

Abstract
Purpose
We report the efficacy and safety of dabrafenib (BRAF inhibitor) and trametinib (MEK inhibitor) combination therapy in BRAF V600E–mutated anaplastic thyroid cancer, a rare, aggressive, and highly lethal malignancy with poor patient outcomes and no systemic therapies with clinical benefit.
Methods
In this phase II, open-label trial, patients with predefined BRAF V600E–mutated malignancies received dabrafenib 150 mg twice daily and trametinib 2 mg once daily until unacceptable toxicity, disease progression, or death. The primary end point was investigator-assessed overall response rate. Secondary end points included duration of response, progression-free survival, overall survival, and safety.
Results
Sixteen patients with BRAF V600E–mutated anaplastic thyroid cancer were evaluable (median follow-up, 47 weeks; range, 4 to 120 weeks). All patients had received prior radiation treatment and/or surgery, and six had received prior systemic therapy. The confirmed overall response rate was 69% (11 of 16; 95% CI, 41% to 89%), with seven ongoing responses. Median duration of response, progression-free survival, and overall survival were not reached as a result of a lack of events, with 12-month estimates of 90%, 79%, and 80%, respectively. The safety population was composed of 100 patients who were enrolled with seven rare tumor histologies. Common adverse events were fatigue (38%), pyrexia (37%), and nausea (35%). No new safety signals were detected.
Conclusion
Dabrafenib plus trametinib is the first regimen demonstrated to have robust clinical activity in BRAF V600E–mutated anaplastic thyroid cancer and was well tolerated. These findings represent a meaningful therapeutic advance for this orphan disease.
INTRODUCTION
Anaplastic thyroid carcinomas (ATCs) are rare, highly aggressive, undifferentiated tumors and patients diagnosed with ATCs have a median survival of 5 to 12 months and a 1-year overall survival of 20% to 40%.1-3 ATCs comprise 1% to 2% of all thyroid cancers in the United States and 1% to 10% of thyroid cancers worldwide.4 They are among the most lethal cancers and all are considered stage IV at diagnosis. Despite multimodality therapy, including surgery, external beam radiation, and systemic chemotherapy, response rates to standard systemic therapies are < 15% (Data Supplement), and long-term outcomes remain dismal,5 with no curative options for patients who have exhausted locoregional therapies. No data conclusively demonstrate that cytotoxic chemotherapy prolongs survival or improves quality of life in patients with ATCs. Effective therapies are urgently needed for this rare and aggressive disease.
Molecular profiling studies have begun to elucidate the molecular drivers associated with ATC tumorigenesis. Between 20% and 50% of ATCs harbor activating B-Raf kinase (BRAF) V600 mutations, with unknown prognostic significance.6-9 Well-differentiated papillary thyroid cancer precedes or coexists with approximately 50% of ATCs.10 BRAF V600 mutations are an early and common driver mutation in these well-differentiated thyroid tumors, with additional late-event mutations that promote progressive dedifferentiation to ATC.7,10 In transgenic mouse models of BRAF V600–mutant ATC, combined inhibition of BRAF and mitogen-activated protein kinase (MAPK) kinase (MEK) kinases enhances antitumor activity compared with single-agent BRAF inhibitors,11 which suggests that the dual inhibition of the MAPK signaling pathway improves treatment response and delays or prevents MAPK pathway reactivation, a known resistance mechanism. This strategy has been successful in treating BRAF V600–mutant melanoma and lung cancer, whereby combined BRAF plus MEK inhibition increased overall response frequency, duration of response, progression-free survival, and overall survival compared with BRAF inhibitor monotherapy.12-15
In a phase II, open-label trial, patients with BRAF V600E–mutated rare malignancies, including ATC, were treated with the BRAF inhibitor dabrafenib (150 mg twice daily) plus the MEK inhibitor trametinib (2 mg once daily). We report the clinical efficacy and safety of dabrafenib and trametinib in patients with locally advanced or metastatic BRAF V600E–mutated ATC.
METHODS
Study Design and Population
The study was a multicenter, open-label, nonrandomized, phase II trial designed to allow the simultaneous evaluation of efficacy and safety in responses to dabrafenib and trametinib combination therapy in patients with BRAF V600E–mutated cancer in prespecified histologies (Data Supplement). The appropriate ethics committee or institutional review board at each study center approved the study protocol. The study was conducted in accordance with Guidelines for Good Clinical Practice and the ethical principles described in the Declaration of Helsinki, following all applicable local regulations.
Patients with seven of nine predetermined tumor histologies—ATC, biliary tract cancer, WHO grade 1 or 2 glioma, WHO grade 3 or 4 glioma, adenocarcinoma of the small intestine, hairy cell leukemia, and multiple myeloma (Data Supplement)—had been enrolled at data cutoff. BRAF V600E tumor mutations were identified by using locally approved assays (Clinical Laboratory Improvement Amendments or Clinical Laboratory Improvement Amendments equivalent) at individual sites or via the designated central reference laboratory (Hematogenix Laboratory Services, Tinley Park, IL), using the THxID-BRAF kit (bioMérieux, Durham, NC). All local mutation results were retrospectively confirmed by the same central reference laboratory. All study patients in the ATC cohort were accrued from subspecialty centers where onsite expertise in head and neck and/or endocrine pathology was present and pathology rereview was standard practice. All patients provided written informed consent before enrollment.
Eligibility criteria included age ≥ 18 years, no standard locally or regionally available treatment options as determined by the treating physician, measurable disease on the basis of Response Evaluation Criteria In Solid Tumors (RECIST) version 1.116 or Response Assessment in Neuro-Oncology criteria for grade 1 to 4 glioma cohorts,17,18 availability of adequate archival or fresh tumor tissue for confirmatory BRAF V600E mutation analysis, Eastern Cooperative Oncology Group performance status19 of 0 to 2, ability to swallow orally administered medication, and adequate baseline organ function (Data Supplement).
Patient exclusion criteria included prior treatment with BRAF and/or MEK inhibitor(s). For the ATC cohort, radiotherapy was not permitted within 7 days and any treatment-related adverse events must have been resolved before enrollment. Patients with ATC who were potentially curable by surgical excision alone, had not received standard-of-care treatment, or had thyroid lymphoma, sarcoma, or metastatic disease from other sites were also excluded (Data Supplement).
Study Assessments
Patients received continuous dabrafenib (150 mg twice daily) and trametinib (2 mg once daily) until disease progression, unacceptable toxicity, death, or discontinuation for any other reason. For solid tumor cohorts, patients underwent local disease assessment every 8 weeks while on study treatment. Follow-up visits for patients who discontinued and/or withdrew from study treatment occurred within 28 days after the last treatment dose, every 4 weeks for the first 6 months, and then every 3 months thereafter.
Enrollment in each primary analysis cohort was capped at 25 patients, and we conducted futility and efficacy analyses quarterly. If a cohort closed early for efficacy, an expansion cohort was opened to accommodate additional patient enrollment. Additional analysis information is provided in the Data Supplement. On November 6, 2015, the study independent data monitoring committee recommended early closure on the basis of the ATC cohort meeting the protocol-specified rules for early efficacy. An ATC expansion cohort was opened; treatment of the first patient began on May 20, 2016, and enrollment continues. We present results from an interim analysis of data that were available as of August 26, 2016. Efficacy results for additional histologic cohorts will be reported as mature data become available.
Outcomes
The study primary end point was investigator-assessed overall response rate. Response determination for ATC and other solid tumors was based on RECIST v1.1.16 Independent radiology review was performed for the ATC cohort by Paraxel Informatics Medical Imaging Service (Waltham, MA). Secondary end points included duration of response, progression-free survival, overall survival, and safety. For the subset of patients with ATC who demonstrated a confirmed complete response or partial response, duration of response was defined as the time (in weeks) from the first documented evidence of complete response or partial response (the first response before confirmation) until the time of documented disease progression or death as a result of any cause, whichever was first. Progression-free survival was defined as the time (in weeks) between the first dose and the date of disease progression or death as a result of any cause. Finally, overall survival was defined as the time (in weeks) from the first dose of study drug until death as a result of any cause.
Safety was monitored throughout the study for all 100 patients across cohorts by physical examinations, laboratory evaluations, vital signs, weight, performance status evaluations, ECGs, echocardiograms, and adverse event collection (characterized and graded per Common Terminology Criteria for Adverse Events, version 4.0).20 Adverse events were recorded by using standard Medical Dictionary for Regulatory Activities coding.
Statistical Analysis
To address the small sample size per histologic cohort, we used an adaptive design with a Bayesian hierarchical model (Data Supplement) that increases the power to detect clinically meaningful differences in overall response rate by borrowing information across histologic cohorts while controlling the type 1 error rate. This design allowed for multiple interim evaluations of the accumulating data to determine if at least one histologic cohort should discontinue enrollment early because of either success or futility. Data from the expansion cohorts did not contribute to the primary analysis of overall response rate using the Bayesian model but did provide additional safety data and supportive efficacy data.
The ATC intent-to-treat population included all 16 enrolled patients. The population of patients with centrally confirmed BRAF V600E consisted of all enrolled patients with ATC for whom positive verification of BRAF V600E mutation was obtained from a certified central reference laboratory. Bayesian modeling (Data Supplement) was performed on the intent-to-treat population and the BRAF V600E centrally confirmed population of the primary analysis cohort. All safety analyses were performed on the safety set (all treated patients). The Bayesian estimate of overall response rate was summarized by using posterior mean and the corresponding 95% credible interval. Overall response rate was also summarized descriptively, along with the exact 95% CI. Kaplan-Meier methodology was used to analyze the duration of response, progression-free survival, and overall survival end points. Adverse events were summarized on the basis of the frequency and proportion of total patients by preferred term.
Study Oversight
This study was designed, conducted, and analyzed by the funder (Novartis Pharmaceuticals, Basel, Switzerland) in conjunction with a steering committee. The study independent data monitoring committee reviewed the safety and efficacy results from interim analyses at regular intervals and provided recommendations to the sponsor. All authors had full access to the study data and share final responsibility for the content of the manuscript and the decision to submit for publication.
RESULTS
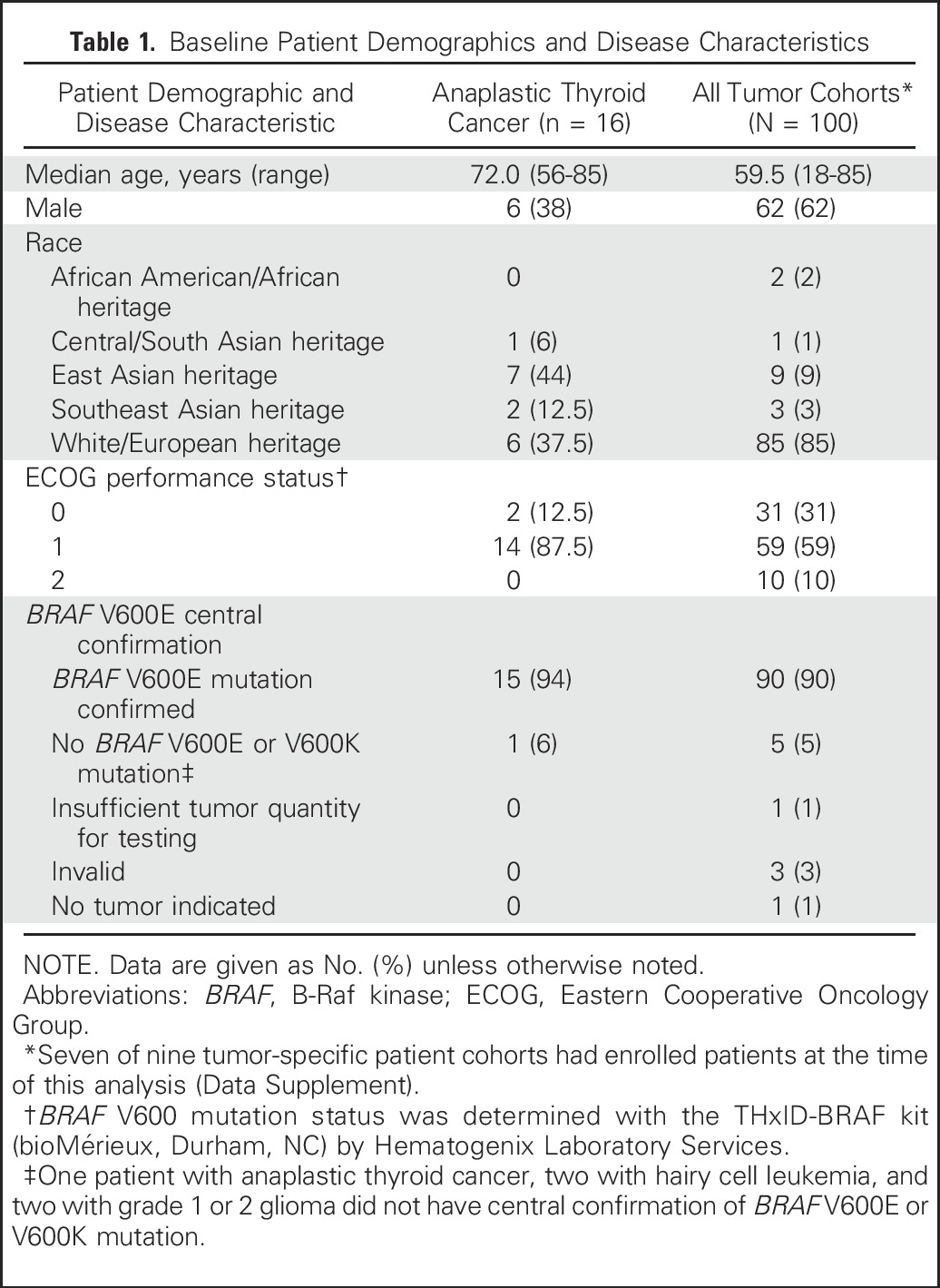
Between March 12, 2014, and August 26, 2016, 100 patients with BRAF V600E–mutated rare cancers in seven of the nine prespecified histologies were enrolled at 47 centers worldwide (Data Supplement). Baseline demographics and disease characteristics for all cohorts and for the ATC cohort are presented in Table 1. At data cutoff, 28 patients with ATC were screened for enrollment, 12 patients were excluded through screening, and 16 patients were enrolled (15 in the primary analysis cohort and one in the expansion cohort). The median patient age was 72 years, 63% were female, and 63% were of Asian heritage. Prior treatments included surgery (88%), external beam radiotherapy (81%), and chemotherapy (38%). Central laboratory testing confirmed tumor BRAF V600E mutations in 15 (94%) of 16 patients with ATC (Table 1).
Table 1.
Baseline Patient Demographics and Disease Characteristics
As of August 26, 2016, median duration of follow-up for the ATC cohort was 47 weeks (range 4 to 120 weeks). Eight (50%) of 16 patients with ATC remained on study treatment and two patients died > 30 days after the last study treatment dose as a result of disease progression (Data Supplement).
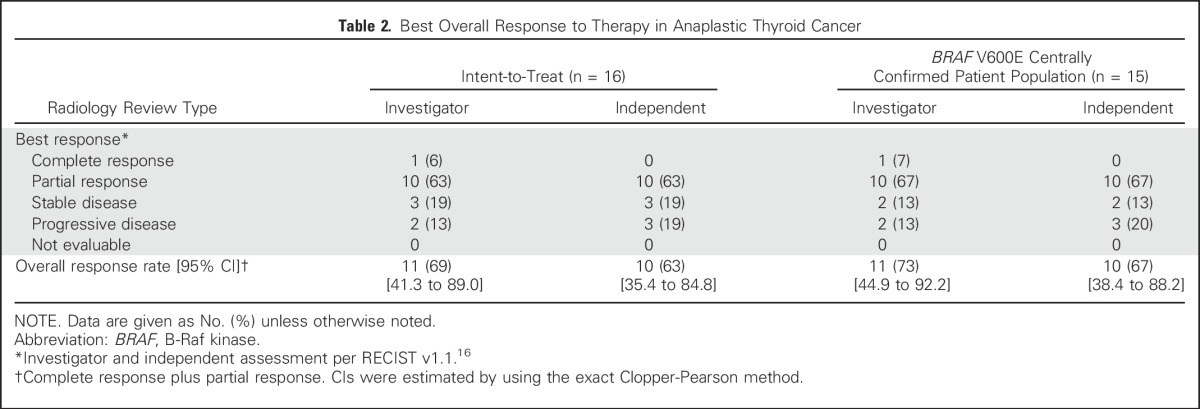
All 16 patients with ATC were evaluable for response. Dabrafenib plus trametinib demonstrated activity in ATC, with a confirmed, investigator-assessed overall response rate (complete response plus partial response as the best overall response) of 69% (complete response [n = 1], partial responses [n = 10]) in the intent-to-treat population (Fig 1A and Table 2). Representative computed tomography scans of primary and metastatic lesions that were collected at baseline and after 8 weeks of dabrafenib and trametinib treatment are presented in Figure 1B. In the population of patients with centrally confirmed BRAF V600E, the investigator-assessed overall response rate was 73% (11 of 15 patients). Independent radiologic review of response demonstrated similar results, with overall response rates of 63% and 67% in the intent-to-treat population and the BRAF V600E centrally confirmed population, respectively (Table 2). For the 15 patients with ATC in the primary analysis cohort, the Bayesian estimate of the primary end point—confirmed overall response rate on the basis of investigator assessment—was 69% (95% credible interval, 46.9% to 86.9%). The posterior probability was 100% that the overall response rate of 69% exceeded the historical control response rate of 15% (Data Supplement), thereby meeting the protocol-specified rules for early stopping for efficacy. This finding was consistent with the results of the four preceding interim analyses in which the posterior probability of the ATC cohort exceeded the threshold for early efficacy on the basis of data accrued at the time of those analyses.
Fig 1.

Maximum percent change from baseline in the sum of target lesion diameters in the anaplastic thyroid cancer intent-to-treat population. (A) Change from baseline in target lesion diameter was determined according to RECIST v1.1.16 The gray horizontal line indicates a 30% decrease, which is the minimum change needed to qualify for partial response according to RECIST. Best confirmed response (bar color) and best unconfirmed response (bar height) are as indicated. One patient experienced progression of disease in the brain at week 1 and, thus, a percent change could not be calculated. (*) An anaplastic thyroid cancer B-Raf kinase (BRAF) V600E mutation identified locally was not centrally confirmed in this patient. (B) Computed tomography scans of a representative patient with anaplastic thyroid cancer collected at baseline and after 8 weeks of treatment with dabrafenib plus trametinib. (Top) A patient presented with anaplastic thyroid cancer and symptomatic metastasis to the lung (grade 2 cough). The patient also had grade 2 dysphagia caused by a paratracheal mass and grade 2 dyspnea secondary to pericardial effusion (arrows indicate lesion locations). Cough and dyspnea both resolved after receiving dabrafenib plus trametinib combination therapy for 2 weeks. (Bottom) The size of all lesions was visibly reduced after 8 weeks of treatment.
Table 2.
Best Overall Response to Therapy in Anaplastic Thyroid Cancer
Confirmed responses in the ATC cohort were durable, with seven of 11 responses ongoing at the time of data cutoff (Fig 2). Median duration of response, progression-free survival, and overall survival were not reached as a result of ongoing responses that resulted in insufficient progression and death events at the time of data cutoff. Kaplan-Meier estimates at 12 months of duration of response, progression-free survival, and overall survival were 90%, 79%, and 80%, respectively.
Fig 2.

Treatment duration and time to events in individual patients with B-Raf kinase (BRAF) V600E–mutated anaplastic thyroid cancer. Data presented are the best investigator-assessed responses in the intent-to-treat population. Each bar denotes an individual patient, with bar length representing the duration of treatment and bar color denoting the best confirmed response. The blue arrowhead is positioned at the time of first radiologic response. The dagger denotes a patient in whom an anaplastic thyroid cancer BRAF V600E mutation that was identified locally was not centrally confirmed. The gray vertical line indicates the 12-month (52-week) time point. AE, adverse event.
Median durations of exposure to dabrafenib and trametinib were 10 and 9 months, respectively, in the ATC cohort, and 6 and 5.5 months, respectively, among all patients (Data Supplement). Across all histologic cohorts, 93% of patients experienced any adverse event and 42% experienced any grade 3 or 4 event. Adverse events that led to dose reduction, dose interruption/delay, or permanent discontinuation were observed in 30 (30%), 38 (38%), and eight (8%) patients, respectively (Data Supplement). The overall safety profile of dabrafenib and trametinib across all histologic cohorts was similar to previous reports in advanced or metastatic melanoma and non–small-cell lung cancer.12-15,21 Furthermore, the safety profile in the ATC cohort was similar to that of all treated patients, although the small size of this cohort limits conclusions.
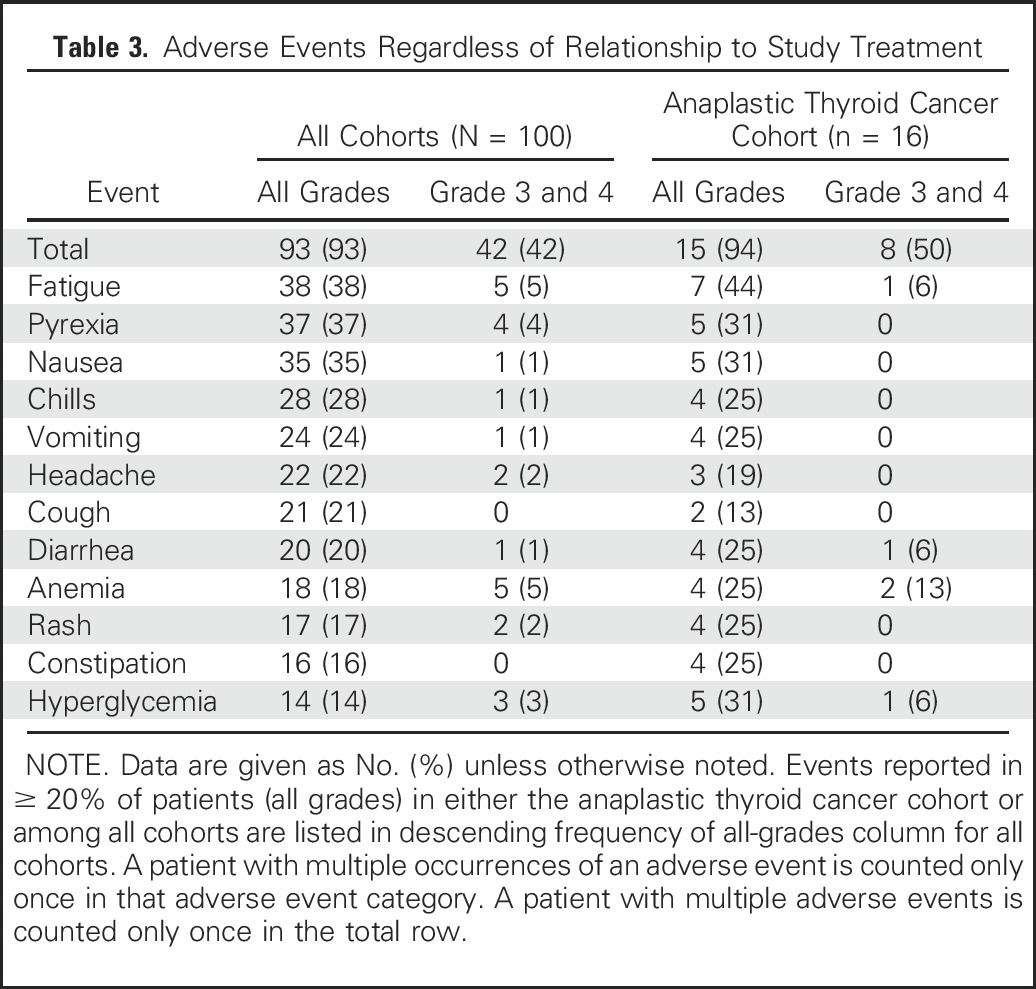
The most frequent adverse events (regardless of relationship to study treatment) reported in ≥ 20% of patients are summarized in Table 3. Among all cohorts, the most common adverse events were fatigue (38%), pyrexia (37%), and nausea (35%), and the most common grade 3 and 4 adverse events were fatigue, anemia, and neutropenia (all 5%). Similarly, in the ATC cohort, the most common adverse events of any grade were fatigue (44%), pyrexia (31%), and nausea (31%), and the most common grade 3 and 4 adverse event was anemia (13%). Three patients with ATC experienced treatment-related serious adverse events (Data Supplement; acute kidney injury and rhabdomyolysis, pyrexia, and hyponatremia). Among all cohorts, 13 of 100 patients experienced a treatment-related serious adverse event, with pyrexia (n = 6) as the only serious adverse event that occurred in more than one patient. Adverse events that led to discontinuation are summarized in the Data Supplement. There were two fatal adverse events, deemed unrelated to the study drug (sepsis, 2%), both of which occurred in patients with biliary tract cancer (Data Supplement).
Table 3.
Adverse Events Regardless of Relationship to Study Treatment
DISCUSSION
This study demonstrated that combination BRAF and MEK inhibition has robust clinical activity in patients with locally advanced or metastatic BRAF V600E–mutant ATC. Dabrafenib plus trametinib combination therapy resulted in a confirmed overall response rate of 69%, with similar results reported after independent review. Responses typically occurred early in the treatment course. Within the first 8 weeks of therapy, the complete responder had achieved the resolution of multiple pulmonary metastases. Responses were also durable, with a 12-month Kaplan-Meier estimate of duration of response of 90%. Furthermore, the Kaplan-Meier 12-month estimate of overall survival was 80%, which is remarkable relative to the historical rate of 20% to 40% for this patient population.1-3 Although this study evaluated a small number of patients and independent confirmation of pathology was not performed as a result of the limited sample availability and prioritization of BRAF mutation analysis, these results are nonetheless noteworthy because of the unmet need for an effective therapy for this rare patient population. These results require confirmation in additional clinical studies, although conducting a randomized trial in this setting is challenging, given the rarity of the patient population and the loss of clinical equipoise.
A disease model of acquired mutations driving ATC tumor progression is supported by transgenic mouse models. Acquisition of the BRAF V600E mutation in thyroid tissue is sufficient to initiate papillary thyroid cancer,11,22-24 with subsequent p53 deletion driving progression to high-grade carcinoma with pathologic features that are consistent with ATC. Transgenic mice that harbor thyroid-specific mutations in BRAF V600E and TP53 developed lethal, poorly differentiated thyroid tumors that exhibited focal necrosis, local invasion, and distant metastasis.11 Treatment of these mice with the BRAF inhibitor PLX4720 improved survival, but with no evidence of tumor regression or the suppression of MAPK pathway signaling. Because ATC resistance to BRAF inhibition likely involves the reactivation of upstream receptor tyrosine kinase signaling, concurrent downstream MEK inhibition was hypothesized to produce a more complete MAPK pathway blockade. Indeed, PLX4720 plus the MEK inhibitor PD0325901 resulted in enhanced tumor regression and improved survival in ATC-bearing mice versus PLX4720 alone, with complete responses in three of four mice. These experiments confirmed that combined BRAF and MEK inhibition resulted in more complete MAPK blockade and an enhanced antitumor effect in mouse ATC tumors.11
Clinical data also indicate that BRAF inhibitor monotherapy has modest clinical activity in BRAF V600E–mutant ATC. In a phase II basket study of BRAF V600–mutant, histology-independent, nonmelanoma cancer, treatment of seven patients with BRAF V600E–mutant ATC with the BRAF inhibitor vemurafenib resulted in one complete response and one partial response, for an overall response rate of 29%.25 Both responses were durable, with a time to progression of 11.5 months and the complete response ongoing for 12.7 months. In contrast to this modest response to BRAF inhibitor monotherapy, dabrafenib plus trametinib therapy for 16 patients with BRAF V600E–mutant ATC produced a markedly higher response rate. These findings are analogous to those noted in patients with BRAF V600–mutant melanoma and non–small-cell lung cancer in which the combined inhibition of BRAF and MEK resulted in substantial increases in response, progression-free survival, and overall survival compared with BRAF inhibitor monotherapy.12-15
Mechanisms of acquired resistance to BRAF inhibitors across multiple tumor types involve alterations that lead to MAPK pathway reactivation, including KRAS and NRAS mutations, RAS amplification, BRAF amplification, MEK1/2 mutations, and MET amplification.26-30 It is currently unknown whether similar mechanisms mediate acquired resistance to dabrafenib and trametinib therapy in patients with BRAF V600E–mutated ATC. Although additional investigation is warranted, tumor biopsies at baseline and progression are difficult to obtain. Analysis of circulating tumor DNA may allow for a minimally invasive evaluation of acquired mechanisms of resistance. Circulating tumor DNA was serially collected in the current study and additional sample acquisition and follow-up is ongoing.
For most rare cancers, conventional drug development programs are not possible because of the low incidence of disease and the infeasibility of conducting a randomized controlled trial. These challenges may reduce the incentive to explore the potential benefit of new targeted therapies for rare cancers. An alternative—and likely more feasible approach—may be based on the detection of BRAF V600E mutations in these rare cancers as a common denominator for the investigation of BRAF plus MEK inhibition in well-defined patient populations. Enriched Bayesian statistical designs and a high target effect size may enable the meaningful clinical investigation of targeted therapies in these small patient groups.
In conclusion, dabrafenib plus trametinib is a highly promising new combination targeted therapy for patients with BRAF V600E–mutated ATC, demonstrating a high overall response rate, prolonged duration of response, and prolonged survival with manageable toxicity. This is the first regimen to demonstrate robust clinical activity in BRAF V600E–mutated ATC. These data indicate that tumor mutation screening should be performed for patients with ATC as it has the potential to transform outcomes for these patients.
ACKNOWLEDGMENT
We thank the patients who took part in the trial and their families, as well as the staff who assisted with the trial at each site. We thank Don Berry, PhD, Kert Viele, PhD, and Liz Krachey, PhD, from Berry Consultants for their statistical consultation during the design of this study. Medical editorial assistance was provided by Chris Barnes, PhD (ArticulateScience, London, United Kingdom; supported by Novartis Pharmaceuticals).
Footnotes
Funded by Novartis Pharmaceuticals and the National Institutes of Health (Grant No. P30-CA016672).
Presented at the Annual Meeting of the American Society of Clinical Oncology, Chicago, IL, June 5, 2017.
Processed as a Rapid Communication manuscript.
Clinical trial information: NCT02034110.
AUTHOR CONTRIBUTIONS
Conception and design: Vivek Subbiah, Maria E. Cabanillas, Gladys Urbanowitz, Bijoyesh Mookerjee, Dazhe Wang, Fatima Rangwala, Bhumsuk Keam
Administrative support: Vivek Subbiah
Provision of study materials or patients: Vivek Subbiah, Maria E. Cabanillas
Collection and assembly of data: Vivek Subbiah, Robert J. Kreitman, Zev A. Wainberg, Jae Yong Cho, Jan H.M. Schellens, Jean Charles Soria, Patrick Y. Wen, Christoph Zielinski, Dazhe Wang, Fatima Rangwala, Bhumsuk Keam
Data analysis and interpretation: Vivek Subbiah, Robert J. Kreitman, Zev A. Wainberg, Jae Yong Cho, Jan H.M. Schellens, Jean Charles Soria, Patrick Y. Wen, Christoph Zielinski, Gladys Urbanowitz, Bijoyesh Mookerjee, Dazhe Wang, Fatima Rangwala, Bhumsuk Keam
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Dabrafenib and Trametinib Treatment in Patients With Locally Advanced or Metastatic BRAF V600–Mutant Anaplastic Thyroid Cancer
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/site/ifc.
Vivek Subbiah
Research Funding: Novartis, GlaxoSmithKline, Nanocarrier, NorthWest Biotherapeutics, Genentech, BergPharma, Bayer, Incyte, FujiFilm, Pharmamar, D3, Pfizer Amgen, AbbVie, Multivir, Bluprint Medicines, LOXO
Travel, Accommodations, Expenses: Novartis, PharmaMar, FujiFilm, Bayer
Robert J. Kreitman
Research Funding: GlaxoSmithKline (Inst), Novartis (Inst), National Cancer Institute (Inst)
Zev A. Wainberg
Consulting or Advisory Role: Sirtex Medical, Amgen, Array BioPharma, Five Prime Therapeutics, Novartis
Speakers' Bureau: Genentech
Research Funding: Novartis (Inst), Plexxikon (Inst), Pfizer (Inst), Biomarin (Inst), Merck (Inst)
Travel, Accommodations, Expenses: Genentech, Amgen
Jae Yong Cho
No relationship to disclose
Jan H.M. Schellens
Stock or Other Ownership: Modra Pharmaceuticals
Jean Charles Soria
Honoraria: Novartis
Patrick Y. Wen
Honoraria: Cavion, Cortice BioSciences, Foundation Medicine, Insys, Genentech, Monteris, Novartis, Vascular Biogenics
Speakers' Bureau: Merck
Research Funding: Agios, Acerta, Angiochem, AstraZeneca, Genentech, GlaxoSmithKline, Karyopharm, Merck, Novartis, Oncoceutics, Sanofi, Vascular Biogenics
Christoph Zielinski
Honoraria: Novartis
Consulting or Advisory Role: Novartis
Maria E. Cabanillas
Consulting or Advisory Role: Elsevier
Research Funding: Kura, Eisai, Exelixis, Genentech
Gladys Urbanowitz
Employment: Novartis
Stock or Other Ownership: Novartis
Research Funding: Novartis
Bijoyesh Mookerjee
Employment: Novartis, GlaxoSmithKline
Stock or Other Ownership: Novartis, GlaxoSmithKline, Incyte, AstraZeneca
Dazhe Wang
Employment: Novartis
Stock or Other Ownership: Novartis
Fatima Rangwala
Employment: Novartis
Stock or Other Ownership: Novartis
Bhumsuk Keam
No relationship to disclose
REFERENCES
- 1.Gilliland FD, Hunt WC, Morris DM, et al. : Prognostic factors for thyroid carcinoma. A population-based study of 15,698 cases from the Surveillance, Epidemiology and End Results (SEER) Program 1973-1991. Cancer 79:564-573, 1997 [DOI] [PubMed] [Google Scholar]
- 2.Kebebew E, Greenspan FS, Clark OH, et al. : Anaplastic thyroid carcinoma. Treatment outcome and prognostic factors. Cancer 103:1330-1335, 2005 [DOI] [PubMed] [Google Scholar]
- 3.Rao SN, Zafereo M, Dadu R, et al. : Patterns of treatment failure in anaplastic thyroid carcinoma. Thyroid 27:672-681, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.National Cancer Institute : SEER Cancer Stat Facts: Thyroid Cancer. https://seer.cancer.gov/statfacts/html/thyro.html
- 5.Lee DY, Won JK, Choi HS, et al. : Recurrence and survival after gross total removal of resectable undifferentiated or poorly differentiated thyroid carcinoma. Thyroid 26:1259-1268, 2016 [DOI] [PubMed] [Google Scholar]
- 6.Guerra A, Di Crescenzo V, Garzi A, et al. : Genetic mutations in the treatment of anaplastic thyroid cancer: A systematic review. BMC Surg 13:S44, 2013. (suppl 2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kunstman JW, Juhlin CC, Goh G, et al. : Characterization of the mutational landscape of anaplastic thyroid cancer via whole-exome sequencing. Hum Mol Genet 24:2318-2329, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Landa I, Ibrahimpasic T, Boucai L, et al. : Genomic and transcriptomic hallmarks of poorly differentiated and anaplastic thyroid cancers. J Clin Invest 126:1052-1066, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sandulache VC, Williams MD, Lai SY, et al. : Real-time genomic characterization utilizing circulating cell-free DNA in patients with anaplastic thyroid carcinoma. Thyroid 27:81-87, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McIver B, Hay ID, Giuffrida DF, et al. : Anaplastic thyroid carcinoma: A 50-year experience at a single institution. Surgery 130:1028-1034, 2001 [DOI] [PubMed] [Google Scholar]
- 11.McFadden DG, Vernon A, Santiago PM, et al. : p53 constrains progression to anaplastic thyroid carcinoma in a Braf-mutant mouse model of papillary thyroid cancer. Proc Natl Acad Sci USA 111:E1600-E1609, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Flaherty KT, Infante JR, Daud A, et al. : Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med 367:1694-1703, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Long GV, Stroyakovskiy D, Gogas H, et al. : Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med 371:1877-1888, 2014 [DOI] [PubMed] [Google Scholar]
- 14.Long GV, Flaherty KT, Stroyakovskiy D, et al. : Dabrafenib plus trametinib versus dabrafenib monotherapy in patients with metastatic BRAF V600E/K-mutant melanoma: Long-term survival and safety analysis of a phase 3 study. Ann Oncol 28:1631-1639, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Planchard D, Besse B, Groen HJM, et al. : Dabrafenib plus trametinib in patients with previously treated BRAF(V600E)-mutant metastatic non-small cell lung cancer: An open-label, multicentre phase 2 trial. Lancet Oncol 17:984-993, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Therasse P, Arbuck SG, Eisenhauer EA, et al. : New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 92:205-216, 2000 [DOI] [PubMed] [Google Scholar]
- 17.Wen PY, Macdonald DR, Reardon DA, et al. : Updated response assessment criteria for high-grade gliomas: Response Assessment in Neuro-Oncology Working Group. J Clin Oncol 28:1963-1972, 2010 [DOI] [PubMed] [Google Scholar]
- 18.van den Bent MJ, Wefel JS, Schiff D, et al. : Response Assessment in Neuro-Oncology (a report of the RANO Group): Assessment of outcome in trials of diffuse low-grade gliomas. Lancet Oncol 12:583-593, 2011 [DOI] [PubMed] [Google Scholar]
- 19.Oken MM, Creech RH, Tormey DC, et al. : Toxicity and response criteria of the Eastern Cooperative Oncology Group. Am J Clin Oncol 5:649-655, 1982 [PubMed] [Google Scholar]
- 20.National Cancer Institute : Common Terminology Criteria for Adverse Events (CTCAE) v4.03. https://evs.nci.nih.gov/ftp1/CTCAE/CTCAE_4.03_2010-06-14_QuickReference_5x7.pdf
- 21.Daud A, Tsai K: Management of treatment-related adverse events with agents targeting the MAPK pathway in patients with metastatic melanoma. Oncologist 22:823-833, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chakravarty D, Santos E, Ryder M, et al. : Small-molecule MAPK inhibitors restore radioiodine incorporation in mouse thyroid cancers with conditional BRAF activation. J Clin Invest 121:4700-4711, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Charles RP, Iezza G, Amendola E, et al. : Mutationally activated BRAF(V600E) elicits papillary thyroid cancer in the adult mouse. Cancer Res 71:3863-3871, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Franco AT, Malaguarnera R, Refetoff S, et al. : Thyrotrophin receptor signaling dependence of Braf-induced thyroid tumor initiation in mice. Proc Natl Acad Sci USA 108:1615-1620, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hyman DM, Puzanov I, Subbiah V, et al. : Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med 373:726-736, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ahronian LG, Sennott EM, Van Allen EM, et al. : Clinical acquired resistance to RAF inhibitor combinations in BRAF-mutant colorectal cancer through MAPK pathway alterations. Cancer Discov 5:358-367, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnson DB, Menzies AM, Zimmer L, et al. : Acquired BRAF inhibitor resistance: A multicenter meta-analysis of the spectrum and frequencies, clinical behaviour, and phenotypic associations of resistance mechanisms. Eur J Cancer 51:2792-2799, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pietrantonio F, Oddo D, Gloghini A, et al. : MET-driven resistance to dual EGFR and BRAF blockade may be overcome by switching from EGFR to MET inhibition in BRAF-mutated colorectal cancer. Cancer Discov 6:963-971, 2016 [DOI] [PubMed] [Google Scholar]
- 29.Wagle N, Van Allen EM, Treacy DJ, et al. : MAP kinase pathway alterations in BRAF-mutant melanoma patients with acquired resistance to combined RAF/MEK inhibition. Cancer Discov 4:61-68, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Danysh BP, Rieger EY, Sinha DK, et al. : Long-term vemurafenib treatment drives inhibitor resistance through a spontaneous KRAS G12D mutation in a BRAF V600E papillary thyroid carcinoma model. Oncotarget 7:30907-30923, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]