

ABSTRACT
Senescence is an irreversible form of growth arrest and is generally considered a favorable outcome of cancer therapies, yet little is known about the molecular events that distinguish this state from readily reversible growth arrest (i.e. quiescence). Recently, we discovered that during therapy induced senescence the chromatin remodeling protein α-thalassemia, mental retardation, X-linked (ATRX) represses Harvey rat sarcoma viral oncogene homolog (HRAS), and repression of HRAS is necessary to establish senescence, suggesting how new clinical combinations might be used to achieve durable senescence.
KEYWORDS: Therapy induced senescence, CDK4 inhibitors, ATRX, geroconversion
Introduction
Senescence is a stable form of cell cycle exit elicited by various types of stress. Senescent cells fail to return to the cell cycle when the stress is removed and become refractory to mitogenic stimuli. These cells also remain metabolically active and elaborate a secretory program referred to as the senescence associated secretory phenotype (SASP).
Senescence has important implications in development, wound healing, aging, disease, and cancer biology.1 Many cancer therapies elicit senescence, in a process termed therapy induced senescence (TIS). TIS is considered a favorable clinical outcome due to the durability of the growth arrest and the capacity of the SASP to stimulate tumor clearance by the immune system. Indeed, indirect evidence from a cyclin dependent kinase 4 (CDK4) inhibitor (CDK4i) clinical trial in well-differentiated/dedifferentiated liposarcoma suggests that senescence and the downregulation of MDM2 proto-oncogene (commonly reffered to as MDM2) instructs a more favorable outcome.2 Remarkably, downregulating MDM2 in a quiescent cell induced by CDK4 inhibition is sufficient to induce senescence. Understanding the molecular drivers of the transition from quiescence into senescence Fig. 1), or gerconversion, is critical to inducing this state in a controlled fashion and exploiting senescence for maximal clinical benefit.
Figure 1.
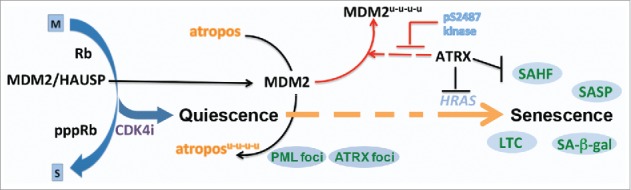
Senescence after growth arrest. The molecular and genetic insights into the mechanism of cyclin dependent kinase 4 (CDK4) inhibition therapy induced senescence in mammalian cell lines. The progression into senescence follows CDK4 inhibitor dependent growth arrest with the dissociation of the HAUSP (herpesvirus associated ubiquitin specific peptidase 7) deubiquitinase from MDM2 proto-oncogene (MDM2), allowing its turnover.2 A yet unidentified MDM2 substrate, here called atropos, is likely allowed to accumulate as MDM2 levels decrease prompting the transition of the cell along a path to senescence. ATRX is a multifaceted regulator of this process playing roles in MDM2 turnover2, HRAS (Harvey rat sarcoma viral oncogene homolog) transcription7 and senescence associated heterochromatic foci (SAHF) formation and maintenance.7 Phenotypes and markers are indicated in the blue circles (SAHF; SA-β-gal, senescence associated beta-galactosidase activity; SASP, senescence associated secretory program; LTC, long term durable stable clonogenic growth arrest upon replating in the absence of drug; PML, promyelocytic leukemia protein foci detection; ATRX, α-thalassemia, mental retardation, X-linked foci detection). Colors indicate different pathways, with broken arrows remaining to be mechanistically elucidated.
Dramatic and evolving chromatin changes occur in senescent cells. These begin with the formation of senescence associated heterochromatic foci (SAHF) approximately 5–7 days after cells are challenged with a senescence inducer.3,4 The SAHF are dense regions of facultative chromatin marked by focal deposition of retinoblastoma (Rb), the repressive histone variant macro H2A (mH2A), trimethylated histone H3 at the K9 residue (H3K9Me3), and the repressive family of heterochromatin proteins (HP1).3–6 While SAHF do not form in every senescent cell, genetic manipulation to prevent their formation in cells that normally accumulate SAHF prevents the cell from exiting the cell cycle and becoming senescent when challenged with an inducing stress. Functionally, SAHF contribute to the repression of E2F transcription factor target genes, conspiring with cell cycle inhibitor proteins to induce stable cell cycle exit.5 However, it is unclear if repression of E2F target genes alone can distinguish senescence from quiescence.
In our recent paper, we demonstrated that the chromatin remodeling protein ATRX (α-thalassemia, mental retardation, X-linked) is also required for the formation and maintenance of the SAHF.7 ATRX has been reported in physical complex with several components of the SAHF and is known to regulate H3.3 deposition and gene expression, but was never previously implicated in senescence.8,9 We found that ATRX is required for signals to drive cells into senescence, during either CDK4i or DNA damage (doxorubicin) TIS. However, unlike other genes which encode proteins that regulate the SAHF3–6, such as Rb, HMGA (high mobility group AT-hook), or HIRA (histone cell cycle regulator), ATRX deficient cells exit the cell cycle upon treatment and do not continue to cycle. This is the first and only protein (to our knowledge) whose role in SAHF is unrelated to cell cycle exit.
In a senescent cell, the elimination of ATRX results in the dissolution of the SAHF; however, these cells do not return to the cell cycle, nor become quiescent as other markers of senescence are not diminished. This demonstrates that SAHF are dynamic and are actively maintained, but probably work redundantly with something else to maintain the cell in a senescent state.
Our current work also showed that ATRX, like the SAHF, accumulates in distinct nuclear foci upon senescence induction. ATRX foci begin to appear very early (∼48 h) after the addition of a senescence inducer, just after the formation of PML (promyelocytic leukemia protein) nuclear foci and well before other classical markers of senescence.7 Unlike MDM2 turnover which is an early CDK4 inhibitor specific effect, ATRX foci formation occurs when cancer cells are challenged with DNA damaging agents or CDK4 inhibitors, suggesting a more conserved role in senescence after growth arrest. Thus, ATRX foci may be a valuable tool to identify cells that have embarked on a path towards irreversible growth arrest.
The genomic location of ATRX binding sites provides insights into therapeutic interventions with CDK4 inhibitors as well. Using chromatin immunoprecipitation followed by sequencing (ChIP-seq), we identified a number of senescence-specific loci where ATRX bound in both the context of CDK4i and DNA damage TIS. By coupling the ChIP-seq with RNA sequencing, we found that ATRX could directly regulate gene expression from some of these. One locus in particular that we found to be bound and repressed by ATRX across a variety of cell lines, derived from different tumor types, was HRAS.7 Moreover, enforced expression of HRAS could prevent senescence induction, while directly reducing HRAS was sufficient to drive cells into senescence.
Other groups have already examined combinations of CDK4i with RAS/MAPK (mitogen-activated protein kinase) inhibitors in cells, and the proposed mechanism of action accounting for the increased efficacy is surmised to be via cooperative suppression of cyclin D and stronger, more durable cell cycle exit.10 However, insight from our analysis of senescence after growth arrest suggest another – albeit not mutually exclusive – mechanistic alternative. Combining CDK4i with RAS/MAPK inhibition may actually drive cells into growth arrest (CDK4i) and then promote senescence. Additional work will be necessary to determine whether such combinatorial therapies have improved efficacy in patients, and whether additional and/or unique ATRX targets exist in response to different TIS treatment regiments.
Literature cited
- 1.Muñoz-Espín D, Serrano M. Cellular senescence: from physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014;15:482–496. [DOI] [PubMed] [Google Scholar]
- 2.Kovatcheva M, Liu DD, Dickson MA, Klein ME, O'Connor R, Wilder FO, Socci ND, Tap WD, Schwartz GK, Singer S, et al.. MDM2 turnover and expression of ATRX determine the choice between quiescence and senescence in response to CDK4 inhibition. Oncotarget. 2015;6:8226–8243. doi: 10.18632/oncotarget.3364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang R, Chen W, Adams PD. Molecular Dissection of Formation of Senescence-Associated Heterochromatin Foci. Mol. Cell. Biol. 2007;27:2343–2358. doi: 10.1128/MCB.02019-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang R, Poustovoitov MV, Ye X, Santos HA, Chen W, Daganzo SM, Erzberger JP, Serebriiskii IG, Canutescu AA, Dunbrack RL, et al.. Formation of MacroH2A-containing senescence-associated heterochromatin foci and senescence driven by ASF1a and HIRA. Dev. Cell. 2005;8:19–30. doi: 10.1016/j.devcel.2004.10.019. [DOI] [PubMed] [Google Scholar]
- 5.Narita M, Nũnez S, Heard E, Narita M, Lin AW, Hearn SA, Spector DL, Hannon GJ, Lowe SW. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell. 2003;113:703–16. doi: 10.1016/S0092-8674(03)00401-X. [DOI] [PubMed] [Google Scholar]
- 6.Narita M, Narita M, Krizhanovsky V, Nuñez S, Chicas A, Hearn SA, Myers MP, Lowe SW. A Novel Role for High-Mobility Group A Proteins in Cellular Senescence and Heterochromatin Formation. Cell. 2006;126:503–514. doi: 10.1016/j.cell.2006.05.052. [DOI] [PubMed] [Google Scholar]
- 7.Kovatcheva M, Liao W, Klein ME, Robine N, Geiger H, Crago AM, Dickson MA, Tap WD, Singer S, Koff A. ATRX is a regulator of therapy induced senescence in human cells. Nat. Commun. 2017;8:386. doi: 10.1038/s41467-017-00540-5. PMID:28855512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Voon HPJ, Wong LH. New players in heterochromatin silencing: histone variant H3.3 and the ATRX/DAXX chaperone. Nucleic Acids Res. 2016;44:1496–501. doi: 10.1093/nar/gkw012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ratnakumar K, Duarte LF, LeRoy G, Hasson D, Smeets D, Vardabasso C, Bönisch C, Zeng T, Xiang B, Zhang DY, et al.. ATRX-mediated chromatin association of histone variant macroH2A1 regulates -globin expression. Genes Dev. 2012;26, 433–438. doi: 10.1101/gad.179416.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sherr CJ, Beach D, Shapiro GI. Targeting CDK4 and CDK6: From Discovery to Therapy. Cancer Discov. 2016;6;353–367. doi: 10.1158/2159-8290.CD-15-0894. [DOI] [PMC free article] [PubMed] [Google Scholar]