

ABSTRACT
TNFR1-mediated cell signaling involves complex molecular pathways leading to inflammation and death. Cytosolic RARγ plays a pivotal role in converting TNF-induced inflammatory responses to RIP1 initiated cell death and this finely regulated function of RARγ serves as a checkpoint to engage death pathways in response to TNF.
KEYWORDS: Cell death, inflammatory signaling, Mechanisms of oncogenesis and tumor progression, Mechanisms of resistance to therapy, necroptosis, RIP1, RARg
Tumor necrosis factor (TNF) plays a critical role in diverse cellular events including inflammation, apoptosis and necroptosis through different signaling complexes. It is known that TNF normally elicits inflammatory responses, but under certain conditions, TNF can trigger cells to undergo apoptosis or necroptosis.1,2 However, little is known about how the transition from inflammatory signaling to death pathways is regulated.
TNF engagement triggers the formation of a TNFR1 signaling complex by recruiting effector molecules such as TRADD (TNFR1-associated death domain protein), RIP1 (receptor interacting protein), and TRAF2 (TNFR-associated factor 2) to mediate the activation of the inflammatory pathways including transcription factor NF-κB and MAP kinases.1 To induce cell death pathways, this TNFR1 signaling complex (complex I) dissociates from the receptor and recruits other proteins to form different secondary complexes for apoptosis and necroptosis.3 The death domain protein FADD (Fas-associated death domain protein) and Casp-8 are recruited to form complex IIa to mediate apoptosis and RIP3 (receptor interacting protein 3) and MLKL (mixed lineage kinase-domain like) are key components of the necrosome to carry out necroptosis.4
Our recent work demonstrated that cytoplasmic RARγ, not the nuclear RARγ, is critical for converting the inflammatory response to death signaling by mediating the formation of cytosolic death complexes and is a key regulator of RIP1-initiated cell death.5 TNF-induced cell death (apoptosis and necroptosis) could be initiated by TRADD when de novo protein synthesis is blocked or by RIP1 when IAP E3 ligases are inhibited. Our study found that TCZ-induced cell death is initiated by TRADD. In the case of IAP E3 ligases inhibition, RIP1-mediated cell death happens. It is thought that the blockage of RIP1 ubiquitination by cIAP1/2 is sufficient for RIP1 to recruit FADD and RIP3 to engage apoptosis and necroptosis respectively.6,7 However, our data indicates that RARγ is essential for the release of RIP1 from TNFR1 because the non-ubiquitinylated RIP1 was stuck in the TNFR1 complex and no complex IIa or necrosome was formed when RARγ was knocked down. In contrast, the release of TRADD from TNFR1 does not require RARγ. Therefore, RARγ is required for RIP1-initiated, but not TRADD-initiated, cell death.
The functions of RARs have been extensively studied as nuclear transcription factors and the RARs are predominantly nuclear even in the absence of their ligands.8 To rule out the contribution of RARγ transcriptional activity in cell death, we show that RARγ without the transcription activation domain, DNA binding domain and the NLS site interacts with the C-terminal of RIP1 and is sufficient to restore the sensitivity of RARγ knockdown cells to TSZ-induced necroptosis. Importantly, we fund that RARγ, but not other RARs such as RARα, was specifically released from the nucleus. Interestingly, our findings showed that Smac mimetic alone is sufficient to induced the release of RARγ from the nucleus. Our data suggests that inhibiting cIAP1/2 function such as in cIAP2H570A MEFs causes the elevated levels of cytoplasmic RARγ. The finding that RARγ localizes in both the cytoplasm and the nucleus in L929 cells provides new insights as to why TNF induces necroptosis in these cells without the presence of Smac mimetic. These observations suggest that cIAPs may play a role in the regulation of RARγ localization.
Our in vivo study with RARγ1-knockout mice further supported that RARγ plays a key role in RIP1-initiated necroptosis, but not in TRADD-initiated cell death. RARγ1-knockout mice are resistant to TZ, but not TG treatment while TRADD-knockout mice are resistant to TG, but not TZ treatment.5,9 These findings further supported our conclusion that RARγ is required for RIP1-initiated cell death.
Our study reveals a novel function for the nuclear receptor RARγ as a key regulator of RIP1-initiated cell death. Because RIP1-initiated cell death is a vital cellular response triggered by death factors, the engagement of this pathway is finely regulated. As shown in Fig. 1, the requirement of RARγ, which is released from the nucleus, provides a critical checkpoint for the transition from inflammatory signaling to death machinery of RIP1-initiated cell death. This safety control by RARγ allows cells to engage death pathways only when they are fully committed to die.
Figure 1.
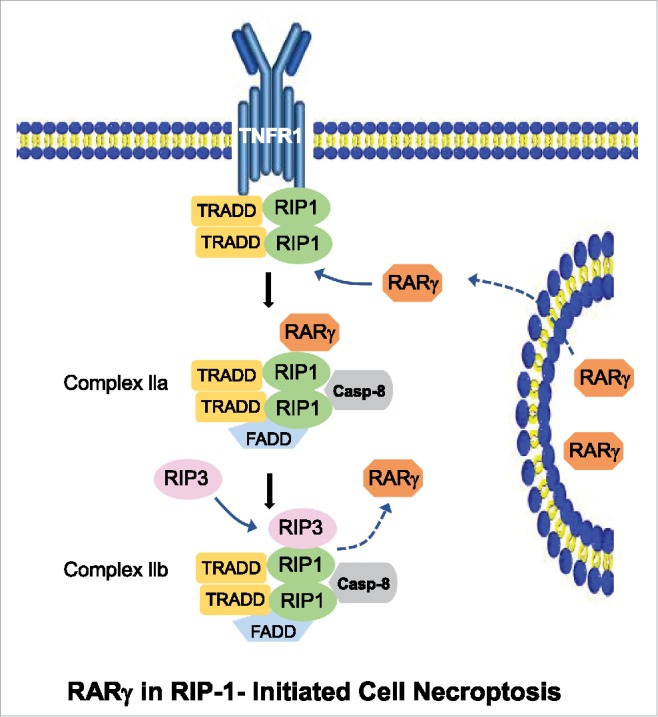
RARγ is essential for the transition from inflammatory signaling to death pathways in TNF-induced RIP1-initiated cell death. When RARγ is released from the nucleus in response to TNF under the conditions of cIAP inhibition, it initiates the formation of death complex IIa by dissociating RIP1 from TNFR1. When RIP1 recruits RIP3 to necrosome/complex IIb, RARγ is replaced from the complex.
Acknowledgements
This research was supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health.
Funding
This research was supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health.
References
- 1.Brenner D, Blaser H, Mak TW. Regulation of tumour necrosis factor signalling: live or let die. Nat Rev Immunol. 2015;15(6):362–74. doi: 10.1038/nri3834. PMID: 26008591. [DOI] [PubMed] [Google Scholar]
- 2.Chen G, Goeddel DV. TNF-R1 signaling: a beautiful pathway. Science. 2002;296(5573):1634–5. doi: 10.1126/science.1071924. PMID: 12040173. [DOI] [PubMed] [Google Scholar]
- 3.Ofengeim D, Yuan J. Regulation of RIP1 kinase signalling at the crossroads of inflammation and cell death. Nat Rev Mol Cell Biol. 2013;14(11):727–36. doi: 10.1038/nrm3683. PMID: 24129419. [DOI] [PubMed] [Google Scholar]
- 4.Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J, Ward Y, Wu LG, Liu ZG. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol. 2014;16(1):55–65. doi: 10.1038/ncb2883. PMID: 24316671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xu Q, Jitkaew S, Choksi S, Kadigamuwa C, Qu J, Choe M, Jang J, Liu C, Liu ZG. The cytoplasmic nuclear receptor RARgamma controls RIP1 initiated cell death when cIAP activity is inhibited. Nat Commun. 2017;8(1):425. doi: 10.1038/s41467-017-00496-6. PMID: 28871172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003;114(2):181–90. doi: 10.1016/S0092-8674(03)00521-X. PMID: 12887920. [DOI] [PubMed] [Google Scholar]
- 7.He S, Wang L, Miao L, Wang T, Du F, Zhao L, Wang X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137(6):1100–11. doi: 10.1016/j.cell.2009.05.021. PMID: 19524512. [DOI] [PubMed] [Google Scholar]
- 8.Kastner P, Mark M, Chambon P. Nonsteroid nuclear receptors: what are genetic studies telling us about their role in real life? Cell. 1995;83(6):859–69. doi: 10.1016/0092-8674(95)90202-3. PMID: 8521510. [DOI] [PubMed] [Google Scholar]
- 9.Pobezinskaya YL, Kim YS, Choksi S, Morgan MJ, Li T, Liu C, Liu Z. The function of TRADD in signaling through tumor necrosis factor receptor 1 and TRIF-dependent Toll-like receptors. Nat Immunol. 2008;9(9):1047–54. doi: 10.1038/ni.1639. PMID: 18641653. [DOI] [PMC free article] [PubMed] [Google Scholar]