic architecture of the trait (e.g., whether trait variance is influenced by many genes of small effect, or a single gene of large effect). Furthermore, heritability estimates may differ somewhat based on the characteristics of the population used for estimation (Falconer & Mackay, 1996); thus slight differences in h2 values are to be expected between samples. Finally, heritability estimates are not informative at the level of a single individual.
What have we learned from family studies?
Twin, family and adoption studies provided an approach for quantifying the genetic contribution to complex traits like cognitive abilities, allowing researchers to disentangle the impact of genetic and environment, both unique (i.e., non-shared) and common (i.e. shared among related individuals), factors and their interactions (J. Blangero et al., 2005; Johnson, Turkheimer, Gottesman, & Bouchard, 2010). Typically, these methods contrast phenotype values for individuals based upon their expected genetic relationship due to Mendelian transmission (e.g., 1 for monozygotic twins; .5 for first degree relatives or 0.25 for second degree relatives) or their expected environmental relationship (e.g. common environment, completely unique environment due to adoption) (Lynch & Walsh, 1998). Additionally, twin and pedigree studies also offer an excellent resource for studying the significance of genotype by environment interactions (J. Blangero et al., 2005; S. Purcell, 2002), such as effects of lifestyle, diet or exposure to toxins. Much of this work has utilized the classical twin design, comparing mono- and dizygotic twin pairs, and large, worldwide registries of twin data have been established to facilitate this work (Boomsma, Busjahn, & Peltonen, 2002).
While an exhaustive review of methods and findings of twin, family and adoption research is outside of the scope of the current manuscript and been comprehensively described elsewhere (Robert Plomin et al., 2013), below we provide a focused discussion and brief summary of advances in knowledge regarding genetic and environmental influences on cognitive abilities offered by these studies over the past several decades.
Adoption Studies
Adopted children provide one of the strongest tests of environmental influences on cognitive ability, as comparing these children to their biological sibling or parents provides a means of disentangling genetic and environmental influences (R. Plomin, Owen, & McGuffin, 1994). For example, in their seminal 1949 paper, Skodak and Skeels reported that adopted children at age 13 years had higher IQs than their biological mothers, suggesting that rearing environment impacts IQ (Skodak & Skeels, 1949); nevertheless, some of this difference may be due to the Flynn effect - that is, the sustained increases in intelligence test scores that have been observed over time in the twentieth century (Flynn, 1993). In an early review of family and adoption studies, Bouchard and McGue (1981) found overwhelming evidence for familial resemblance in intelligence. However, the authors also noted that a number of factors appear to influence within-family correlations, including sex, test used and family socioeconomic status (Bouchard & McGue, 1981). More recently, Kendler and colleagues investigated a national Swedish sample of separated male siblings, finding that adoption into improved socioeconomic circumstances was associated with a mean 4.4-point increase in IQ at age 18 (Kendler, Turkheimer, Ohlsson, Sundquist, & Sundquist, 2015). Higher education of the rearing parents was also associated with increased IQ in the offspring. This finding was then replicated in a sample of over 2000 half-siblings, collectively indicating that rearing environment has an impact on IQ in late adolescence, and that educational level of adoptive parents explains a portion of IQ variance in adopted siblings (Kendler et al., 2015). Given the close connection between educational level and socioeconomic status (White, 1982; Winkleby, Jatulis, Frank, & Fortmann, 1992), these results are consistent with reports of substantial impact of parental socioeconomic status on the heritability of child intelligence (i.e., more of the variance in IQ is attributable to genetics in affluent families; (E Turkheimer et al., 2003).
Heritability of Intelligence: Generalist Genes
Most early twin and family work focused on general intellectual functioning, or IQ, and today the heritability of general cognitive abilities is well established (Devlin et al., 1997; Finkel, Pedersen, McGue, et al., 1995; Knowles et al., 2014; McClearn et al., 1997b; McGue & Christensen, 2001). Indeed, twin and family studies have determined that 30–60% of the individual differences in adult intelligence test performance are due to genetic factors (Bouchard & McGue, 1981; Bouchard, Segal, & Lykken, 1990; Devlin et al., 1997; McClearn et al., 1997a). While numerous factors, including sampling strategy, analytic assumptions and assessments employed, could influence differences in heritability estimates, there is overwhelming evidence that intelligence is under substantial genetic control (R. Plomin, DeFries, Knopik, & Neiderhiser, 2016). Meta- analytic heritability estimates for measures of global intelligence are typically about 50% (Haworth et al., 2010; Polderman et al., 2015).
One intriguing observation is that heritability estimates for intellectual functioning appear to change over the lifespan, possibly inconsistent with the substantial evidence for the stability of IQ measures from childhood to old age (Deary, Whiteman, Starr, Whalley, & Fox, 2004; Gow et al., 2011; Haworth et al., 2010). In a cohort of 11,000 twin pairs from four countries, Haworth and colleagues (2010) reported that the heritability of general cognitive ability increases significantly and linearly from 41% in childhood (9 years) to 55% in adolescence (12 years) and to 66% in young adulthood (17 years). Similarly, in a large meta-analysis of longitudinal twin studies, Tucker-Drob and Briley (2014) report that cross-time correlations for genetic and shared environmental components were low during early childhood, increased sharply over child development, and remained relatively high from adolescence through late adulthood. In contrast, while cross-time correlations for non-shared environmental effects were also low across childhood, they increased gradually up to adulthood (Tucker-Drob & Briley, 2014). Increasing phenotypic stability over childhood development was almost entirely mediated by genetic factors. In a cross-sectional study, McGue and Christensen (2013) found no evidence of declining twin similarity between the ages of 46–96, which is consistent with longitudinal twin studies (Tucker-Drob & Briley, 2014), but at odds with commonly held assumptions that genetic influences tend to diminish over time (Haworth et al., 2010; McGue & Christensen, 2013).
Recent studies of genetic influences on cognitive abilities have highlighted the role of g, or general intelligence, based on findings of phenotypic and genetic correlations across different cognitive domains that are present even in cognitive disorders (Davis, Haworth, & Plomin, 2009; R. Plomin et al., 2016; R. Plomin & Kovas, 2005). For example, Davis and colleagues (2009) performed a multivariate genetic analysis of intelligence, reading, mathematics, and language in over 5,000 pairs of 12-year-old twins, reporting that more than half of the phenotypic correlations between cognitive tests is attributable to genetic factors. Knowles and colleagues (2014) reported similar results in a sample of 1269 individuals from extended pedigrees and similar results were observed using common variants in 2500 unrelated individuals (Trzaskowski et al., 2013). Together, these results suggest that a set of “generalist genes” appear to influence multiple cognitive domains, implying both substantial pleiotropy and polygenicity of normal and abnormal cognitive ability (Kovas & Plomin, 2006).
The ‘g’ phenotype is an alternative means of indexing overall IQ, as it is a statistically derived index of overall neuropsychological test performance, typically the first principal component extracted from a principal component analysis of individual measures. g has advantages for large-scale studies that combine data from multiple cohorts, as it is relatively insensitive to the specific cognitive tasks/abilities assessed (Knowles et al., 2014; Lencz et al., 2013). Furthermore, the long-term stability of g is well-established, and it is predictive of later educational and occupational function (Gottfredson, 1997). However, some critics feel that the emphasis on g is misguided as it leads to a devaluation of other important abilities (e.g., (S. J. Gould, 1981)), and clearly all of cognition cannot be explained by a single factor. Nonetheless, its use in genetics studies is supported by the very high genetic correlations among specific cognitive abilities (Knowles et al., 2014; Petrill, 1997). g explains about 40% of the variance among diverse cognitive measures (R. Plomin, 1999), and is also correlated with brain size, suggesting a biological underpinning (McDaniel, 2004; van Leeuwen M et al., 2009; Vuoksimaa et al., 2015). Thus, g can be considered a rough index of intellectual ability and, given that it can be calculated in almost any study that included 5 or more neuropsychological tests, its use has facilitated large scale GWAS meta analyses (Davies et al., 2015; Lencz et al., 2013).
Heritability of Specific Cognitive Abilities
The heritabilities of individual cognitive traits, such as working and declarative memory and processing speed, have been investigated, although not to the same extent as general intellectual function or g. Nevertheless, data from twin and family studies indexing a wide variety of cognitive measures, including processing speed (heritability, h2=26–76; Luciano et al., 2001; Posthuma, de Geus, & Boomsma, 2001; Swan & Carmelli, 2002), attention/vigilance (h2=16–89; Fan, Wu, Fossella, & Posner, 2001), executive functioning or cognitive control (h2=33–68; Swan & Carmelli, 2002), working memory (h2=2–60; Ando, Ono, & Wright, 2001; Jacob et al., 2001; Neubauer, Spinath, Riemann, & Angleitner, 2000) and declarative memory (h2=50–65; Swan et al., 1999), indicate that these abilities are strongly influenced by genetics. The greater variability observed in some domains, such as working memory, may be at least partially attributable to psychometric properties of the measures used. Notably, recent evidence from an ethnically and socioeconomically diverse sample of over 500 twins found that a common executive function factor was 100% heritable, suggesting that correlations among four domains of executive function (Inhibition, Switching, Working Memory, and Updating) are entirely due to shared underlying genetic etiology (Engelhardt, Briley, Mann, Harden, & Tucker-Drob, 2015). It is important to note that a heritability estimate of 100% implies that environmental factors do not influence executive functioning (as operationalized by the particular measures applied in this study, and with the specific analytic approach taken), in this population at this particular stage of development (e.g. ages 7–15 years). However, at other developmental stages or in the presence of other environmental factors, this heritability estimate may be lower. Indeed, complete genetic control of behavioral or cognitive abilities is almost never observed (R. Plomin et al., 2016; E. Turkheimer, 2000), making it likely that any replication of this finding will find high, but not perfect, heritability.
As noted above, while roughly half of individual differences in long-term memory are attributable to genetic factors (Alarcon, Plomin, Fulker, Corley, & DeFries, 1998; Finkel & McGue, 1993; Finkel, Pedersen, & McGue, 1995; Swan et al., 1999; Volk, McDermott, Roediger, & Todd, 2006), distinct components of learning and memory may have different genetic determinants. For instance, in middle-aged twins from the Vietnam Era Twin Study of Aging, shared genetic influences on learning ability and retrieval were noted, as well as unique genetic influences on learning ability, likely reflecting a distinct genetic contribution to information acquisition (Panizzon et al., 2011). Finally, factors such as processing speed may also limit the ability to perform memory tasks, thus accounting for some of the genetic influence on episodic memory, particularly as related to heritability of age-associated changes (Finkel, Reynolds, McArdle, Hamagami, & Pedersen, 2009).
Genetic Correlations
The genetic correlation between two traits is an informative metric regarding the proportion of variance shared by two traits that is due to genetic causes (Lynch & Walsh, 1998). In other words, while individual cognitive abilities are moderately heritable, the genetic correlation between them may range from 0 (indicating the abilities are influenced by completely different sets of genes) to 1 (indicating that the traits are pleiotropic, i.e., influenced by the same genes). Genetic correlations among specific cognitive abilities tend to be very high (Petrill, 1997). For example, in our investigation of large pedigrees genetically enriched for bipolar disorder (Fears et al., 2014), we found strong genetic correlations among multiple domains of cognitive function (Figure 1). The network structure of the genetic correlations (right) was sparser than, but qualitatively similar to, that of phenotypic correlations (left). Traits mainly clustered within phenotypic domains, but some genetic correlations across domains were observed, such as interference errors on the Stroop task with inferior parietal cortical surface area, a brain region involved in maintaining attention.
Figure 1.
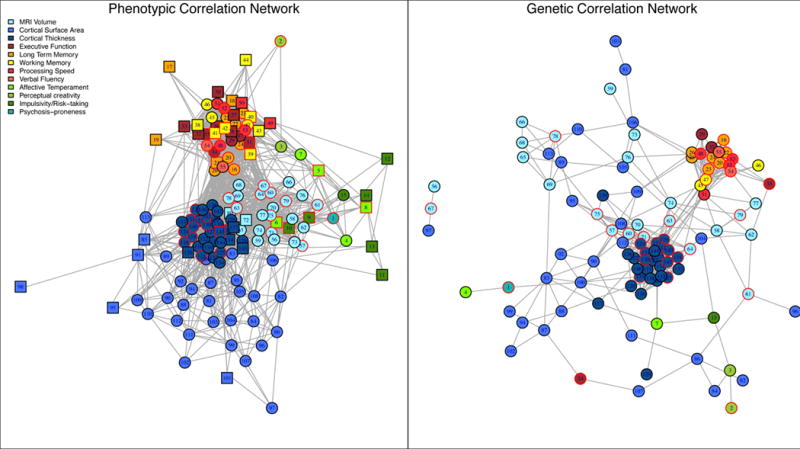
Network structure across domains. Figure indicates phenotypic (left) and genetic (right) correlations. We found strong genetic correlations among multiple domains of cognitive function; the network structure of the genetic correlations was sparser than, but qualitatively similar to, that of phenotypic correlations. Reprinted with permission from (Fears et al., 2014).
Using different methods for evaluating genetic correlations in unrelated individuals, Marioni et al. (2016) recently found that body mass index and cognitive functions showed significant inverse genetic correlations; in other words, genetic variants associated with increased cognitive function scores were associated with lower BMI. These findings suggest shared biological pathways for these two key health outcomes.
Endophenotypes and Choosing Between Endophenotypes
Identifying genes that influence a cognitive or brain imaging trait alone would represent an intrinsic scientific advance. However, many cognitive genomic experiments are conducted in order to discover the causes of psychiatric or neurological diseases. In this context, the neuropsychological index is often described as an endophenotype (Gottesman & Gould, 2003; Gottesman & Shields, 1972). Although there are subtle distinctions between theorists when defining an endophenotype (Lenzenweger, 2013), it may be easiest to conceptualize an endophenotype as a biomarker that is influenced by the same genes that confer risk for an illness (T. Gould & Gottesman, 2006) but that is not a sign or symptom of the disease or its progression (Gottesman & Gould, 2003). While most early models of endophenotypes argue that these traits will be closer to gene action than disease phenotypes (e.g. Gottesman and Gould, 2003), empirical evidence supporting this claim has been limited (C. Bearden & Freimer, 2006; Flint, Timpson, & Munafo, 2014), partially because of the highly polygenetic nature of many commonly investigated endophenotypes, e.g., intelligence (Sniekers et al., 2017) or neuroanatomy (Hibar et al., 2017; Hibar et al., 2015; Stein et al., 2012). However, endophenotypes may be helpful for localizing risk genes (J Blangero, 2004), characterizing previously identified genetic loci (D. C. Glahn et al., 2014; Hall & Smoller, 2010), or potentially by providing genetically informed data to refine our diagnostic nosology (Cuthbert & Insel, 2013; Insel & Cuthbert, 2009). Yet, in order to take full advantage of an endophenotype for unlocking the genetic architecture of an illness, one much choose an appropriate endophenotype.
Recently, Glahn and colleagues (2012) developed the Endophenotype Ranking Value (ERV), an empirical metric for ranking endophenotypes based upon their genetic similarity to the studied illness. The ERV is the standardized genetic covariance between an endophenotype (e) and illness (i), and is defined as ERVie=√hi2√he2ρg. Heritability (h2) represents the portion of the phenotypic variance accounted for by additive genetic variance (h2 = σ2g/σ2p). Genetic correlation (ρg) represents the common genetic covariance between two traits, or pleiotropy. The ERV statistic, which is conceptually similar to the co-heritability between traits, is used to rank putative endophenotypes, as higher values indicate stronger shared genetic influence between the endophenotype and the illness (D. C. Glahn et al., 2012). Traditionally, calculating the ERV requires family or twin data (D. C. Glahn & Blangero, 2011). However, as described below, recent techniques for estimating heritability and genetic correlation from genome-wide SNP data allows one to estimate the ERV in unrelated individuals.
As defined above, the ERV statistic is dependent upon the genetic correlation between the illness and the endophenotype. However, if the illness is relatively rare in the studied population, it may be difficult to have sufficient numbers of cases to calculate a genetic correlation. Thus, Glahn and colleagues (2015) developed a mean-based fixed-effects test that indexes each family member’s risk as a function of shared genetic kinship with an affected individual. To demonstrate the utility of this coefficient of relatedness approach, we searched for neurocognitive and neuroanatomic endophenotypes for schizophrenia in large unselected multigenerational pedigrees with only 6 affected individuals but with 233 of their unaffected relatives. We identified three neurocognitive measures (digit-symbol substitution, facial memory, and emotion recognition) and six medial temporal and prefrontal cortical surfaces associated with liability for schizophrenia (D. C. Glahn et al., 2015).
Genetic Linkage Studies/Identification of QTL
In the context of family studies, considerable effort has been dedicated to the detection of quantitative trait loci (QTL) for cognitive traits and disorders of cognition; i.e., a genetic locus or chromosomal region that contributes to variability in complex quantitative traits. Initially, most investigators utilized genetic linkage analysis, an analytic technique designed to detect the chromosomal location of influencing a phenotype (Terwilliger & Ott, 1994). This method requires families and is based on the observation that genes that reside physically close on a chromosome remain linked during meiosis. While linkage analysis is quite powerful for localizing QTL, additional molecular genetic analyses, often termed fine mapping, is required for gene identification (E. Lander & Kruglyak, 1995).
One of the most compelling and well-replicated linkage findings for specific cognitive abilities is in the area of dyslexia (i.e., specific impairment in learning to read). An early linkage study identified a genome-wide significant linkage for dyslexia to a segment of chromosome 6p (S. E. Fisher et al., 1999), in a sample of 181 sibling pairs selected on the basis of a dyslexic proband. This study was innovative in its investigation of both the categorical phenotype of dyslexia, based on IQ/reading discrepancy score, as well as quantitative measures of word recognition, irregular-word reading, and non-word reading, hypothesized to correlate with different aspects of the phenotype. The authors identified a QTL that was significantly associated with both phonological and orthographic skills, suggesting that developmental dyslexia is not specific to phoneme awareness, as was previously believed, and in fact shares genetic underpinnings with written language abilities. Thus, this study provides an example of the way that genetic information can provide new insights into the pathophysiological basis of a disorder, which has important treatment implications.
Fisher and colleagues (1999) conducted the first systematic search for genetic variants associated with g, using what was at the time a dense map of DNA markers on chromosome 4, in a sample of just 51 children of high g and 51 controls (average g). They identified 11 significant QTL associations, three of which replicated in independent samples (P. J. Fisher et al., 1999). Unfortunately, the genes responsible for these QTLs have yet to be localized.
Some investigators have postulated that identifying genes that impact specific cognitive domains may be more tractable than finding genes for g, given the cognitive neuroscience view that relatively distinct brain circuits are implicated in specific cognitive domains (van der Sluis, Verhage, Posthuma, & Dolan, 2010). Taking this approach, Knowles and colleagues (2014) successfully localized a QTL for working memory at chromosome 8q21.11–13 and 8q24.22 in 1,269 participants in the Genetics of Brain Structure (GOBS) sample, a cohort of large, randomly ascertained Mexican-American families. Association analysis conducted in the region of these QTLs identified 5 SNPs that were significantly associated with memory ability, one of which showed a nominal association with schizophrenia risk in the Psychiatrics Genomics Consortium.
The identification of the APOE locus on chromosome 19q13.2 that is associated with late-onset familial Alzheimer disease (Pericak-Vance et al., 1991; Strittmatter et al., 1993) was clearly one of the first definitive associations of specific genetic loci with cognition. In addition to being one of the strongest genetic risk factors for a brain-related disorder, the APOE locus has been consistently linked to impaired cognitive functioning in non-demented individuals. Indeed, several meta-analyses with samples of ~40,000 participants indicate that the carriers of APOE allele 4 (ε4) perform significantly worse on measures of episodic memory, executive functioning, and overall global cognitive ability (Small, Rosnick, Fratiglioni, & Backman, 2004; Wisdom, Callahan, & Hawkins, 2011). Interestingly, older non-demented APOE ε4 allele carriers show more brain activation in Alzheimer’s related brain regions relative to individuals homozygous of the ε3 allele during episodic memory performance and this activation level correlated with degree of memory decline over two years (Bookheimer et al., 2000), suggesting neurobiological correlates of APOE-related cognitive decline. To date, the APOE ε4 allele remains one of the only examples of a common variant of modest penetrance that exerts adverse, albeit small effects on a range of neurocognitive functions in cognitively healthy adults (Wisdom et al., 2011). Furthermore, as discussed below, this chromosome 19q13.2 locus has recently been associated with cognitive ability in a large scale GWAS study (Davies et al., 2014), suggesting that other genes or variants in that region may influence cognitive performance as well.
Movement to Genome-Wide Association Studies (GWAS): General Cognitive Abilities
With the complete mapping of the human genome (E. S. Lander et al., 2001), genome-wide association studies (GWAS) began to proliferate. In a GWAS, associations between single nucleotide polymorphisms (SNPs) and human diseases or traits are assessed across the genome, to determine whether any variant is associated with the trait of interest (Risch & Merikangas, 1996). Given the large number of SNPs being tested (typically several million), large sample sizes and multiple testing correction are critical. Further, while GWAS can identify variants that are statistically associated with a disease or trait, this kind of analysis alone cannot determine which gene(s) are causal (Manolio, 2010). Thus, like linkage analysis, GWAS is useful for localizing a genetic signal but additional work must be conducted to identify the specific gene or genes driving that association. Although this may appear to be a subtle analytic point, the implications of mistaking a GWAS QTL for a gene identification are substantial. For example, in an influential article, Dina and colleagues (2007) performed a GWAS comparing ~3000 overweight individuals to ~5,000 comparison subjects, localizing a set of SNPs in the first intron (i.e., an intragenic region removed by RNA splicing before translation) of the fat mass and obesity-associated (FTO) gene that were strongly associated with early- onset, severe obesity (experiment wise p=1.67×10−26). One of these SNPs (rs1121980) was strongly associated with severe class III adult obesity (BMI>40; odds ratio (OR) = 1.55, p=3.5×10−16), and the authors concluded that FTO contributes to human obesity and hence may be “a target for subsequent functional analyses” (Dina et al., 2007). Based upon this evidence, Church and colleagues quickly developed an FTO knockout mouse and reported that a point mutation in the mouse Fto gene resulted in reduced fat mass, increased energy expenditure, and unchanged physical activity (Church et al., 2009). Gene expression profiling further revealed increased expression of some fat and carbohydrate metabolism genes and an improved inflammatory profile in white adipose tissue of mutant mice. Although these striking results fostered intensive study of FTO biology (Tung & Yeo, 2011), no direct connection between the obesity-associated variants and FTO expression or function was identified. In 2014, Smemo and colleagues reassessed the obesity-associated noncoding variants within FTO, reporting that these variants are functionally connected with the homeobox gene IRX3. The obesity-associated FTO region directly interacts with the promoters of IRX3 as well as FTO in the human, mouse and zebrafish genomes. Furthermore, long-range enhancers within this region recapitulate aspects of IRX3 expression, suggesting that the obesity-associated interval belongs to the regulatory landscape of IRX3 (Smemo et al., 2014). Smemo and colleagues further demonstrated a direct connection between the variants in the intron of FTO and IRX3 function and with body mass and composition. This set of findings thus offers a cautionary tale for the simple interpretation of GWAS results and the subsequent biological inferences, as they highlight that other loci may be responsible for regulation of a statistically associated variant.
GWAS of complex quantitative phenotypes such as height have successfully discovered and replicated associations with hundreds of genome-wide significant common variants (Wood et al., 2014). However, despite the substantial evidence from family and twin studies reviewed above, indicating that cognitive abilities are highly heritable, identification of replicable loci associated with individual differences in cognitive ability has proven difficult. Early GWAS meta-analyses of general cognitive ability did not identify any genome-wide significant hits, in samples of ~3500 to 5000 adults (Davies et al., 2011; Lencz et al., 2013) or in 18,000 youth (Benyamin et al., 2014). The latter study found that the aggregate effects of common SNPs explained 22–46% of phenotypic variation in childhood IQ, supporting the highly polygenic nature of cognition. Further, the FNBP1L (formin binding protein 1-like) gene, which is involved in a pathway that links cell surface signals to the actin cytoskeleton (Ho et al., 2004) and was reported to be the strongest association for adult intelligence in the Davies et al. (2011) study, was also associated with childhood intelligence in a gene-based analysis in the youth study (Liu et al., 2010). Recently, Sniekers and colleagues (2017) published a meta-analysis for intelligence in 78,308 individuals, in which 336 genome-wide significant associated SNPs are identified in 18 genomic loci, 15 of which are new, suggesting that prior studies have been underpowered to pick up on the highly polygenic signal for IQ. The identified genes are mostly highly expressed in brain tissue, and several have been previously implicated in either metabolic traits or neuropsychiatric disorders associated with cognitive impairment (schizophrenia, Alzheimer’s Disease). Moreover, pathway analysis indicated enrichment of genes regulating cell development.
In a meta-analysis of 31 CHARGE cohorts (n=53,949), Davies and colleagues (2015) localized three genomic regions (6q16.1, 14q12 and 19q13.32) that influenced several cognitive variables. Notably, the 19q13.32 region, which includes the APOE and TOMM40 genes (Davies et al., 2015), was previously associated with cognitive phenotypes in old age (De Jager et al., 2012; Deary et al., 2002; Wisdom et al., 2011), age-related decline (Davies et al., 2014; Zhang & Pierce, 2014), and Alzheimer’s dementia (Corder et al., 1993; Strittmatter et al., 1993).
In a recent very large study using data collected as part of the UK Biobank study, three genomic regions were significantly associated with performance on a test of verbal numerical reasoning (N~ 36K), and two independent loci were significantly associated with performance on a reaction time task obtained on an even larger sample (N = 111,483) (Davies et al., 2016). Although a brief memory measure (‘pairs matching’ of symbols on a touch-screen computer) was investigated in over 112,000 individuals, no genome-wide significant variants were detected. Unfortunately, this task had very poor psychometric properties (test-retest correlations for this measure were only 0.15) which likely limited its utility for gene discovery (Davies et al., 2016).
Debette and colleagues (2015) examined 29,076 dementia- and stroke-free individuals of European ancestry (>45 years of age) and identified a single locus on chromosome 19 near the APOE locus. This locus is associated with poorer delayed recall performance in discovery and replication samples (Debette et al., 2015).
A recent paper from the Cognitive Genomics Consortium (COGENT) (Lencz et al., 2013) an international multisite effort to investigate the molecular genetics of cognitive function, applied a GWAS meta-analysis to examine general cognitive function in 35,298 healthy individuals of European ancestry, across 24 cohorts (Trampush et al., 2017). The primary analysis identified two novel associated SNPs, on chromosomes 1 and 2, that reached genomewide significance. The top GWAS hit in this study was an intronic variant in the CENPO gene on chromosome 2, which is highly expressed in the thalamus and basal ganglia (Trampush et al., 2017). Individual SNP results were also aggregated to conduct a gene-based analysis, which revealed three additional novel gene-based loci, at chromosomes 17q21.31 (a region containing a known inversion polymorphism previously associated with neurobehavioral phenotypes (Zollino et al., 2012)), as well as 17p13.1 and 1p13.3. This study also offered additional support for previously identified associations in the CHARGE consortium. While the identification of these replicable findings is encouraging, it must be noted that the top two SNPs in this study individually accounted for ~ 0.1% of the variance in cognitive performance, effect sizes that are quite a bit smaller than those observed for the top individual loci associated with other quantitative traits such as height (Wood et al., 2014). In GWAS of over 200,000 individuals, Wood and colleagues (2014) show that additive contributions of fewer than 10K SNPS can account for 36% of the variability in adult height. It is possible that human intelligence is even more polygenic/genetically complex.
GWAS of Educational Attainment
Given that educational attainment tends to be highly correlated, both phenotypically and genetically, with cognitive abilities (Calvin et al., 2012; Wainwright, Wright, Geffen, Luciano, & Martin, 2005), and given that is its regularly assessed in medical genetics studies, multiple recent GWAS have used educational attainment as a proxy for cognitive ability (Okbay et al., 2016; Trampush et al., 2015). Rietveld and colleagues (2013) identified three genome-wide significant loci in a discovery sample of 101,069 individuals, which were replicated in an additional sample of 25,000 individuals. In follow-up exploratory gene co-expression analyses, the authors found that several implicated genes were involved in pathways related to learning and long-term memory, as well as neuronal development, providing (at least tentative) biological plausibility for the findings (Rietveld et al., 2013). Despite these large sample sizes, and the insights this study provided into the genetic architecture of cognition, the identified loci explain only a small fraction of the genetic variance associated with cognitive ability (roughly .02% of the variance). In a follow-up study, using a two-stage (‘proxy phenotype’) approach, Rietveld and colleagues (2014) identified a set of 69 ‘education-associated’ SNPs in a large sample (over 100,000 individuals), which they then investigated in relationship to cognitive performance in independent, somewhat smaller samples (n=24,189). This analysis yielded 3 SNPs that surpassed genome-wide significance; each additional reference allele for each SNP was associated with an ~0.02 SD increase in cognitive performance, corresponding to a 0.3 point IQ increase (Rietveld et al., 2014). In a follow-up bioinformatics analysis, the authors identified four genes associated with the identified loci that are predicted to be involved in glutamatergic neurotransmission, which is relevant to synaptic plasticity.
Although educational attainment has offered a useful proxy given that it is readily available on very large numbers of individuals, some would argue that the use of educational attainment to represent cognitive abilities is a biased approach, as it is influenced by many known environmental factors, including public policies. Nevertheless, the promising findings of Rietveld and colleagues described above would suggest that systematic application of this proxy- phenotype method to genetic studies of other complex traits could be a useful complement to traditional gene discovery methods, if it provides larger samples, and thus greater statistical power.
GWAS of Specific Cognitive Abilities
Much like the studies of heritability in twins, substantially less is known regarding variants associated with specific cognitive abilities. There have been a handful of GWAS of mathematical ability: the first of these (applying top-performing SNPs in a study of 1200 children with high and low mathematics ability to a validation sample of ~2300 individuals) did not find any genome-wide significant variants (Docherty et al., 2010). Another GWAS in monozygotic vs. dizygotic twin pairs confirmed that children’s ability in reading and mathematics is under substantial genetic influence, and estimated that about 50% of the observed correlation in these traits is due to shared genetic effects (i.e., ‘Generalist Genes’). Davis and colleagues (2014) found several trend-level associations with mathematical abilities in this study, but again none reached genome-wide significance (Davis et al., 2014). Recently, in a combined meta-analysis of 3 cohorts of Chinese elementary school students, Chen and colleagues (2017) identified the first genome-wide significant SNPs associated with mathematical ability. In particular, they identified four intronic SNPs in the SPOCK1 gene, which has been implicated in various types of cancer and may play a key role during neurogenesis. De novo mutations of this gene may be protein-damaging, although this study offers the first evidence that it may also play a role in normal variation in cognitive abilities (Dhamija, Graham, Smaoui, Thorland, & Kirmani, 2014).
An early genome-wide screen in a relatively small Swiss cohort of healthy young adults (n=351) found that a locus encoding the kidney and brain expressed protein (KIBRA) gene - known to act as a binding partner for dendrin, a putative modulator of synaptic plasticity (Schneider et al., 2010) - was significantly associated with verbal learning and memory performance (Papassotiropoulos et al., 2006). This finding was replicated in a second Swiss cohort, using a visual episodic memory task, and in a US sample of cognitively healthy adults, using two other verbal memory tasks. No significant associations were found for control tasks of executive function, attention or working memory, suggesting that the action of the KIBRA protein is specific to hippocampal-dependent memory. The investigators further validated the association findings by fine-mapping the genomic region harboring the WWC1 gene which expresses the KIBRA protein and neighboring genes, to ensure that the observed association was not due to linkage disequilibrium with surrounding genes. Additionally, they found that expression levels of the truncated WWC1 gene were higher in brain regions critical for memory, the hippocampal formation and dentate gyrus, relative to brain structures not implicated in memory, in both human and murine brain tissue. Finally, functional magnetic resonance imaging (fMRI) studies of an associative memory task were conducted in a subset of Swiss participants. Although there were no genotype differences in encoding, nor in behavioral performance, T allele carries of the rs17070145 variant showed reduced neural activity in the medial temporal lobe and frontal cortex during retrieval relative to those without the T allele, suggesting that healthy individuals who do not carry the T allele require more activation in memory-related brain structures to achieve comparable performance.
Although some studies have failed to replicate this association (Need et al., 2008), other subsequent studies have found that KIBRA is associated with memory in healthy elderly adults (Schaper, Kolsch, Popp, Wagner, & Jessen, 2008), and have also found a modest role for this gene’s involvement in memory and risk for Alzheimer’s Disease (Corneveaux et al., 2010). In a large but ethnically diverse sample (N>5400), Burgess et al. (Burgess et al., 2011) found a suggestive protective effect of the KIBRA T allele on AD risk, but no significant associations with episodic memory. A recent meta-analysis of ~9000 individuals with episodic memory data and ~5000 with working memory performance reported a significant association of rs17070145 with both episodic (r =0.068, p = 0.001) and working memory (r = 0.035, p = 0.018) (Milnik et al., 2012), suggesting that the SNP rs17070145 explains 0.5% of the variance for episodic memory performance and 0.1% of the variance for working memory performance. Recently Witte et al. further pursued these findings, investigating verbal learning as well as structural and functional neural connectivity of the hippocampus in 140 individuals (Witte, Kobe, Kerti, Rujescu, & Floel, 2016). They found a non-significant trend for better memory performance in KIBRA T-allele carriers vs. non-T-allele carriers. T-allele carriers also showed significantly larger total hippocampal volume, differences in some metrics of white matter integrity in hippocampal subfields, and ‘more selective’ functional connectivity of the hippocampus. While these findings need to be replicated, they are interesting in that they suggest some potential biological mechanisms associated with KIBRA genotype. Thus, the multi-tiered approach taken by Papassotiropoulos and colleagues offers a compelling example of the value of going beyond a statistical association in a GWAS to achieve further insights into mechanisms of gene function.
The Candidate Gene Controversy
In contrast to a GWAS, candidate gene studies focus on associations between genetic variation within particular genes of interest and quantitative traits and/or disease states. These are typically conducted as case-control studies, with pre-specified candidate genes selected based on a priori knowledge of the physiological, functional or biochemical aspects of the gene. While these types of studies proliferated in the field of cognitive genomics, investigating a select set of candidate genes in relationship to a vast number of phenotypes, many of these findings have failed to replicate (Carter et al., 2016; Flint & Munafo, 2013). Investigators have pointed to a ‘fatal information bottleneck’ as being a major limitation for candidate gene studies (i.e., akin to looking for one’s keys under the lamp-post because “that’s where the light is”) (Zhu & Zhao, 2007). The fact that many existing candidate gene findings of significant associations with complex traits (e.g., cognitive functions or brain structure) have often reported effect sizes with R2 values more than one hundred times larger than those identified in GWAS induces further skepticism about the validity of such findings (Benjamin et al., 2012). Estimates from large- scale GWAS that common variants account for <1 % of the variance in a complex trait like g can serve as a useful benchmark for evaluating the plausibility of such findings in the literature (Rietveld et al., 2013).
Polygenic Risk, Cognition and Overlap with Cognitive Disorders
Although even the largest effect sizes for associations between specific common variants and behavioral or cognitive traits are very small, it is possible to aggregate the effects of thousands of SNP associations into a single score for a particular trait. This liability distribution is based on the selection of alleles from the results of large scale case-control GWAS conducted in a discovery sample, and involves creation of a weighted sum of these disease-associated alleles, referred to as a polygenic risk score (PRS) (SM Purcell et al.). The PRS can then be constructed for any individual in an independent target sample. PRS for a disorder thus reflects the individual-level genetic ‘burden’ for a particular disorder that is attributable to common genetic variation, and can be applied to assess associations with additional intermediate phenotypes. An association between PRS and a trait in the target sample (e.g., an illness or quantitative measure) implies that the genetic signal associated with the illness studied in the discovery sample can be used for prediction of an individual’s trait values in the target sample (Wray et al., 2014). An advantage of this approach is that one can analyze genetic overlap between cognitive abilities and distinct neuropsychiatric disorders in a population sample, i.e., avoiding the confounding effects of disease state. In addition, given that PRS models the aggregate effect of large numbers of SNPs associated with disease status, it can explain a great deal more of the variance than can any individual SNP. However, there are also a number of limitations to the PRS approach. For example, the utility of the polygenic score method is dependent upon the absolute level of genetic variance explained by common variants in the discovery sample (D. Glahn & McIntosh, 2017). Thus, for illnesses like schizophrenia where a large set of common SNPs have been identified (Schizophrenia Working Group of the Psychiatric Genomics, 2014), the method may work well. In contrast, for disorders like Attention Deficit Hyperactivity Disorder (ADHD), where we have yet to identify a genomewide significant locus [although see (Middeldorp et al., 2016)], the method may be less useful at present. Additionally, it should be noted that substantial sample sizes are still needed for stable results with the PRS approach. While there is a strong mathematical argument that the size of the discovery and target samples should be approximately equal (Dudbridge, 2013), others have suggested that a sample size of approximately 2000 individuals is sufficient to detect a significant proportion of variance in a target sample (Wray et al., 2013).
Several valuable insights have been gained via the investigation of polygenic profiles of variants collectively associated with cognitive abilities. For example, in their GWAS analysis of educational attainment, in an independent sample of over 8000 older Americans, Rietveld and colleagues (2014) found that a polygenic score obtained from the education-associated SNPs was associated with memory performance and absence of dementia. Clarke and colleagues (2016) assessed the relationship between polygenic risk for autism spectrum disorder (ASD) and ADHD with general cognitive abilities and other specific cognitive measures (memory, attention and fluency) in a large population-based Scottish cohort, as well as two replication samples. This study revealed that polygenic risk for ASD was positively associated with multiple cognitive functions, although effect sizes were small (ASD polygenic scores explained less than 0.5% of the variance in cognitive performance). In contrast, genetic risk for ADHD was negatively correlated with IQ at age 11 in one of the cohorts, although this finding didn’t replicate in the other cohorts (Clarke et al., 2016). Notably, in a population-based sample in Iceland, Stefansson and colleagues (2013) found that rare copy number variants (de novo mutations) that increase risk for ASD are associated with poorer cognitive function in healthy individuals; that is, clinically unaffected carriers of a chromosome 16p11.2 deletion were significantly impaired on measures of verbal intelligence, working memory and executive function (Stefansson et al., 2014). These somewhat contrasting findings suggest that the relationship between ASD and cognition may be complicated by distinct classes of genetic risk associated with ASD (common vs. rare variation, as discussed in more detail below).
Given that cognitive dysfunction is a hallmark feature of schizophrenia (Green, 1996) and of genetic liability for the illness (Mark & Toulopoulou, 2016; Van Erp et al., 2002), several studies have investigated the overlap of schizophrenia PRS with cognitive traits. For example, in the Avon Longitudinal Study of Parents and Children (ALSPAC) cohort, a prospective population-based cohort study of over 14,000 children, schizophrenia PRS was significantly associated with lower performance IQ, poorer language fluency and intelligibility, and lower social understanding at ages 7–9 (Riglin et al., 2017). Schizophrenia PRS also predicted social and behavioral impairment as early as age 4, suggesting that subtle behavioral anomalies in early childhood, which have been previously implicated in high-risk and epidemiologic studies, may reflect early manifestations of broad genetic liability to psychosis. A follow-up study of this sample found that about 14% of the genetic contribution to schizophrenia was shared with that for performance IQ (but not Verbal IQ or other cognitive measures), which implicates fluid rather than crystallized intelligence in the genetic liability for schizophrenia (Hubbard et al., 2016). Using data from the COGENT consortium, Lencz and colleagues (2013) examined the relationship between the schizophrenia PRS and g in ~5000 individuals, reporting that polygenic risk scores for SCZ were associated with lower global cognitive ability in the general population. Together these studies replicate prior findings of genetic correlations between schizophrenia and cognitive ability in family-based studies (Cannon et al., 2000; D. Glahn et al., 2007; Toulopoulou et al., 2007).
Estimating Heritability and Genetic Correlations in Unrelated Individuals
In general, the total variance explained by identified variants in GWAS is much smaller than the reported trait heritability, leading many investigators to speculate about causes of ‘missing heritability’ (Eichler et al., 2010; Zuk, Hechter, Sunyaev, & Lander, 2012). Some conclude that non-additive variation must make a substantial contribution (Zuk et al., 2012). However, typical GWAS studies focus exclusivly on common genetic variation and it is more than possible that rare variation may be critical for understanding the genetic arcetecture of individual differecnes in cognition in the normative range.
A new method called genome-wide complex-trait analysis (GCTA) allows estimation of the trait variance explained by SNPs (i.e., common variants) in unrelated individuals, commonly referred to as SNP-based heritability (Yang et al., 2010; Yang, Lee, Goddard, & Visscher, 2011). In the recent analysis of UK Biobank data from over 100,000 individuals, this approach identified significant SNP-based heritabilities of 31% (s.e.m.=1.8%) for verbal-numerical reasoning, 5% (s.e.m.=0.6%) for memory, 11% (s.e.m.=0.6%) for reaction time and 21% (s.e.m.=0.6%) for educational attainment (Davies et al., 2016). Using the same basic methodology, Robinson and colleagues (2015) estimated that SNP-based heritability for several cognitive measures using 3689 members of the Philadelphia Neurodevelopmental Cohort, a general population sample comprising those aged 8–21 years. The investigators reported a strong influence of common genetic variants on measures like reading ability (0.43, p=4×10−6), emotion identification (0.36, p=1×10−5), and verbal memory (0.24, p=0.005) in this developmental sample (Robinson et al., 2015).
Notably, SNP-derived heritability estimates tend to be lower than estimates from twin or family studies. This may reflect uncaptured non-additive influences (e.g., dominance, genegene interactions), and possibly gene-environment (GE) correlations that are captured in heritability estimates derived from twin studies (Reynolds & Finkel, 2015), or may reflect the impact of rare variation that is ostensibly present in twin or family studies but less prominent in the GCTA method.
Linkage disequilibrium (LD) score regression is a method of using GWAS summary-level statistics to quantify the extent to which two phenotypes share genetic etiology (at least with respect to common variants), by deriving genetic correlations between different phenotypes (B. Bulik-Sullivan et al., 2015; B. K. Bulik-Sullivan et al., 2015). This method yielded intriguing results in the UK Biobank sample, identifying robust genetic correlations between cognitive test scores and several mental and physical health related traits and disorders (Hagenaars et al., 2016). Cognitive performance was also strongly associated with polygenic profiles scores for coronary artery disease, stroke, Alzheimer's disease, psychiatric disorders including schizophrenia, autism, major depressive disorder, as well as body mass index and intracranial volume. These results would suggest genetic pleiotropy between cognition and many aspects of human health, both mental and physical. Resources such as LD Hub - a centralized database of summary-level GWAS results for hundreds of diseases/traits from different publicly available resources - have been recently developed, greatly facilitating analysis of hundreds of traits that potentially share common genetic etiologies (Zheng et al., 2017).
Continuum Model - or Not?
It may be that common behavioral and cognitive disorders represent variation at the extremes of the same factors (both genetic and environmental) associated with normal variation (R. Plomin, 1999). Given that intellectual disability (ID) is a highly heritable trait (Robert Plomin et al., 2013), many cognitive researchers assume ID (typically defined as IQ below 70, two standard deviations below the population mean) to represent the extreme low end of the normal distribution. In other words, the same genes that contribute to the normal range of individual differences in intelligence may be responsible for causing a disorder, at the extreme end of the continuum. Supporting this notion, Paracchini and colleagues (2006) followed up their initial important findings of a significant QTL for reading disability on chromosome 6p by identifying a haplotype in this region of chromosome 6p22 that was both associated with dyslexia in two independent family cohorts, and was also associated with reduced expression of KIAA0319, a gene in the region implicated in brain development via regulation of neuronal migration and cell adhesion (Paracchini et al., 2006). In a subsequent study in the ALSPAC cohort, the authors genotyped the 4 significant SNPs in the locus (Paracchini et al., 2008). One SNP in particular- part of the 3-SNP risk haplotype identified previously – was significantly associated with variation in reading ability at age 7–9, whereas the risk haplotype was associated with poor reading. These results implicate the KIAA0319 gene in dyslexia and also suggest that this gene may influence reading ability in the general population (Paracchini et al., 2008).
At the same time, advances in our understanding of the contribution of rare genomic structural variation to intellectual disability and other developmental neuropsychiatric disorders indicate that non-inherited de novo mutations play an important role in the etiology of severe intellectual disability (de Ligt et al., 2012; Ellison, Rosenfeld, & Shaffer, 2013; Veltman & Brunner, 2012). Increased copy number burden, as well as other rare mutations, have been found to increase the rate of ID, as well as neurodevelopmental psychiatric disorders (schizophrenia and ASD (Sebat et al., 2007)). As postulated in 1938 by Lionel Penrose, it may be the case that most ID is caused by the same genetic and enviromental influences as those that influence the normal IQ distribution, but that severe ID is etiologically distinct (Penrose, 1938). This hypothesis was recently tested by Reichenberg and colleagues (2016) in a sample of over 1 million sibling pairs and 9000 male twin pairs. These findings revealed that mild ID (ie, the lowest 3% of IQ scores) is highly familial, as siblings of individuals with mild ID have substantially lower IQs than the general population (Reichenberg et al., 2016). Twin analyses further indicated that about half the variance in IQ scores throughout the entire distribution is attributable to genetic influences. In contrast, severe ID was not familial as IQs of siblings of individuals with severe ID are indistinguishable from the rest of the population. These findings offer important evidence for discontinuity of genetic influences on ID; namely that mild ID represents the extreme end of the normal distribution, and is thus likely to be highly polygenic, reflecting the contribution of many genes of small effect. However, severe ID appears to be etiologically distinct; the non-inherited effects could reflect de novo mutations, although it is not yet known what proportion of ID is accounted for by these genomic events (Reichenberg et al., 2016). It is also important to note that several environmental factors, such as pregnancy and birth complications, and environmental exposures (such as lead) could also cause severe ID. Further research is warranted on the etiology of severe LD, as well as the spectrum of cognitive effects of de novo mutations.
Translational Studies
Identification of genes associated with individual differences in human cognitive abilities will provide insights into the way in which brain pathways leading from genes to complex cognitive processes can be understood and experimentally manipulated in animal models. Findings regarding g (and high genetic correlations between cognitive abilities) would suggest that in fact, even at a cellular level, most genes are likely to have broad influence on cognitive abilities, as opposed to isolated effects on specific domains (R. Plomin, 1999). Nevertheless, much of the work on molecular mechanisms of cognition has focused on processes of learning and memory, as the underlying cellular processes are now well understood (Bear et al., 2004; Kauer, Malenka, & Nicoll, 1988a, 1988b; Malenka, 1991; Silva, Elgersma, & Costa, 2000). Such foundational studies have led to major insights regarding the cellular and circuit bases of learning deficits in the context of specific knockout models of human genes that cause cognitive disorders; translational studies of Neurofibromatosis I (Nf1) provide a compelling example.
Nf1 is one of the most common single-gene disorders affecting brain function in humans. It results from a mutation in the neurofibromin gene on chromosome 17, which is critical for orderly differentiation of neurons in the central nervous system (North, 2000). Nf1 is associated with serious and debilitating learning disabilities, particularly in the areas of visuospatial learning and executive function, which are present in the vast majority of patients, even in the absence of ostensible neural pathology. Mouse models have provided invaluable insights into the etiology of cognitive disabilities associated with Nf1. As the mouse and human neurofibromin are highly homologous (98% sequence similarity), as are the promoter sequences of the gene, its biochemistry is highly conserved across species (Silva, Elgersma, Friedman, Stern, & Kogan, 1998). Mice carrying NF1 mutations share important characteristics of the NF1 cognitive profile, including prominent working memory deficits. In 2002, Costa and colleagues made the key discovery that the learning deficits of Nf1 mice appear due to excessive Ras signaling activity which leads to impairments in long-term potentiation. Furthermore, these defects could be rescued by genetic and pharmacological manipulations that decrease function of the p21 Ras signaling pathway(Costa et al., 2002). Given these important findings, therapeutic interventions designed to inhibit Ras function have been proposed as treatments for NF1. Lovastatin, a specific inhibitor of the rate-limiting enzyme in cholesterol biosynthesis, has been shown to inhibit p21 Ras activity, and to rescue the learning and electrophysiological deficits in Nf1 mice (Li et al., 2005). This finding has generated a great deal of interest in randomized clinical trials to assess whether statin treatment may also be able to ameliorate cognitive deficits in human patients with Nf1. Thus far, the results of these clinical trials are mixed (C. E. Bearden et al., 2016; Payne et al., 2016), highlighting the challenges of translating findings in mouse models into the clinic. It is likely that the NF1 mutation affects cognition via downstream effects on the expression of multiple other genes, which further emphasizes the complexity of developing treatments for cognitive disability in humans.
Future Directions
The field of cognitive genomics has advanced by leaps and bounds just in the past few years. In particular, this has been facilitated by increasingly large-scale, collaborative studies. For other complex traits, including metabolic, neuroanatomic, and anthropomorphic traits, the use of comparable measures across studies has allowed for meta-analytic genetic analyses (and the identification of replicable findings) (Wood et al., 2014). It is imperative that cognitive researchers develop tools for high-throughput cognitive phenotyping that can be used across a broad range of populations. For example, the development of computerized adaptive testing strategies (Wainer & Dorans, 2000) with adequate psychometric properties for the rapid and efficient assessment of cognitive abilities would greatly facilitate such efforts.
Although genetic variants that influence cognitive performance have been identified, the path from variant discovery (which tend to be of very small effect) to a mechanistic understanding is slow. This level of understanding is needed for the development of treatments for ID and specific cognitive impairments, which is imperative for improving quality of life for millions of individuals suffering from these disorders. Furthermore, as cognitive deficits have been proposed as key intermediate phenotypes or endophenotypes for a number of neuropsychiatric disorders, notably schizophrenia, cognitive endophenotypes may offer a window into the neurobiological signature of schizophrenia and other developmental or neurodegenerative brain disorders (D. C. Glahn et al., 2014). By examining the impact of schizophrenia-associated loci on cognitive phenotypes, researchers could determine relevant brain circuitry or other biological mechanisms by which risk variants may act to produce the complex and heterogeneous outcome of psychosis (Hall & Smoller, 2010).
In addition, the advent of sequencing technologies (both exome and whole genome) is likely to reveal a host of novel rare variants associated with cognitive function. For example, a recent study of African American individuals focusing on coding variants associated with Alzheimer’s Disease (AD) identified two missense variants associated with AD risk, as well as suggestive evidence for coding variants that may be protective by modulating cognition (N’Songo et al., 2017). As noted above, rare variants may be more relevant to larger, deleterious effects on cognition (Reichenberg et al., 2016); however, it is also possible that rare variants associated with enhanced cognitive function may be discovered.
We have gained major insights into the molecular underpinnings of cognition, in both health and disease, via pre-clinical studies in animal models. Studies of cognitive disorders in mouse models to date have focused on knockout models and specific mutations, but in order to more accurately reflect human variability, it is important to examine these phenotypes in the context of a variety of genetic backgrounds. Further, examination of correlation structure between multiple cognitive measures would allow investigators to determine whether there is a g phenotype in animal models. Finally, studies of learning and memory to date focused on mouse models which do not recapitulate aspects of the primate brain very well.
Summary
With the sequencing of the human genome, increasingly sophisticated analytic tools, and ever-larger sample sizes via collaborative studies, progress identifying the genetic underpinnings of cogntive abalities has greatly accelerated. Indeed, we may now be moving towards pinpointing the actual genetic and molecular basis of cognitive abilities, leading to optimism regarding possibilities for novel treatments for illnesses related to cogntive function. Identification of genetic variants associated with cognitive abilities, both general and specific, can lead to insights regarding the underlying biological pathways, which can be further interrogated in order to link biological mechanisms to behavior.
Significance.
The genetic contribution to human intelligence has long been a topic of considerable controversy. Recently, new analytic methods and dramatically increased sample sizes have led to major advances in our understanding of the genetic architecture of cognition. Here, we review this literature, including the latest developments in the field, which are now leading to new insights regarding the genetic underpinnings of normal and abnormal cognitive abilities.
Acknowledgments
We wish to thank Dr. Jennifer Forsyth for insightful and very helpful feedback on the manuscript draft. This work was supported in part by NIH grants R01MH085953 and U54EB020403.
GLOSSARY OF TERMS
- Allele
Variant form of a gene. Most multi-cellular organisms are diploid, meaning they have two sets of genes. If both alleles at a locus are the same, that organism is homozygous at that particular locus; it is heterozygous if it has two different alleles at that locus.
- Genome Wide Association Study (GWAS)
Also known as a whole genome association study, this is an analytic approach that examines a genome wide set of variants across individuals to determine whether any variants are associated with a particular trait of interest. Genetic correlation: Also denoted as ρg (rho g), the proportion of variance shared by two traits that is due to genetic causes
- Linkage disequilibrium (LD)
non-random association of alleles at two different loci (i.e., when the transmission of two alleles is not independent)
- Intron
Segment of a DNA molecule that does not code for proteins and is removed by RNA splicing during maturation of the final RNA product
- Single Nucleotide Polymorphism (SNP)
Variation in a single nucleotide that occurs at a specific base position in the genome. SNPs may fall within coding or non-coding regions of genes, or in intergenic regions
Footnotes
The authors report no relevant financial conflicts of interest.
Contributor Information
Carrie E. Bearden, University of California at Los Angeles.
David C. Glahn, Yale University.
References
- Alarcon M, Plomin R, Fulker DW, Corley R, DeFries JC. Multivariate path analysis of specific cognitive abilities data at 12 years of age in the Colorado Adoption Project. Behav Genet. 1998;28(4):255–264. doi: 10.1023/a:1021667213066. [DOI] [PubMed] [Google Scholar]
- Almasy L. Quantitative risk factors as indices of alcoholism susceptibility. Ann Med. 2003;35(5):337–343. doi: 10.1080/07853890310004903. [DOI] [PubMed] [Google Scholar]
- Almasy L, Blangero J. Endophenotypes as quantitative risk factors for psychiatric disease: rationale and study design. American Journal of Medical Genetics. 2001;105(1):42–44. [PubMed] [Google Scholar]
- Ando J, Ono Y, Wright MJ. Genetic structure of spatial and verbal working memory. Behav Genet. 2001;31(6):615–624. doi: 10.1023/a:1013353613591. [DOI] [PubMed] [Google Scholar]
- Bear MF, Huber KM, Warren ST. The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004;27(7):370–377. doi: 10.1016/j.tins.2004.04.009. [DOI] [PubMed] [Google Scholar]
- Bearden C, Freimer N. Endophenotypes for psychiatric disorders: ready for primetime? Trends Genet. 2006;22(6):306–313. doi: 10.1016/j.tig.2006.04.004. [DOI] [PubMed] [Google Scholar]
- Bearden CE, Hellemann GS, Rosser T, Montojo C, Jonas R, Enrique N, Silva AJ. A randomized placebo-controlled lovastatin trial for neurobehavioral function in neurofibromatosis I. Ann Clin Transl Neurol. 2016;3(4):266–279. doi: 10.1002/acn3.288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamin DJ, Cesarini D, Chabris CF, Glaeser EL, Laibson DI, Guethnason V, Lichtenstein P. The Promises and Pitfalls of Genoeconomics*. Annu Rev Econom. 2012;4:627–662. doi: 10.1146/annurev-economics-080511-110939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benyamin B, Pourcain B, Davis OS, Davies G, Hansell NK, Brion MJ, Visscher PM. Childhood intelligence is heritable, highly polygenic and associated with FNBP1L. Mol Psychiatry. 2014;19(2):253–258. doi: 10.1038/mp.2012.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blangero J. Localization and identification of human quantitative trait loci: king harvest has surely come. Curr Opin Genet Dev. 2004;14(3):233–240. doi: 10.1016/j.gde.2004.04.009. [DOI] [PubMed] [Google Scholar]
- Blangero J, Goring HH, Kent JW, Jr, Williams JT, Peterson CP, Almasy L, Dyer TD. Quantitative trait nucleotide analysis using Bayesian model selection. Hum Biol. 2005;77(5):541–559. doi: 10.1353/hub.2006.0003. [DOI] [PubMed] [Google Scholar]
- Bookheimer SY, Strojwas MH, Cohen MS, Saunders AM, Pericak-Vance MA, Mazziotta JC, Small GW. Patterns of brain activation in people at risk for Alzheimer’s disease. N Engl J Med. 2000;343(7):450–456. doi: 10.1056/NEJM200008173430701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boomsma D, Busjahn A, Peltonen L. Classical twin studies and beyond. Nat Rev Genet. 2002;3(11):872–882. doi: 10.1038/nrg932. [DOI] [PubMed] [Google Scholar]
- Bouchard TJ, Jr, McGue M. Familial studies of intelligence: A review. Science. 1981;212:1055–1059. doi: 10.1126/science.7195071. [DOI] [PubMed] [Google Scholar]
- Bouchard TJ, Jr, Segal NL, Lykken DT. Genetic and environmental influences on special mental abilities in a sample of twins reared apart. Acta Geneticae Medicae et Gemellologiae. 1990;39(2):193–206. doi: 10.1017/s0001566000005420. [DOI] [PubMed] [Google Scholar]
- Bulik-Sullivan B, Finucane HK, Anttila V, Gusev A, Day FR, Loh PR, Neale BM. An atlas of genetic correlations across human diseases and traits. Nat Genet. 2015;47(11):1236–1241. doi: 10.1038/ng.3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulik-Sullivan BK, Loh PR, Finucane HK, Ripke S, Yang J, Schizophrenia Working Group of the Psychiatric Genomics, C. Neale BM. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet. 2015;47(3):291–295. doi: 10.1038/ng.3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvin CM, Deary IJ, Webbink D, Smith P, Fernandes C, Lee SH, Visscher PM. Multivariate genetic analyses of cognition and academic achievement from two population samples of 174,000 and 166,000 school children. Behav Genet. 2012;42(5):699–710. doi: 10.1007/s10519-012-9549-7. [DOI] [PubMed] [Google Scholar]
- Cannon T, Huttunen M, Lonnqvist J, Tuulio-Henriksson A, Pirkola T, Glahn D, Koskenvuo M. The inheritance of neuropsychological dysfunction in twins discordant for schizophrenia. Am J Hum Genet. 2000;67(2):369–382. doi: 10.1086/303006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter CS, Bearden CE, Bullmore ET, Geschwind DH, Glahn DC, Gur RE, Weinberger DR. Enhancing the Informativeness and Replicability of Imaging Genomics Studies. Biol Psychiatry. 2016 doi: 10.1016/j.biopsych.2016.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Gu XH, Zhou Y, Ge Z, Wang B, Siok WT, Sun Y. A GenomeWide Association Study Identifies Genetic Variants Associated with Mathematics Ability. Sci Rep. 2017;7:40365. doi: 10.1038/srep40365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Church C, Lee S, Bagg EA, McTaggart JS, Deacon R, Gerken T, Cox RD. A mouse model for the metabolic effects of the human fat mass and obesity associated FTO gene. PLoS Genet. 2009;5(8):e1000599. doi: 10.1371/journal.pgen.1000599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke TK, Lupton MK, Fernandez-Pujals AM, Starr J, Davies G, Cox S, McIntosh AM. Common polygenic risk for autism spectrum disorder (ASD) is associated with cognitive ability in the general population. Mol Psychiatry. 2016;21(3):419–425. doi: 10.1038/mp.2015.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261(5123):921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- Corneveaux JJ, Liang WS, Reiman EM, Webster JA, Myers AJ, Zismann VL, Huentelman MJ. Evidence for an association between KIBRA and late-onset Alzheimer’s disease. Neurobiol Aging. 2010;31(6):901–909. doi: 10.1016/j.neurobiolaging.2008.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa RM, Federov NB, Kogan JH, Murphy GG, Stern J, Ohno M, Silva AJ. Mechanism for the learning deficits in a mouse model of neurofibromatosis type 1. Nature. 2002;415(6871):526–530. doi: 10.1038/nature711. [DOI] [PubMed] [Google Scholar]
- Cuthbert BN, Insel TR. Toward the future of psychiatric diagnosis: the seven pillars of RDoC. BMC Med. 2013;11:126. doi: 10.1186/1741-7015-11-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies G, Armstrong N, Bis JC, Bressler J, Chouraki V, Giddaluru S, Deary IJ. Genetic contributions to variation in general cognitive function: a meta-analysis of genome-wide association studies in the CHARGE consortium (N=53949) Mol Psychiatry. 2015;20(2):183–192. doi: 10.1038/mp.2014.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies G, Harris SE, Reynolds CA, Payton A, Knight HM, Liewald DC, Deary IJ. A genome-wide association study implicates the APOE locus in nonpathological cognitive ageing. Mol Psychiatry. 2014;19(1):76–87. doi: 10.1038/mp.2012.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies G, Marioni RE, Liewald DC, Hill WD, Hagenaars SP, Harris SE, Deary IJ. Genome-wide association study of cognitive functions and educational attainment in UK Biobank (N=112 151) Mol Psychiatry. 2016;21(6):758–767. doi: 10.1038/mp.2016.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies G, Tenesa A, Payton A, Yang J, Harris SE, Liewald D, Deary IJ. Genome-wide association studies establish that human intelligence is highly heritable and polygenic. Mol Psychiatry. 2011;16(10):996–1005. doi: 10.1038/mp.2011.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis OS, Band G, Pirinen M, Haworth CM, Meaburn EL, Kovas Y, Spencer CC. The correlation between reading and mathematics ability at age twelve has a substantial genetic component. Nat Commun. 2014;5:4204. doi: 10.1038/ncomms5204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis OS, Haworth CM, Plomin R. Learning abilities and disabilities: generalist genes in early adolescence. Cogn Neuropsychiatry. 2009;14(4–5):312–331. doi: 10.1080/13546800902797106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Jager PL, Shulman JM, Chibnik LB, Keenan BT, Raj T, Wilson RS, Evans DA. A genome-wide scan for common variants affecting the rate of age-related cognitive decline. Neurobiol Aging. 2012;33(5):1017 e1011–1015. doi: 10.1016/j.neurobiolaging.2011.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Ligt J, Willemsen MH, van Bon BW, Kleefstra T, Yntema HG, Kroes T, Vissers LE. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med. 2012;367(20):1921–1929. doi: 10.1056/NEJMoa1206524. [DOI] [PubMed] [Google Scholar]
- Deary IJ, Johnson W, Houlihan LM. Genetic foundations of human intelligence. Hum Genet. 2009;126(1):215–232. doi: 10.1007/s00439-009-0655-4. [DOI] [PubMed] [Google Scholar]
- Deary IJ, Whiteman MC, Pattie A, Starr JM, Hayward C, Wright AF, Whalley LJ. Cognitive change and the APOE epsilon 4 allele. Nature. 2002;418(6901):932. doi: 10.1038/418932a. [DOI] [PubMed] [Google Scholar]
- Deary IJ, Whiteman MC, Starr JM, Whalley LJ, Fox HC. The impact of childhood intelligence on later life: following up the Scottish mental surveys of 1932 and 1947. J Pers Soc Psychol. 2004;86(1):130–147. doi: 10.1037/0022-3514.86.1.130. [DOI] [PubMed] [Google Scholar]
- Debette S, Ibrahim Verbaas CA, Bressler J, Schuur M, Smith A, Bis JC, Aging Research in Genomic Epidemiology, C Genome-wide studies of verbal declarative memory in nondemented older people: the Cohorts for Heart and Aging Research in Genomic Epidemiology consortium. Biol Psychiatry. 2015;77(8):749–763. doi: 10.1016/j.biopsych.2014.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devlin B, Daniels M, Roeder K. The heritability of IQ. Nature. 1997;388:468–471. doi: 10.1038/41319. [DOI] [PubMed] [Google Scholar]
- Dhamija R, Graham JM, Jr, Smaoui N, Thorland E, Kirmani S. Novel de novo SPOCK1 mutation in a proband with developmental delay, microcephaly and agenesis of corpus callosum. Eur J Med Genet. 2014;57(4):181–184. doi: 10.1016/j.ejmg.2014.02.009. [DOI] [PubMed] [Google Scholar]
- Dina C, Meyre D, Gallina S, Durand E, Korner A, Jacobson P, Froguel P. Variation in FTO contributes to childhood obesity and severe adult obesity. Nat Genet. 2007;39(6):724–726. doi: 10.1038/ng2048. [DOI] [PubMed] [Google Scholar]
- Docherty SJ, Davis OS, Kovas Y, Meaburn EL, Dale PS, Petrill SA, Plomin R. A genome-wide association study identifies multiple loci associated with mathematics ability and disability. Genes Brain Behav. 2010;9(2):234–247. doi: 10.1111/j.1601-183X.2009.00553.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudbridge F. Power and predictive accuracy of polygenic risk scores. PLoS Genet. 2013;9(3):e1003348. doi: 10.1371/journal.pgen.1003348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichler EE, Flint J, Gibson G, Kong A, Leal SM, Moore JH, Nadeau JH. Missing heritability and strategies for finding the underlying causes of complex disease. Nat Rev Genet. 2010;11(6):446–450. doi: 10.1038/nrg2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellison JW, Rosenfeld JA, Shaffer LG. Genetic basis of intellectual disability. Annu Rev Med. 2013;64:441–450. doi: 10.1146/annurev-med-042711-140053. [DOI] [PubMed] [Google Scholar]
- Engelhardt LE, Briley DA, Mann FD, Harden KP, Tucker-Drob EM. Genes Unite Executive Functions in Childhood. Psychol Sci. 2015;26(8):1151–1163. doi: 10.1177/0956797615577209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falconer DS, Mackay TFC. Introduction to quantitative genetics. 4th. Harlow, England; New York: Prentice Hall; 1996. [Google Scholar]
- Fan J, Wu Y, Fossella J, Posner M. Assessing the heritability of attentional networks. BMC Neuroscience. 2001;2(14):1–7. doi: 10.1186/1471-2202-2-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fears SC, Service SK, Kremeyer B, Araya C, Araya X, Bejarano J, Bearden CE. Multisystem component phenotypes of bipolar disorder for genetic investigations of extended pedigrees. JAMA Psychiatry. 2014;71(4):375–387. doi: 10.1001/jamapsychiatry.2013.4100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel D, McGue M. The origins of individual differences in memory among the elderly: a behavior genetic analysis. Psychol Aging. 1993;8(4):527–537. doi: 10.1037//0882-7974.8.4.527. [DOI] [PubMed] [Google Scholar]
- Finkel D, Pedersen N, McGue M. Genetic influences on memory performance in adulthood: comparison of Minnesota and Swedish twin data. Psychol Aging. 1995;10(3):437–446. doi: 10.1037//0882-7974.10.3.437. [DOI] [PubMed] [Google Scholar]
- Finkel D, Pedersen NL, McGue M, McClearn GE. Heritability of cognitive abilities in adult twins: comparison of Minnesota and Swedish data. Behavior Genetics. 1995;25(5):421–431. doi: 10.1007/BF02253371. [DOI] [PubMed] [Google Scholar]
- Finkel D, Reynolds CA, McArdle JJ, Hamagami F, Pedersen NL. Genetic variance in processing speed drives variation in aging of spatial and memory abilities. Dev Psychol. 2009;45(3):820–834. doi: 10.1037/a0015332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher PJ, Turic D, Williams NM, McGuffin P, Asherson P, Ball D, Owen MJ. DNA pooling identifies QTLs on chromosome 4 for general cognitive ability in children. Hum Mol Genet. 1999;8(5):915–922. doi: 10.1093/hmg/8.5.915. [DOI] [PubMed] [Google Scholar]
- Fisher SE, Marlow AJ, Lamb J, Maestrini E, Williams DF, Richardson AJ, Monaco AP. A quantitative-trait locus on chromosome 6p influences different aspects of developmental dyslexia. Am J Hum Genet. 1999;64(1):146–156. doi: 10.1086/302190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint J, Munafo MR. Candidate and non-candidate genes in behavior genetics. Curr Opin Neurobiol. 2013;23(1):57–61. doi: 10.1016/j.conb.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint J, Timpson N, Munafo M. Assessing the utility of intermediate phenotypes for genetic mapping of psychiatric disease. Trends Neurosci. 2014;37(12):733–741. doi: 10.1016/j.tins.2014.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn JR. Skodak and Skeels: The inflated mother-child IQ gap. Intelligence. 1993;17(4):557–561. [Google Scholar]
- Galton F. Hereditary genius: an inquiry into its laws and consequences. London: Macmillan and co; 1869. [Google Scholar]
- Glahn D, Almasy L, Blangero J, Burk G, Estrada J, Peralta J, Escamilla M. Adjudicating neurocognitive endophenotypes for schizophrenia. Am J Med Genet B Neuropsychiatr Genet. 2007;144B(2):242–249. doi: 10.1002/ajmg.b.30446. [DOI] [PubMed] [Google Scholar]
- Glahn D, McIntosh A. Using Polygenic Risk Scores to Establish Endophenotypes: Considerations and Current Constraints. Biological Psychiatry: Cognitive Neuroscience and Neuroimaging. 2017;2(2):113–114. doi: 10.1016/j.bpsc.2017.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glahn DC, Blangero J. Why endophenotype development requires families. Chinese Science Bulletin. 2011;56:3382–3384. [Google Scholar]
- Glahn DC, Curran JE, Winkler AM, Carless MA, Kent JW, Charlesworth JC, Blangero J. High dimensional endophenotype ranking in the search for major depression risk genes. Biol Psychiatry. 2012;71(1):6–14. doi: 10.1016/j.biopsych.2011.08.022. doi:S0006-3223(11)00857-2 [pii] 10.1016/j.biopsych.2011.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glahn DC, Knowles EE, McKay DR, Sprooten E, Raventos H, Blangero J, Almasy L. Arguments for the sake of endophenotypes: Examining common misconceptions about the use of endophenotypes in psychiatric genetics. Am J Med Genet B Neuropsychiatr Genet. 2014 doi: 10.1002/ajmg.b.32221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glahn DC, Williams JT, McKay DR, Knowles EE, Sprooten E, Mathias SR, Blangero J. Discovering schizophrenia endophenotypes in randomly ascertained pedigrees. Biol Psychiatry. 2015;77(1):75–83. doi: 10.1016/j.biopsych.2014.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottesman II, Gould TD. The Endophenotype Concept in Psychiatry: Etymology and Strategic Intentions. American Journal of Psychiatry. 2003;160:636–645. doi: 10.1176/appi.ajp.160.4.636. [DOI] [PubMed] [Google Scholar]
- Gottesman II, Shields J. Schizophrenia and genetics: A twin study vantage point. New York: Academic Press; 1972. [Google Scholar]
- Gottfredson LS. Why g matters: The complexity of everyday life. Intelligence. 1997;24(1):79–132. [Google Scholar]
- Gould SJ. The mismeasure of man. 1st. New York: Norton; 1981. [Google Scholar]
- Gould T, Gottesman I. Psychiatric endophenotypes and the development of valid animal models. Genes Brain Behav. 2006;5(2):113–119. doi: 10.1111/j.1601-183X.2005.00186.x. [DOI] [PubMed] [Google Scholar]
- Gow AJ, Johnson W, Pattie A, Brett CE, Roberts B, Starr JM, Deary IJ. Stability and change in intelligence from age 11 to ages 70, 79, and 87: the Lothian Birth Cohorts of 1921 and 1936. Psychol Aging. 2011;26(1):232–240. doi: 10.1037/a0021072. [DOI] [PubMed] [Google Scholar]
- Green MF. What are the functional consequences of neurocognitive deficits in schizophrenia? American Journal of Psychiatry. 1996;153(3):321–330. doi: 10.1176/ajp.153.3.321. [DOI] [PubMed] [Google Scholar]
- Hagenaars SP, Harris SE, Davies G, Hill WD, Liewald DC, Ritchie SJ, Deary IJ. Shared genetic aetiology between cognitive functions and physical and mental health in UK Biobank (N=112 151) and 24 GWAS consortia. Mol Psychiatry. 2016;21(11):1624–1632. doi: 10.1038/mp.2015.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall MH, Smoller JW. A new role for endophenotypes in the GWAS era: functional characterization of risk variants. Harv Rev Psychiatry. 2010;18(1):67–74. doi: 10.3109/10673220903523532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haworth CM, Wright MJ, Luciano M, Martin NG, de Geus EJ, van Beijsterveldt CE, Plomin R. The heritability of general cognitive ability increases linearly from childhood to young adulthood. Mol Psychiatry. 2010;15(11):1112–1120. doi: 10.1038/mp.2009.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibar DP, Adams HH, Jahanshad N, Chauhan G, Stein JL, Hofer E, Ikram MA. Novel genetic loci associated with hippocampal volume. Nat Commun. 2017;8:13624. doi: 10.1038/ncomms13624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibar DP, Stein JL, Renteria ME, Arias-Vasquez A, Desrivieres S, Jahanshad N, Medland SE. Common genetic variants influence human subcortical brain structures. Nature. 2015;520(7546):224–229. doi: 10.1038/nature14101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho HY, Rohatgi R, Lebensohn AM, Le M, Li J, Gygi SP, Kirschner MW. Toca-1 mediates Cdc42-dependent actin nucleation by activating the N-WASP-WIP complex. Cell. 2004;118(2):203–216. doi: 10.1016/j.cell.2004.06.027. [DOI] [PubMed] [Google Scholar]
- Hubbard L, Tansey KE, Rai D, Jones P, Ripke S, Chambert KD, Zammit S. Evidence of Common Genetic Overlap Between Schizophrenia and Cognition. Schizophr Bull. 2016;42(3):832–842. doi: 10.1093/schbul/sbv168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insel T, Cuthbert B. Endophenotypes: bridging genomic complexity and disorder heterogeneity. Biol Psychiatry. 2009;66(11):988–989. doi: 10.1016/j.biopsych.2009.10.008. [DOI] [PubMed] [Google Scholar]
- Jacob N, Van Gestel S, Derom C, Thiery E, Vernon P, Derom R, Vlietinck R. Heritability estimates of intelligence in twins: effect of chorion type. Behavior Genetics. 2001;31(2):209–217. doi: 10.1023/a:1010257512183. [DOI] [PubMed] [Google Scholar]
- Johnson W, Turkheimer E, Gottesman II, Bouchard TJ., Jr Beyond Heritability: Twin Studies in Behavioral Research. Curr Dir Psychol Sci. 2010;18(4):217–220. doi: 10.1111/j.1467-8721.2009.01639.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauer JA, Malenka RC, Nicoll RA. NMDA application potentiates synaptic transmission in the hippocampus. Nature. 1988a;334(6179):250–252. doi: 10.1038/334250a0. [DOI] [PubMed] [Google Scholar]
- Kauer JA, Malenka RC, Nicoll RA. A persistent postsynaptic modification mediates long-term potentiation in the hippocampus. Neuron. 1988b;1(10):911–917. doi: 10.1016/0896-6273(88)90148-1. [DOI] [PubMed] [Google Scholar]
- Kendler KS, Turkheimer E, Ohlsson H, Sundquist J, Sundquist K. Family environment and the malleability of cognitive ability: a Swedish national home-reared and adopted-away cosibling control study. Proc Natl Acad Sci U S A. 2015;112(15):4612–4617. doi: 10.1073/pnas.1417106112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles EE, Carless MA, de Almeida MA, Curran JE, McKay DR, Sprooten E, Glahn DC. Genome-wide significant localization for working and spatial memory: Identifying genes for psychosis using models of cognition. Am J Med Genet B Neuropsychiatr Genet. 2014;165(1):84–95. doi: 10.1002/ajmg.b.32211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovas Y, Plomin R. Generalist genes: implications for the cognitive sciences. Trends Cogn Sci. 2006;10(5):198–203. doi: 10.1016/j.tics.2006.03.001. [DOI] [PubMed] [Google Scholar]
- Lander E, Kruglyak L. Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet. 1995;11(3):241–247. doi: 10.1038/ng1195-241. [DOI] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, International Human Genome Sequencing, C Initial sequencing and analysis of the human genome. Nature. 2001;409(6822):860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Lencz T, Knowles E, Davies G, Guha S, Liewald DC, Starr JM, Malhotra AK. Molecular genetic evidence for overlap between general cognitive ability and risk for schizophrenia: a report from the Cognitive Genomics consorTium (COGENT) Mol Psychiatry. 2013 doi: 10.1038/mp.2013.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenzenweger MF. Endophenotype, intermediate phenotype, biomarker: definitions, concept comparisons, clarifications. Depress Anxiety. 2013;30(3):185–189. doi: 10.1002/da.22042. [DOI] [PubMed] [Google Scholar]
- Li W, Cui Y, Kushner SA, Brown RA, Jentsch JD, Frankland PW, Silva AJ. The HMG-CoA reductase inhibitor lovastatin reverses the learning and attention deficits in a mouse model of neurofibromatosis type 1. Curr Biol. 2005;15(21):1961–1967. doi: 10.1016/j.cub.2005.09.043. [DOI] [PubMed] [Google Scholar]
- Liu JZ, McRae AF, Nyholt DR, Medland SE, Wray NR, Brown KM, Macgregor S. A versatile gene-based test for genome-wide association studies. Am J Hum Genet. 2010;87(1):139–145. doi: 10.1016/j.ajhg.2010.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luciano M, Wright M, Smith GA, Geffen GM, Geffen LB, Martin NG. Genetic covariance among measures of information processing speed, working memory, and IQ. Behavior Genetics. 2001;31(6):581–592. doi: 10.1023/a:1013397428612. [DOI] [PubMed] [Google Scholar]
- Lynch M, Walsh B. Genetics and analysis of quantitative traits. Sunderland, Mass: Sinauer; 1998. [Google Scholar]
- Malenka RC. The role of postsynaptic calcium in the induction of long-term potentiation. Mol Neurobiol. 1991;5(2–4):289–295. doi: 10.1007/BF02935552. [DOI] [PubMed] [Google Scholar]
- Manolio TA. Genomewide association studies and assessment of the risk of disease. N Engl J Med. 2010;363(2):166–176. doi: 10.1056/NEJMra0905980. [DOI] [PubMed] [Google Scholar]
- Mark W, Toulopoulou T. Cognitive intermediate phenotype and genetic risk for psychosis. Curr Opin Neurobiol. 2016;36:23–30. doi: 10.1016/j.conb.2015.08.008. [DOI] [PubMed] [Google Scholar]
- McClearn GE, Johansson B, Berg S, Pedersen NL, Ahern F, Petrill SA, Plomin R. Substantial genetic influence on cognitive abilities in twins 80 or more years old. Science. 1997a;276(5318):1560–1563. doi: 10.1126/science.276.5318.1560. [DOI] [PubMed] [Google Scholar]
- McClearn GE, Johansson B, Berg S, Pedersen NL, Ahern F, Petrill SA, Plomin R. Substantial genetic influence on cognitive abilities in twins 80 or more years old. Science. 1997b;276:1560–1563. doi: 10.1126/science.276.5318.1560. [DOI] [PubMed] [Google Scholar]
- McDaniel MA. Big-brained people are smarter: A meta-analysis of the relationship between in vivo brain volume and intelligence. Intelligence. 2004;33(4):337–346. [Google Scholar]
- McGue M, Christensen K. The heritability of cognitive functioning in very old adults: evidence from Danish twins aged 75 years and older. Psychol Aging. 2001;16(2):272–280. doi: 10.1037//0882-7974.16.2.272. [DOI] [PubMed] [Google Scholar]
- McGue M, Christensen K. Growing old but not growing apart: twin similarity in the latter half of the lifespan. Behav Genet. 2013;43(1):1–12. doi: 10.1007/s10519-012-9559-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middeldorp CM, Hammerschlag AR, Ouwens KG, Groen-Blokhuis MM, Pourcain BS, Greven CU, Boomsma DI. A Genome-Wide Association MetaAnalysis of Attention-Deficit/Hyperactivity Disorder Symptoms in Population-Based Pediatric Cohorts. J Am Acad Child Adolesc Psychiatry. 2016;55(10):896–905 e896. doi: 10.1016/j.jaac.2016.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milnik A, Heck A, Vogler C, Heinze HJ, de Quervain DJ, Papassotiropoulos A. Association of KIBRA with episodic and working memory: a meta-analysis. Am J Med Genet B Neuropsychiatr Genet. 2012;159B(8):958–969. doi: 10.1002/ajmg.b.32101. [DOI] [PubMed] [Google Scholar]
- Need AC, Attix DK, McEvoy JM, Cirulli ET, Linney KN, Wagoner AP, Goldstein DB. Failure to replicate effect of Kibra on human memory in two large cohorts of European origin. Am J Med Genet B Neuropsychiatr Genet. 2008;147B(5):667–668. doi: 10.1002/ajmg.b.30658. [DOI] [PubMed] [Google Scholar]
- Neubauer A, Spinath F, Riemann R, Angleitner A. Genetic and Environmental Influences on Two Measures of Speed of Information Processing and Their Relation to Psychometric Intelligence: Evidence from the German Observational Study of Adult Twins. Intelligence. 2000;28(4):267–289. [Google Scholar]
- Nisbett RE, Aronson J, Blair C, Dickens W, Flynn J, Halpern DF, Turkheimer E. Intelligence: new findings and theoretical developments. Am Psychol. 2012;67(2):130–159. doi: 10.1037/a0026699. [DOI] [PubMed] [Google Scholar]
- North K. Neurofibromatosis type 1. Am J Med Genet. 2000;97(2):119–127. doi: 10.1002/1096-8628(200022)97:2<119::aid-ajmg3>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Okbay A, Beauchamp JP, Fontana MA, Lee JJ, Pers TH, Rietveld CA, Benjamin DJ. Genome-wide association study identifies 74 loci associated with educational attainment. Nature. 2016;588(7604):539–542. doi: 10.1038/nature17671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panizzon MS, Lyons MJ, Jacobson KC, Franz CE, Grant MD, Eisen SA, Kremen WS. Genetic architecture of learning and delayed recall: a twin study of episodic memory. Neuropsychology. 2011;25(4):488–498. doi: 10.1037/a0022569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papassotiropoulos A, Stephan DA, Huentelman MJ, Hoerndli FJ, Craig DW, Pearson JV, de Quervain DJ. Common Kibra alleles are associated with human memory performance. Science. 2006;814(5798):475–478. doi: 10.1126/science.1129837. doi:314/5798/475 [pii] 10.1126/science. 1129837. [DOI] [PubMed] [Google Scholar]
- Paracchini S, Steer CD, Buckingham LL, Morris AP, Ring S, Scerri T, Monaco AP. Association of the KIAA0319 dyslexia susceptibility gene with reading skills in the general population. Am J Psychiatry. 2008;165(12):1576–1584. doi: 10.1176/appi.ajp.2008.07121872. [DOI] [PubMed] [Google Scholar]
- Paracchini S, Thomas A, Castro S, Lai C, Paramasivam M, Wang Y, Monaco AP. The chromosome 6p22 haplotype associated with dyslexia reduces the expression of KIAA0319, a novel gene involved in neuronal migration. Hum Mol Genet. 2006;15(10):1659–1666. doi: 10.1093/hmg/ddl089. [DOI] [PubMed] [Google Scholar]
- Payne JM, Barton B, Ullrich NJ, Cantor A, Hearps SJ, Cutter G, Consortium, N. F. C. T Randomized placebo-controlled study of lovastatin in children with neurofibromatosis type 1. Neurology. 2016;87(24):2575–2584. doi: 10.1212/WNL.0000000000003435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penrose L. A Clinical and Genetic Study of 1,280 Cases of Mental Defect. Special Report Series. Medical Research Council. 1938;229:159. [Google Scholar]
- Pericak-Vance MA, Bebout JL, Gaskell PC, Jr, Yamaoka LH, Hung WY, Alberts MJ, et al. Linkage studies in familial Alzheimer disease: evidence for chromosome 19 linkage. Am J Hum Genet. 1991;48(6):1034–1050. [PMC free article] [PubMed] [Google Scholar]
- Peters N, Opherk C, Danek A, Ballard C, Herzog J, Dichgans M. The pattern of cognitive performance in CADASIL: a monogenic condition leading to subcortical ischemic vascular dementia. Am J Psychiatry. 2005;162(11):2078–2085. doi: 10.1176/appi.ajp.162.11.2078. [DOI] [PubMed] [Google Scholar]
- Petrill SA. Molarity Versus Modularity of Cognitive Functioning? A Behavioral Genetic Perspective. Current Directions in Psychological Science. 1997;6(4):96–99. [Google Scholar]
- Plomin R. Genetics and general cognitive ability. Nature. 1999;402(6761 Suppl):C25–29. doi: 10.1038/35011520. [DOI] [PubMed] [Google Scholar]
- Plomin R, DeFries JC, Knopik VS, Neiderhiser JM. Behavioral genetics : a primer. Sixth. New York: Worth Publishers; 2013. [Google Scholar]
- Plomin R, DeFries JC, Knopik VS, Neiderhiser JM. Top 10 Replicated Findings From Behavioral Genetics. Perspect Psychol Sci. 2016;11(1):3–23. doi: 10.1177/1745691615617439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plomin R, Kosslyn SM. Genes, brain and cognition. Nat Neurosci. 2001;4(12):1153–1154. doi: 10.1038/nn1201-1153. [DOI] [PubMed] [Google Scholar]
- Plomin R, Kovas Y. Generalist genes and learning disabilities. Psychol Bull. 2005;131(4):592–617. doi: 10.1037/0033-2909.131.4.592. [DOI] [PubMed] [Google Scholar]
- Plomin R, Owen MJ, McGuffin P. The genetic basis of complex human behaviors. Science. 1994;264(5166):1733–1739. doi: 10.1126/science.8209254. [DOI] [PubMed] [Google Scholar]
- Plomin R, Spinath FM. Intelligence: genetics, genes, and genomics. J Pers Soc Psychol. 2004;86(1):112–129. doi: 10.1037/0022-3514.86.1.112. [DOI] [PubMed] [Google Scholar]
- Polderman TJ, Benyamin B, de Leeuw CA, Sullivan PF, van Bochoven A, Visscher PM, Posthuma D. Meta-analysis of the heritability of human traits based on fifty years of twin studies. Nat Genet. 2015;47(7):702–709. doi: 10.1038/ng.3285. [DOI] [PubMed] [Google Scholar]
- Posthuma D, de Geus EJ, Boomsma DI. Perceptual speed and IQ are associated through common genetic factors. Behav Genet. 2001;31(6):593–602. doi: 10.1023/a:1013349512683. [DOI] [PubMed] [Google Scholar]
- Purcell S. Variance components models for gene-environment interaction in twin analysis. Twin Res. 2002;5(6):554–571. doi: 10.1375/136905202762342026. [DOI] [PubMed] [Google Scholar]
- Purcell S, Wray N, Stone J, Visscher P, O’Donovan M, Sullivan P, Consortium, I.S Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460(7256):748–752. doi: 10.1038/nature08185. doi:nature08185 [pii] 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramakers GJ. Rho proteins, mental retardation and the cellular basis of cognition. Trends Neurosci. 2002;25(4):191–199. doi: 10.1016/s0166-2236(00)02118-4. [DOI] [PubMed] [Google Scholar]
- Reichenberg A, Cederlof M, McMillan A, Trzaskowski M, Kapara O, Fruchter E, Lichtenstein P. Discontinuity in the genetic and environmental causes of the intellectual disability spectrum. Proc Natl Acad Sci U S A. 2016;113(4):1098–1103. doi: 10.1073/pnas.1508093112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds CA, Finkel D. A meta-analysis of heritability of cognitive aging: minding the “missing heritability” gap. Neuropsychol Rev. 2015;25(1):97–112. doi: 10.1007/s11065-015-9280-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rietveld CA, Esko T, Davies G, Pers TH, Turley P, Benyamin B, Koellinger PD. Common genetic variants associated with cognitive performance identified using the proxy-phenotype method. Proc Natl Acad Sci U S A. 2014;111(38):13790–13794. doi: 10.1073/pnas.1404623111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rietveld CA, Medland SE, Derringer J, Yang J, Esko T, Martin NW, Study, L.C GWAS of 126,559 individuals identifies genetic variants associated with educational attainment. Science. 2013;340(6139):1467–1471. doi: 10.1126/science.1235488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riglin L, Collishaw S, Richards A, Thapar AK, Maughan B, O’Donovan MC, Thapar A. Schizophrenia risk alleles and neurodevelopmental outcomes in childhood: a population-based cohort study. Lancet Psychiatry. 2017;4(1):57–62. doi: 10.1016/S2215-0366(16)30406-0. [DOI] [PubMed] [Google Scholar]
- Risch N, Merikangas K. The future of genetic studies of complex human diseases. Science. 1996;273(5281):1516–1517. doi: 10.1126/science.273.5281.1516. [DOI] [PubMed] [Google Scholar]
- Robinson EB, Kirby A, Ruparel K, Yang J, McGrath L, Anttila V, Hakonarson H. The genetic architecture of pediatric cognitive abilities in the Philadelphia Neurodevelopmental Cohort. Mol Psychiatry. 2015;20(4):454–458. doi: 10.1038/mp.2014.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseau F, Heitz D, Biancalana V, Blumenfeld S, Kretz C, Boue J, et al. Direct diagnosis by DNA analysis of the fragile X syndrome of mental retardation. N Engl J Med. 1991;325(24):1673–1681. doi: 10.1056/NEJM199112123252401. [DOI] [PubMed] [Google Scholar]
- Schaper K, Kolsch H, Popp J, Wagner M, Jessen F. KIBRA gene variants are associated with episodic memory in healthy elderly. Neurobiol Aging. 2008;29(7):1123–1125. doi: 10.1016/j.neurobiolaging.2007.02.001. [DOI] [PubMed] [Google Scholar]
- Schizophrenia Working Group of the Psychiatric Genomics, C. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511(7510):421–427. doi: 10.1038/nature13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider A, Huentelman MJ, Kremerskothen J, Duning K, Spoelgen R, Nikolich K. KIBRA: A New Gateway to Learning and Memory? Front Aging Neurosci. 2010;2:4. doi: 10.3389/neuro.24.004.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, Wigler M. Strong association of de novo copy number mutations with autism. Science. 2007;316(5823):445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva AJ, Elgersma Y, Costa RM. Molecular and cellular mechanisms of cognitive function: implications for psychiatric disorders. Biol Psychiatry. 2000;47(3):200–209. doi: 10.1016/s0006-3223(99)00294-2. [DOI] [PubMed] [Google Scholar]
- Silva AJ, Elgersma Y, Friedman E, Stern J, Kogan J. A mouse model for learning and memory defects associated with neurofibromatosis type I. Pathol Biol (Paris) 1998;46(9):697–698. [PubMed] [Google Scholar]
- Skodak M, Skeels HM. A final follow-up study of 100 adopted children. J Genet Psychol. 1949;75(1):85–125. doi: 10.1080/08856559.1949.10533511. [DOI] [PubMed] [Google Scholar]
- Small BJ, Rosnick CB, Fratiglioni L, Backman L. Apolipoprotein E and cognitive performance: a meta-analysis. Psychol Aging. 2004;19(4):592–600. doi: 10.1037/0882-7974.19.4.592. [DOI] [PubMed] [Google Scholar]
- Smemo S, Tena JJ, Kim KH, Gamazon ER, Sakabe NJ, Gomez-Marin C, Nobrega MA. Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature. 2014;507(7492):371–375. doi: 10.1038/nature13138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sniekers S, Stringer S, Watanabe K, Jansen PR, Coleman JRI, Krapohl E, Posthuma D. Genome-wide association meta-analysis of 78,308 individuals identifies new loci and genes influencing human intelligence. Nat Genet. 2017;49(7):1107–1112. doi: 10.1038/ng.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefansson H, Meyer-Lindenberg A, Steinberg S, Magnusdottir B, Morgen K, Arnarsdottir S, Stefansson K. CNVs conferring risk of autism or schizophrenia affect cognition in controls. Nature. 2014;505(7483):361–366. doi: 10.1038/nature12818. [DOI] [PubMed] [Google Scholar]
- Stein JL, Medland SE, Vasquez AA, Hibar DP, Senstad RE, Winkler AM, Enhancing Neuro Imaging Genetics through Meta-Analysis, C Identification of common variants associated with human hippocampal and intracranial volumes. Nat Genet. 2012;44(5):552–561. doi: 10.1038/ng.2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, Roses AD. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90(5):1977–1981. doi: 10.1073/pnas.90.5.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swan GE, Carmelli D. Evidence for genetic mediation of executive control: a study of aging male twins. Journals of Gerontology Series B-Psychological Sciences & Social Sciences. 2002;57(2):P133–143. doi: 10.1093/geronb/57.2.p133. [DOI] [PubMed] [Google Scholar]
- Swan GE, Reed T, Jack LM, Miller BL, Markee T, Wolf PA, Carmelli D. Differential genetic influence for components of memory in aging adult twins. Archives of Neurology. 1999;56(9):1127–1132. doi: 10.1001/archneur.56.9.1127. [DOI] [PubMed] [Google Scholar]
- Terwilliger JD, Ott J. Handbook of human genetic linkage. Baltimore: Johns Hopkins University Press; 1994. [Google Scholar]
- Toulopoulou T, Picchioni M, Rijsdijk F, Hua-Hall M, Ettinger U, Sham P, Murray R. Substantial genetic overlap between neurocognition and schizophrenia: genetic modeling in twin samples. Arch Gen Psychiatry. 2007;64(12):1348–1355. doi: 10.1001/archpsyc.64.12.1348. [DOI] [PubMed] [Google Scholar]
- Trampush JW, Lencz T, Knowles E, Davies G, Guha S, Pe’er I, Malhotra AK. Independent evidence for an association between general cognitive ability and a genetic locus for educational attainment. Am J Med Genet B Neuropsychiatr Genet. 2015;168B(5):363–373. doi: 10.1002/ajmg.b.32319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trampush JW, Yang ML, Yu J, Knowles E, Davies G, Liewald DC, Lencz T. GWAS meta-analysis reveals novel loci and genetic correlates for general cognitive function: a report from the COGENT consortium. Mol Psychiatry. 2017;22(3):336–345. doi: 10.1038/mp.2016.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trzaskowski M, Davis OS, DeFries JC, Yang J, Visscher PM, Plomin R. DNA evidence for strong genome-wide pleiotropy of cognitive and learning abilities. Behav Genet. 2013;43(4):267–273. doi: 10.1007/s10519-013-9594-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker-Drob EM, Briley DA. Continuity of genetic and environmental influences on cognition across the life span: a meta-analysis of longitudinal twin and adoption studies. Psychol Bull. 2014;140(4):949–979. doi: 10.1037/a0035893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tung YC, Yeo GS. From GWAS to biology: lessons from FTO. Ann N Y Acad Sci. 2011;1220:162–171. doi: 10.1111/j.1749-6632.2010.05903.x. [DOI] [PubMed] [Google Scholar]
- Turkheimer E. Three laws of behavior genetics and what they mean. Current Directions in Psychological Science. 2000;9:160–164. [Google Scholar]
- Turkheimer E, Haley A, Waldron M, D’Onofrio B, Gottesman II. Socioeconomic status modifies heritability of IQ in young children. Psychological Science. 2003;14(6):623–628. doi: 10.1046/j.0956-7976.2003.psci_1475.x. [DOI] [PubMed] [Google Scholar]
- van der Sluis S, Verhage M, Posthuma D, Dolan CV. Phenotypic complexity, measurement bias, and poor phenotypic resolution contribute to the missing heritability problem in genetic association studies. PLoS One. 2010;5(11):e13929. doi: 10.1371/journal.pone.0013929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Erp TG, Saleh PA, Rosso IM, Huttunen M, Leonnqvist J, Pirkola T, Cannon TD. Contributions of genetic risk and fetal hypoxia to hippocampal volume in patients with schizophrenia or schizoaffective disorder, their unaffected siblings, and healthy unrelated volunteers. The American Journal of Psychiatry. 2002;159(9):1514–1520. doi: 10.1176/appi.ajp.159.9.1514. [DOI] [PubMed] [Google Scholar]
- van Leeuwen M, Peper JS, van den Berg SM, Brouwer RM, Hulshoff Pol HE, Kahn RS, Boomsma DI. A genetic analysis of brain volumes and IQ in children. Intelligence. 2009;37:181–191. [Google Scholar]
- Veltman JA, Brunner HG. De novo mutations in human genetic disease. Nat Rev Genet. 2012;13(8):565–575. doi: 10.1038/nrg3241. [DOI] [PubMed] [Google Scholar]
- Volk HE, McDermott KB, Roediger HL, 3rd, Todd RD. Genetic influences on free and cued recall in long-term memory tasks. Twin Res Hum Genet. 2006;9(5):623–631. doi: 10.1375/183242706778553462. [DOI] [PubMed] [Google Scholar]
- Vuoksimaa E, Panizzon MS, Chen CH, Fiecas M, Eyler LT, Fennema-Notestine C, Kremen WS. The Genetic Association Between Neocortical Volume and General Cognitive Ability Is Driven by Global Surface Area Rather Than Thickness. Cereb Cortex. 2015;25(8):2127–2137. doi: 10.1093/cercor/bhu018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wainer H, Dorans NJ. Computerized adaptive testing : a primer. 2nd. Mahwah, N.J.: Lawrence Erlbaum Associates; 2000. [Google Scholar]
- Wainwright MA, Wright MJ, Geffen GM, Luciano M, Martin NG. The genetic basis of academic achievement on the Queensland Core Skills Test and its shared genetic variance with IQ. Behav Genet. 2005;35(2):133–145. doi: 10.1007/s10519-004-1014-9. [DOI] [PubMed] [Google Scholar]
- White K. The relation between socioeconomic status and academic achievement. Psychological Bulletin. 1982;91(3):461–481. [Google Scholar]
- Winkleby MA, Jatulis DE, Frank E, Fortmann SP. Socioeconomic status and health: how education, income, and occupation contribute to risk factors for cardiovascular disease. Am J Public Health. 1992;82(6):816–820. doi: 10.2105/ajph.82.6.816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisdom NM, Callahan JL, Hawkins KA. The effects of apolipoprotein E on nonimpaired cognitive functioning: a meta-analysis. Neumbiol Aging. 2011;32(1):63–74. doi: 10.1016/j.neurobiolaging.2009.02.003. [DOI] [PubMed] [Google Scholar]
- Wood AR, Esko T, Yang J, Vedantam S, Pers TH, Gustafsson S, Frayling TM. Defining the role of common variation in the genomic and biological architecture of adult human height. Nat Genet. 2014;46(11):1173–1186. doi: 10.1038/ng.3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray NR, Lee SH, Mehta D, Vinkhuyzen AA, Dudbridge F, Middeldorp CM. Research review: Polygenic methods and their application to psychiatric traits. J Child Psychol Psychiatry. 2014;55(10):1068–1087. doi: 10.1111/jcpp.12295. [DOI] [PubMed] [Google Scholar]
- Wray NR, Yang J, Hayes BJ, Price AL, Goddard ME, Visscher PM. Pitfalls of predicting complex traits from SNPs. Nat Rev Genet. 2013;14(7):507–515. doi: 10.1038/nrg3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Benyamin B, McEvoy BP, Gordon S, Henders AK, Nyholt DR, Visscher PM. Common SNPs explain a large proportion of the heritability for human height. Nat Genet. 2010;42(7):565–569. doi: 10.1038/ng.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet. 2011;88(1):76–82. doi: 10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Pierce BL. Genetic susceptibility to accelerated cognitive decline in the US Health and Retirement Study. Neurobiol Aging. 2014;35(6):1512 e1511–1518. doi: 10.1016/j.neurobiolaging.2013.12.021. [DOI] [PubMed] [Google Scholar]
- Zheng J, Erzurumluoglu AM, Elsworth BL, Kemp JP, Howe L, Haycock PC, Neale BM. LD Hub: a centralized database and web interface to perform LD score regression that maximizes the potential of summary level GWAS data for SNP heritability and genetic correlation analysis. Bioinformatics. 2017;33(2):272–279. doi: 10.1093/bioinformatics/btw613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu M, Zhao S. Candidate gene identification approach: progress and challenges. Int J Biol Sci. 2007;3(7):420–427. doi: 10.7150/ijbs.3.420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zollino M, Orteschi D, Murdolo M, Lattante S, Battaglia D, Stefanini C, Marangi G. Mutations in KANSL1 cause the 17q21.31 microdeletion syndrome phenotype. Nat Genet. 2012;44(6):636–638. doi: 10.1038/ng.2257. [DOI] [PubMed] [Google Scholar]
- Zuk O, Hechter E, Sunyaev SR, Lander ES. The mystery of missing heritability: Genetic interactions create phantom heritability. Proc Natl Acad Sci U S A. 2012;109(4):1193–1198. doi: 10.1073/pnas.1119675109. [DOI] [PMC free article] [PubMed] [Google Scholar]