

ABSTRACT
Here, we discuss the role of Brf2, an RNA Polymerase III core transcription factor, as a master switch of the oxidative stress response. We highlight the interplay of Brf2 with the Nrf2/Keap1 pathway, as well as the role of Brf2 in cancer and other possible regulations.
KEYWORDS: Brf2, Nrf2, redox stress, RNA polymerase III, transcription
Introduction
Reactive oxygen species (ROS) are defined as chemical compounds containing highly reactive oxygen. They include hydrogen peroxide (H2O2) as well as radicals such as ·O2− (superoxide) or HO·.1 They can be produced by external factors (xenobiotics or ionising radiation) or by the cells themselves (by the respiratory chain for example). Despite being implicated in many fundamental cellular processes (i.e. proliferation, differentiation, signal transduction, defense against pathogens), excessive amounts of ROS can be deleterious for the cells. They can induce DNA damage, thereby providing favorable settings for carcinogenesis and tumor progression. Oxidation of proteins may lead to ageing and neurodegenerative diseases,2 whereas lipid peroxidation damages cell membranes.
Since it is not possible to avoid ROS formation in aerobic conditions, their accumulation must be minimized in order to maintain a redox equilibrium and any damage needs to be repaired to preserve cellular integrity. Complex pathways have evolved to cope with oxidative stress.3 Among them, the Nrf2/Keap1 axis is considered to be the principal regulator, being responsible for sensing the redox levels and organizing the cellular defense.4 However, upon prolonged ROS exposure, damage accumulates despite the antioxidant response and sustaining cell viability can become hazardous.
We recently identified an unanticipated layer of regulation in the oxidative stress response. Brf2, an RNA Polymerase (Pol) III core transcription factor found exclusively in vertebrates, encompasses a redox-sensing module and controls the expression of a very small subset of genes in a redox-dependent manner.5 At least one of them, the selenocysteine (Secys) tRNA is paramount for cell survival under oxidative stress conditions. In this respect, the Brf2 redox-sensing capability acts as a safety mechanism, by setting the limit of stress that can be tolerated by the cells before triggering apoptosis. Cancer cells experiencing high levels of ROS as by-product of their higher metabolic rate can hijack this mechanism to thrive in conditions that would normally be lethal.5
In this short point of view, we wish to address the crosstalk between Nrf2, the detoxification system, and Brf2. An emphasis will be put on the possible regulations of Brf2 and their implications, especially in cancer.
Nrf2/Keap1 pathway, the detoxification process and selenoproteins
The Nrf2/Keap1 pathway is arguably the main regulator of redox homeostasis.6 Keap1 acts as a substrate adaptor between an E3 ubiquitin ligase (Cul3) and the transcription factor Nrf2. Under basal conditions, the latter is constitutively targeted for proteosomal degradation following a hinge and latch model.7 However, under oxidative stress conditions, the structure of Keap1 is altered and the binding of Nrf2 is modified in a way that impairs the ubiquitination and poisons the ligase complex.8 Newly synthesized Nrf2 is then stabilized and translocated into the nucleus. Once imported, Nrf2 associates with the obligate partner sMAF to form an active transcription factor. The heterodimer can then bind the Antioxidant/Electrophile Response Elements (ARE/EpRE) in the promoter regions of the target genes (Fig. 1). ChIP-seq experiments have shown that more than 500 genes are under the control of Nrf2.9,10 Furthermore, this pathway controls the expression of proteins involved in many cellular processes including phase II detoxification enzymes.9
Figure 1.
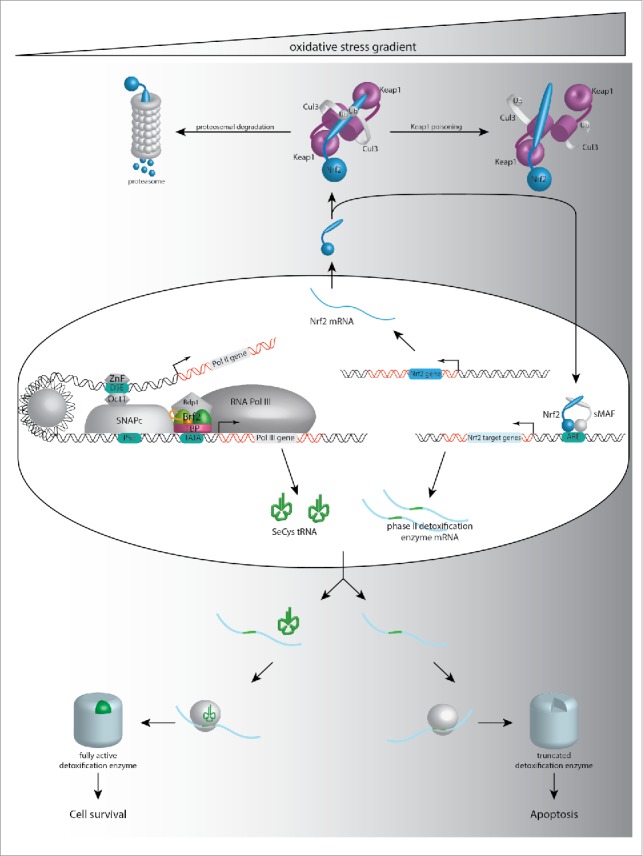
Interplay between Nrf2 and Brf2 pathways. The Nrf2 pathway is activated through the redox stress: under basal conditions, Nrf2 is ubiquitinylated by Keap1 and Cul3 and targeted for proteosomal degradation. Under stress, the structure of Keap1 is altered and the newly synthesized Nrf2 molecules can translocate into the nucleus to form a complex with its binding partner sMAF and transcribe target genes. TFIIIB, composed of TBP, Bdp1 and Brf2, recruits the RNA polymerase III machinery to transcribe a small set of genes, including the SeCys tRNA. Brf2 is redox sensitive and, as a consequence, controls the transcriptional output in function of the redox state of the cell. Efficient translation of the phase II detoxification enzymes containing selenocysteine requires both high levels of their mRNA and high levels of SeCys tRNA. At low levels of stress, the proteins can be synthesized due to availability of both components, ensuring cell survival. At high levels of redox stress, however, the lack of SeCys tRNA results in impaired detoxification enzymes and redox homeostasis regulators, triggering apoptosis to protect the cells from further accumulation of damage.
As a result of Keap1/Nrf2 activation, the redox stress is addressed by the detoxification system. This relies on the fact that glutathione (GSH) and thioredoxin 1 (Trx1), molecules whose synthesis and/or recycling is under Nrf2 transcriptional control,11 can cycle between a reduced and an oxidized form. Reduced GSH scavenges the hydrogen peroxide in a reaction catalyzed by the glutathione peroxidase family of proteins (GPx) and protects cysteines of cellular proteins by S-glutathionylation.12 Trx1 possesses a different spectrum of action: it can scavenge ROS in cooperation with Peroxiredoxin,13 reduce oxidized proteins,13 modulate redox stress response pathways (i.e. Nrf214 or NF-kB13) or even trigger apoptosis.15 Once GSH and Trx1 are oxidized, they become inactive and need to be recycled for another round of detoxification; a role ensured by the Glutathione Reductases and the Thioredoxin Reductase family (TrxR).
As a consequence, TrxR and GPx, both under the control of Nrf2, play a pivotal role in the protection mechanism as they are in charge of quenching ROS. Interestingly, they both contain a SeCys in their active site. SeCys, often called the 21st amino acid, is encoded by the opal stop codon (UGA) and thereby requires a specific machinery to recode it into SeCys (see Ref. [16] for a review). Any impairment of the recoding system leads to either a loss of the protein (if the STOP codon is at the N-terminal as in GPx), a truncated form (if the STOP codon is at the C-terminal as in TrxR) or eventually a functionally compromised isoform.17 Rats fed with diets deficient in Selenium show decreased levels of GPx and TrxR, leading to the activation of the Nrf2-ARE pathway due to higher oxidant levels.17,18 More strikingly, it was shown that TrxR1 acquires an oxidant function when the Selenium is compromised by electrophiles or simply absent due to a truncation. Instead of contributing to the redox stress response, the enzyme becomes a powerful oxidative agent that is able to induce cell death.19
These studies highlight the importance of SeCys incorporation for redox homeostasis. The loss of selenoproteins results in a higher oxidative state, and eventually cellular death in a feed forward mechanism. As described below, the observation that SeCys tRNA levels are downregulated under prolonged oxidative stress conditions sheds new light on the role of Pol III in the redox stress response.
Brf2, a new key player in the redox stress response
Pol III is responsible for the transcription of short and untranslated RNAs, such as the entire pool of tRNAs and the U6 snRNA, the RNA molecule harboring the active site of the spliceosome. Synthesis of SeCys tRNA is under the control of Pol III type III promoter that is characterized by a unique architecture when compared to the bulk of Pol III transcriptional units (see20 for an overview of the architecture of Pol III promoters). This extragenic promoter is composed of a Distal Sequence Element (DSE, −200 base pairs upstream of the transcriptional start site), an enhancer that recruits Znf143 and Oct-1, and the Proximal Sequence Element (PSE, −50 base pairs upstream of the transcriptional start site), which recruits the SNAPc complex. A positioned nucleosome brings the DSE and the PSE in close proximity, allowing direct interaction between SNAPc and Oct1.21 SNAPc enhances the recruitment of a TFIIIB complex on a TATA-Box located at −20. Finally, TFIIIB physically bridges Pol to the transcription start site. At type III promoters, TFIIIB is composed of Bdp1, TBP and Brf2 (Fig. 1). Brf2 shares structural and functional features with TFIIB, the canonical Pol II factor, and Brf1, the TFIIIB component present at most Pol III promoters. However, Brf2 uniquely contains a redox sensitive module that regulates TFIIIB assembly on the DNA.5 Under oxidative stress conditions, formation of Brf2-containing TFIIIB complexes is impaired, resulting in lower Pol III transcriptional output at the type III promoters. As a consequence, the levels of precursors and mature SeCys tRNAs decrease, which impacts the translation of selenoproteins5 (Fig. 1). The detoxification process therefore relies solely on the pre-existing pool of SeCys tRNAs, which displays a higher turn-over rate compared to other tRNAs.22 This ultimately modulates the redox stress response. Indeed, overexpression of Brf2 allows cells to tolerate higher levels of ROS before triggering apoptosis.5 We postulated that Brf2 acts as safety mechanism to set the limit of stress that cells can sustain.
Additional layers of Brf2 regulation?
Regulation of Brf2 upon redox stress might not be all black or white but rather modulated by the levels of ROS (Fig. 1). In fact, we have observed a quick recovery of SeCys tRNA transcription upon removal of the oxidative agent. This can be due to the reactivation of Brf2 by a rapid reduction of the redox sensitive C361 residue. Furthermore, we observed that the oxidation of C361 is almost fully reversible by addition of reducing agents in vitro, suggesting that the cysteine is protected from irreversible modifications. A possible explanation for this could be the formation of an intramolecular disulfide bond with C370 since both cysteines were shown to be S-gluthationylated in vitro.5 Additionally, the nuclear localization signal (NLS) of Brf2 is predicted to be bipartite and located around C361 (score 11.8, NLSMapper.23) A disulfide bridge between C361 and 370 might therefore hide the NLS and sequester Brf2 in the cytoplasm under oxidative stress.
We also noted that Brf2 and the SeCys tRNA precursors levels are upregulated in situation of low oxidative stress (short exposure and/or low concentration of stressor), suggesting the existence of a positive regulatory mechanism in these conditions. Concomitantly, as expected, we observed the activation of the Nrf2 pathway. Despite the fact that Brf2 gene was not found to be a target of Nrf2 in ChIP-seq experiments,9,10 we interrogated the eukaryotic promoter database (EPDnew24) and found a Nrf2 binding site in the 5′-UTR of Brf2 (Chr8: 37849845–37849855, GRCh38/hg38). This putative binding site is a perfect EpRE (GTGAGGCAGCA compared to (G/A)TGA(G/C)NNNGC(G/A).8) It is tantalizing to envisage a model in which Nrf2 might positively regulate Brf2 expression levels at the onset of the oxidative stress response.
Brf2 and cancer
Brf2 has cytoprotective effects by transcribing the SeCys tRNA required by the detoxifying enzymes. Redox regulation by Brf2 allows the cells to have a precise threshold for the amount of stress that they can tolerate. However, cancer cells, which often have elevated levels of ROS, have bypassed this safety mechanism to proliferate in otherwise lethal conditions.
Brf2 has been identified as a putative oncogenic driver by different groups over the years25-27 and is often amplified and overexpressed in many types of cancer.28 The mechanistic link, however, remained elusive. The newly discovered role of Brf2 as a master switch of the antioxidant defense opened new avenues to dissect the role of Brf2 in cancer. Increased levels of Brf2 help cells to evade apoptosis under high ROS levels. Conversely, cancer cells that naturally overexpress Brf2 can be sensitized by siRNA knockdown of the protein, leading to rapid induction of apoptosis.5 Thus, this transcription factor can play an important role during tumorigenesis; Brf2 overexpression allows the cells to tolerate high ROS levels, allowing accumulation of DNA damage and, as a consequence, enhancing their mutagenic rate.
Conclusion and perspectives
The antioxidant response possesses an innate safety mechanism that governs the level of stress that can be tolerated before allowing the cells to trigger apoptosis. It relies on a Pol III core transcription factor, Brf2, that is redox sensitive. Cancer cells have hijacked this process to survive in conditions that would normally be lethal. However, they subsequently become addicted to the cytoprotective effects of Brf2. This constitutes an opportunity to treat cancer: several therapies aim at increasing the redox state of cancer cells to promote their death.29,30 In this regard, targeting Brf2 represents an attractive option, especially considering the very small number of genes affected by the Brf2-dependent transcriptional program.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Dr H. King and Dr A. Leonidou for fruitful discussions while writing this review.
References
- [1].Lushchak VI. Free radicals, reactive oxygen species, oxidative stress and its classification. Chem Biol Interact 2014; 224:164–175; PMID:25452175; https://doi.org/10.1016/j.cbi.2014.10.016 [DOI] [PubMed] [Google Scholar]
- [2].Davies MJ. Protein oxidation and peroxidation. Biochem J 2016; 473:805–825; PMID:27026395; https://doi.org/10.1042/BJ20151227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Trachootham D, Lu W, Ogasawara MA, Valle NR-D, Huang P. Redox regulation of cell survival. Antioxid Redox Signal 2008; 10:1343–1374; PMID:18522489; https://doi.org/10.1089/ars.2007.1957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Canning P, Sorrell FJ, Bullock AN. Structural basis of Keap1 interactions with Nrf2. Free Radic Biol Med 2015; 88:101–107; PMID:26057936; https://doi.org/10.1016/j.freeradbiomed.2015.05.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Gouge J, Satia K, Guthertz N, Widya M, Thompson AJ, Cousin P, Dergai O, Hernandez N, Vannini A. Redox signaling by the RNA polymerase III TFIIB-related factor Brf2. Cell 2015; 163:1375–1387; PMID:26638071; https://doi.org/10.1016/j.cell.2015.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Krajka-Kuźniak V, Paluszczak J, Baer-Dubowska W. The Nrf2-ARE signaling pathway: an update on its regulation and possible role in cancer prevention and treatment. Pharmacol Rep 2017; 69:393–402; PMID:28267640; https://doi.org/10.1016/j.pharep.2016.12.011 [DOI] [PubMed] [Google Scholar]
- [7].Tong KI, Kobayashi A, Katsuoka F, Yamamoto M. Two-site substrate recognition model for the Keap1-Nrf2 system: a hinge and latch mechanism. Biol Chem 2006; 387:1311–1320; PMID:17081101; https://doi.org/10.1515/BC.2006.164 [DOI] [PubMed] [Google Scholar]
- [8].Hayes JD, Chowdhry S, Dinkova-Kostova AT, Sutherland C. Dual regulation of transcription factor Nrf2 by Keap1 and by the combined actions of β-TrCP and GSK-3. Biochem Soc Trans 2015; 43:611–620; PMID:26551701; https://doi.org/10.1042/BST20150011 [DOI] [PubMed] [Google Scholar]
- [9].Malhotra D, Portales-Casamar E, Singh A, Srivastava S, Arenillas D, Happel C, Shyr C, Wakabayashi N, Kensler TW, Wasserman WW et al.. Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res 2010; 38:5718–5734; PMID:20460467; https://doi.org/10.1093/nar/gkq212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Chorley BN, Campbell MR, Wang X, Karaca M, Sambandan D, Bangura F, Xue P, Pi J, Kleeberger SR, Bell DA. Identification of novel NRF2-regulated genes by ChIP-Seq: influence on retinoid X receptor alpha. Nucleic Acids Res 2012; 40:7416–7429; PMID:22581777; https://doi.org/10.1093/nar/gks409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Brigelius-Flohé R, Flohé L. Basic principles and emerging concepts in the redox control of transcription factors. Antioxid Redox Signal 2011; 15:2335–2381; PMID:21194351; https://doi.org/10.1089/ars.2010.3534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Pastore A, Piemonte F. S-Glutathionylation signaling in cell biology: progress and prospects. Eur J Pharm Sci 2012; 46:279–292; PMID:22484331; https://doi.org/10.1016/j.ejps.2012.03.010 [DOI] [PubMed] [Google Scholar]
- [13].Lu J, Holmgren A. The thioredoxin antioxidant system. Free Radic Biol Med 2014; 66:75–87; PMID:23899494; https://doi.org/10.1016/j.freeradbiomed.2013.07.036 [DOI] [PubMed] [Google Scholar]
- [14].Cebula M, Schmidt EE, Arnér ESJ. TrxR1 as a potent regulator of the Nrf2-Keap1 response system. Antioxid Redox Signal 2015; 23:823–853; PMID:26058897; https://doi.org/10.1089/ars.2015.6378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y, Kawabata M, Miyazono K, Ichijo H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J 1998; 17:2596–2606; PMID:9564042; https://doi.org/10.1093/emboj/17.9.2596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Labunskyy VM, Hatfield DL, Gladyshev VN. Selenoproteins: molecular pathways and physiological roles. Physiol Rev 2014; 94:739–777; PMID:24987004; https://doi.org/10.1152/physrev.00039.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Lu J, Zhong L, Lönn ME, Burk RF, Hill KE, Holmgren A. Penultimate selenocysteine residue replaced by cysteine in thioredoxin reductase from selenium-deficient rat liver. FASEB J 2009; 23:2394–2402; PMID:19351701; https://doi.org/10.1096/fj.08-127662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Burk RF, Hill KE, Nakayama A, Mostert V, Levander XA, Motley AK, Freeman ML, Austin LM. Selenium deficiency activates mouse liver Nrf2-ARE but vitamin E deficiency does not. Free Radic Biol Med 2008; 44:1617–1623; PMID:18279678; https://doi.org/10.1016/j.freeradbiomed.2008.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Anestål K, Prast-Nielsen S, Cenas N, Arnér ESJ. Cell death by SecTRAPs: thioredoxin reductase as a prooxidant killer of cells. PLoS ONE 2008; 3:e1846; PMID:18382651; https://doi.org/10.1371/journal.pone.0001846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Schramm L, Hernandez N. Recruitment of RNA polymerase III to its target promoters. Genes Dev 2002; 16:2593–2620; PMID:12381659; https://doi.org/10.1101/gad.1018902 [DOI] [PubMed] [Google Scholar]
- [21].Zhao X, Pendergrast PS, Hernandez N. A positioned nucleosome on the human U6 promoter allows recruitment of SNAPc by the Oct-1 POU domain. Mol Cell 2001; 7:539–549; PMID:11463379; https://doi.org/10.1016/S1097-2765(01)00201-5 [DOI] [PubMed] [Google Scholar]
- [22].Jameson RR, Carlson BA, Butz M, Esser K, Hatfield DL, Diamond AM. Selenium influences the turnover of selenocysteine tRNA[Ser]Sec in Chinese hamster ovary cells. J Nutr 2002; 132:1830–1835; PMID:12097655; https://doi.org/10.4172/2157-2518.1000205 [DOI] [PubMed] [Google Scholar]
- [23].Kosugi S, Hasebe M, Tomita M, Yanagawa H. Systematic identification of cell cycle-dependent yeast nucleocytoplasmic shuttling proteins by prediction of composite motifs. Proc Natl Acad Sci USA 2009; 106:10171–10176; PMID:19520826; https://doi.org/10.1073/pnas.0900604106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Dreos R, Ambrosini G, Périer RC, Bucher P. The eukaryotic promoter database: expansion of EPDnew and new promoter analysis tools. Nucleic Acids Res 2015; 43:D92–6; PMID:25378343; https://doi.org/10.1093/nar/gku1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Lockwood WW, Chari R, Coe BP, Thu KL, Garnis C, Malloff CA, Campbell J, Williams AC, Hwang D, Zhu CQ et al.. Integrative genomic analyses identify BRF2 as a novel lineage-specific oncogene in lung squamous cell carcinoma. PLoS Med [Internet] 2010. [cited 2017May5]; 7:e1000315. Available at http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2910599/; PMID:20668658; https://doi.org/10.1371/journal.pmed.1000315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Cabarcas S, Schramm L. RNA polymerase III transcription in cancer: the BRF2 connection. Mol Cancer 2011; 10:47; PMID:21518452; https://doi.org/10.1186/1476-4598-10-47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Sanchez-Garcia F, Villagrasa P, Matsui J, Kotliar D, Castro V, Akavia UD, Chen BJ, Saucedo-Cuevas L, Rodriguez Barrueco R, Llobet-Navas D et al.. Integration of genomic data enables selective discovery of breast cancer drivers. Cell 2014; 159:1461–1475; PMID:25433701; https://doi.org/10.1016/j.cell.2014.10.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Koo J, Cabarcas-Petroski S, Schramm L. BRF2, a biomarker in cancer? J Carcinog Mutagen 2014; 6:1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov 2013; 12:931–947; PMID:24287781; https://doi.org/10.1038/nrd4002 [DOI] [PubMed] [Google Scholar]
- [30].Galadari S, Rahman A, Pallichankandy S, Thayyullathil F. Reactive oxygen species and cancer paradox: to promote or to suppress? Free Radic Biol Med 2017; 104:144–164; PMID:28088622; https://doi.org/10.1016/j.freeradbiomed.2017.01.004 [DOI] [PubMed] [Google Scholar]