

Abstract
Purpose
The TAXYNERGY trial (ClinicalTrials.gov identifier: NCT01718353) evaluated clinical benefit from early taxane switch and circulating tumor cell (CTC) biomarkers to interrogate mechanisms of sensitivity or resistance to taxanes in men with chemotherapy-naïve, metastatic, castration-resistant prostate cancer.
Patients and Methods
Patients were randomly assigned 2:1 to docetaxel or cabazitaxel. Men who did not achieve ≥ 30% prostate-specific antigen (PSA) decline by cycle 4 (C4) switched taxane. The primary clinical endpoint was confirmed ≥ 50% PSA decline versus historical control (TAX327). The primary biomarker endpoint was analysis of post-treatment CTCs to confirm the hypothesis that clinical response was associated with taxane drug-target engagement, evidenced by decreased percent androgen receptor nuclear localization (%ARNL) and increased microtubule bundling.
Results
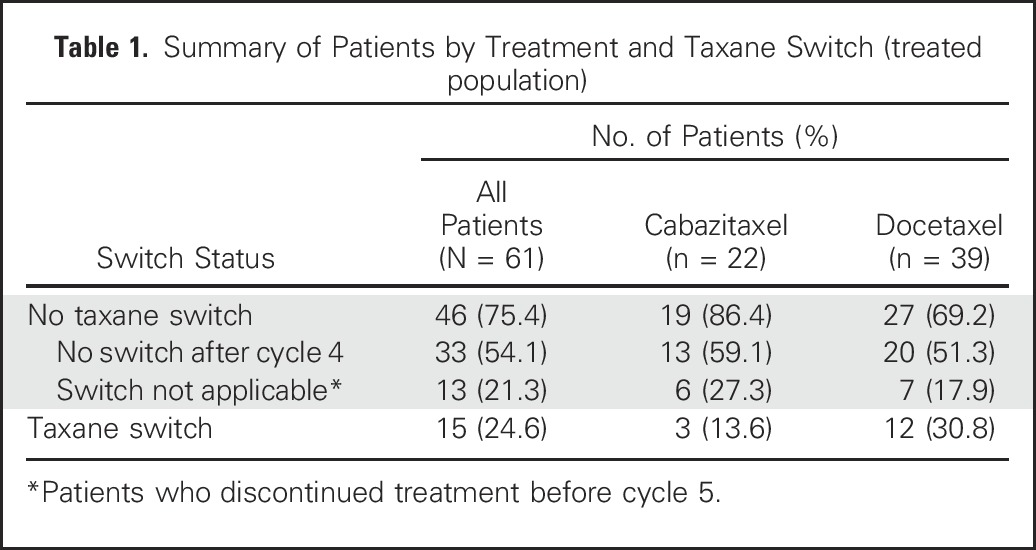
Sixty-three patients were randomly assigned to docetaxel (n = 41) or cabazitaxel (n = 22); 44.4% received prior potent androgen receptor–targeted therapy. Overall, 35 patients (55.6%) had confirmed ≥ 50% PSA responses, exceeding the historical control rate of 45.4% (TAX327). Of 61 treated patients, 33 (54.1%) had ≥ 30% PSA declines by C4 and did not switch taxane, 15 patients (24.6%) who did not achieve ≥ 30% PSA declines by C4 switched taxane, and 13 patients (21.3%) discontinued therapy before or at C4. Of patients switching taxane, 46.7% subsequently achieved ≥ 50% PSA decrease. In 26 CTC-evaluable patients, taxane-induced decrease in %ARNL (cycle 1 day 1 v cycle 1 day 8) was associated with a higher rate of ≥ 50% PSA decrease at C4 (P = .009). Median composite progression-free survival was 9.1 months (95% CI, 4.9 to 11.7 months); median overall survival was not reached at 14 months. Common grade 3 or 4 adverse events included fatigue (13.1%) and febrile neutropenia (11.5%).
Conclusion
The early taxane switch strategy was associated with improved PSA response rates versus TAX327. Taxane-induced shifts in %ARNL may serve as an early biomarker of clinical benefit in patients treated with taxanes.
INTRODUCTION
Taxanes are the only class of chemotherapy agents that extend survival in men with advanced prostate cancer.1-4 These drugs bind β-tubulin and stabilize cellular microtubules, leading to inhibition of microtubule-dependent intracellular trafficking and signaling, mitotic arrest, and apoptotic cell death.5-7 Although taxanes are generally considered antimitotic agents, they also inhibit tumor growth via several different mechanisms.5 Prostate cancer cells rely heavily on sustained androgen receptor (AR) nuclear signaling, which drives progression despite androgen-deprivation therapy.8 It was recently discovered that AR binds to cellular microtubules via the microtubule-associated motor protein dynein to facilitate its nuclear translocation.9 Taxanes can therefore inhibit AR nuclear trafficking via stabilization of microtubules; taxane-induced microtubule stabilization (termed drug-target engagement [DTE]) results in microtubule bundling (MTB), cytoplasmic sequestration of AR, inhibition of AR transcriptional activity, and inhibition of prostate cancer cell growth.6,7 In summary, the effectiveness of taxanes in prostate cancer can, at least in part, be attributed to the inhibition of AR signaling.9
Overcoming primary (intrinsic) and secondary (acquired) resistance to taxane therapy remains a challenge in prostate cancer treatment, and several different mechanisms of taxane resistance have been proposed (many of which may operate simultaneously).10-13 However, because cabazitaxel retains activity in many docetaxel-refractory patients with metastatic castration-resistant prostate cancer (mCRPC), there is evidence to suggest that not all of the same resistance mechanisms apply to all taxanes.14-17 Therefore, the central clinical hypothesis of this study was that some patients with mCRPC with a suboptimal initial prostate-specific antigen (PSA) decline with their first taxane can subsequently achieve a PSA response by an early switch to a second taxane before clinical progression.
Circulating tumor cells (CTCs) isolated from the blood of patients with mCRPC can serve as a powerful tool to study mechanisms of taxane sensitivity and resistance and can provide a liquid biopsy for serial tumor analysis.18,19 Recently developed microfluidic capture techniques enable reliable isolation of CTCs from peripheral blood, which can be analyzed using functional and molecular assays.20 Our central biomarker hypothesis was that CTCs can be used to interrogate AR localization and MTB to determine an association with response in men treated with a taxane.
In the current study (TAXYNERGY; ClinicalTrials.gov identifier: NCT01718353), we prospectively evaluated the benefit of an early switch from docetaxel to cabazitaxel or vice versa using early PSA changes (in the first 12 weeks of therapy) as a predictor of clinical efficacy or therapeutic resistance. In parallel, CTCs were analyzed at the single-cell level, using DTE criteria to assess whether CTC-specific DTE correlated with sensitivity to taxane treatment on an individual basis. The study met its primary objectives by demonstrating a benefit for early taxane switch as evaluated by confirmed PSA responses and an association of response with early changes in AR nuclear localization (ARNL) in CTCs.
PATIENTS AND METHODS
Patient Population and Study Design
TAXYNERGY enrolled chemotherapy-naïve patients with progressive mCRPC and an Eastern Cooperative Oncology Group performance score (ECOG PS) of 0 to 2, with no prior β isotope therapy, whole pelvic radiotherapy, or radiotherapy to > 30% of bone marrow. Patients with neuropathy grade > 2 were excluded. Prior hormonal therapy (including potent CYP17 inhibitors and AR signaling inhibitors) and immunotherapy were allowed.
In this noncomparative randomized study, patients were randomly assigned 2:1 to initiate docetaxel 75 mg/m2 or cabazitaxel 25 mg/m2 every 3 weeks plus daily prednisone 10 mg. The study was initially designed shortly after the US Food and Drug Administration approval of cabazitaxel, and conservatively, it was decided to have more patients receive docetaxel followed by cabazitaxel (the approved sequence) with the primary analysis in that cohort. With five patients enrolled, the protocol was amended, under the hypothesis that the two taxanes had similar activity in the first-line setting, to reduce the sample size and to pool both random assignment arms in the primary clinical and biomarker analyses while maintaining adequate power. Enrollment continued for 10 months after the amendment up to a total of 63 patients; study data and random assignment remained blinded during this time.
Patients achieving a ≥ 30% PSA decline from baseline by cycle 4 (C4; week 12) continued to receive the same chemotherapy agent, whereas those who did not achieve a ≥ 30% PSA decline were switched to the other taxane (Fig 1A). Patients could also switch taxanes for radiologic progression by Response Evaluation Criteria in Solid Tumors (RECIST) 1.121 in measurable lesions, regardless of PSA change. Switch could only occur at cycle 5 (C5). Treatment continued until disease progression, death, unacceptable toxicity, or withdrawal of consent.
Fig 1.

(A) Study design. (B) CONSORT diagram. C5, cycle 5; CTC, circulating tumor cell; mCRPC, metastatic castration-resistant prostate cancer; PI, principal investigator; PSA, prostate-specific antigen.
The study was approved by the institutional review board at each participating center and conducted in compliance with guidelines for Good Clinical Practice. Patients provided written informed consent before participation.
Efficacy Assessments
The primary clinical endpoint was PSA response rate, defined as the proportion of patients who achieved a ≥ 50% PSA decrease from baseline, confirmed 3 weeks later,22 whether a treatment switch occurred. Secondary efficacy assessments included progression-free survival (PFS; defined as the time from random assignment to the first of the composite endpoints: PSA, radiographic, or clinical progression or death) and overall survival (OS; defined as the time from random assignment to death). PSA measurements were taken every 3 weeks, and radiologic evaluations (chest, abdomen, and pelvis computed tomography; whole-body bone scan) were performed every 12 weeks until radiologic tumor progression or study cutoff.
The coprimary efficacy endpoint was DTE. CTCs were isolated at specified time points (Fig 1A) using geometrically enhanced differential immunocapture,20 immunostained, and analyzed for CTC-specific biomarkers using multiplex confocal microscopy (Data Supplement). CTCs at baseline (cycle 1 day 1 [C1D1]) were compared with CTCs isolated after 1 week of treatment (cycle 1 day 8 [C1D8]) for percent ARNL (%ARNL) and MTB. %ARNL was determined by quantification of integrated AR staining intensity in the total cell and nucleus areas. MTB was qualitatively assessed by three independent operators for increase compared with C1D1 on a scale of 0 to 3 from no to most MTB increase. Findings were correlated with clinical parameters for all CTC-evaluable patients.
Safety Assessments
Safety assessments included ongoing analysis of adverse events and serious adverse events (National Cancer Institute Common Terminology Criteria for Adverse Events v.4.03),23 vital signs, physical examinations, ECOG PS, and laboratory findings (hematology, serum biochemistry, coagulation, and urinalysis).
Statistical Considerations
Sample size determination was based on a historical PSA response rate of 45.4% from the intent-to-treat population in the TAX327 study.1 In TAXYNERGY, we reasoned that a 25% relative improvement in PSA response rate to 58% using the early-switch strategy would be clinically meaningful, and an estimate of the response rate with a one-sided 90% CI was calculated. The trial would be considered successful if the lower bound of the one-sided 90% CI did not contain the TAX327 PSA response rate of 45.4% (with 60 patients, the width of the one-sided 90% CI was 8.2%).
The main focus of the biomarker analyses was the change between C1D1 and C1D8 in %ARNL and MTB and its association with PSA (≥ 50%) response. Additional exploratory analyses examined other intervals (including change between pretreatment and C5 and PSA decline by ≥ 30%). Absolute differences, and differences separated into categories, were examined by analysis of variance and waterfall plots.
Secondary clinical endpoints were analyzed by descriptive statistics and survival techniques. PSA response rates were reported. Time-to-event endpoints (PFS and OS) were evaluated using Kaplan-Meier curves and 95% CIs. All statistical tests were two-sided, with a significance level of P < .05. Analyses were performed using SAS 9.3 software (SAS Institute, Cary, NC).
RESULTS
Patient and Treatment Characteristics
Between November 2012 and June 2014, 63 patients were randomly assigned to initial docetaxel (n = 41) or cabazitaxel (n = 22), and 61 patients received treatment (Fig 1B). Two patients initially randomly assigned to receive docetaxel did not receive treatment as a result of withdrawal of consent.
Baseline patient and disease characteristics are provided in the Data Supplement. Median age was 71 years; most patients had an ECOG PS ≥ 1 (66.7% and 4.8% had ECOG PS of 1 and 2, respectively). Median PSA was 82.30 ng/mL, and 34.9% of patients had visceral metastases. In total, 28 patients (44.4%) received prior treatment with potent AR-targeted therapy. The mean duration of study treatment was 26.1 weeks (median, 27 weeks; range, 3.1 to 60 weeks); the mean number of cycles was 8.4. The most common reasons for treatment discontinuation were disease progression (n = 21, 34.4%), adverse event (n = 17, 27.9%), and investigator decision (n = 14, 23.0%; Data Supplement).
Primary Efficacy EndPoint and PSA Changes From Baseline
The primary endpoint of this study was PSA response rate, defined as the proportion of randomly assigned patients who achieved a confirmed ≥ 50% PSA response during the whole treatment continuum, before treatment switch and after treatment switch, if applicable. Across the entire treatment continuum by intent-to-treat analysis, 35 (55.6%) of 63 patients achieved a ≥ 50% PSA response; 25 patients (39.7%) achieved the response on or before C4, and 10 patients (15.9%) achieved the response after C4. The lower limit of the 90% one-sided CI was 47.5%, which did not overlap with the rate of 45.4% in TAX327.1 Therefore, the primary clinical endpoint was met. The PSA response rates in men who had and had not received prior potent AR-targeted therapy were 44% (11 of 25 patients) and 68% (24 of 35 patients), respectively (P = .069).
Of 61 treated patients, 15 (24.6%) switched treatment after C4 as a result of an early PSA reduction < 30% (n = 14) or radiographic progression (n = 1) and were treated with the alternative taxane. Thirty-three patients (54.1%) achieved early ≥ 30% PSA reduction and remained on their initial taxane, and 13 patients (21.3%) discontinued treatment before C5 (Table 1). Of the 15 patients who switched, seven (46.7%) subsequently achieved a PSA response. Changes in PSA from baseline per patient are shown in the Data Supplement.
Table 1.
Summary of Patients by Treatment and Taxane Switch (treated population)
Secondary Efficacy Endpoints
Median composite PFS was 9.1 months (95% CI, 4.93 to 11.70 months; Fig 2A). Median radiographic PFS and OS were not reached; after a median follow-up of 14 months, > 50% of patients were still alive (Fig 2B). PSA PFS is shown in the Data Supplement.
Fig 2.

(A) Progression-free survival (all patients). (B) Overall survival (all patients). NR, not reached.
Biomarker Analysis: Nuclear AR Localization and Tubulin Bundling in CTCs
Of 60 patients who had PSA assessment, 44 had evaluable CTCs at C1D1, 31 had evaluable CTCs at C1D8, and 25 had evaluable CTCs at both C1D1 and C1D8. This analysis was based on mechanistic studies showing that taxanes impair ARNL downstream of microtubule stabilization.6,7 Therefore, we calculated %ARNL at C1D1 and C1D8 and assessed whether a decrease in %ARNL at C1D8 was associated with PSA response. There were no significant correlations between baseline biomarker parameters and any clinical outcomes (data not shown). Figure 3 shows representative high-resolution images of CTCs captured by geometrically enhanced differential immunocapture with high and low %ARNL. After 1 week of taxane therapy (C1D8), mean %ARNL was significantly lower in patients who subsequently achieved a ≥ 50% PSA reduction at C4 versus those without PSA response (44.0% v 64.1%, respectively; P = .004; Data Supplement). PSA responses were more common in patients with %ARNL at C1D8 that was in the lower three quartiles than in patients with %ARNL in the upper quartile (Fig 4A). By C1D8, mean %ARNL decreased by 17.6% in patients with ≥ 50% PSA decrease and increased by 2.3% in patients without a ≥ 50% PSA decrease (P = .020). A taxane-induced decrease in mean %ARNL (C1D8 v C1D1) was associated with a higher rate of ≥ 50% PSA decrease (72.7% v 12.5% of patients with no decrease in mean %ARNL; P = .009). PSA responses were also more common in patients with decreasing mean %ARNL at C1D8 compared with C1D1 than in men with increasing mean %ARNL (Fig 4B). In exploratory analysis of %ARNL at C5 day 1 to C5 day 8 after taxane switch, mean %ARNL decreased (74.26 at C5 day 1 to 63.84 at C5 day 8; P = .05).
Fig 3.

Representative high-resolution images of circulating tumor cells captured by geometrically enhanced differential immunocapture with (A) high and (B) low percent androgen receptor (AR) nuclear localization as assessed by quantitative image analysis. Immunofluorescence staining for AR is indicated in green. 4',6-Diamidino-2-phenylindole staining for DNA (nucleus) is indicated in blue (arrow).
Fig 4.

Greatest prostate-specific antigen (PSA) change from baseline at any time on study according to mean percent androgen receptor nuclear localization (%ARNL) at cycle 1 day 8 (C1D8; after 1 week of therapy). (A) Comparison of the lower three quartiles versus the upper quartile of mean %ARNL at C1D8 (n = 31); PSA ≥ 50% decrease from baseline was more common in patients with %ARNL in the lower three quartiles (71%) than in patients with %ARNL in the upper quartile (29%). (B) Comparison of increase versus decrease in %ARNL from cycle 1 day 1 (C1D1) to C1D8 (n = 25); PSA change from baseline was 40% in patients with increased mean %ARNL compared with 67% in patients with decreased mean %ARNL. Thirty-one patients had PSA change and evaluable circulating tumor cells (CTCs) at C1D8; 25 patients had PSA change and evaluable CTCs at both C1D1 and C1D8.
At C1D8, mean increase in MTB was numerically higher in patients who achieved an initial ≥ 30% PSA decrease compared with nonresponders, although this did not achieve statistical significance (0.69 v 0.09, respectively; P = .093). Taxane-induced increase in mean MTB score trended toward an association with response and was observed in patients who did not require a taxane switch after C4, but this did not achieve statistical significance (0.75 v 0.09 in those who required a switch; P = .059; Data Supplement).
Safety
Treatment-emergent adverse events (TEAE) leading to permanent treatment discontinuation were reported in 17 patients (27.9%) and were possibly related to the study drugs in 13 patients (21.3%). Dose modification as a result of a TEAE was reported in 27 patients (44.3%). The most frequently reported TEAEs in all patients were fatigue, diarrhea, and nausea (67.2%, 55.7%, and 41.0% overall, respectively; Table 2).
Table 2.
Summary of Key TEAEs
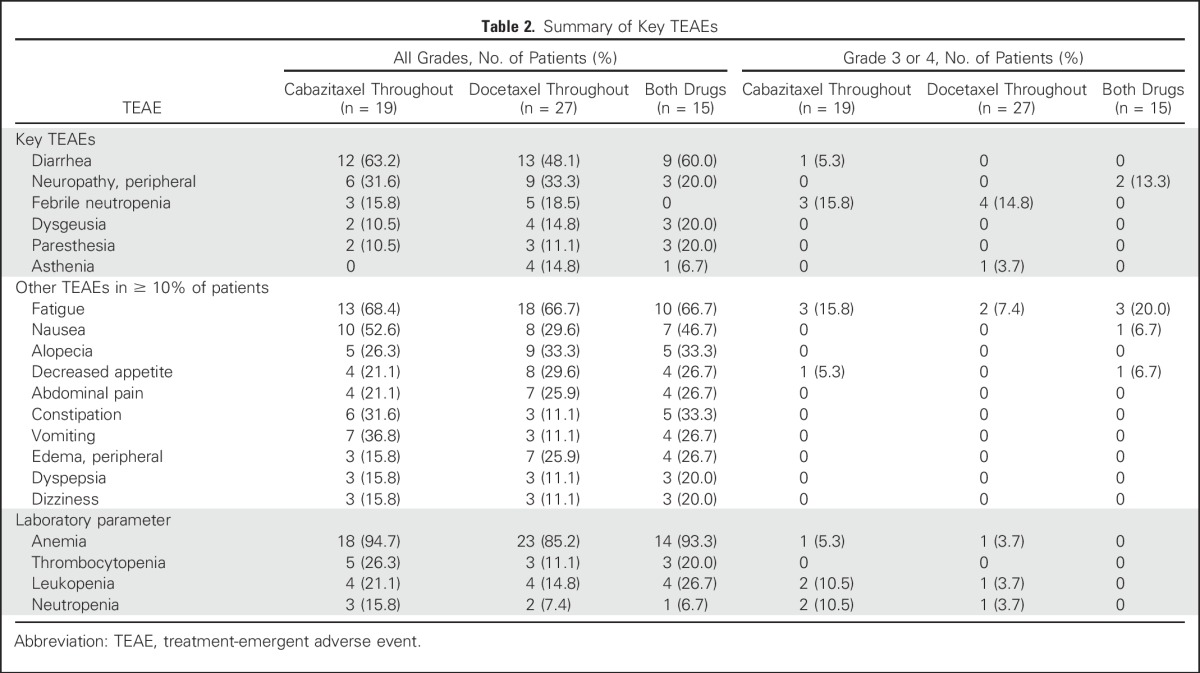
The most frequent grade 3 or 4 TEAEs were febrile neutropenia and fatigue. There were two deaths as a result of TEAEs, both in docetaxel-treated patients; one death was a result of an unrelated septic shock, and the other was reported as a possibly related myocardial infarction. Serious adverse events and dose modifications are summarized in the Data Supplement.
DISCUSSION
TAXYNERGY was conducted to test the clinical hypothesis that switching taxane therapy in men who do not achieve an optimal PSA reduction in the first 12 weeks of therapy may improve clinical outcomes and subvert resistance. Our primary biomarker hypothesis was that ARNL and MTB could be assessed reliably from CTCs and would be associated with outcome in patients treated with taxanes. The confirmed PSA response rate observed using the switch strategy (55.6%) was superior to the prespecified historical control trial (TAX327).1 Importantly, 46.7% of men who did not achieve a ≥ 30% PSA reduction in the first 12 weeks of treatment subsequently achieved a ≥ 50% PSA response by switching taxane therapy.
In addition, biomarker analyses from CTCs corroborated emerging preclinical data that taxanes may mediate some of their activity in prostate cancer by impairing AR trafficking along the microtubules from the cytoplasm into the nucleus, whereas persistent nuclear AR localization despite taxane therapy may be a marker of primary or early acquired resistance.6,7,9 To our knowledge, this is the first prospective trial to report changes in ARNL and MTB in CTCs in patients with prostate cancer receiving taxane therapy, and this trial has demonstrated the feasibility of conducting a biomarker-rich trial across multiple US and Canadian sites. Initial exploratory analyses of %ARNL and MTB in CTCs as a marker of DTE suggest a potential association with sensitivity to taxane therapy. To this end, the totality of the data from TAXYNERGY suggest that although these baseline biomarker parameters are not prognostic for clinical outcomes to chemotherapy, dynamic taxane-induced changes in ARNL (and perhaps MTB) may be indicative of benefit. More specifically, a decrease in nuclear-localized AR (which may be evaluable in CTCs as early as 1 week after therapy initiation) is prognostic for better outcomes. This clinical experience corroborates previous laboratory studies showing that taxanes can inhibit AR nuclear trafficking via stabilization of microtubules.6,7,9 How prostate cancer cells manipulate this mechanism of action to resist DTE and the differences between sensitivity to docetaxel versus cabazitaxel have yet to be determined. Interestingly, β-tubulin point mutations have been shown to confer resistance to paclitaxel, but not docetaxel, suggesting that single point mutations may affect one taxane but not others, despite a shared binding site.24-26
There are several clinical implications of this study. First, although this study is not sufficient to change the standard of care among patients with mCRPC receiving taxane therapy, it suggests that a treatment switch from one taxane to the other may be worthy of further investigation in patients who do not achieve a ≥ 30% PSA reduction within the first 12 weeks. Importantly, prior studies have shown that men who do not achieve a 30% PSA decline by week 12 of treatment have a poorer survival with docetaxel and cabazitaxel than those who do.27,28 Second, it suggests that changes in CTC-specific ARNL observed as early as 1 week after therapy initiation may be a potentially more sensitive and specific biomarker of subsequent clinical response than 12-week PSA changes. Future prospective studies should evaluate whether switching taxane therapy early on the basis of a CTC biomarker may improve outcomes compared with switching therapy on the basis of PSA trends (or not switching therapy at all).
This study has several limitations. First, on the basis of its design and relatively small sample size, it used an assumption that the activity of docetaxel and cabazitaxel is similar in chemotherapy-naïve patients with mCRPC. The limited sample size affected follow-up biomarker time points even more profoundly, especially with respect to paired samples. For example, not all patients with evaluable baseline CTCs had paired samples available at C1D8, possibly as a result of elimination of tumor cells from the circulation by effective chemotherapy. Second, the length of follow-up was relatively short, and we were not able to assess radiographic PFS or OS reliably or determine whether any biomarker signature was associated with improved survival. Third, the trial was not designed to definitively answer the question of whether a taxane switch (based on either PSA criteria or CTC-derived ARNL criteria) was superior with respect to OS than no switch at all. Such questions should form the basis for future studies randomly assigning patients to a switch strategy versus no switch. Finally, our ability to draw firm conclusions about the value of the early-switch strategy was limited by the small number of patients who underwent a taxane switch (n = 15), as a result of the significant activity of both agents in the chemotherapy-naïve mCRPC setting. Therefore, the study was unable to definitively prove that the taxane switch was responsible for subsequent PSA responses in those switching therapy or that biomarker modulation after switch induced those PSA responses.
All patients in the safety population experienced at least one TEAE. Grade 3 or 4 TEAEs were reported in 51% of patients; of these, the most frequently reported were febrile neutropenia and fatigue. No grade 3 or 4 febrile neutropenia events were reported in patients who switched taxane, although it should be noted that there was only a small number of patients in this subgroup. In general, the safety profiles of cabazitaxel and docetaxel were consistent with previous studies, and no new safety concerns were identified.
In conclusion, to our knowledge, this is the first prospective study to evaluate the benefit of an early switch in taxane therapy on the basis of the absence of a ≥ 30% PSA response after 12 weeks of therapy, and it suggests improved outcomes compared with historical controls with this approach. To our knowledge, it is also the first study to incorporate real-time on-treatment measurements of %ARNL and MTB from CTCs, indicating that these early measures may be associated with benefit in men treated with taxanes. Further dedicated prospective randomized trials focusing on a taxane switch using integral biomarkers are warranted.
ACKNOWLEDGMENT
Editorial support was provided by Olga Ucar and Paul Scutt of MediTech Media, funded by Sanofi Genzyme.
Footnotes
Supported by Sanofi. This work was also partially supported by the National Institutes of Health (NIH) Grants R01 CA177165 (to PG) and R01 CA179100 (to PG), and by the Clinical and Translational Science Center at Weill Cornell NIH/NCATS grant UL1TR00457 (to GG).
Presented, in part, at the 2015 European Cancer Conference, Vienna, Austria, September 25-29, 2015; 52nd Annual Meeting of the American Society of Clinical Oncology, Chicago, IL, June 3-7, 2016; and 2017 Genitourinary Cancers Symposium, Orlando, FL, February 16-18, 2017.
Clinical trial information: NCT01718353.
See accompanying Editorial on page 3175
AUTHOR CONTRIBUTIONS
Conception and design: Emmanuel S. Antonarakis, Scott T. Tagawa, John Stewart, Atef Zaher, Ted Szatrowski, Mario A. Eisenberger, David M. Nanus, Paraskevi Giannakakou
Provision of study materials or patients: Emmanuel S. Antonarakis, Scott T. Tagawa, Marie Vanhuyse, Guru Sonpavde, Scott North, Costantine Albany, Mario A. Eisenberger, David M. Nanus, Fred Saad
Collection and assembly of data: Emmanuel S. Antonarakis, Scott T. Tagawa, Giuseppe Galletti, Daniel Worroll, Karla Ballman, Marie Vanhuyse, Guru Sonpavde, Scott North, Costantine Albany, Che-Kai Tsao, Ada Gjyrezi, Shinsuke Tasaki, Luigi Portella, Yang Bai, Timothy B. Lannin, Shalu Suri, Conor N. Gruber, Erica D. Pratt, Brian J. Kirby, Mario A. Eisenberger, David M. Nanus, Fred Saad, Paraskevi Giannakakou
Data analysis and interpretation: All authors
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Randomized, Noncomparative, Phase II Trial of Early Switch From Docetaxel to Cabazitaxel or Vice Versa, With Integrated Biomarker Analysis, in Men With Chemotherapy-Naïve, Metastatic, Castration-Resistant Prostate Cancer
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/site/ifc.
Emmanuel S. Antonarakis
Honoraria: Sanofi, Dendreon, Medivation, Janssen Biotech, ESSA, Astellas Pharma
Consulting or Advisory Role: Sanofi, Dendreon, Medivation, Janssen Biotech, ESSA, Astellas Pharma
Research Funding: Janssen Biotech (Inst), Johnson & Johnson (Inst), Sanofi (Inst), Dendreon (Inst), Aragon Pharmaceuticals (Inst), Exelixis (Inst), Millennium (Inst), Genentech (Inst), Novartis (Inst), Astellas Pharma (Inst), Tokai Pharmaceuticals (Inst)
Travel, Accommodations, Expenses: Sanofi, Dendreon, Medivation
Scott T. Tagawa
Consulting or Advisory Role: Medivation, Astellas Pharma, Dendreon, Janssen, Bayer, Genentech, Sanofi, Endocyte, Immunomedics
Research Funding: Eli Lilly (Inst), Sanofi (Inst), Janssen (Inst), Astellas Pharma (Inst), Progenics (Inst), Millennium (Inst), Amgen (Inst), Bristol-Myers Squibb (Inst), Dendreon (Inst), Rexahn Pharmaceuticals (Inst), Bayer (Inst), Genentech (Inst), Newlink Genetics (Inst), Inovio Pharmaceuticals (Inst), AstraZeneca (Inst), Immunomedics (Inst), Novartis (Inst), AVEO (Inst), Rexahn Pharmaceuticals (Inst), Boehringer Ingelheim (Inst), Merck (Inst), Stem CentRx (Inst)
Travel, Accommodations, Expenses: Sanofi
Giuseppe Galletti
No relationship to disclose
Daniel Worroll
No relationship to disclose
Karla Ballman
Consulting or Advisory Role: ARIAD, Hospira
Patents, Royalties, Other Intellectual Property: Prostate cancer signature patent (Inst), Immune enrichment signature for response to trastuzumab in HER2-positive breast cancer (Inst)
Marie Vanhuyse
Consulting or Advisory Role: Bristol-Myers Squibb
Research Funding: Sanofi (Inst)
Guru Sonpavde
Honoraria: UpToDate
Consulting or Advisory Role: Merck, Genentech, Bayer, Sanofi, Novartis, Pfizer, Argos Therapeutics, Agensys, Eisai, AstraZeneca, Janssen, Amgen, Bristol-Myers Squibb, Exelixis
Speakers' Bureau: Clinical Care Options
Research Funding: Onyx (Inst), Bayer (Inst), Boehringer Ingelheim (Inst), Celgene (Inst), Merck (Inst), Pfizer
Other Relationship: Boehringer Ingelheim
Scott North
Honoraria: Janssen-Ortho, Astellas Pharma, Novartis, Pfizer, Sanofi Canada, Roche Canada, GlaxoSmithKline/Novartis, AstraZeneca/MedImmune
Consulting or Advisory Role: Janssen Oncology, Astellas Pharma, Novartis, Sanofi Canada, Pfizer, Roche Canada, Merck, AstraZeneca
Costantine Albany
Honoraria: Sanofi
Speakers' Bureau: Sanofi
Che-Kai Tsao
Consulting or Advisory Role: Genentech, Boehringer Ingelheim, Pfizer, AstraZeneca (I)
John Stewart
Employment: Sanofi Canada
Stock or Other Ownership: Sanofi
Atef Zaher
Consulting or Advisory Role: Sanofi, Bristol-Myers Squibb
Travel, Accommodations, Expenses: Sanofi, Bristol-Myers Squibb
Ted Szatrowski
Employment: Sanofi
Stock or Other Ownership: Sanofi, Roche
Consulting or Advisory Role: Eli Lilly (I), EMD Serono (I), Janssen (I), Natera (I)
Wei Zhou
Employment: Sanofi, Chiltern
Ada Gjyrezi
No relationship to disclose
Shinsuke Tasaki
No relationship to disclose
Luigi Portella
No relationship to disclose
Yang Bai
No relationship to disclose
Timothy B. Lannin
Research Funding: Sanofi
Patents, Royalties, Other Intellectual Property: Sagittal Saw (with figure-8 path for orthopedic surgery): WO2012170459 A2, US9579105, US20140243832, WO2012170459A3
Shalu Suri
Stock or Other Ownership: Medtronic
Consulting or Advisory Role: Dexcom (I)
Patents, Royalties, Other Intellectual Property: Georgia Tech (I), Georgia Tech
Conor N. Gruber
Employment: Advance care physician (I)
Stock or Other Ownership: Radius Health
Consulting or Advisory Role: Amgen (I), Merck (I)
Speakers' Bureau: Bristol-Myers Squibb (I), Johnson & Johnson (I), Amgen (I), Celgene (I), Genentech (I), Sanofi Genzyme Regeneron (I), Hansen Medical (I)
Erica D. Pratt
No relationship to disclose
Brian J. Kirby
Research Funding: Sanofi (Inst)
Mario A. Eisenberger
Honoraria: Sanofi, Pfizer
Consulting or Advisory Role: Bayer, Sanofi, Pfizer
Research Funding: Sanofi, Tokai Pharmaceuticals, Genentech
Travel, Accommodations, Expenses: Bayer, Astellas Pharma, Sanofi, Pfizer
David M. Nanus
Consulting or Advisory Role: Genentech
Fred Saad
Honoraria: Astellas Pharma, Abbvie, Amgen, Janssen Oncology, Sanofi, Bayer
Consulting or Advisory Role: Astellas Pharma, Janssen Oncology, Amgen, Sanofi, Bayer
Research Funding: Astellas Pharma (Inst), Bayer (Inst), Amgen (Inst), Janssen Oncology (Inst), Bristol-Myers Squibb (Inst), Oncogenex (Inst), Sanofi (Inst), AstraZeneca (Inst)
Paraskevi Giannakakou
Employment: Novartis (I)
Stock or Other Ownership: Novartis (I)
Research Funding: Sanofi
REFERENCES
- 1.Tannock IF, de Wit R, Berry WR, et al. : Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med 351:1502-1512, 2004 [DOI] [PubMed] [Google Scholar]
- 2.de Bono JS, Oudard S, Ozguroglu M, et al. : Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: A randomised open-label trial. Lancet 376:1147-1154, 2010 [DOI] [PubMed] [Google Scholar]
- 3.Sweeney CJ, Chen YH, Carducci M, et al. : Chemohormonal therapy in metastatic hormone-sensitive prostate cancer. N Engl J Med 373:737-746, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.James ND, Sydes MR, Clarke NW, et al. : Addition of docetaxel, zoledronic acid, or both to first-line long-term hormone therapy in prostate cancer (STAMPEDE): Survival results from an adaptive, multiarm, multistage, platform randomised controlled trial. Lancet 387:1163-1177, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jordan MA, Wilson L: Microtubules as a target for anticancer drugs. Nat Rev Cancer 4:253-265, 2004 [DOI] [PubMed] [Google Scholar]
- 6.Darshan MS, Loftus MS, Thadani-Mulero M, et al. : Taxane-induced blockade to nuclear accumulation of the androgen receptor predicts clinical responses in metastatic prostate cancer. Cancer Res 71:6019-6029, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhu ML, Horbinski CM, Garzotto M, et al. : Tubulin-targeting chemotherapy impairs androgen receptor activity in prostate cancer. Cancer Res 70:7992-8002, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nelson PS: Molecular states underlying androgen receptor activation: A framework for therapeutics targeting androgen signaling in prostate cancer. J Clin Oncol 30:644-646, 2012 [DOI] [PubMed] [Google Scholar]
- 9.Thadani-Mulero M, Nanus DM, Giannakakou P: Androgen receptor on the move: Boarding the microtubule expressway to the nucleus. Cancer Res 72:4611-4615, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mezynski J, Pezaro C, Bianchini D, et al. : Antitumour activity of docetaxel following treatment with the CYP17A1 inhibitor abiraterone: Clinical evidence for cross-resistance? Ann Oncol 23:2943-2947, 2012 [DOI] [PubMed] [Google Scholar]
- 11.Pezaro CJ, Omlin AG, Altavilla A, et al. : Activity of cabazitaxel in castration-resistant prostate cancer progressing after docetaxel and next-generation endocrine agents. Eur Urol 66:459-465, 2014 [DOI] [PubMed] [Google Scholar]
- 12.Diamond E, Garcias Mdel C, Karir B, et al. : The evolving role of cytotoxic chemotherapy in the management of patients with metastatic prostate cancer. Curr Treat Options Oncol 16:9, 2015 [DOI] [PubMed] [Google Scholar]
- 13.Kavallaris M: Microtubules and resistance to tubulin-binding agents. Nat Rev Cancer 10:194-204, 2010 [DOI] [PubMed] [Google Scholar]
- 14. doi: 10.1053/j.seminoncol.2008.02.010. Chien AJ, Moasser MM: Cellular mechanisms of resistance to anthracyclines and taxanes in cancer: Intrinsic and acquired. Semin Oncol 35:S1-S14, 2008 (suppl 2) [DOI] [PubMed] [Google Scholar]
- 15.Duran GE, Wang YC, Francisco EB, et al. : Mechanisms of resistance to cabazitaxel. Mol Cancer Ther 14:193-201, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Leeuw R, Berman-Booty LD, Schiewer MJ, et al. : Novel actions of next-generation taxanes benefit advanced stages of prostate cancer. Clin Cancer Res 21:795-807, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van Soest RJ, de Morrée ES, Kweldam CF, et al. : Targeting the androgen receptor confers in vivo cross-resistance between enzalutamide and docetaxel, but not cabazitaxel, in castration-resistant prostate cancer. Eur Urol 67:981-985, 2015 [DOI] [PubMed] [Google Scholar]
- 18.Gleghorn JP, Pratt ED, Denning D, et al. : Capture of circulating tumor cells from whole blood of prostate cancer patients using geometrically enhanced differential immunocapture (GEDI) and a prostate-specific antibody. Lab Chip 10:27-29, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Galletti G, Portella L, Tagawa ST, et al. : Circulating tumor cells in prostate cancer diagnosis and monitoring: An appraisal of clinical potential. Mol Diagn Ther 18:389-402, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kirby BJ, Jodari M, Loftus MS, et al. : Functional characterization of circulating tumor cells with a prostate-cancer-specific microfluidic device. PLoS One 7:e35976, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eisenhauer EA, Therasse P, Bogaerts J, et al. : New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur J Cancer 45:228-247, 2009 [DOI] [PubMed] [Google Scholar]
- 22.Scher HI, Halabi S, Tannock I, et al. : Design and end points of clinical trials for patients with progressive prostate cancer and castrate levels of testosterone: Recommendations of the Prostate Cancer Clinical Trials Working Group. J Clin Oncol 26:1148-1159, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. National Cancer Institute: NCI Common Terminology Criteria for Adverse Events (CTCAE) version 4.03, 03/03/2016 update. http://ctep.cancer.gov/protocolDevelopment/electronic_applications/ctc.htm.
- 24.Galletti E, Magnani M, Renzulli ML, et al. : Paclitaxel and docetaxel resistance: Molecular mechanisms and development of new generation taxanes. ChemMedChem 2:920-942, 2007 [DOI] [PubMed] [Google Scholar]
- 25.Giannakakou P, Sackett DL, Kang YK, et al. : Paclitaxel-resistant human ovarian cancer cells have mutant beta-tubulins that exhibit impaired paclitaxel-driven polymerization. J Biol Chem 272:17118-17125, 1997 [DOI] [PubMed] [Google Scholar]
- 26.Giannakakou P, Poy G, Zhan Z, et al. : Paclitaxel selects for mutant or pseudo-null p53 in drug resistance associated with tubulin mutations in human cancer. Oncogene 19:3078-3085, 2000 [DOI] [PubMed] [Google Scholar]
- 27.Armstrong AJ, Garrett-Mayer ES, Yang YC, et al. : A contemporary prognostic nomogram for men with hormone-refractory metastatic prostate cancer: A TAX327 study analysis. Clin Cancer Res 13:6396-6403, 2007 [DOI] [PubMed] [Google Scholar]
- 28.Halabi S, Armstrong AJ, Sartor O, et al. : Prostate-specific antigen changes as surrogate for overall survival in men with metastatic castration-resistant prostate cancer treated with second-line chemotherapy. J Clin Oncol 31:3944-3950, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]