

Abstract
Objective
To investigate whether chronic traumatic encephalopathy (CTE) and CTE with amyotrophic lateral sclerosis (CTE-ALS) exhibit features previously observed in other tauopathies of pathologic phosphorylation of microtubule-associated protein tau at Thr175 (pThr175 tau) and Thr231 (pThr231 tau), and glycogen synthase kinase–3β (GSK3β) activation, and whether these pathologic features are a consequence of traumatic brain injury (TBI).
Methods
Tau isoform expression was assayed by western blot in 6 stage III CTE cases. We also used immunohistochemistry to analyze 5 cases each of CTE, CTE-ALS, and 5 controls for expression of activated GSK3β, pThr175 tau, pThr231 tau, and oligomerized tau within spinal cord tissue and hippocampus. Using a rat model of moderate TBI, we assessed tau pathology and phospho-GSK3β expression at 3 months postinjury.
Results
CTE and CTE-ALS are characterized by the presence of all 6 tau isoforms in both soluble and insoluble tau isolates. Activated GSK3β, pThr175 tau, pThr231 tau, and oligomerized tau protein expression was observed in hippocampal neurons and spinal motor neurons. We observed tau neuronal pathology (fibrillar inclusions and axonal damage) and increased levels of pThr175 tau and activated GSK3β in moderate TBI rats.
Conclusions
Pathologic phosphorylation of tau at Thr175 and Thr231 and activation of GSK3β are features of the tauopathy of CTE and CTE-ALS. These features can be replicated in an animal model of moderate TBI.
Chronic traumatic encephalopathy (CTE) is a fatal neurodegenerative disease that is closely associated with traumatic brain injury (TBI).1 Although TBI is typically associated with elite athletes, it also occurs as a result of both recreational and non-sport-related accidents.2,3 While there remains some controversy, a relationship between TBI and amyotrophic lateral sclerosis (ALS) has been observed, with between 4% and 6% of patients with CTE demonstrating either clinical or neuropathologic features consistent with a diagnosis of ALS.4–7 CTE is an affective, behavioral, or cognitive disorder associated with repetitive head trauma that has a characteristic pattern of widespread neuronal and glial microtubule-associated tau (tau protein) deposition.8 In patients with concomitant ALS (CTE-ALS), motor neuron degeneration is also observed, including the presence of TDP-43 neuronal cytoplasmic inclusions.9
The presence of a tauopathy in CTE and CTE-ALS raises the potential of a shared pathophysiology with ALS with cognitive impairment (ALSci), where tau is pathologically phosphorylated at threonine 175 (pThr175 tau) and threonine 231 (pThr231 tau), yielding pathogenic tau oligomers that subsequently assemble into insoluble pathologic tau fibrils.1,10–15 In this process, pThr175 tau induces phosphorylation and activation of glycogen synthase kinase–3β (GSK3β), which in turn promotes tau phosphorylation at Thr231.16 Supporting the key role of pThr175, pThr175 tau has only been observed in pathologic states, including a broad range of tauopathies.10,13,17
To date, there have been no detailed studies of the phosphorylation state of tau in either CTE or CTE-ALS. Given this, we examined postmortem archival tissues from patients with CTE and patients with CTE-ALS for pThr175, pThr 231, the active isoform of GSK3β (phospho-GSK3β [pGSK3β]), and tau oligomerization. We also examined whether this process can be triggered by moderate TBI in a rodent.
Methods
Standard protocol approvals, registrations, and patient consents
All studies on human tissues were conducted in accordance with the institutional ethics board standards at University Hospital (London, Canada) and Boston University School of Medicine (Massachusetts). All animal protocols were approved by the University of Western Ontario Animal Care Committee in accordance with the Canadian Council on Animal Care.
CTE and CTE-ALS studies
Microscope slides with 6-μm-thick hippocampal and spinal cord sections from neuropathologically confirmed cases of CTE (n = 5) and CTE-ALS (n = 5) in addition to 5 control cases with no neuropathologic evidence of a neurodegenerative disease state (cases 7–19) were used for immunohistochemical studies. An additional 6 cases (cases 1–6) were obtained as frozen tissue from anterior cingulate and temporal pole. All tissue and slides were obtained from the Veterans Affairs–Boston University–Concussion Legacy Foundation brain bank and were neuropathologically diagnosed as stage III CTE.8,18 Demographic data are summarized in table e-1 (http://links.lww.com/WNL/A99).
Tau fractionation and western blot
Tau protein was isolated from the anterior cingulate gyrus and the temporal pole of 6 stage III CTE cases and a single Alzheimer disease (AD) case as a control. Tau isolation, fractionation, dephosphorylation, and western blot were conducted as previously reported (e-Methods, http://links.lww.com/WNL/A100).19,20
After transfer to nitrocellulose membranes, samples were probed for total tau with rabbit anti-tau T14/T46 antibodies (Thermo-Fisher, Burlington, Canada). After horseradish peroxidase–conjugated secondary antibody labeling, blots were visualized using enhanced chemiluminescence (Perkin-Elmer, Waltham, MA).
Immunohistochemistry
Six-micrometer paraffin-embedded sections from the hippocampus and spinal cord were analyzed for all cases. Immunohistochemistry was conducted using a series of antibodies (complete details in table e-2, http://links.lww.com/WNL/A99) that recognized pThr175 tau (21st Century, Marlboro, MA), pThr231 tau (Thermo-Fisher), and oligomeric tau (T22; EMD Millipore, Billerica, CA), and activated GSK3β (pTyr216; BD Biosciences, Mississauga, Canada). Antigen retrieval (10 mM sodium citrate, 0.05% Tween 20 pH 6.0) was conducted for all antibodies using a pressure cooker (2100 Retriever; Aptum Biologics, Southampton, UK, table e-2). Endogenous peroxidase was quenched with 3% hydrogen peroxide (VWR, Mississauga, Canada). Primary antibody incubation was performed at 4°C overnight in blocking buffer (5% bovine serum albumin, 0.3% Triton-X 100 in 1 × phosphate-buffered saline). After washing, secondary antibody (1:200 biotinylated immunoglobulin G) incubation was performed for 1 hour at room temperature (RT) in blocking buffer.
Antigen
Antibody complex was visualized with horseradish peroxidase according to the manufacturer's instructions (Vectastain ABC kit, Vector Laboratories, Burlingame, CA), followed by substrate development with 3,3′-diaminobenzidine (DAB). Counterstaining was performed using Harris hematoxylin.
The extent of pathology was described semiquantitatively as previously reported using visualization with a 20× objective under light microscopy (Olympus BX45; Center Valley, PA).10,21 The semiquantitative scale was applied as follows: − = none; ± = fewer than 5; + = fewer than 10; ++ = more than 20 with scattered distribution; +++ = more than 20 but with locally dense distribution; ++++ = more than 20 with a diffuse distribution as observed for each 20× objective field. In addition, the case positive ratio was defined for each antibody as the number of cases showing any pathology (± or more) compared to the total number of cases stained. Spinal cord pathology was assessed by a binary scale due to the sparse nature of pathology where + = pathology present and − = pathology absent.
Colocalizations and fluorescence staining
Double labeling was performed on sections from the hippocampus from one case per double-label experiment. Tau protein was probed with pThr175 or oligomeric tau (T22) rabbit primary antibody overnight at 4°C and Alexa Fluor goat anti-rabbit 488 nm secondary (Thermo-Fisher) for 1 hour at RT. Rabbit anti-tau pThr231 antibody was labeled using a Zenon primary antibody labeling kit with Alexa Fluor 555 nm dye (Thermo-Fisher) and probed for 1 hour at RT. Slides were visualized within 24 hours by confocal imaging on a Zeiss (Oberkochen, Germany) LSM 510 Meta NLO multiphoton confocal microscope. For colocalizations with pGSK3β, staining was performed with mouse anti-pTyr216 GSK3β antibody (BD Biosciences) followed by secondary labeling with goat anti-mouse Alexa Fluor 633 nm (Invitrogen, Carlsbad, CA).
In vivo studies
Twelve adult female Sprague-Dawley rats were subjected to a 5-mm-diameter craniotomy followed by a single moderate head trauma (3.5 m/s, 2 mm deep, dwell time of 500 ms) using a cortical impactor (Precision Systems [Horsham, PA] model TBI 0310) (moderate TBI). After 3 months, all rats were killed by transcardiac perfusion with ice-cold saline after intraperitoneal injection with a lethal dose of Euthanyl. Six brains were drop-fixed in ice cold Bouin fixative (Thermo-Fisher) for immunohistochemical analysis while 6 were frozen on dry ice for neurochemical analysis. Bouin fixative was used to reduce artefactual tau pathology.22,23 After 24 hours of fixation, tissue was embedded in paraffin.
Western blots
Immunoblots were also performed using isolates from 6 moderate TBI rats and 4 age-matched controls (e-Methods, links.lww.com/WNL/A100). Briefly, brain tissue was homogenized in RIPA buffer containing protease and phosphatase inhibitors followed by concentration determination by modified Bradford assay (Bio-Rad, Hercules, CA). After immunoprecipitation of brain lysate for total tau (T46 antibody), the entire immunoprecipitation yield was run as a western blot (e-Methods). After transfer to nitrocellulose membrane, blots were probed with rabbit anti-pThr175 tau. Gels were stripped for 30 minutes at 50°C and reprobed with rabbit anti–total tau (Abcam, Cambridge, UK). pGSK3β studies were performed on total brain lysate with mouse anti-GSK3β pTyr216 followed by reprobing with mouse anti-total GSK3β (BD Biosciences). Blots were visualized digitally by enhanced chemiluminescence (Perkin-Elmer) (Bio-Rad Chemidoc MP imaging system and acquired with ImageLab 5.2.1 software). Densitometry was conducted in ImageJ.
Immunohistochemistry
Six moderate TBI and 3 age-matched control rat brains were cut to 5–6 μm thickness and stained for pThr175 tau, pThr231 tau, and pTyr216 GSK3β using the same antibodies and protocol used in human cases.
Statistical analysis
Statistical analyses were conducted using SigmaPlot 10.0 software. A one-way analysis of variance (ANOVA) was conducted following a Shapiro-Wilk test for normality. Post hoc Tukey test was conducted and a p value of 0.05 or lower was considered significant.
Results
Western blot of human CTE
Insoluble tau protein isolated from CTE cases contained all 6 isoforms in both phosphorylated and dephosphorylated isolates. This was in contrast to the 3 isoforms constituting the paired helical filament motif in the insoluble fraction observed in a control AD case (figure 1).20
Figure 1. Tau isoform characterization.

Representative Western blot of chronic traumatic encephalopathy (CTE)–derived fractionated tau protein shows all 6 tau isoforms in the insoluble fraction in distinction to the 3-isoform, paired helical filament (PHF) motif observed in Alzheimer disease (AD). (Probed with mouse anti-T14/T46 total tau antibody; blots shown are from the same case on 2 separate blots.)
Immunohistochemistry in CTE cases
pThr175, pThr231, and T22 immunoreactivity was observed in the hippocampal formation in all cases of CTE and CTE-ALS (table 1, figure 2A). This included tau-immunoreactive neurofibrillary tangles (NFTs) and dystrophic neurites throughout the CA1–4 regions and extending into the entorhinal cortex in all cases. Consistent with the limited pThr175 tau non-neuronal pathology observed previously,10 oligodendroglial tau immunoreactivity was observed to a limited extent in 5 cases only.10 When present in control cases (3/5), tau immunoreactivity was observed only as faint immunoreactive punctate neuronal staining in the absence of NFTs (figure 2B). No neuritic pathology was observed in controls.
Table 1.
Hippocampal and spinal cord pathology summary
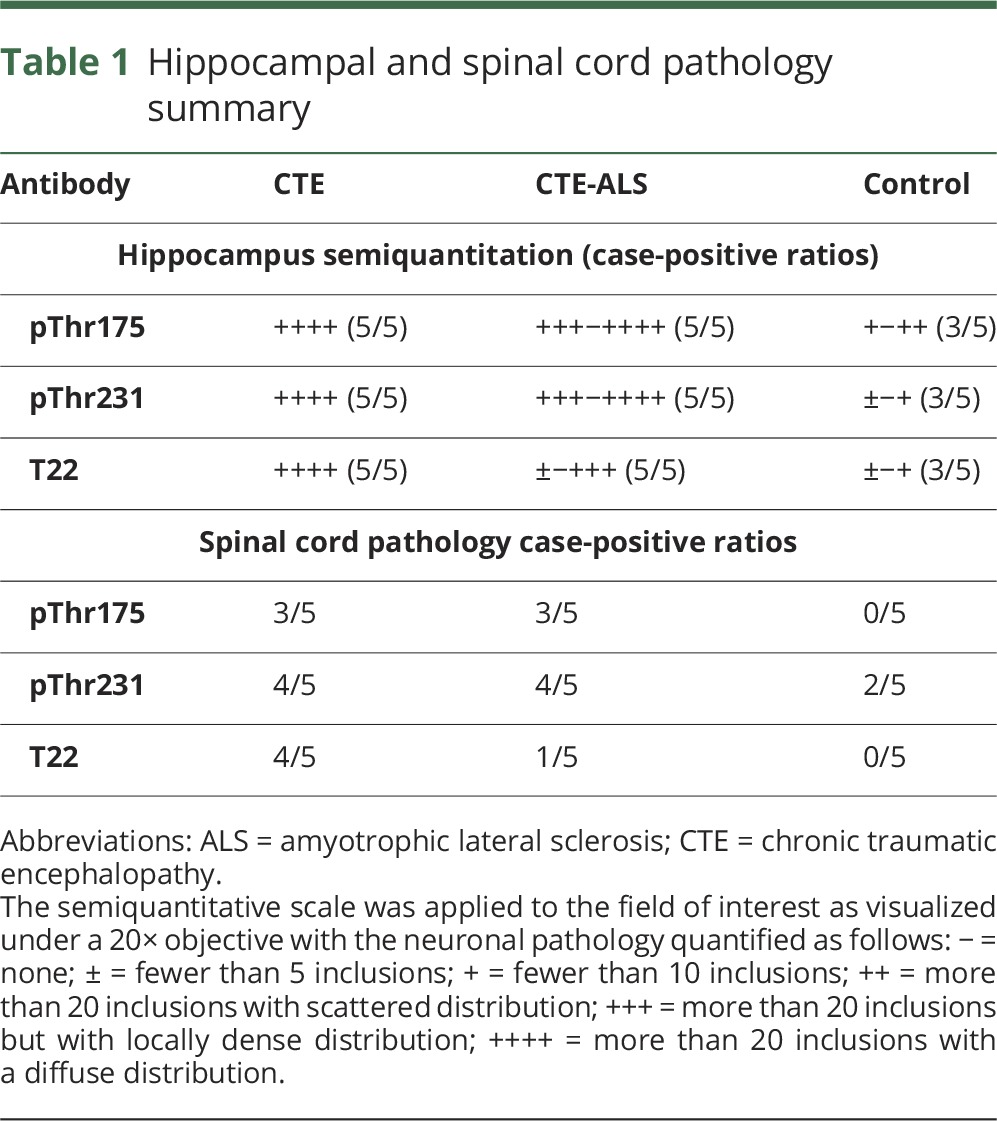
Figure 2. Tau pathology in hippocampus and ventral horn in chronic traumatic encephalopathy (CTE) and CTE–amyotrophic lateral sclerosis (ALS).

(A) Low-magnification (4× objective) composites of whole hippocampus stained for tau phosphorylated at threonine 175 (pThr175 tau) in CTE and control (inlay is CA2 region taken with 40× objective, scale bar = 50 μm). CA1 = CA1 region; CA3 = CA3 region; DG = dentate gyrus; sub = subiculum (composite image based on images taken with a 4× objective). Note the presence of prominent tau immunoreactivity in CA1 through CA4, extending into the entorhinal cortex. (B) Representative tau pathology observed in both the hippocampus (CTE case) and ventral horn of the spinal cord (CTE-ALS) when probed by rabbit anti-pThr175 tau, rabbit anti–tau phosphorylated at threonine 231 (pThr231 tau), or rabbit anti-T22 (tau oligomer) antibodies. Images taken with 100× objective (scale bar = 20 μm).
We also observed pathologic tau deposition within the spinal cord regardless of the tau phosphoepitope studied (figure 2B). The presence of tau pathology was independent of whether the underlying pathologic diagnosis was CTE or CTE-ALS (table 1) and consisted of sparsely distributed NFTs in motor neurons and dystrophic neurites. In all cases, pathologic inclusions were minimal in number relative to a more diffuse immunoreactivity to pThr231 tau and pThr175 tau. No pThr175 tau staining was observed in control motor neurons, whereas pThr231 tau was observed, but when present, only as diffuse perikaryal staining of motor neurons. Lipofuscin staining was observed in some controls.
We observed a shift in the pattern of immunoreactivity of pGSK3β from primarily nuclear as observed in the control cases to a diffusely cytosolic pattern (figure e-1, http://links.lww.com/WNL/A98). This pattern was observed in hippocampal CA2 and spinal cord motor neurons in all CTE cases while only being present in occasional isolated cells in controls.
Double-immunolabeling of the hippocampus for both pThr175 tau and pThr231 tau consistently demonstrated colocalization of immunoreactivity to pThr175 with pThr231 immunoreactivity. However, the converse was not observed in that not all pThr231 tau immunoreactive hippocampal neurons were also pThr175-positive, suggesting that only a subset of pThr231 tau pathology is also pThr175-positive.10 This is consistent with our previous observations.10 We also observed colocalization of pThr231 tau and T22 (oligomerized tau), suggesting that oligomeric tau was a component of pThr231 tau pathology. Due to the nature of the antibodies, it was not possible to test for colocalization of pThr175 with T22 as they were raised in the same species, not purified and not compatible with the primary antibody labeling system available to us. While we can therefore only infer that pThr175 tau protein colocalizes with oligomeric tau as well, this inference is supported by our previous studies of a range of tauopathies in which serial sections were available for analysis.10 The inference is further supported by our finding that pThr175 tau immunoreactivity consistently colocalized with active GSK3β (figure 3).
Figure 3. Colocalization of tau phosphorylated at threonine 175 (pThr175 tau) and tau phosphorylated at threonine 231 (pThr231 tau).

Hippocampal neurons in chronic traumatic encephalopathy (CTE) (upper panels) demonstrate colocalization of pThr175 tau and pThr231 tau. The presence of pathologic tau oligomers (T22 immunoreactivity) was colocalized to pThr231 tau-immunoreactive neurons in CTE (middle panels). Consistent with a role in activation of glycogen synthase kinase–3β (GSK3β) in inducing pathologic tau deposition, we observed the colocalization of pThr175tau with the active pGSK3β immunoreactivity (lower panel). Tissues were immunolabeled with rabbit anti-pThr175 tau, rabbit anti-pThr231 tau, rabbit anti-T22 antibody, and mouse anti-GSK3β pTyr216 antibody. For double-labeled tissue, red channel antibodies were labeled directly with Alexa Fluor 555. Scale bar = 5 μm.
pThr175 and pThr231 expression in moderate TBI
Both pThr175 and pThr231 tau neuronal immunoreactivity was observed in moderate TBI rat brains (figure 4). Of note, pThr175-positive neuronal staining was observed in regions distant to the injury site mainly as axonal staining; however, no fibrillar inclusion-type pathology was observed in regions distant from the injury site. While pThr175 tau expression was elevated within the hemisphere ipsilateral to the injury, pThr175 tau expression in the contralateral hemisphere, while appearing to be increased, was not different from controls when normalized against total tau (p = 0.05 and 0.065, respectively, one-way ANOVA with p = 0.008 and F = 5.757). Diffuse pThr231 staining was observed in healthy neurons. In contrast, pThr231 tau immunoreactive pathology was only observed near the site of injury.
Figure 4. Tau phosphorylated at threonine 175 (pThr175 tau) pathology in moderate traumatic brain injury (TBI).

At 3 months following moderate TBI, pThr175 tau pathology was observed. (A) Composite images of whole brain sections stained for pThr175 tau. Images were taken with a 4× objective. Inlay image taken with 40× objective. Arrow denotes site of injury. CA1 = CA1 region; CA3 = CA3 region; DG = dentate gyrus. Scale bar = 50 μm. (B) High-magnification images with moderate TBI show neuronal and neuritic pathology. Images were taken with 100× objective. Scale bar = 20 μm. (C) Western blots of pThr175 tau and total tau in ipsilateral and contralateral brain injuries. (D) Densitometry of western blots probed for pThr175 tau and total tau (pThr175/total tau). (E) pTyr216 glycogen synthase kinase–3β (GSK3β) and total GSK3β (pTyr216/total GSK3β) in ipsilateral and contralateral brain injuries. (F) Densitometry of western blots probed for pTyr216 GSK3β and total GSK3β. *p < 0.05. Contra = contralateral injury hemisphere; Ipsi = ipsilateral injury hemisphere.
We investigated tau protein phosphorylation and GSK3β activation by western blot (figure 4C). pTyr216 GSK3β was elevated in ipsilateral and contralateral hemispheres relative to uninjured controls (p = 0.005 and 0.001, respectively, one-way ANOVA with p < 0.001 and F = 13.928) when normalized against total GSK3β (figure 4). Finally, we observed the same change in localization of pGSK3β in moderate TBI rat brains as in CTE cases. This was quantified by blinded counts in which we observed an increase in diffuse expression of pTyr216 GSK3β in the injured hemisphere compared to uninjured control rats (p = 0.01 by Tukey post hoc test after one-way ANOVA p = 0.004 and F = 7.559; figure e-2, http://links.lww.com/WNL/A98).
Discussion
We have observed that both CTE and CTE-ALS are tauopathies in which pathologic tau aggregates contain aberrantly phosphorylated tau with immunoreactivity to both pThr175 tau and pThr231 tau. The presence of T22 immunoreactivity (recognizing oligomeric tau) is consistent with a pivotal role for phosphorylation at Thr175 in the pathogenesis of CTE and CTE-ALS. The inference can be made on the basis of our previous study, which showed that pThr175 tau pathology only occurs in pathologic conditions and that the expression of pThr175 tau coincides with oligomerized tau in the same neuronal populations, as does the presence of pThr231.10 Of note, this pathologic process of tau phosphorylation can be triggered experimentally in an in vivo model of moderate TBI and is consistent with a previously reported tauopathy induced by repeated trauma in a murine model of TBI, although this latter study did not investigate the physicochemical properties of the tauopathy.24 The finding that the tauopathy of CTE and CTE-ALS consists of the expression of all 6 tau isoforms in both the soluble and insoluble tau isolates confirms that this process is biochemically distinct from the tauopathy of AD.
Consistent with our previous reports, pThr175 tau staining was only observed when other pathologic tau markers were also present10 and as such, pThr175 tau-positive staining in controls was restricted to those individuals with advanced age, where some tau pathology is expected in the hippocampal formation.25 Therefore, pThr175 tau appears to also be an indicator of toxicity and neuronal damage across a broad range of tauopathies and should be considered in future studies as a CSF or blood biomarker in the diagnosis of CTE26 either alone or in combination with other markers of neuronal injury such as 14-3-327 or neurofilament proteins.28
The tau isoform composition profile observed in CTE in this study and previously in ALSci20 in which all 6 tau isoforms are observed in the soluble and insoluble tau isolates is in distinction to that observed in a number of tauopathies, including AD (in which the pathogenic tau marker is the PHF triplet), corticobasal degeneration and progressive supranuclear palsy (4R tauopathies), and Pick disease (3R tauopathy).29–31 This could be interpreted as indicative that the tauopathy of CTE, CTE-ALS, and ALSci is a secondary event triggered in response to a primary neuronal injury. In both CTE and CTE-ALS, this can be postulated to be directly due to the TBI itself, a hypothesis that is supported by the in vivo moderate TBI experimental paradigm. While the trigger for the tauopathy of ALSci remains unknown, it is clear that once initiated, the presence of pThr175 leads to a cascade of events that culminates in neuronal death in vitro.16,32
In relationship to our understanding of the pathophysiology of ALS, 4%–6% of patients with CTE also develop ALS (CTE-ALS),7 a rate much higher than the incidence of ALS (2–3 per 100,000) in the general population.9 Unique to motor neuron degeneration associated with CTE-ALS is tau pathology in the spinal cord. The only other instance of a disseminated tauopathy in association with motor neuron degeneration is that observed in the previously hyperendemic Western Pacific variant of ALS, a variant of ALS recognized to be at the intersection of an environmental insult in an at-risk population.33,34
While we have observed pathologic tau phosphorylation in the spinal cords of both patients with CTE and patients with CTE-ALS in this study, the variability observed warrants investigation on a larger cohort of cases with detailed rostrocaudal analysis to discern whether there is a correlation between motor symptom progression and regional tau deposition. We could not undertake such an analysis given the nature of tissue selection (cervical, thoracic, or lumbar for different cases). However, the absence of tau deposition in the spinal cords of patients with sporadic ALS suggests that the spinal motor neuron tauopathy of CTE-ALS is not an incidental finding or secondary to the primary neuronal injury of ALS. It is possible that tau protein phosphorylation and pathology begins early in the neurodegenerative process in CTE or CTE-ALS, in which case spinal cord pathology would be expected to precede symptom onset in patients who would otherwise develop motor impairment. In addition, given previous reports of TDP-43 deposition in the spinal cord of CTE-ALS,9 a much larger study investigating the correlation of TDP-43-positive neuronal cytoplasmic inclusions is required to bring clarity to the contributing pathologies concomitant to motor neuron death.
In these studies, we have observed pThr175 tau, in conjunction with pThr231 tau, oligomerized tau, and changes in active GSK3β localization consistent with a pathologic tauopathy driven by the aberrant phosphorylation of Thr175 tau in CTE and CTE-ALS. While we have shown that moderate TBI can directly drive this process, understanding how this induces pThr175 is the topic of current studies. Given our previous findings that this pathologic cascade of tau phosphorylation can be fully inhibited, and that this inhibition abolishes pThr175 tau-induced neuronal death,16 our work suggests that both CTE and CTE-ALS may be amenable to pharmacologic inhibition of GSK3β activation.
Glossary
- AD
Alzheimer disease
- ALS
amyotrophic lateral sclerosis
- ALSci
amyotrophic lateral sclerosis with cognitive impairment
- ANOVA
analysis of variance
- CTE
chronic traumatic encephalopathy
- GSK3β
glycogen synthase kinase–3β
- NFT
neurofibrillary tangle
- pGSK3β
phospho–glycogen synthase kinase–3β
- pThr175 tau
tau phosphorylated at threonine 175
- pThr231 tau
tau phosphorylated at threonine 231
- RT
room temperature
- TBI
traumatic brain injury
Author contributions
Alexander J. Moszczynski: contributed to design of all studies, conducted or assisted with all experimental procedures, performed all quantification and interpretation of data, wrote manuscript. Wendy Strong: performed tau fractionation experiments and western blot. Kathy Xu: performed animal surgeries. Ann McKee: contributed tissue to the study, performed pathologic diagnoses, edited manuscript. Arthur Brown: supervised and designed animal studies, edited manuscript. Michael J. Strong: supervised and designed all elements of the studies, edited manuscript.
Study funding
Research supported by an operating grant from the Ontario Neurodegenerative Disease Research Initiative and the Windsor-Essex ALS Society.
Disclosure
The authors report no disclosures relevant to the manuscript. Go to Neurology.org/N for full disclosures.
References
- 1.McKee AC, Cantu RC, Nowinski CJ, et al. Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J Neuropathol Exp Neurol 2009;68:709–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Coronado VG, Haileyesus T, Cheng TA, et al. Trends in sports- and recreation-related traumatic brain injuries treated in US emergency departments: the National Electronic Injury Surveillance System-All Injury Program (NEISS-AIP) 2001–2012. J Head Trauma Rehabil 2015;30:185–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Langlois JA, Rutland-Brown W, Wald MM. The epidemiology and impact of traumatic brain injury: a brief overview. J Head Trauma Rehabil 2006;21:375–378. [DOI] [PubMed] [Google Scholar]
- 4.Bazarian JJ, Cernak I, Noble-Haeusslein L, Potolicchio S, Temkin N. Long-term neurologic outcomes after traumatic brain injury. J Head Trauma Rehabil 2009;24:439–451. [DOI] [PubMed] [Google Scholar]
- 5.Chen H, Richard M, Sandler DP, Umbach DM, Kamel F. Head injury and amyotrophic lateral sclerosis. Am J Epidemiol 2007;166:810–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Perry DC, Sturm VE, Peterson MJ, et al. Traumatic brain injury is associated with subsequent neurologic and psychiatric disease: a meta-analysis. J Neurosurg 2016;2:511–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mez J, Daneshvar DH, Kiernan PT, et al. Clinicopathological evaluation of chronic traumatic encephalopathy in players of American football. JAMA 2017;318:360–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McKee AC, Cairns NJ, Dickson DW, et al. The first NINDS/NIBIB consensus meeting to define neuropathological criteria for the diagnosis of chronic traumatic encephalopathy. Acta Neuropathol 2016;131:75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McKee AC, Gavett BE, Stern RA, et al. TDP-43 proteinopathy and motor neuron disease in chronic traumatic encephalopathy. J Neuropathol Exp Neurol 2010;69:918–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moszczynski AJ, Yang W, Hammond R, Ang LC, Strong MJ. Threonine175, a novel pathological phosphorylation site on tau protein linked to multiple tauopathies. Acta Neuropathol Commun 2017;5:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tartaglia MC, Hazrati LN, Davis KD, et al. Chronic traumatic encephalopathy and other neurodegenerative proteinopathies. Front Hum Neurosci 2014;8:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lasagna-Reeves CA, Castillo-Carranza DL, Sengupta U, et al. Identification of oligomers at early stages of tau aggregation in Alzheimer's disease. FASEB J 2012;26:1946–1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin YT, Cheng JT, Liang LC, Ko CY, Lo YK, Lu PJ. The binding and phosphorylation of Thr231 is critical for tau's hyperphosphorylation and functional regulation by glycogen synthase kinase 3beta. J Neurochem 2007;103:802–813. [DOI] [PubMed] [Google Scholar]
- 14.Nakamura K, Zhen ZX, Ping LK. Cis phosphorylated tau as the earliest detectable pathogenic conformation in Alzheimer disease, offering novel diagnostic and therapeutic strategies. Prion 2013;7:117–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ward SM, Himmelstein DS, Lancia JK, Binder LI. Tau oligomers and tau toxicity in neurodegenerative disease. Biochem Soc Trans 2012;40:667–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moszczynski AJ, Gohar M, Volkening K, Leystra-Lantz C, Strong W, Strong MJ. Thr175-phosphorylated tau induces pathologic fibril formation via GSK3beta-mediated phosphorylation of Thr231 in vitro. Neurobiol Aging 2015;36:1590–1599. [DOI] [PubMed] [Google Scholar]
- 17.Schwalbe M, Kadavath H, Biernat J, et al. Structural impact of tau phosphorylation at threonine 231. Structure 2015;23:1448–1458. [DOI] [PubMed] [Google Scholar]
- 18.McKee AC, Stern RA, Nowinski CJ, et al. The spectrum of disease in chronic traumatic encephalopathy. Brain 2013;136:43–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hanger DP, Betts JC, Loviny TL, Blackstock WP, Anderton BH. New phosphorylation sites identified in hyperphosphorylated tau (paired helical filament-tau) from Alzheimer's disease brain using nanoelectrospray mass spectrometry. J Neurochem 1998;71:2465–2476. [DOI] [PubMed] [Google Scholar]
- 20.Strong MJ, Yang W, Strong WL, Leystra-Lantz C, Jaffe H, Pant HC. Tau protein hyperphosphorylation in sporadic ALS with cognitive impairment. Neurology 2006;66:1770–1771. [DOI] [PubMed] [Google Scholar]
- 21.Yang W, Strong MJ. Widespread neuronal and glial hyperphosphorylated tau deposition in ALS with cognitive impairment. Amyotroph Lateral Scler 2012;13:178–193. [DOI] [PubMed] [Google Scholar]
- 22.Planel E, Miyasaka T, Launey T, et al. Alterations in glucose metabolism induce hypothermia leading to tau hyperphosphorylation through differential inhibition of kinase and phosphatase activities: implications for Alzheimer's disease. J Neurosci 2004;24:2401–2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Trojanowski JQ, Schuck T, Schmidt ML, Lee VM. Distribution of tau proteins in the normal human central and peripheral nervous system. J Histochem Cytochem 1989;37:209–215. [DOI] [PubMed] [Google Scholar]
- 24.Thomsen GM, Ma AM, Harada MY, et al. A model of recurrent concussion that leads to long-term motor deficits, CTE-like tauopathy and exacerbation of an ALS phenotype. Trauma Acute Care Surg 2016;81:1070–1079. [DOI] [PubMed] [Google Scholar]
- 25.Yang W, Ang LC, Strong MJ. Tau protein aggregation in the frontal and entorhinal cortices as a function of aging. Brain Res Dev Brain Res 2005;156:127–138. [DOI] [PubMed] [Google Scholar]
- 26.Mattsson N, Zetterberg H, Hansson O, et al. CSF biomarkers and incipient Alzheimer disease in patients with mild cognitive impairment. JAMA 2009;302:385–393. [DOI] [PubMed] [Google Scholar]
- 27.Foote M, Zhou Y. 14-3-3 proteins in neurological disorders. Int J Biochem Mol Biol 2012;3:152–164. [PMC free article] [PubMed] [Google Scholar]
- 28.Li D, Shen D, Tai H, Cui L. Neurofilaments in CSF as diagnostic biomarkers in motor neuron disease: a meta-analysis. Front Aging Neurosci 2016;8:290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Buee SV, Hof PR, Buee L, et al. Hyperphosphorylated tau proteins differentiate corticobasal degeneration and Pick's disease. Acta Neuropathol 1996;91:351–359. [DOI] [PubMed] [Google Scholar]
- 30.Delacourte A, Robitaille Y, Sergeant N, et al. Specific pathological tau protein variants characterize Pick's disease. J Neuropathol Exp Neurol 1996;55:159–168. [DOI] [PubMed] [Google Scholar]
- 31.Delacourte A, Sergeant N, Wattez A, Gauvreau D, Robitaille Y. Vulnerable neuronal subsets in Alzheimer's and Pick's disease are distinguished by their tau isoform distribution and phosphorylation. Ann Neurol 1998;43:193–204. [DOI] [PubMed] [Google Scholar]
- 32.Gohar M, Yang W, Strong W, Volkening K, Leystra-Lantz C, Strong MJ. Tau phosphorylation at threonine-175 leads to fibril formation and enhanced cell death: implications for amyotrophic lateral sclerosis with cognitive impairment. J Neurochem 2009;108:634–643. [DOI] [PubMed] [Google Scholar]
- 33.Garruto RM. Pacific paradigms of environmentally-induced neurological disorders: clinical, epidemiological and molecular perspectives. Neurotoxicology 1991;12:347–377. [PubMed] [Google Scholar]
- 34.Hirano A, Malamud N, Kurland LT. Parkinsonism-dementia complex, an endemic disease on the island of Guam. II. Pathological features. Brain 1961;84:662–679. [DOI] [PubMed] [Google Scholar]